
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigns and understanding of small molecule-based non-fullerene acceptors for realizing commercially viable organic photovoltaics
Minjun
Kim
b,
Seung Un
Ryu
 a,
Sang Ah
Park
a,
Yong-Jin
Pu
b and
Taiho
Park
*a
a,
Sang Ah
Park
a,
Yong-Jin
Pu
b and
Taiho
Park
*a
aDepartment of Chemical Engineering, Pohang University of Science and Technology (POSTECH), 77 Cheongam-ro, Nam-gu, Pohang, Gyeongsangbuk-do 37673, Republic of Korea. E-mail: taihopark@postech.ac.kr
bRIKEN Center for Emergent Matter Science (CEMS), 2-1 Hirosawa, Wako, Saitama, 351-0198, Japan. E-mail: minjun.kim@riken.jp; yongjin.pu@riken.jp
First published on 12th October 2021
Abstract
Organic photovoltaics (OPVs) have emerged as a promising next-generation technology with great potential for portable, wearable, and transparent photovoltaic applications. Over the past few decades, remarkable advances have been made in non-fullerene acceptor (NFA)-based OPVs, with their power conversion efficiency exceeding 18%, which is close to the requirements for commercial realization. Novel molecular NFA designs have emerged and evolved in the progress of understanding the physical features of NFA-based OPVs in relation to their high performance, while there is room for further improvement. In this review, the molecular design of representative NFAs is described, and their blend characteristics are assessed via statistical comparisons. Meanwhile, the current understanding of photocurrent generation is reviewed along with the significant physical features observed in high-performance NFA-based OPVs, while the challenging issues and the strategic perspectives for the commercialization of OPV technology are also discussed.
 Minjun Kim | Minjun Kim is a post-doctoral researcher in RIKEN, CEMS, Japan, under the supervision of Dr Yong-Jin Pu since 2019. He received his Ph.D. degree from the Department of Chemical Engineering at POSTECH, South Korea, in 2019, under the supervision of Prof. Taiho Park. His current research interests are the development of organic-based electronic materials and the photophysics of optoelectronic materials. |
 Taiho Park | Taiho Park is a Namgo endowed chair professor of POSTECH. He received his Ph.D. degree from the University of Cambridge, UK, under the supervision of Prof. Andrew B. Holmes in 2003, and then worked as a post-doctoral researcher under Prof. Steven C. Zimmerman at the University of Illinois (Urbana-Champaign), USA. He joined the faculty in the Department of Chemical Engineering at POSTECH in March of 2007. His current research interests include the material properties and device functions of optoelectronic devices. |
1. Introduction
The photovoltaic (PV) technology for converting sunlight into electricity is one of the sustainable solutions for overcoming the energy crisis and addressing climate change and is a rapidly growing field with increasing efficiencies.1,2 While various other PV types are being actively developed, organic photovoltaic (OPV) technology is emerging as the next-generation PV technology with great potential for portable, flexible/stretchable, and transparent applications.3–7 With significant advances in material designs, film processing techniques, and device architectures,8–12 the power conversion efficiency (PCE) of OPVs has reached around 18%, which is close to the requirements for commercialization.13–17Two major breakthroughs in the history of OPV technology have significantly boosted device efficiency. While early-stage OPV devices adopted a single-component material for the photoactive layer and the PCE was only ∼1%, the advent of the bulk-heterojunction (BHJ) system18–20 blended with an electron donor (D) and an acceptor (A) has allowed for improving the PCE by more than 10%, the first significant breakthrough in the OPV field.21,22 The BHJ system has demonstrated efficient photocurrent generation via increasing the D/A interfacial area and is widely adopted in most OPV devices.18–20 Specifically, the BHJ blend OPVs consisting of a polymer donor and a fullerene acceptor (FA) (e.g., PC61BM and PC71BM) have received a great deal of attention, providing a comprehensive understanding of the photo-conversion process in the BHJ blend system as well as valuable insights into the design of high-performance OPVs. The PCE of FA-based binary blend OPVs has reached up to ∼12% (ref. 23) through the development of numerous polymer donors (e.g., 11.7% for PffBT4T-C9C13:PC71BM); however, further improvements have been hindered by various critical drawbacks, including narrow and weak light absorption, large voltage loss, and thermal and/or photo-chemical instability.24–26 Meanwhile, chemically modifiable non-fullerene acceptors (NFAs) have emerged as an alternative to FAs for overcoming the aforementioned optical and photophysical issues.8,27–35 In practice, employing an NFA as a host acceptor in binary blends17,24,31,36 or as a secondary acceptor in ternary blends37–41 has proven to exceed the theoretical efficiency (∼15%) of FA-based OPVs,42 which presents the second major breakthrough in OPV technology. As the molecular design of NFAs continues to evolve, NFA-based binary blend OPVs have recently demonstrated a high PCE of over 18%,15,17 indicating their potential for meeting the commercial requirements (e.g., 18–22% for silicon solar cells). Moreover, NFA-based OPVs have the technological capacity for wearable and transparent PV applications owing to their excellent mechanical stability4 and near-infrared (NIR) light absorption.6,7,43–48
The distinctive features of NFA-based OPVs become clear following statistical comparisons with FA-based OPVs (Fig. 1). One significant feature of NFA-based OPVs is the high short-circuit current density (JSC) with a small optical bandgap (Eg), i.e., the smaller optical bandgap of the donor or acceptor (Fig. 1a and b). In fact, FA-based OPVs have a Eg distribution concentrated at 1.5–1.8 eV, with the JSC being less than 20 mA cm−2. Meanwhile, in terms of NFA-based OPVs, the Eg is mainly distributed in the 1.2–1.6 eV range, while they exhibit a higher JSC (>20 mA cm−2) than FA-based OPVs at Eg < 1.5 eV. The other important feature is the low voltage loss (Vloss) with a small energy offset (Fig. 1c–f), where the energy offset defines the lowest unoccupied molecular orbital (LUMO) offset (ΔELUMO) or the highest occupied molecular orbital (HOMO) offset (ΔEHOMO). Here, while FA-based OPVs have a Vloss of 0.6–1.2 V at a ΔEHOMO or ΔELUMO of <1.0 eV, NFA-based OPVs exhibit a lower Vloss (<0.8 V) in the same offset region. In fact, comparatively low Vloss (≈0.5 V) values can be observed in many NFA-based OPVs with a ΔEHOMO of <0.3 eV. These statistical comparisons indicate that NFA-based OPVs generate a photocurrent with different operating mechanisms compared with FA-based OPVs; however, the origin of the significant improvements in the performance of NFA-based OPVs has, as yet, not been fully understood. In addition to the features of Eg and energy offset (i.e., ΔELUMO and ΔEHOMO), various updated materials and physical discoveries related to NFA-based OPVs are being regularly reported, thus providing a better fundamental understanding for designing high-performance OPVs.
 | ||
| Fig. 1 Statistical data collected from ≈2000 pairs of FA- and NFA-based binary blend OPVs over the last 10 years. (a) Short-circuit current density (JSC) and (b) power conversion efficiency (PCE) versus Eg,min. (c) Voltage loss (Vloss) and (d) PCE versus the lowest unoccupied molecular orbital (LUMO) offset (ΔELUMO). (e) Vloss and (f) PCE versus the highest occupied molecular orbital (HOMO) offset (ΔEHOMO). The each color means data of polymer donor:FA (grey dots), polymer donor:PDI-based NFA (orange dots), polymer donor:IDT-based NFA (navy dots), polymer donor:Y6-based NFA (pink dots) and polymer donor:others (NFAs which are not included in PDI, IDT, and Y6 categories) (green dots). | ||
In this review, we first cover the existing knowledge regarding the operating principles of OPVs (Section 2) and describe the molecular designs of representative NFAs in relation to the statistically collected PV parameters (Section 3). Following this, we review the current understanding and the significant features observed in NFA-based OPVs, with a specific focus on the photophysical, electrical, and morphological characteristics (Section 4). Finally, we provide a concise summary and discuss the various perspectives on realizing high-performance and functional OPVs for technological commercialization (Section 5).
2. OPV operation
The OPV operation involves photocurrent generation via a sequential process in the BHJ active layer (Fig. 2),20,26,49,50 which consists of the following stages: (1) Frenkel excitons (i.e., tightly bound electron–hole pairs) are formed following light absorption, (2) the excitons diffuse to the D/A interfaces and form charge transfer (CT) excitons (i.e., weakly bound electron–hole pairs), (3) the CT excitons are dissociated into free charge carriers by the built-in potential in devices, (4) the free charges transport to each electrode (i.e., holes to anode, electrons to cathode), and (5) the charges are collected at the electrodes, thus generating an electric current. The overall efficiency (η) of photocurrent generation is defined as follows:| η = ηabsηedηCTηCC | (1) |
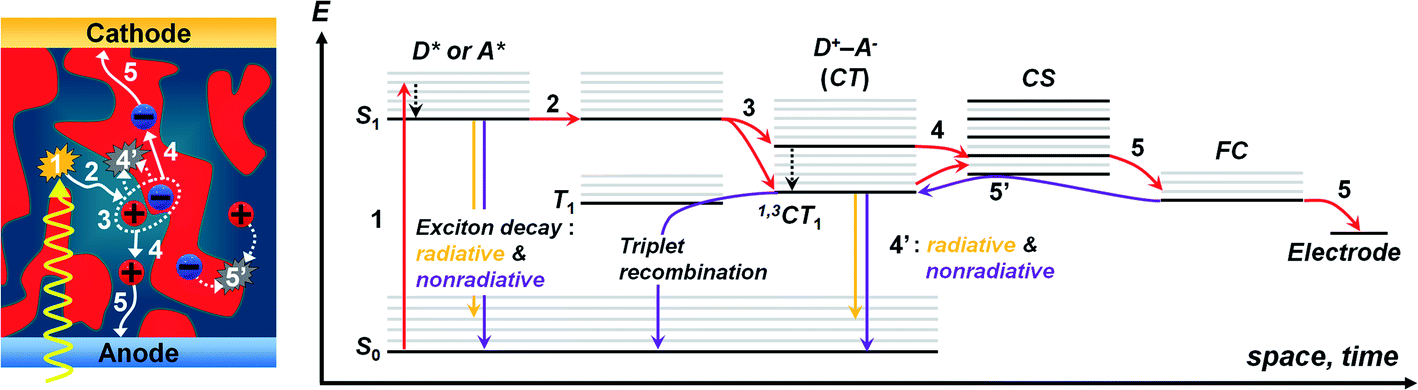 | ||
| Fig. 2 Illustration of OPV operation according to a sequential process: (1) light absorption and exciton generation, (2) exciton diffusion, (3) charge separation, (4) charge transport, (4′) geminate recombination, (5) charge collection, (5′) non-geminate recombination (left), excited-state Jablonski diagram for description of charge generation and recombination processes in OPVs based on the BHJ active layer (S0: ground state; S1: the lowest singlet state; T1: the lowest triplet state; CT: charge transfer state; CS: charge separation state; FC: free charge carrier) (right). | ||
In practice, the PV characteristics of OPVs are evaluated in terms of current–voltage (I–V) measurements, where the output current is obtained at each step of an applied voltage under solar illumination. The efficiency in converting sunlight to electricity (i.e., PCE) is estimated from the measured I–V curve and is defined as follows:51,52
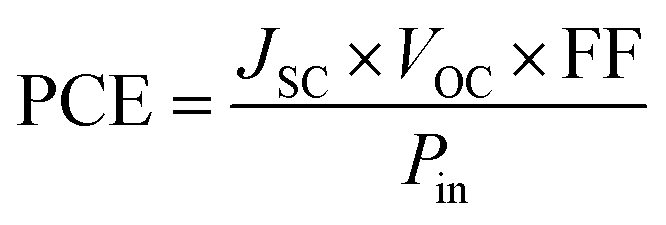 | (2) |
3. Molecular designs of NFAs
Among the various NFAs, three types of molecular design, perylenediimide (PDI)-based, indacenodithiophene (IDT)-based, and Y6-based, have demonstrated outstanding PV performances (see Fig. 1).32,36,53 In this section, we describe the molecular design for these three types of NFA in terms of the optoelectronic and structural properties required for high-performance OPVs, while the device characteristics observed in various blend systems are also discussed (the chemical structures of polymer donors, FAs, PDI-based NFAs, IDT-based NFAs, and Y6-based NFAs are shown in Fig. 3, and OPV performance of representative FA- and NFA-based OPVs is shown in Table 1).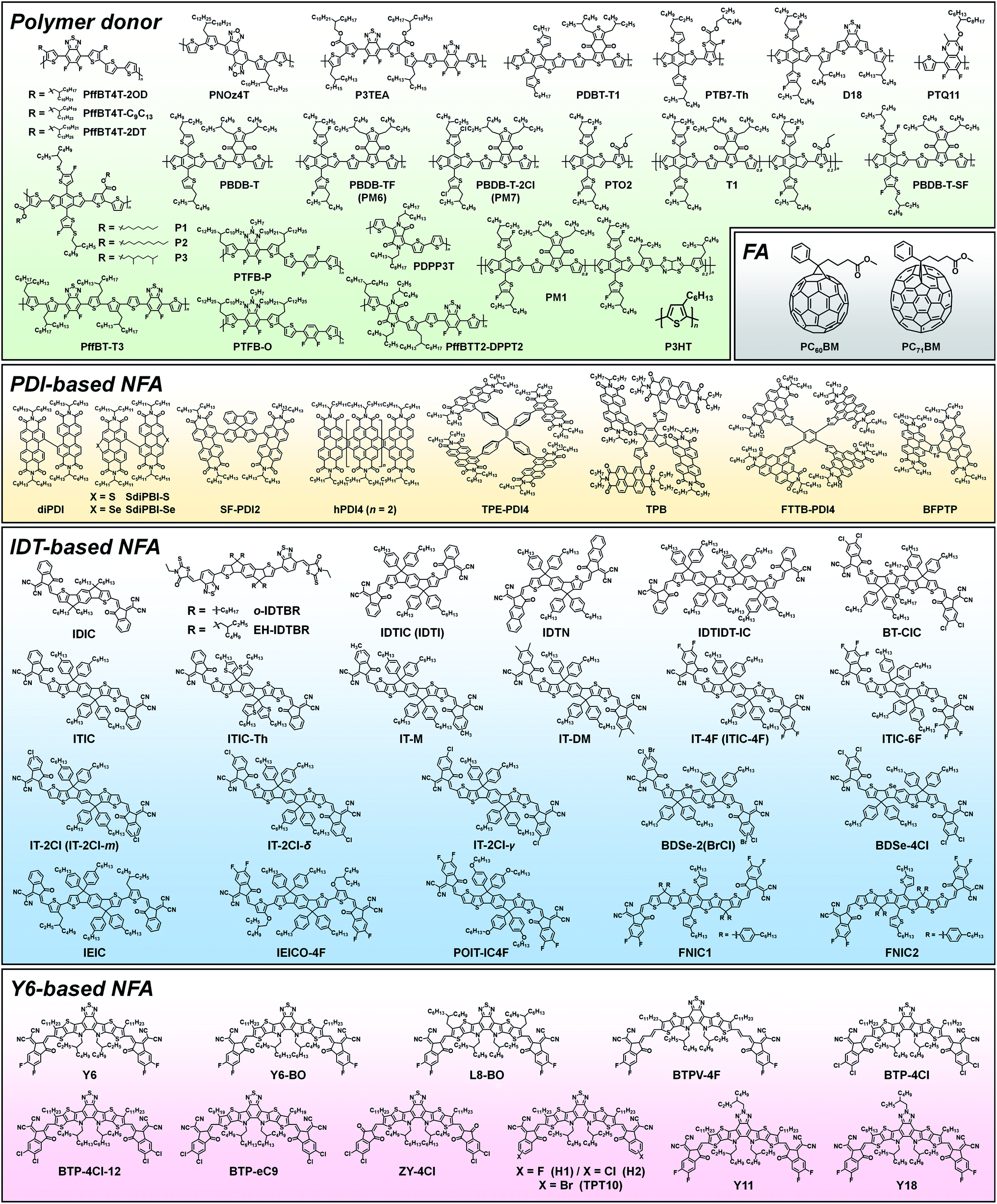 | ||
| Fig. 3 Chemical structures of polymer donors, FAs, and NFAs (PDI-based NFA, IDT-based NFA, and Y6-based NFA) discussed in this review. | ||
| Acceptor | Donor | V OC [V] | J SC [mA cm−2] | FF [%] | PCE [%] | V loss [V] | Ref. |
|---|---|---|---|---|---|---|---|
| FAs | |||||||
| PC71BM | PffBT4T-C9C13 | 0.78 | 19.8 | 73 | 11.7 | 0.87 | 23 |
| PC71BM | PffBT4T-2OD | 0.77 | 18.4 | 74 | 10.5 | 0.88 | 22 |
| PC71BM | PNOz4T | 0.96 | 14.5 | 64 | 8.9 | 0.56 | 74 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||
| PDI-based NFAs | |||||||
| SdiPBI-S | PDBT-T1 | 0.90 | 11.7 | 66 | 7.2 | 0.95 | 66 |
| SdiPBI-Se | PDBT-T1 | 0.95 | 12.5 | 70 | 8.4 | 0.90 | 67 |
| SF-PDI2 | P3TEA | 1.11 | 13.3 | 64 | 9.5 | 0.61 | 69 |
| hPDI4 | PTB7-Th | 0.80 | 15.2 | 68 | 8.3 | 0.78 | 64 |
| TPB | PTB7-Th | 0.79 | 17.9 | 58 | 8.5 | 0.79 | 70 |
| FTTB-PDI4 | P3TEA | 1.13 | 13.9 | 66 | 10.6 | 0.53 | 72 |
|
|||||||
| IDT-based NFAs | |||||||
| IT-4F | T1 | 0.90 | 21.5 | 78 | 15.1 | 0.64 | 75 |
| IT-4F | PTO2 | 0.91 | 21.5 | 75 | 14.7 | 0.67 | 76 |
| IT-4F | PBDB-T-SF | 0.88 | 20.9 | 71 | 13.1 | 0.66 | 77 |
| BDSe-4Cl | PM7 | 0.83 | 22.5 | 74 | 13.8 | 0.56 | 78 |
| BDSe-2(BrCl) | PM7 | 0.83 | 22.9 | 77 | 14.5 | 0.56 | 78 |
| POIT-IC4F | PM6 | 0.91 | 20.9 | 73 | 13.8 | 0.58 | 79 |
|
|||||||
| Y6-based NFAs | |||||||
| Y6 | D18 | 0.86 | 27.7 | 77 | 18.2 | 0.47 | 15 |
| Y6-BO | PM6 | 0.86 | 25.3 | 76 | 16.4 | 0.48 | 80 |
| L8-BO | PM6 | 0.87 | 25.7 | 82 | 18.3 | 0.55 | 17 |
| BTP-4Cl-12 | PM6 | 0.86 | 25.6 | 78 | 17.0 | 0.53 | 81 |
| BTP-eC9 | PM6 | 0.84 | 26.2 | 81 | 17.8 | 0.56 | 16 |
| Y11 | PM6 | 0.83 | 26.7 | 74 | 16.5 | 0.48 | 82 |
3.1. PDI-based NFAs
The PDI unit has a large planar structure with tetracarboxylic diimide groups, which means PDI-based molecules have strong intermolecular interaction and high electron affinity (LUMO ≈ −3.8 eV).32,54 Hence, PDI-based molecules are currently employed as electron-accepting54–57 and/or transporting materials57–60 in organic electronic devices. However, the self-aggregation induced by the strong intermolecular interactions among the PDI units lowers the solubility in common solvents and forms particularly large domains in BHJ blends, thus limiting the exciton dissociation and diffusion.32,61,62 The molecular design of PDI-based materials appears to suppress the self-aggregation through structural torsion63 or ring fusion.64 Here, the structure of PDI dimers or tetramers linked through C–C single bonds65–67 or π-conjugated units68–71 (e.g., spirofluorene and benzene) exhibit highly twisted conformation with torsional angles of 40°–80°, which effectively inhibits the self-aggregation in blend films. Nonetheless, low device performance remains an issue in twisted PDI-based blend OPVs because of their low electron mobility (μe) and narrow light absorption (400–600 nm). Here, the ring fusion of PDI-based NFAs can lower the torsional angles between the units (≈25°),64 resulting in closer intermolecular packing than that in PDI dimers and tetramers. These structural features have been reported to be effective in improving the μe (≈10−3 cm2 V−1 s−1) without generating large domains in blend films. For example, ring fusion to PDI tetramer (FTTB-PDI4)72,73 builds a “double-decker” geometry, where adjacent PDI units exhibit close to parallel conformation for close intermolecular packing.The high-performance blend system of PDI-based NFAs and polymer donors (Eg ≈ 1.6 eV) exhibits a light absorption in the 300–800 nm range and a JSC of 15–20 mA cm−2, which is similar to FA-based OPVs (Fig. 1a). Meanwhile, the Vloss of PDI-based OPVs is mainly distributed in the 0.6–0.8 V range at a ΔELUMO of <0.5 eV, and is around 0.4 V smaller than that of FA-based OPVs (Fig. 1c).
3.2. IDT-based NFAs
The IDT unit, a pentacyclic ladder-type arene, has a five-fused aromatic structure with high planarity and strong rigidity.83,84 The introduction of IDT as a core unit in organic semiconductors (e.g., small molecules or polymers) enhances the intermolecular interaction and structural resilience, demonstrating high charge carrier mobility with a low degree of energetic disorder.85 Most IDT-based NFAs adopt the A–D–A type structure, with two strong electron-withdrawing end-groups (e.g., indanone or rhodanine derivatives) attached to the IDT core unit. The A–D–A structure in IDT-based NFAs effectively reduces the Eg to a great extent through efficient intramolecular charge transfer (ICT), providing NIR light absorption with an onset of 800–1100 nm (Eg = 1.2–1.6 eV). In addition, IDT-based NFAs have low crystallinity, exhibiting a well-mixed blend morphology with polymer donors.86–88The molecular design of IDT-based NFAs has evolved through chemical modifications of the backbone, side chains, and end-groups.32,89 Increasing the number of fused rings or inserting additional electron-donating units has proven to be effective in reducing the Eg of IDT-based NFAs through extending the conjugation length.44,90–92 The bulky side chains on the sp3-hybridized carbon (C) atoms of the IDT unit play an important role in determining the solubility of IDT-based NFAs, but cause some steric hindrance among the IDT units. Hence, for the most part, IDT-based NFAs have a linear stacked packing structure formed through end-group π–π stacking,36,93 and the modification of the alkyl side chains (e.g., shape, length, and branch position) has been exploited to modulate the intermolecular interactions and the blend miscibility with the polymer donors.94,95 Chemically substituting hydrogen (H) atoms of the end-group with strong electronegative atoms (e.g., fluorine [F] and chlorine [Cl]) has also proven to be an effective molecular design strategy for improving the μe (≈10−4 cm2 V−1 s−1) of IDT-based NFAs through enhancing the intermolecular interactions.77,96–98
The blend system of IDT-based NFAs and polymer donors (e.g., PBDB-T analogs, PTB7-Th) covers the light absorption in the range of 400–1000 nm, and IDT-based devices with an Eg of <1.5 eV tend to exhibit a high JSC of >20 mA cm−2 (Fig. 1a). Specifically, the PTB7-Th:IEICO-4F device99 achieves the highest JSC of 27.3 mA cm−2 among all the IDT-based blend OPVs. The Vloss of IDT-based blends tends to be sensitive to the ΔEHOMO and has a narrow distribution of 0.4–0.8 V at a ΔEHOMO of <0.5 eV (Fig. 1e).
3.3. Y6-based NFAs
Unlike IDT-based NFAs, the molecular design feature of Y6-based NFAs revolves around the adoption of an electron-deficient polycyclic arene as a core unit, in which benzothiadiazole and dithienothiophene[3,2-b]-pyrrolo are fused (i.e., D–A′–D-type structure).7,100–102 With two end-groups linked to the core unit (i.e., A–D–A′–D–A structure), Y6-based NFAs exhibit a crescent shape and an axisymmetric structure.100,101 The A–D–A′–D–A structure in Y6-based NFAs enables a smaller Eg (1.2–1.4 eV) than do IDT-based NFAs through a strong ICT effect, absorbing light with an onset of 900–1100 nm. Another feature of Y6-based NFAs is the multiple packing between the core units or end-groups,103,104 since the alkyl side chains are attached to the sp2-hybridized nitrogen (N) atoms of the core unit, less bulky than the sp3-hybridized C atoms (i.e., one alkyl chain per N atom and two alkyl chains per C atom). The multiple packing results in the efficient electron transport of Y6-based NFAs with a μe of ≈10−4 cm2 V−1 s−1.101,103,104Similar to IDT-based NFAs, the molecular modification of Y6-based NFAs has been focused on the electronic and structure properties. The extension of the fused-ring core and the insertion of a π-linker have been the main approaches in terms of backbone modification in view of increasing the effective conjugation length of Y6-based NFAs (i.e., smaller Eg),105–107 while modifications of the alkyl side chains have also been employed to control the intermolecular packing and solubility in this type of NFA.16,17,80,108 Interestingly, it has recently been demonstrated that the alkyl side chains adjacent to the end-groups prevent the rotating end-groups (i.e., conformational locking), thereby lowering the energetic disorders to improve the charge transport.109,110 Meanwhile, the end-group of Y6-based NFAs has largely been modified through chemical substitutions using electronegative atoms (e.g., F and Cl) to control the LUMO levels and molecular packing, much like in IDT-based NFAs.101,111,112
The Y6-based blend system exhibits a complementary light absorption of 400–1100 nm with polymer donors (e.g., PBDB-T analogs or PTB7-Th). The Y6-based devices with an Eg of <1.4 eV attain a high JSC of >23 mA cm−2 (Fig. 1a). The Vloss of the Y6-based blends is distributed in the 0.4–0.6 V range with an average of 0.52 V at a ΔEHOMO of <0.3 eV, which is much lower than those of the other two types of NFA-based OPV (Fig. 1e). A significant number of Y6-based blends have exhibited excellent device performance, with a PCE of 15–18%, while most recently, the highest PCE of 18.32% has been reported in a Y6 analog-based device (PBDB-TF:L8-BO).17
4. Key physical features in NFA-based OPVs
The increase in the JSC and VOC of NFA-based OPVs can be largely attributed to the specific molecular design of the NFA, which creates distinctive physical features that differ from those of FA-based OPVs. In this section, we describe the physical features observed in NFA-based OPVs in relation to those of FA-based OPVs and review the current understanding of their fundamental aspects in terms of light harvesting, voltage loss, charge transport, triplet-exciton utilization, and blend morphology.4.1. Light harvesting
| EQE = ηabsIQE | (3) |
The internal quantum efficiency (IQE) is the ratio of the number of charge carriers collected by the device to the number of photons from the device (i.e., photons absorbed by the device). The JSC, one of the main PV parameters, is expressed as follows:49
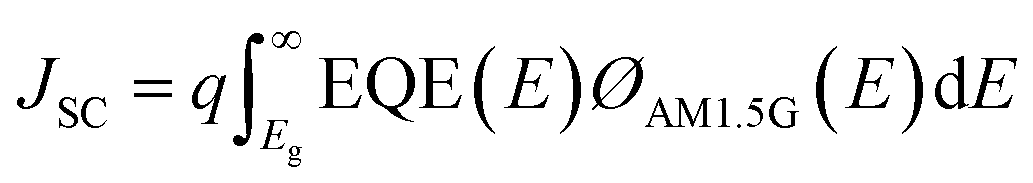 | (4) |
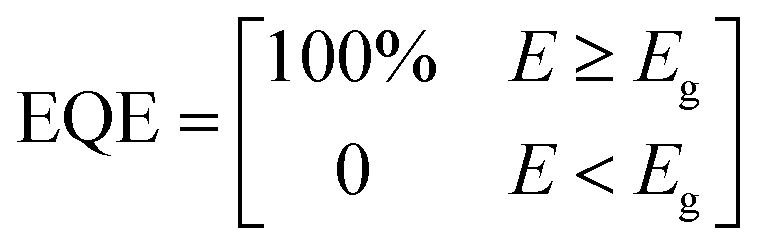 | (5) |
In theory, when photons with E ≥ Eg are absorbed, they are completely converted to electricity, and the JSC can be maximized to JSC,SQ (Fig. 4a).
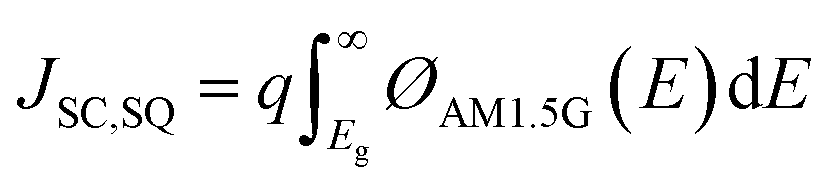 | (6) |
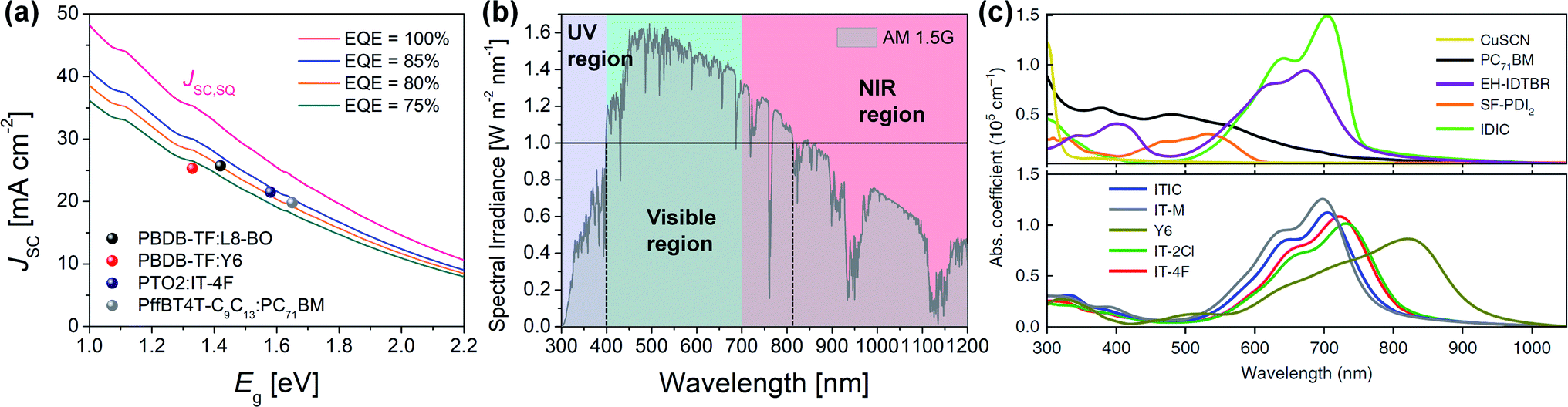 | ||
| Fig. 4 (a) Calculated JSC as a function of Eg according to the external quantum efficiency (EQE) assumed to be a constant value above the Eg. JSC of representative FA- and NFA-based OPVs (i.e., PBDB-TF:L8-BO,17 PBDB-TF:Y6,101 PTO2:IT-4F,76 and PffBT4T-C9C13:PC71BM23). (b) Solar spectrum under AM 1.5G; purple (≤400 nm), green (400–700 nm), and red (≥700 nm) area mean UV, visible, and near-infrared (NIR) region, respectively. Black solid line represents spectral irradiance of 1.0 W m−2 nm−1 and black dotted lines provide the region over spectral irradiance of 1.0 W m−2 nm−1 (400–800 nm). (c) Absorption coefficient (α) of PC71BM and NFAs (EH-IDTBR, SF-PDI2, IDIC, ITIC, IT-M, Y6, IT-2Cl, and IT-4F). Reproduced with permission.116 Copyright 2020, Springer Nature. | ||
In addition, the solar irradiance in the range of 400–800 nm is over 1.0 W m−2 nm−1 (Fig. 4b), indicating that a large number of photons are provided in this region. Therefore, a desirable D/A blend should have a strong absorption coefficient (α) while absorbing visible and NIR regions (>400 nm).
Conventional FAs (e.g., PC61BM or PC71BM) exhibit a relatively short absorption range of 300–500 nm with weak α values;114 hence, the light harvesting of FA-based OPVs is largely determined by the absorption of polymer donors. For example, the PffBT4T-C9C13:PC71BM blend OPV absorbs up to 750 nm of light and exhibits a JSC of 19.8 mA cm−2.23 Meanwhile, the complementary absorption of medium-Eg polymer donors and narrow-Eg NFAs covers a wide range of light absorption, enabling a high JSC of >20 mA cm−2. This is also related to the extinction coefficient (k), an optical constant for how strongly a material absorbs light at a specific wavelength (α = 4πk/λ, λ is wavelength).115 Most NFAs have higher α and k values (α > 1.0 × 105 cm−1; k > 1.0) than PC71BM (α ≈ 0.9 × 105 cm−1; k ≈ 0.4) in the region from visible to NIR (Fig. 4c).115,116
The approach to designing D–A-type conjugated molecules is effective in terms of adjusting the Egvia the ICT effect and provides variable light absorption. Furthermore, the more planar the molecular backbone, the stronger the ICT effect. In recent years, it has been demonstrated that the molecular design of an A–D–A-type NFA is effective in terms of NIR light absorption because of its high planarity and the strong electron-withdrawing ability of the A units (e.g., ![[E with combining low line]](https://www.rsc.org/images/entities/i_char_0045_0332.gif) g = 1.59, 1.51, and 1.24 eV for ITIC, IT-4F, and IEICO-4F, respectively).77,117–119 The crescent-shaped A–D–A′–D–A-type structure (Y6 analogs) has also demonstrated an effective design approach to NIR light absorption >900 nm (e.g., g = 1.33 eV for Y6 and g = 1.21 eV for BTPV-4F),101,107 while devices with Y6 analogs exhibit a high JSC of >23 mA cm−2, which is comparable to that of perovskite solar cells (e.g., 27.7 mA cm−2 for D18:Y6 (ref. 15) and 28.3 mA cm−2 for PTB7-Th:BTPV-4F107).
g = 1.59, 1.51, and 1.24 eV for ITIC, IT-4F, and IEICO-4F, respectively).77,117–119 The crescent-shaped A–D–A′–D–A-type structure (Y6 analogs) has also demonstrated an effective design approach to NIR light absorption >900 nm (e.g., g = 1.33 eV for Y6 and g = 1.21 eV for BTPV-4F),101,107 while devices with Y6 analogs exhibit a high JSC of >23 mA cm−2, which is comparable to that of perovskite solar cells (e.g., 27.7 mA cm−2 for D18:Y6 (ref. 15) and 28.3 mA cm−2 for PTB7-Th:BTPV-4F107).
4.2. Voltage loss
| ECT = |HOMOD − LUMOA| − Eb | (7) |
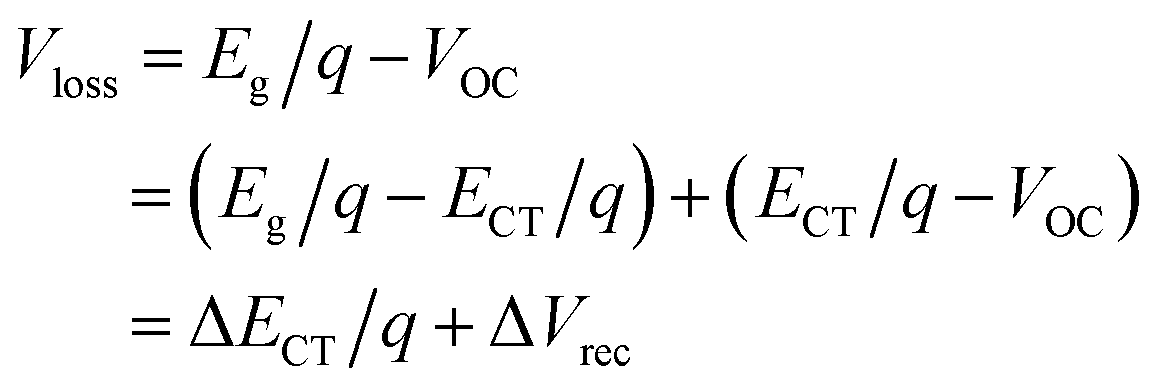 | (8) |
 | (9) |
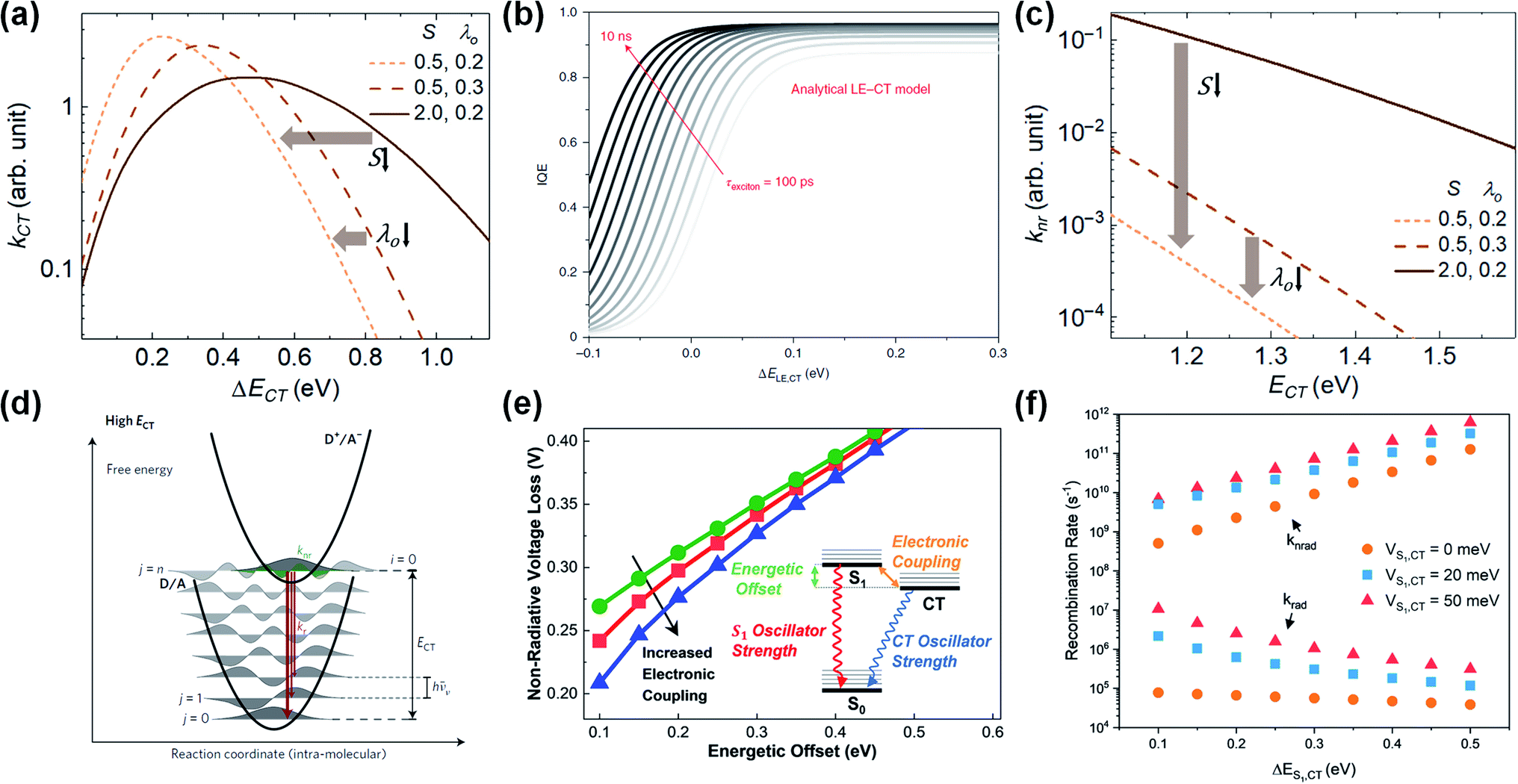 | ||
| Fig. 5 (a) CT rate (kCT) versus energy offset (ΔECT). The peak kCT occurs as intermolecular reorganization energy (λO) and intramolecular coupling strength (S) decrease. S is correlated with intramolecular reorganization energy (λI) and is expressed as S = λI/ℏ〈ωI〉. Reproduced with permission.129 Copyright 2019, Elsevier. (b) Calculation of IQE (consistent with exciton-splitting efficiency) based on the energy difference between the lowest excited state (LE is consistent with S1) and CT state (ΔELE,CT), and exciton lifetime (τexc). Reproduced with permission.139 Copyright 2020, Springer Nature. (c) Non-radiative CT recombination rate (knr) versus ECT as a function of λO and S. The knr decreases as λO and S decreases and ECT increases. Reproduced with permission.129 Copyright 2019, Elsevier Inc. (d) The potential energy of the ground state (D/A) and the CT state (D+/A−). Reproduced with permission.132 Copyright 2017, Springer Nature. (e) Simulated non-radiative recombination voltage loss (ΔVnon-radOC) versus ΔECT as a function of electronic coupling (hybridization). Inset schematic diagram represents three-state model. (f) Recombination rate versus ΔECT as a function of hybridization. Reproduced with permission.141 Copyright 2019, American Chemical Society. | ||
In terms of molecular structure, increasing the planarity and/or the effective conjugation length of the molecular backbone has proven to be effective in reducing the λI. Most NFAs have a lower λI (e.g., λI = 0.179, 0.101, and 0.157 for IDT-IC, IDTIDT-IC, and BT-CIC, respectively) than FAs (e.g., λI = 0.180 for PC71BM).134 The molecular reorientation (i.e., intermolecular reorganization) occurs at the D/A interface during the CT process, changing the charge distribution of the donor and the acceptor. In this regard, the electronic polarization, or the dielectric response, influences the λO in CT exciton splitting, which can be simplified in terms of the spherical cavity model established by Marcus:134–136
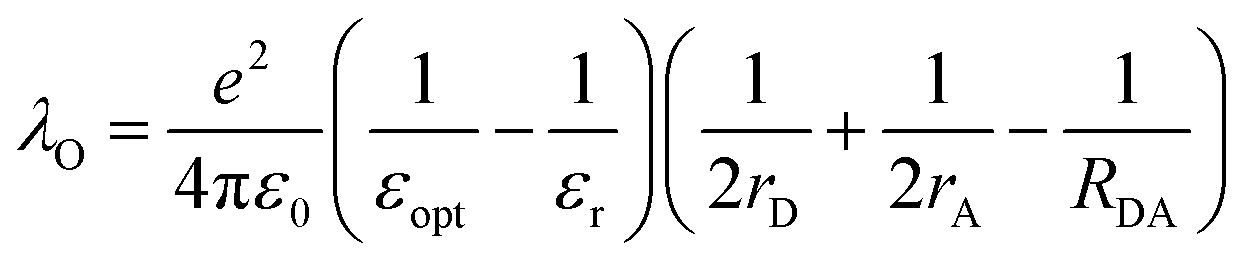 | (10) |
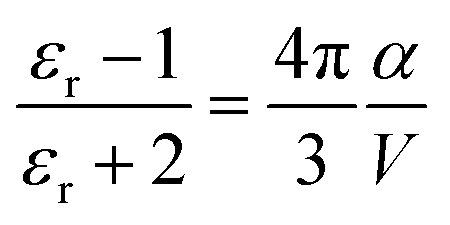 | (11) |
In short, a larger εopt and εr and a smaller RDA can lead to a lower λO in the CT state. Furthermore, a large εr and an average electron–hole distance (r) will reduce the Eb, resulting in a small ΔECT. The Eb can be expressed as follows:138
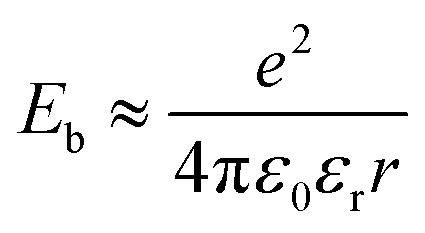 | (12) |
The difference in the n of acceptor materials was reported by Kerremans et al., with the ellipsometry measurement revealing higher n values (i.e., εopt) in NFAs than in conventional FAs (e.g., n = 2.7 for IT-4F and n = 2.2 for PC71BM).115 Meanwhile, in terms of the εr of acceptors, Zhu et al. performed a computational study using density functional theory (DFT) and found that planar and rigid A–D–A structures have a higher α (e.g., α = 247.5 Å3, εr = 5.11 for IT-4F, and α = 114.5 Å3 and εr = 3.83 for PC71BM) and a smaller Eb (e.g., Eb = 0.35 and 0.90, 0.63 for IT-4F, PC71BM, SF-PDI2, respectively) than bulky or spherical structures.138 In addition, grazing-incidence wide-angle X-ray scattering measurement has been used to reveal that the RDA between the polymer donor and the NFA is close enough for effective intermolecular reorganization (e.g., RDA = 3.61 Å for PM6:Y6).101
The long exciton lifetime (τ) also plays a critical role in improving the exciton-splitting efficiency in low ΔECT systems (Fig. 5b). Here, Classen et al. examined the lifetime and efficiency of exciton splitting using polymer donor:NFA blends with a ΔEHOMO of 0–0.3 eV, with the NFA-based OPVs exhibiting slow exciton-splitting lifetimes (τsplitting > 20 ps) and high exciton-splitting efficiencies (ηsplitting > 90%) owing to the long τ of NFAs (305.3, 561.5, 898.3, and 1016 ps for ITIC, o-IDTBR, EH-IDTBR, and Y6, respectively).139 This demonstrates that NFAs with a long τ allow for efficient exciton splitting even with a negligible driving force (i.e., ΔECT < 0.2 eV). In practice, NFA-based OPVs have recently been reported to exhibit high PCEs even with a negligible ΔECT or ΔEHOMO. For example, the PM6:Y11-based OPV exhibits a PCE of 16.54% despite the ΔECT of 0 eV,82 while the PTQ11:TPT10-based OPV exhibits a PCE of 16.32% even at a ΔEHOMO of 0 eV.140
In addition to lowering the λI and λO (Fig. 5c),129,134 increasing the ECT level (i.e., decreasing the ΔECT) can reduce the knr (i.e., a smaller ΔVnon-radOC) because of the smaller wave-function overlap between the CT and the ground state (S0) (Fig. 5d).132 Here, Benduhn et al. reported a linear regression with a slope of ≈−0.2 V eV−1 between ΔVnon-radOC and ECT,132 while Classen et al. reported a sharp decrease in ΔVnon-radOC with a slope of ≈−0.6 V eV−1 at a ΔEHOMO of less than 0.2 eV.139 Eisner et al. proposed a three-state model in which a small ΔECT enables electronic coupling (hybridization) between the S1 and the CT state to enhance the oscillator strength from CT to S0.141 As shown in Fig. 5e, the ΔVnon-radOC decreases with an increase in hybridization at a given ΔECT. Consequently, the knr slows down as the ΔECT decreases in the absence of hybridization, while the kr remains almost constant (Fig. 5f). However, the hybridization affects the kr much more than the knr, thus increasing the oscillator strength of the CT exciton in low-ΔECT systems (i.e., higher EQEEL). For example, the PM6:Y11 device has a ΔECT of 0 eV and exhibits a higher EQEEL (EQEEL ≈ 1.4 × 10−3, ΔVnon-radOC = 0.17 V) than conventional high-performance FA-based OPVs. Moreover, the overall Vloss of the PM6:Y11 device is 0.43–0.51 V, which is very close to that of c-Si solar cells (0.38 V).82
4.3. Charge transport
Aldrich et al. studied the change in μe with a number of F substituents at the end-groups of A–D–A-type NFAs (ITIC, ITIC-4F, and ITIC-6F).98 Here, the DFT calculation revealed that all NFAs have similar electron delocalization but that the LUMO and LUMO+1 are predominantly localized on the end-groups. In the single-crystal analysis, the ITIC and ITIC-6F were found to exhibit face-to-face π–π stacking between the end-groups (≈3.4 Å for ITIC and 3.95, 3.28 Å for ITIC-6F), while the ITIC-4F had a coexisting packing of face-to-face and edge-to-face stacking (3.35 Å) (Fig. 6a). Meanwhile, in addition to the π–π stacking, lone-pair⋯π interactions were observed in the ITIC-4F (S⋯π) and ITIC-6F (CN⋯π), contributing to closer intermolecular packing distances (3.21 Å for ITIC-4F and 3.16 Å for ITIC-6F). Finally, the |Je| calculated from the crystal dimers was 11.4, 17.1 and 56.8 meV for the ITIC, ITIC-4F and ITIC-6F, respectively, with the ITIC-6F consequently exhibiting a higher μe (5.7 × 10−4 cm2 V−1 s−1) than the other two NFAs (3.1 × 10−4 and 5.1 × 10−4 cm2 V−1 s−1 for ITIC and ITIC-4F, respectively).
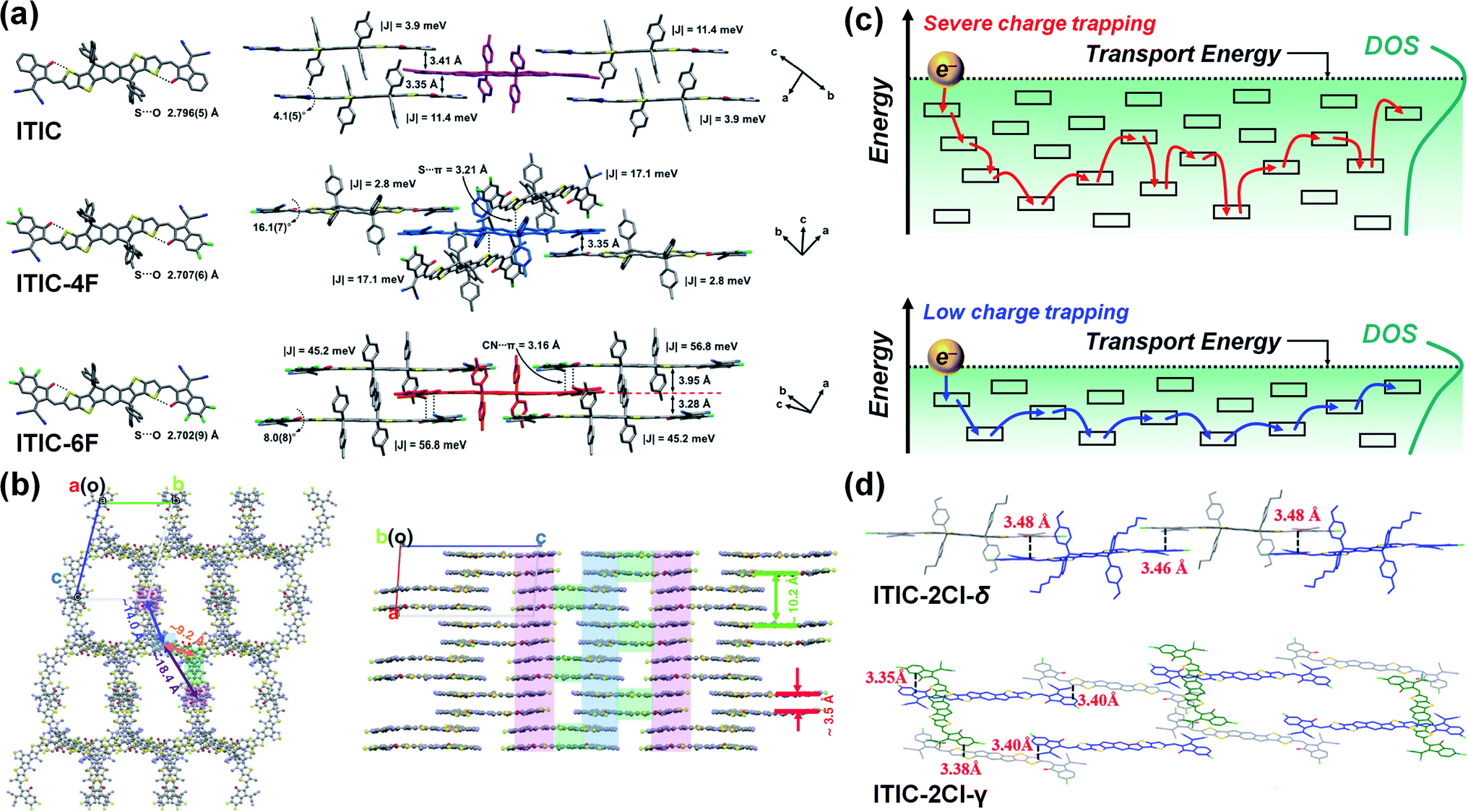 | ||
| Fig. 6 (a) Single crystal and intermolecular packing structure of ITIC, ITIC-4F, and ITIC-6F. Reproduced with permission.98 Copyright 2019, American Chemical Society. (b) Crystal packing structure of Y6. Reproduced with permission.103 Copyright 2020, Springer Nature. (c) Schematic illustration of electron transport for organic semiconductors governed by hopping mechanism (top: large DOS, bottom: small DOS). (d) Molecular packing structure of ITIC-2Cl-δ (top) and ITIC-2Cl-γ (bottom). Reproduced with permission.152 Copyright 2019, Elsevier. | ||
Zhang et al. identified the multiple packings in a Y6 single crystal and obtained the |Je| from the Y6 dimer packings.103 Here, the Y6 had a longer intermolecular distance (≈3.5 Å) than had the ITIC-based NFAs but exhibited a larger |Je| (81 meV) and μe (1.8 × 10−4 cm2 V−1 s−1) because of its multiple packing structure. In addition, the core unit packing was found to enhance the overlap between the HOMO and HOMO-1, exhibiting high hole transfer integral (|Jh|) (74 meV) and hole mobility (μh) (5.6 × 10−4 cm2 V−1 s−1). These features enable the ambipolar charge transport of Y6, which is beneficial for a balanced charge transport in polymer donor:NFA blends. Elsewhere, Hamada et al. examined the μe and μh of PBDB-T based blends,145 where the PBDB-T:NFA blends exhibited a more balanced charge transport than the PBDB-T:PC71BM blend (μh/μe = 0.057, 0.95, 1.14 for PBDB-T:PC71BM, PBDB-T:Y6 and PBDB-T:ITIC, respectively).
Kupgan et al. quantified the MD and DFT approaches and found that the NFA had a lower σ than the FA through a combination of molecular dynamics (σ = 54, 52, 77 meV for FNIC1, FNIC2, PC71BM, respectively).147,150 The low σ values of NFAs are largely due to the significant decrease in σS (24, 25, and 46 meV for FNIC1, FNIC2, and PC71BM, respectively), indicating that coplanar and rigid backbone structures are more effective in reducing σS than are spherical structures.
Modulating the intermolecular interaction of the NFA can further facilitate cross-stacking, constructing an interconnected network structure that provides 3D charge transport pathways in OPV blends.93,151 Lai et al. introduced Cl atoms to the delta (ITIC-2Cl-δ) or gamma (ITIC-2Cl-γ) position of ITIC end-groups.152 Here, the ITIC-2Cl-γ crystal exhibited an interconnected network structure containing both linear- and cross-stackings, while the ITIC-2Cl-δ crystal mainly adopted a linear stacked structure (Fig. 6d). Meanwhile, when blended with PBDB-TF, the PBDB-TF:ITIC-2Cl-γ exhibited a higher μe (2.6 × 10−4 cm2 V−1 s−1) than did the PBDB-TF:ITIC-2Cl-δ (μe = 1.2 × 10−4 cm2 V−1 s−1). Elsewhere, Zhang et al. investigated the energetic disorder of representative polymer-based blends by measuring the Urbach energy (EU), which represents the trap distribution.110 Here, the polymer donor:FA blends exhibited a higher EU (e.g., EU = 36.0 meV for PffBT4T-2OD:PC71BM) than did the polymer donor:NFA blends (e.g., EU = 34.4 and 22.4 meV for PBDB-T:ITIC and PM6:Y18, respectively). This result demonstrated that the interconnected structure of Y18 efficiently reduces the trap density of OPV blends.
4.4. Triplet-exciton harvesting
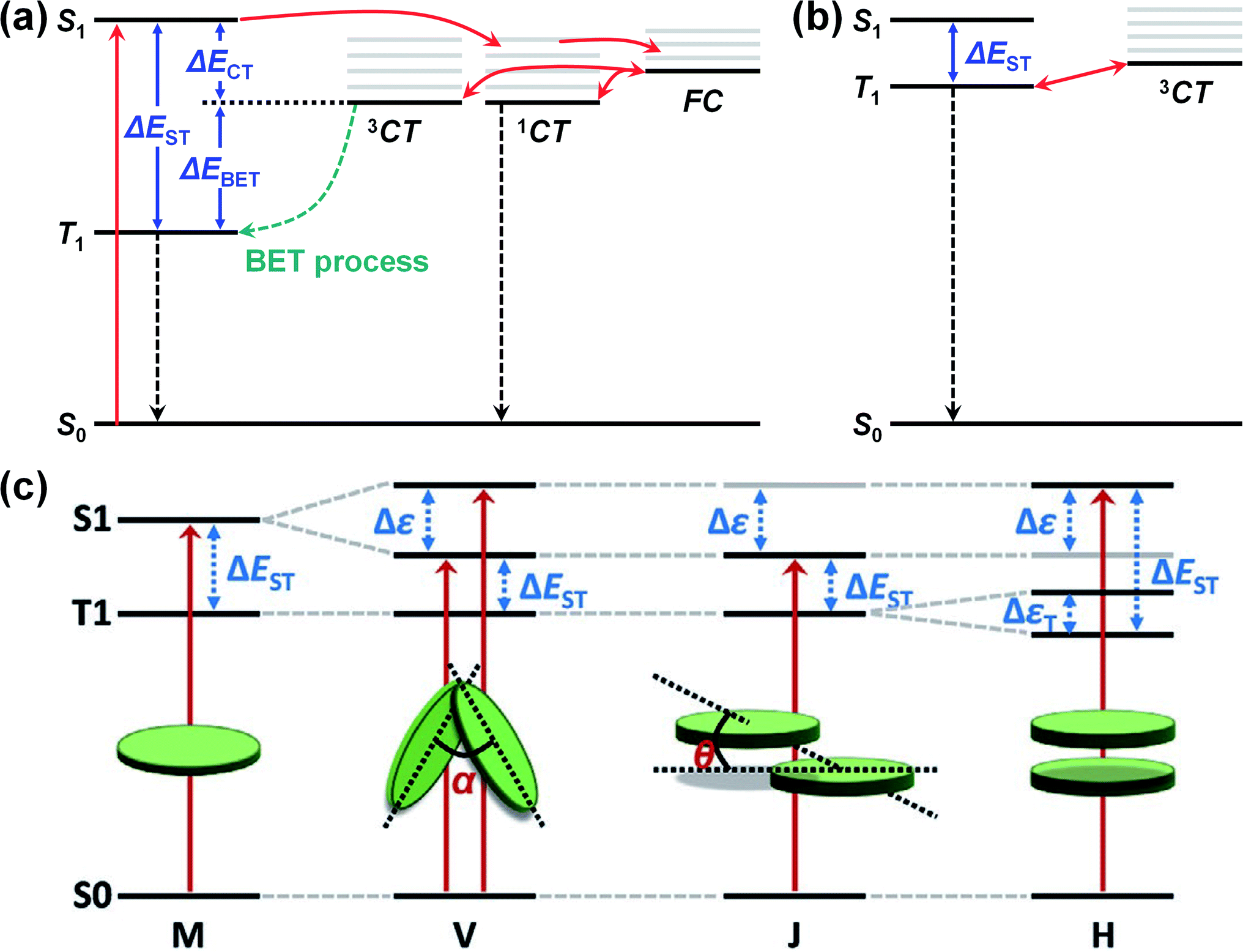 | ||
| Fig. 7 (a) Schematic illustration of triplet-recombination through back electron transfer (BET) process. (b) Energy diagram with small ΔECT, ΔEST, and ΔEBET. (c) Schematic energy diagram of S1 and T1 splitting according to molecular packing modes (V-type, J-type, and H-type molecular packing structures). Reproduced with permission.153 Copyright 2020, WILEY-VCH. | ||
Meanwhile, Yang et al. reported a PDI-based triplet acceptor in which tellurophene was fused with two PDI units (BFPTP).164 The triplet excitons of the BFPTP were confirmed via the photoluminescence (PL) emission with a long lifetime of 24.41 ms at 77 K. The energy level of the T1 (ET1) of the BFPTP was 1.50 eV, which is higher than the ECT of PBDB-T:BFPTP (1.45 eV), indicating that triplet excitons can be converted into 3CT excitons. The PBDB-T/BFPTP bilayer device demonstrated that the BFPTP had a long LD of 34 nm (similar to the LD of triplet FAs), and, as a result, the PBDB-T:BFPTP-based OPV exhibited a PCE of 7.52%, which was a meaningful result that demonstrated, for the first time, the feasibility of utilizing a triplet NFA.
Among the NFA molecules, Y6-derivatives have a slightly twisted structure owing to the sp2-hybridization of the N atoms in the main backbone and are good candidates for triplet acceptors.168 Qin et al. reported two triplet acceptors, both analogs of Y6 (H1 and H2),168 and observed long PL decays on the μs scale in both acceptors (8.15 μs for H1 and 7.66 μs for H2 at 77 K). The two NFAs exhibited a relatively small ΔEST (≈0.35 eV) and had an ET1 (≈1.07 eV) close to the ECT (≈1.37 eV) of blend films employing PBDB-T as a polymer donor. Meanwhile, the transient absorption decay was 65 ns for the PBDB-T:H1 and 35 ns for the PBDB-T:H2, which was much longer than the S1 decay of each component. The existence of triplet excitons in the PBDB-T:H1 and PBDB-T:H2 devices was confirmed via magneto-photocurrent measurement, with both devices exhibiting a high PCE of over 14%.
In addition, the ΔEST can be controlled via the molecular packing mode. In V-type or J-type packing modes (i.e., small intermolecular overlap), the T1 splitting is negligible, while the S1 splitting is significant because of the small intermolecular overlap. As shown in Fig. 7c, this splitting difference can reduce the ΔEST. In contrast, an H-type packing mode (i.e., large intermolecular overlap) can increase the ΔEST since both the S1 and the T1 will be significantly split.169,170 Elsewhere, Han et al. conducted a theoretical study using time-dependent DFT (TD-DFT) calculations in combination with MD simulations and found that the ΔEST could be reduced by end-group π–π stacking in A–D–A- or A–D–A′–D–A-type NFAs.153 The calculated ΔEST of the J-type dimer for IT-4F and Y6 were 0.35 and 0.32 eV, respectively, with the two V-type dimers possessing a similar ΔEST to the J-type dimers. Compared with the ΔEST of the monomers (0.44 eV for IT-4F, 0.42 eV for Y6), the J-type and V-type dimers exhibited a decrease in ΔEST of around 0.1 eV because of the end-group π–π stacking. Meanwhile, because of the reduced ΔEST, the ΔEBET for PM6:IT-4F and PM6:Y6 was calculated to be 0.13 and 0.23 eV, respectively, indicating that T1 excitons can assist the regeneration of free charge carriers. Finally, the transfer rate from T1 to 3CT (kT1→CT) for both blends was estimated to be 107–109 s−1, while the rate of triplet recombination (kT1→S0) was calculated to be 4–7 × 103 s−1, around 4–6 orders lower than the kT1→CT.
4.5. Blend morphology
The phase behavior of the amorphous mixed phase is controlled by the molecular interaction of the D and A. Here, the Flory–Huggins model has been widely used to quantify the degree of mixing between the blend components and has been extended quantitatively to describe the thermodynamic behavior of D:A blends.173,176,177 The change in the Gibbs free energy of mixing (ΔGmix) in a polymer-containing blend can be expressed by the following equation:177
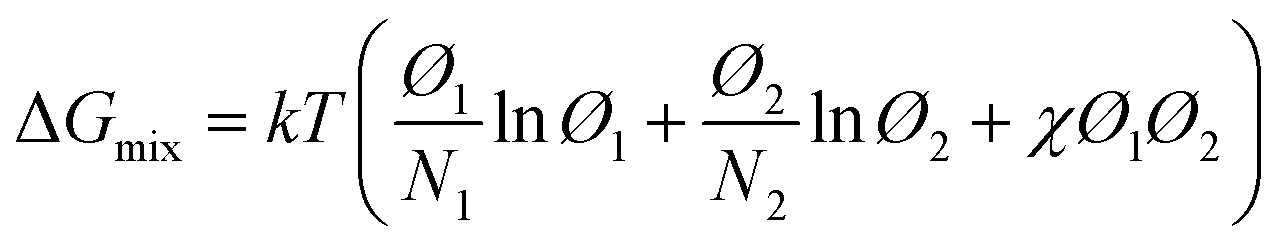 | (13) |
The mixed phase can be divided into three regions in the binodal curve of the χ–Ø phase diagram (Fig. 8a).174,182 Here, the blend in the low χ region is too miscible and has a low domain purity, while large phase separation occurs when the domains are too pure (i.e., high χ region). These two cases result in performance degradation due to inefficient photocurrent generation paths (e.g., increased charge recombination and low charge collection efficiency). The ideal χ with a binary composition close to the percolation threshold can lead to optimal amorphous miscibility and stable morphology in a thermodynamic equilibrium state; however, most OPV blends exhibit a higher or lower χ that deviates from the ideal χ region.
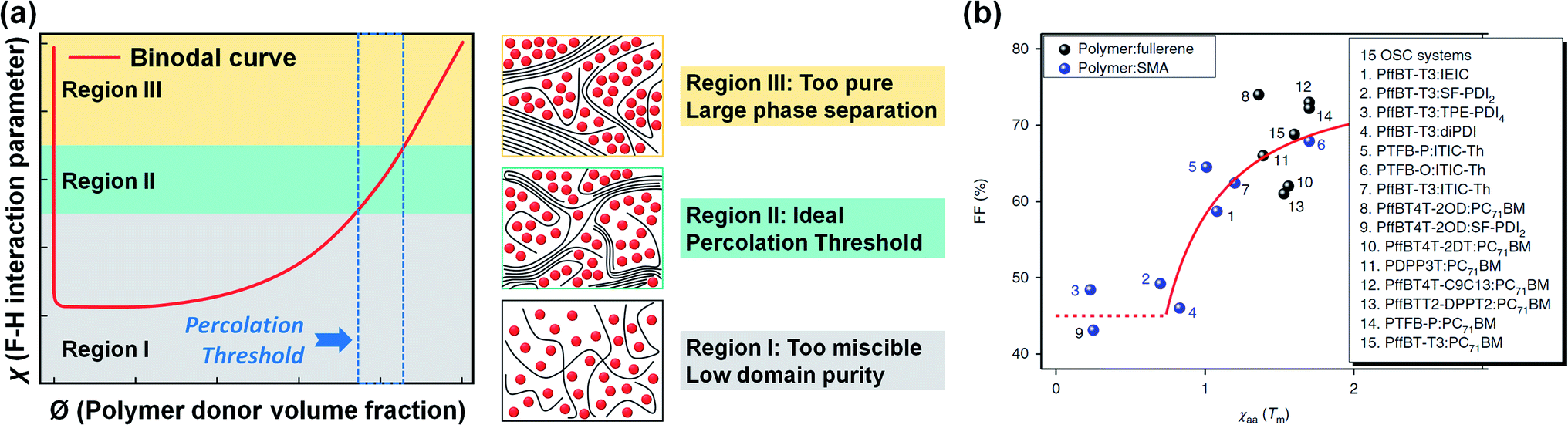 | ||
| Fig. 8 (a) Schematic illustration of three regions in the binodal curve of the χ–Ø phase diagram; low χ (region I), medium χ (region II), and high χ (region III), respectively. (b) χ parameter estimated from DSC and its relation to FF. Reproduced with permission.174 Copyright 2018, Springer Nature. | ||
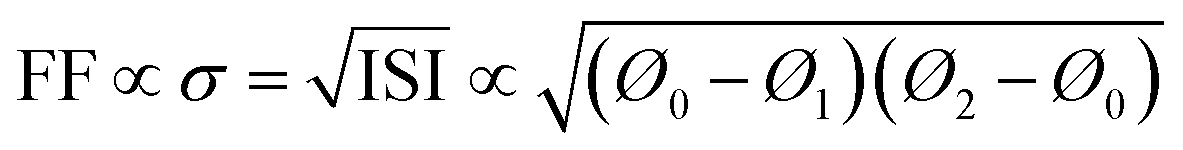 | (14) |
| Acceptor | Donor | V OC [V] | J SC [mA cm−2] | FF [%] | PCE [%] | χ aa | ISI | Ref. |
|---|---|---|---|---|---|---|---|---|
| a The value obtained from DSC. b HSP. c Contact angle measurement. | ||||||||
| FAs | ||||||||
| PC71BM | PDPP3T | 0.66 | 15.4 | 66 | 6.6 | 1.39 (Tm)a | 1 | 174 and 183 |
| PC71BM | PTFB-P | 0.81 | 12.9 | 72 | 7.5 | 1.70 (Tm)a | — | 174 and 184 |
| PC71BM | PffBT4T-2DT | 0.76 | 16.2 | 62 | 7.6 | 1.56 (Tm)a | — | 174 and 185 |
| PC71BM | PffBTT2-DPPT2 | 0.81 | 17.1 | 61 | 8.6 | 1.53 (Tm)a | — | 174 and 186 |
| PC71BM | PffBT4T-2OD | 0.77 | 18.4 | 74 | 10.5 | 1.36 (Tm)a | 1 | 22 and 174 |
| PC71BM | PffBT-T3 | 0.82 | 18.9 | 69 | 10.7 | 1.60 (Tm)a | — | 174 and 187 |
| PC71BM | PffBT4T-C9C13 | 0.78 | 19.8 | 73 | 11.7 | 1.70 (Tm)a | 1 | 23 and 174 |
|
||||||||
| PDI-based NFAs | ||||||||
| SF-PDI2 | PffBT4T-2OD | 0.97 | 6.9 | 43 | 2.9 | 0.25 (Tm)a | — | 174 |
| diPDI | PffBT-T3 | 0.86 | 12.8 | 46 | 5.1 | 0.83 (Tm)a | 0.56 | 174 |
| TPE-PDI4 | PffBT-T3 | 1.02 | 11.5 | 48 | 5.7 | 0.23 (Tm)a | 0.32 | 174 |
| SF-PDI2 | PffBT-T3 | 1.03 | 12.3 | 49 | 6.2 | 0.70 (Tm)a | 0.52 | 174 |
|
||||||||
| IDT-based NFAs | ||||||||
| IEIC | PffBT-T3 | 1.04 | 12.6 | 59 | 7.7 | 1.08 (Tm)a | 0.83 | 174 |
| ITIC-Th | PTFB-P | 0.92 | 15.5 | 65 | 9.2 | 1.01 (Tm)a | 0.91 | 174 |
| ITIC-Th | PTFB-O | 0.92 | 17.1 | 68 | 10.9 | 1.70 (Tm)a | 1 | 174 |
| IT-DM | PBDB-T | 0.96 | 16.3 | 69 | 11.3 | 2.0 (HSP)b | 0.83 | 188 |
| IT-M | PBDB-T | 0.94 | 17.3 | 71 | 12.1 | 2.7 (HSP)b | 0.97 | 188 |
| IT-4F | P1 | 0.90 | 20.1 | 64 | 11.5 | 0.15 Kc | — | 189 |
| IT-4F | P2 | 0.90 | 20.7 | 76 | 14.2 | 0.25 Kc | — | 189 |
| IT-4F | P3 | 0.90 | 20.3 | 61 | 11.2 | 0.19 Kc | — | 189 |
|
||||||||
| Y6-based NFAs | ||||||||
| Y6 | PM6 | 0.86 | 25.5 | 72 | 15.8 | 0.20 Kc | — | 190 |
| Y6 | PM1 | 0.87 | 25.9 | 78 | 17.6 | 0.26 Kc | — | 190 |
| BTP-4Cl | P3HT | 0.50 | 5.16 | 39 | 1.01 | 2.52 (HSP)b | — | 191 |
| ZY-4Cl | P3HT | 0.88 | 16.5 | 65 | 9.46 | 2.56 (HSP)b | — | 191 |
Nevertheless, most of the FA-based OPVs suffer from the so-called “burn-in loss” issue, which causes an initial loss of up to 30–40% of the PCE.193 These burn-in losses originate from the photoinduced degradation of FAs (i.e., dimerization) and from crystallization, leading to a macroscopic phase separation of the blend film.194,195 Li et al. investigated the de-mixing in polymer donor:PC71BM blends under aging conditions (5 days).196 Here, using grazing-incidence small-angle X-ray scattering, the PC71BM domain size was found to have increased from 43 nm (the initial blend film) to 78 nm (the aged blend film; 80% increase). As the size of pure the PC71BM domains increased in the aged sample, the volume fraction and size distribution of the PC71BM domains also increased. In other words, the growth of larger clusters and phase separation occurred in the polymer donor:PC71BM blend under the aging condition when the PC71BM molecules diffused out of the amorphous mixed phases.
On the other hand, it has been reported that NFAs suppress the burn-in loss to light and heat owing to their higher chemical stability and miscibility compared with FAs.86–88,197 Zhu et al. characterized the change in the IEICO-4F domain size in the amorphous mixed phase (PTB7-Th:IEICO-4F) under aging conditions (90 days), as measured via R-SoXs.88 The size of the IEICO-4F domains increased from 38 nm (initial blend film) to 43 nm (aged blend film; 13% increase). This indicates that the NFA-based blend system has a more stable film morphology than the FA-based blend system. Furthermore, Du et al. reported a promising long-term stability system of NFA blend films with an extrapolated T80 lifetime (80% of the initial PCE) of over 11000 h under one-sun illumination.197
In terms of NFA design, Li et al. extended the π-conjugation of an IDTI by replacing the phenyls with naphthyls (IDTN) to enhance the intermolecular packing.198 Here, the PBDB-TF:IDTN blend film had a much longer π–π coherence length (9.4 nm) in the out-of-plane direction than the reported PBDB-T-based blend film (1–5 nm),188,199 while a larger domain size was observed in the PBDB-TF:IDTN blend (65.5 nm) than in the PBDB-TF:IDTI blend, which was due to the more ordered molecular packing of IDTN. The PBDB-TF:IDTN device also exhibited an improved FF (78%) compared with the PBDB-TF:IDTI device (57%).
Chemical substitutions at the end-group of NFAs have also been reported to be effective in increasing the intermolecular interactions. Here, Yang et al. substituted the cyano groups in BTP-4Cl-12 with oxygen atoms (ZY-4Cl) and confirmed that ZY-4Cl lowered the miscibility with P3HT through the weak endo/exothermic peak in the DSC curve (no peak in P3HT:BTP-4Cl-12).191 The χ values obtained from the Hansen solubility parameters (HSPs) were 2.52 and 2.56 for P3HT:BTP-4Cl-12 and P3HT:ZY-4Cl, respectively, indicating the enhanced phase separation in P3HT:ZY-4Cl. Meanwhile, the P3HT:ZY-4Cl blend morphology contributed a higher efficient exciton probability (81% for P3HT:ZY-4Cl, 34% for P3HT:BTP-4Cl-12) and an improved device FF of 65% (FF = 39% in the P3HT:BTP-4Cl-12 device).
5. Summary and outlook
Largely because of the advances in NFA material design, NFA-based OPVs have demonstrated significant improvements in device performance. Specifically, NFA-based OPVs have achieved remarkable increases in the JSC and VOC parameters, which far exceed those of FA-based OPVs. This comprehensive review of the studies on NFAs conducted over the past decade revealed the optimal photophysical, electrical, and morphological characteristics for high-performance OPVs. The distinctive physical features and the current understandings in the field can be summarized as follows:(1) Light harvesting is the key factor in determining the JSC. Overall, NFAs have smaller Eg and larger α values compared with FAs, which enhances the photon absorption. Meanwhile, NFA-based OPVs with an Eg of 1.2–1.6 eV achieve a high JSC of >20 mA cm−2 by covering a wide region (400–1100 nm) through complementary light absorption with the polymer donors. In addition, the long LD of the NFA offers the feasibility of photocurrent generation even in thick photoactive layers (i.e., almost equal to a micrometer scale).
(2) The CT state of a blend system promotes exciton dissociation, but a large ΔECT generates substantial Vloss. Here, NFAs with a low λI and λO provide effective charge-separation, even in blend systems with a small ΔECT (<0.2 eV). In addition, the small ΔECT of NFA-based blends entails a decrease in ΔVnon-radOC through the hybridization between the S1 and CT states in the blends, efficiently lowering the Vloss (<0.8 V) compared with that in FA-based blends (0.6–1.2 V).
(3) Overall, NFAs effectively improve the |J| through end-group π–π stacking (i.e., A–D–A-type NFAs) or multiple stacking (i.e., A–D–A′–D–A-type NFAs). Moreover, the planar and rigid conformation of NFAs enhances the structural resilience and facilitates close intermolecular packing, enabling efficient charge transport with a low degree of energetic disorder (i.e., narrow trap distribution).
(4) The NFAs with heavy atoms or a small ΔEST can become strong candidates for photocurrent generation by harvesting triplet excitons with a long LD; however, they can also present a loss source for photo-excited excitons when the T1 of the donor or acceptor is lower than the 3CT (i.e., the BET process). Engineering the electronic structure of NFAs to increase the T1 or move it closer to the 3CT effectively exploits the triplet excitons.
(5) The majority of NFAs are more miscible with polymer donors than are FAs (i.e., lower χ), while blends tend to have low domain purity. Molecular designs aimed at enhancing the intermolecular packing of the polymer donors or NFAs can increase the domain purity of the blend, thus improving the device FF. In terms of device degradation, NFA-based OPVs exhibit superior operation under photo and thermal stress due to the high chemical stability of the NFAs.
Advances in material design and device fabrications have led to significant improvements in OPV performance, often exceeding 18%, which is comparable to the panel efficiency of polycrystalline silicon PV cells, i.e., 18–20% efficiency for a standard size 60-cell panel (1 × 1.65 m). Along with the design of NFAs, multi-faceted studies covering the development of other layer materials (e.g., donor materials, electron/hole transport materials) and device fabrication (e.g., optimization of device structure, film processing techniques) are still essential to maximize OPV performance. However, many challenges remain in the commercialization of OPV technology with regard to the mass production of photoactive materials and the manufacture of large-scale PV cells with long-term stability.
In terms of the materials for mass production, the current commercially available photoactive materials involve high costs ($2000–3000 g−1), largely owing to the multi-step synthesis that consists of more than 10 steps.200–202 In fact, the synthesis of most photoactive materials involves palladium-catalyzed cross-coupling reactions (e.g., Suzuki or Stille coupling) and low-temperature reactions at −78 °C, which is undesirable both in terms of productivity and the environment. Therefore, a short-step synthesis using precious-metal-free catalysts and moderate reaction conditions is required for the mass production of photoactive materials. Another consideration is processability through the use of green solvents, given that most photoactive materials are highly soluble in toxic, halogenated solvents (e.g., chloroform, chlorobenzene).
The large-scale fabrication of OPVs is also a challenging issue. Numerous high-performance OPVs have been demonstrated in small-area devices, mainly in the order of mm2, using spin-coating methods. However, spin-coating is a non-scalable deposition method involving high material waste, and is not suitable for large-scale fabrication using a continuous deposition process.201 Among the current methods, slot-die coating has demonstrated great potential in terms of an upscaling deposition via roll-to-roll fabrication; however, the low efficiency, small cell area, brittle substrate (e.g., ITO-coated PET sheets), and thermal evaporation are limiting a further increase in the manufacturing scale. Hence, the development of novel upscaling deposition techniques (high efficiency of >10% and large cell area of >1 cm2) and fully roll-to-roll process designs (vacuum-free process, ITO-free device architecture) is crucial for the large-scale fabrication of OPVs. Moreover, the long-term stability of the devices is an essential consideration for commercial manufacturing. Given that the performance warranty of conventional silicon solar panels is 80% of the initial performance over 25 years, a performance lifetime of >10 years is recommended for OPVs.
Aside from the material and cell fabrication perspectives, the origin of the photocurrent generation in OPVs has not yet been fully understood, largely because of the physical complexity of the organic-based blend system. Thus, advanced analytical techniques with high temporal and spatial resolutions are required to allow for clearly and accurately characterizing the physical features of OPV operations. In addition, computational studies using machine learning have recently demonstrated powerful techniques for understanding and predicting the physical properties of the relevant materials and blends while reducing both the time and the cost.203,204 Therefore, a combination of these two techniques is expected to provide a better understanding of the fundamentals in view of designing more efficient and stable OPVs.
Author contributions
All authors contributed to bibliographic research, discussion, writing, and revision of the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Ministry of Science and ICT (MSIT) (Code No. 2021R1A2C3004420, 2021R1A5A1084921, 2020M3H4A1A02084908).Notes and references
- K. A. Mazzio and C. K. Luscombe, Chem. Soc. Rev., 2015, 44, 78–90 RSC.
- K. Wang, C. Liu, T. Meng, C. Yi and X. Gong, Chem. Soc. Rev., 2016, 45, 2937–2975 RSC.
- K. Fukuda, K. Yu and T. Someya, Adv. Energy Mater., 2020, 10, 2000765 CrossRef CAS.
- J. Qin, L. Lan, S. Chen, F. Huang, H. Shi, W. Chen, H. Xia, K. Sun and C. Yang, Adv. Funct. Mater., 2020, 30, 2002529 CrossRef CAS.
- Y. Li, G. Xu, C. Cui and Y. Li, Adv. Energy Mater., 2018, 8, 1701791 CrossRef.
- S.-Y. Chang, P. Cheng, G. Li and Y. Yang, Joule, 2018, 2, 1039–1054 CrossRef CAS.
- P. Cheng and Y. Yang, Acc. Chem. Res., 2020, 53, 1218–1228 CrossRef CAS PubMed.
- G. Zhang, J. Zhao, P. C. Y. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Chem. Rev., 2018, 118, 3447–3507 CrossRef CAS PubMed.
- Y. Wang, J. Lee, X. Hou, C. Labanti, J. Yan, E. Mazzolini, A. Parhar, J. Nelson, J. S. Kim and Z. Li, Adv. Energy Mater., 2021, 11, 2003002 CrossRef CAS.
- M. Wright, R. Lin, M. J. Y. Tayebjee and G. Conibeer, Sol. RRL, 2017, 1, 1700035 CrossRef.
- M. D. M. Faure and B. H. Lessard, J. Mater. Chem. C, 2021, 9, 14–40 RSC.
- W. Cao and J. Xue, Energy Environ. Sci., 2014, 7, 2123 RSC.
- L. Zhan, S. Li, X. Xia, Y. Li, X. Lu, L. Zuo, M. Shi and H. Chen, Adv. Mater., 2021, 33, 2007231 CrossRef CAS PubMed.
- Y. Lin, Y. Firdaus, F. H. Isikgor, M. I. Nugraha, E. Yengel, G. T. Harrison, R. Hallani, A. El-Labban, H. Faber, C. Ma, X. Zheng, A. Subbiah, C. T. Howells, O. M. Bakr, I. McCulloch, S. D. Wolf, L. Tsetseris and T. D. Anthopoulos, ACS Energy Lett., 2020, 5, 2935–2944 CrossRef CAS.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma, C. An, C. He, Z. Wei, F. Gao and J. Hou, Adv. Mater., 2020, 32, 1908205 CrossRef CAS PubMed.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- G. Yu and A. J. Heeger, J. Appl. Phys., 1995, 78, 4510–4515 CrossRef CAS.
- A. Wadsworth, Z. Hamid, J. Kosco, N. Gasparini and I. McCulloch, Adv. Mater., 2020, 32, 2001763 CrossRef CAS PubMed.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174–179 CrossRef CAS.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- J. Huang, H. Tang, C. Yan and G. Li, Cell Rep. Phys. Sci., 2021, 2, 100292 CrossRef CAS.
- R. S. Gurney, D. G. Lidzey and T. Wang, Rep. Prog. Phys., 2019, 82, 036601 CrossRef CAS PubMed.
- P. Cheng, G. Li, X. Zhan and Y. Yang, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS.
- J. Hou, O. Inganas, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed.
- Z. Zhang, J. Yuan, Q. Wei and Y. Zou, Front. Chem., 2018, 6, 414 CrossRef PubMed.
- A. Armin, W. Li, O. J. Sandberg, Z. Xiao, L. Ding, J. Nelson, D. Neher, K. Vandewal, S. Shoaee, T. Wang, H. Ade, T. Heumüller, C. Brabec and P. Meredith, Adv. Energy Mater., 2021, 11, 20003570 Search PubMed.
- A. Karki, A. J. Gillett, R. H. Friend and T. Q. Nguyen, Adv. Energy Mater., 2020, 11, 2003441 CrossRef.
- J. Zhao, C. Yao, M. U. Ali, J. Miao and H. Meng, Mater. Chem. Front., 2020, 4, 3487–3504 RSC.
- J. Zhang, H. S. Tan, X. Guo, A. Facchetti and H. Yan, Nat. Energy, 2018, 3, 720–731 CrossRef CAS.
- H. Sun, F. Chen and Z.-K. Chen, Mater. Today, 2019, 24, 94–118 CrossRef CAS.
- Y. Lin and X. Zhan, Mater. Horiz., 2014, 1, 470–488 RSC.
- A. Wadsworth, M. Moser, A. Marks, M. S. Little, N. Gasparini, C. J. Brabec, D. Baran and I. McCulloch, Chem. Soc. Rev., 2019, 48, 1596–1625 RSC.
- S. Li, C.-Z. Li, M. Shi and H. Chen, ACS Energy Lett., 2020, 5, 1554–1567 CrossRef CAS.
- R. Yu, H. Yao, Y. Cui, L. Hong, C. He and J. Hou, Adv. Mater., 2019, 31, 1902302 CrossRef PubMed.
- J. Gao, J. Wang, Q. An, X. Ma, Z. Hu, C. Xu, X. Zhang and F. Zhang, Sci. China: Chem., 2019, 63, 83–91 CrossRef.
- J. Song, C. Li, L. Zhu, J. Guo, J. Xu, X. Zhang, K. Weng, K. Zhang, J. Min, X. Hao, Y. Zhang, F. Liu and Y. Sun, Adv. Mater., 2019, 31, 1905645 CrossRef CAS PubMed.
- Q. An, J. Wang, W. Gao, X. Ma, Z. Hu, J. Gao, C. Xu, M. Hao, X. Zhang, C. Yang and F. Zhang, Sci. Bull., 2020, 65, 538–545 CrossRef CAS.
- L. Chang, M. Sheng, L. Duan and A. Uddin, Org. Electron., 2021, 90, 106063 CrossRef CAS.
- M. C. Scharber and N. S. Sariciftci, Prog. Polym. Sci., 2013, 38, 1929–1940 CrossRef CAS PubMed.
- Y. Cui, C. Yang, H. Yao, J. Zhu, Y. Wang, G. Jia, F. Gao and J. Hou, Adv. Mater., 2017, 29, 1703080 CrossRef PubMed.
- Z. Xiao, X. Jia, D. Li, S. Wang, X. Geng, F. Liu, J. Chen, S. Yang, T. P. Russell and L. Ding, Sci. Bull., 2017, 62, 1494–1496 CrossRef CAS.
- Y. Xie, R. Xia, T. Li, L. Ye, X. Zhan, H. L. Yip and Y. Sun, Small Methods, 2019, 3, 1900424 CrossRef CAS.
- S. Dai and X. Zhan, Adv. Energy Mater., 2018, 8, 1800002 CrossRef.
- W. Wang, C. Yan, T. K. Lau, J. Wang, K. Liu, Y. Fan, X. Lu and X. Zhan, Adv. Mater., 2017, 29, 1701308 CrossRef PubMed.
- T. Li, S. Dai, Z. Ke, L. Yang, J. Wang, C. Yan, W. Ma and X. Zhan, Adv. Mater., 2018, 30, 1705969 CrossRef PubMed.
- G. Han and Y. Yi, Adv. Theory Simul., 2019, 2, 1900067 CrossRef.
- T. Fukuhara, Y. Tamai and H. Ohkita, Sustainable Energy Fuels, 2020, 4, 4321–4351 RSC.
- H. Hoppe and N. S. Sariciftci, J. Mater. Res., 2011, 19, 1924–1945 CrossRef.
- O. Almora, D. Baran, G. C. Bazan, C. Berger, C. I. Cabrera, K. R. Catchpole, S. Erten-Ela, F. Guo, J. Hauch, A. W. Y. Ho-Baillie, T. J. Jacobsson, R. A. J. Janssen, T. Kirchartz, N. Kopidakis, Y. Li, M. A. Loi, R. R. Lunt, X. Mathew, M. D. McGehee, J. Min, D. B. Mitzi, M. K. Nazeeruddin, J. Nelson, A. F. Nogueira, U. W. Paetzold, N. G. Park, B. P. Rand, U. Rau, H. J. Snaith, E. Unger, L. Vaillant-Roca, H. L. Yip and C. J. Brabec, Adv. Energy Mater., 2020, 11, 2002774 CrossRef.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K.-Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- N. Zink-Lorre, E. Font-Sanchis, A. Sastre-Santos and F. Fernandez-Lazaro, Chem. Commun., 2020, 56, 3824–3838 RSC.
- V. Sharma, J. D. B. Koenig and G. C. Welch, J. Mater. Chem. A, 2021, 9, 6775–6789 RSC.
- J. Wang and X. Zhan, Trends Chem., 2019, 1, 869–881 CrossRef CAS.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed.
- D. Zou, F. Yang, Q. Zhuang, M. Zhu, Y. Chen, G. You, Z. Lin, H. Zhen and Q. Ling, ChemSusChem, 2019, 12, 1155–1161 CrossRef CAS PubMed.
- K. Jiang, F. Wu, H. Yu, Y. Yao, G. Zhang, L. Zhu and H. Yan, J. Mater. Chem. A, 2018, 6, 16868–16873 RSC.
- D. Wang, T. Ye and Y. Zhang, J. Mater. Chem. A, 2020, 8, 20819–20848 RSC.
- A. Sharenko, C. M. Proctor, T. S. van der Poll, Z. B. Henson, T. Q. Nguyen and G. C. Bazan, Adv. Mater., 2013, 25, 4403–4406 CrossRef CAS PubMed.
- Y. Lin and X. Zhan, Adv. Energy Mater., 2015, 5, 1501063 CrossRef.
- R. Singh, J. Lee, M. Kim, P. E. Keivanidis and K. Cho, J. Mater. Chem. A, 2017, 5, 210–220 RSC.
- Y. Zhong, M. T. Trinh, R. Chen, G. E. Purdum, P. P. Khlyabich, M. Sezen, S. Oh, H. Zhu, B. Fowler, B. Zhang, W. Wang, C. Y. Nam, M. Y. Sfeir, C. T. Black, M. L. Steigerwald, Y. L. Loo, F. Ng, X. Y. Zhu and C. Nuckolls, Nat. Commun., 2015, 6, 8242 CrossRef CAS PubMed.
- Y. Zang, C. Z. Li, C. C. Chueh, S. T. Williams, W. Jiang, Z. H. Wang, J. S. Yu and A. K.-Y. Jen, Adv. Mater., 2014, 26, 5708–5714 CrossRef CAS PubMed.
- D. Sun, D. Meng, Y. Cai, B. Fan, Y. Li, W. Jiang, L. Huo, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2015, 137, 11156–11162 CrossRef CAS PubMed.
- D. Meng, D. Sun, C. Zhong, T. Liu, B. Fan, L. Huo, Y. Li, W. Jiang, H. Choi, T. Kim, J. Y. Kim, Y. Sun, Z. Wang and A. J. Heeger, J. Am. Chem. Soc., 2016, 138, 375–380 CrossRef CAS PubMed.
- J. Zhao, Y. Li, H. Lin, Y. Liu, K. Jiang, C. Mu, T. Ma, J. Y. Lin Lai, H. Hu, D. Yu and H. Yan, Energy Environ. Sci., 2015, 8, 520–525 RSC.
- J. Liu, S. Chen, D. Qian, B. Gautam, G. Yang, J. Zhao, J. Bergqvist, F. Zhang, W. Ma, H. Ade, O. Inganäs, K. Gundogdu, F. Gao and H. Yan, Nat. Energy, 2016, 1, 16089 CrossRef CAS.
- Q. Wu, D. Zhao, A. M. Schneider, W. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7248–7251 CrossRef CAS PubMed.
- Y. Duan, X. Xu, H. Yan, W. Wu, Z. Li and Q. Peng, Adv. Mater., 2017, 29, 1605115 CrossRef PubMed.
- J. Zhang, Y. Li, J. Huang, H. Hu, G. Zhang, T. Ma, P. C. Y. Chow, H. Ade, D. Pan and H. Yan, J. Am. Chem. Soc., 2017, 139, 16092–16095 CrossRef CAS PubMed.
- H. Hu, Y. Li, J. Zhang, Z. Peng, L. k. Ma, J. Xin, J. Huang, T. Ma, K. Jiang, G. Zhang, W. Ma, H. Ade and H. Yan, Adv. Energy Mater., 2018, 8, 1800234 CrossRef.
- K. Kawashima, Y. Tamai, H. Ohkita, I. Osaka and K. Takimiya, Nat. Commun., 2015, 6, 10085 CrossRef CAS PubMed.
- Y. Cui, H. Yao, L. Hong, T. Zhang, Y. Xu, K. Xian, B. Gao, J. Qin, J. Zhang, Z. Wei and J. Hou, Adv. Mater., 2019, 31, 1808356 CrossRef PubMed.
- H. Yao, Y. Cui, D. Qian, C. S. Ponseca Jr, A. Honarfar, Y. Xu, J. Xin, Z. Chen, L. Hong, B. Gao, R. Yu, Y. Zu, W. Ma, P. Chabera, T. Pullerits, A. Yartsev, F. Gao and J. Hou, J. Am. Chem. Soc., 2019, 141, 7743–7750 CrossRef CAS PubMed.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- S.-S. Wan, X. Xu, Z. Jiang, J. Yuan, A. Mahmood, G.-Z. Yuan, K.-K. Liu, W. Ma, Q. Peng and J.-L. Wang, J. Mater. Chem. A, 2020, 8, 4856–4867 RSC.
- Q. Fan, W. Su, M. Zhang, J. Wu, Y. Jiang, X. Guo, F. Liu, T. P. Russell, M. Zhang and Y. Li, Sol. RRL, 2019, 3, 1900169 CrossRef CAS.
- L. Hong, H. Yao, Z. Wu, Y. Cui, T. Zhang, Y. Xu, R. Yu, Q. Liao, B. Gao, K. Xian, H. Y. Woo, Z. Ge and J. Hou, Adv. Mater., 2019, 31, 1903441 CrossRef PubMed.
- Y. Cui, H. Yao, L. Hong, T. Zhang, Y. Tang, B. Lin, K. Xian, B. Gao, C. An, P. Bi, W. Ma and J. Hou, Natl. Sci. Rev., 2020, 7, 1239–1246 CrossRef CAS.
- S. Liu, J. Yuan, W. Deng, M. Luo, Y. Xie, Q. Liang, Y. Zou, Z. He, H. Wu and Y. Cao, Nat. Photonics, 2020, 14, 300–305 CrossRef CAS.
- G. Forti, A. Nitti, P. Osw, G. Bianchi, R. Po and D. Pasini, Int. J. Mol. Sci., 2020, 21, 8085 CrossRef CAS PubMed.
- Y. Lin and X. Zhan, Acc. Chem. Res., 2016, 49, 175–183 CrossRef CAS PubMed.
- D. Venkateshvaran, M. Nikolka, A. Sadhanala, V. Lemaur, M. Zelazny, M. Kepa, M. Hurhangee, A. J. Kronemeijer, V. Pecunia, I. Nasrallah, I. Romanov, K. Broch, I. McCulloch, D. Emin, Y. Olivier, J. Cornil, D. Beljonne and H. Sirringhaus, Nature, 2014, 515, 384–388 CrossRef CAS PubMed.
- H. Cha, J. Wu, A. Wadsworth, J. Nagitta, S. Limbu, S. Pont, Z. Li, J. Searle, M. F. Wyatt, D. Baran, J. S. Kim, I. McCulloch and J. R. Durrant, Adv. Mater., 2017, 29, 1701156 CrossRef PubMed.
- N. Gasparini, M. Salvador, S. Strohm, T. Heumueller, I. Levchuk, A. Wadsworth, J. H. Bannock, J. C. de Mello, H.-J. Egelhaaf, D. Baran, I. McCulloch and C. J. Brabec, Adv. Energy Mater., 2017, 7, 17007700 Search PubMed.
- Y. Zhu, A. Gadisa, Z. Peng, M. Ghasemi, L. Ye, Z. Xu, S. Zhao and H. Ade, Adv. Energy Mater., 2019, 9, 1900376 CrossRef.
- J. Wang and X. Zhan, Acc. Chem. Res., 2021, 54, 132–143 CrossRef CAS PubMed.
- S. Dai, T. Li, W. Wang, Y. Xiao, T. K. Lau, Z. Li, K. Liu, X. Lu and X. Zhan, Adv. Mater., 2018, 30, 1706571 CrossRef PubMed.
- B. Jia, J. Wang, Y. Wu, M. Zhang, Y. Jiang, Z. Tang, T. P. Russell and X. Zhan, J. Am. Chem. Soc., 2019, 141, 19023–19031 CrossRef CAS PubMed.
- C. Cui, Front. Chem., 2018, 6, 404 CrossRef CAS PubMed.
- D. Li, X. Zhang, D. Liu and T. Wang, J. Mater. Chem. A, 2020, 8, 15607–15619 RSC.
- Y. Lin, F. Zhao, Q. He, L. Huo, Y. Wu, T. C. Parker, W. Ma, Y. Sun, C. Wang, D. Zhu, A. J. Heeger, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2016, 138, 4955–4961 CrossRef CAS PubMed.
- Y. Lin, Q. He, F. Zhao, L. Huo, J. Mai, X. Lu, C. J. Su, T. Li, J. Wang, J. Zhu, Y. Sun, C. Wang and X. Zhan, J. Am. Chem. Soc., 2016, 138, 2973–2976 CrossRef CAS PubMed.
- S. Dai, F. Zhao, Q. Zhang, T. K. Lau, T. Li, K. Liu, Q. Ling, C. Wang, X. Lu, W. You and X. Zhan, J. Am. Chem. Soc., 2017, 139, 1336–1343 CrossRef CAS PubMed.
- H. Zhang, H. Yao, J. Hou, J. Zhu, J. Zhang, W. Li, R. Yu, B. Gao, S. Zhang and J. Hou, Adv. Mater., 2018, 30, 1800613 CrossRef PubMed.
- T. J. Aldrich, M. Matta, W. Zhu, S. M. Swick, C. L. Stern, G. C. Schatz, A. Facchetti, F. S. Melkonyan and T. J. Marks, J. Am. Chem. Soc., 2019, 141, 3274–3287 CrossRef CAS PubMed.
- X. Song, N. Gasparini, L. Ye, H. Yao, J. Hou, H. Ade and D. Baran, ACS Energy Lett., 2018, 3, 669–676 CrossRef CAS.
- J. Yuan, Y. Zhang, L. Zhou, C. Zhang, T. K. Lau, G. Zhang, X. Lu, H. L. Yip, S. K. So, S. Beaupre, M. Mainville, P. A. Johnson, M. Leclerc, H. Chen, H. Peng, Y. Li and Y. Zou, Adv. Mater., 2019, 31, 1807577 CrossRef PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- J. Yuan, H. Zhang, R. Zhang, Y. Wang, J. Hou, M. Leclerc, X. Zhan, F. Huang, F. Gao, Y. Zou and Y. Li, Chem, 2020, 6, 2147–2161 CAS.
- G. Zhang, X. K. Chen, J. Xiao, P. C. Y. Chow, M. Ren, G. Kupgan, X. Jiao, C. C. S. Chan, X. Du, R. Xia, Z. Chen, J. Yuan, Y. Zhang, S. Zhang, Y. Liu, Y. Zou, H. Yan, K. S. Wong, V. Coropceanu, N. Li, C. J. Brabec, J. L. Bredas, H. L. Yip and Y. Cao, Nat. Commun., 2020, 11, 3943 CrossRef CAS PubMed.
- W. Zhu, A. P. Spencer, S. Mukherjee, J. M. Alzola, V. K. Sangwan, S. H. Amsterdam, S. M. Swick, L. O. Jones, M. C. Heiber, A. A. Herzing, G. Li, C. L. Stern, D. M. DeLongchamp, K. L. Kohlstedt, M. C. Hersam, G. C. Schatz, M. R. Wasielewski, L. X. Chen, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2020, 142, 14532–14547 CrossRef CAS PubMed.
- Z. Zhou, W. Liu, G. Zhou, M. Zhang, D. Qian, J. Zhang, S. Chen, S. Xu, C. Yang, F. Gao, H. Zhu, F. Liu and X. Zhu, Adv. Mater., 2020, 32, 1906324 CrossRef CAS PubMed.
- W. Gao, H. Fu, Y. Li, F. Lin, R. Sun, Z. Wu, X. Wu, C. Zhong, J. Min, J. Luo, H. Y. Woo, Z. Zhu and A. K.-Y. Jen, Adv. Energy Mater., 2021, 11, 2003177 CrossRef CAS.
- Z. Jia, S. Qin, L. Meng, Q. Ma, I. Angunawela, J. Zhang, X. Li, Y. He, W. Lai, N. Li, H. Ade, C. J. Brabec and Y. Li, Nat. Commun., 2021, 12, 178 CrossRef CAS PubMed.
- S. Dong, T. Jia, K. Zhang, J. Jing and F. Huang, Joule, 2020, 4, 2004–2016 CrossRef CAS.
- J. Wu, J. Lee, Y.-C. Chin, H. Yao, H. Cha, J. Luke, J. Hou, J.-S. Kim and J. R. Durrant, Energy Environ. Sci., 2020, 13, 2422–2430 RSC.
- C. Zhang, J. Yuan, K. L. Chiu, H. Yin, W. Liu, G. Zheng, J. K. W. Ho, S. Huang, G. Yu, F. Gao, Y. Zou and S. K. So, J. Mater. Chem. A, 2020, 8, 8566–8574 RSC.
- Y. Cui, H. Yao, J. Zhang, T. Zhang, Y. Wang, L. Hong, K. Xian, B. Xu, S. Zhang, J. Peng, Z. Wei, F. Gao and J. Hou, Nat. Commun., 2019, 10, 2515 CrossRef PubMed.
- Z. Luo, R. Ma, Z. Chen, Y. Xiao, G. Zhang, T. Liu, R. Sun, Q. Zhan, Y. Zou, C. Zhong, Y. Chen, H. Sun, G. Chai, K. Chen, X. Guo, J. Min, X. Lu, C. Yang and H. Yan, Adv. Energy Mater., 2020, 10, 2002649 CrossRef CAS.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- Y. He and Y. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- R. Kerremans, C. Kaiser, W. Li, N. Zarrabi, P. Meredith and A. Armin, Adv. Opt. Mater., 2020, 8, 2000319 CrossRef CAS.
- Y. Firdaus, V. M. Le Corre, S. Karuthedath, W. Liu, A. Markina, W. Huang, S. Chattopadhyay, M. M. Nahid, M. I. Nugraha, Y. Lin, A. Seitkhan, A. Basu, W. Zhang, I. McCulloch, H. Ade, J. Labram, F. Laquai, D. Andrienko, L. J. A. Koster and T. D. Anthopoulos, Nat. Commun., 2020, 11, 5220 CrossRef CAS PubMed.
- H. Yao, Y. Cui, R. Yu, B. Gao, H. Zhang and J. Hou, Angew. Chem., Int. Ed., 2017, 56, 3045–3049 CrossRef CAS PubMed.
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- W. Zhao, D. Qian, S. Zhang, S. Li, O. Inganas, F. Gao and J. Hou, Adv. Mater., 2016, 28, 4734–4739 CrossRef CAS PubMed.
- W. Li, K. H. Hendriks, W. S. Roelofs, Y. Kim, M. M. Wienk and R. A. Janssen, Adv. Mater., 2013, 25, 3182–3186 CrossRef CAS PubMed.
- Y. Jin, Z. Chen, M. Xiao, J. Peng, B. Fan, L. Ying, G. Zhang, X.-F. Jiang, Q. Yin, Z. Liang, F. Huang and Y. Cao, Adv. Energy Mater., 2017, 7, 1700944 CrossRef.
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736–6767 CrossRef CAS PubMed.
- B. Siegmund, M. T. Sajjad, J. Widmer, D. Ray, C. Koerner, M. Riede, K. Leo, I. D. Samuel and K. Vandewal, Adv. Mater., 2017, 29, 1604424 CrossRef PubMed.
- Y. Lin, F. Zhao, S. K. K. Prasad, J. D. Chen, W. Cai, Q. Zhang, K. Chen, Y. Wu, W. Ma, F. Gao, J. X. Tang, C. Wang, W. You, J. M. Hodgkiss and X. Zhan, Adv. Mater., 2018, 30, 1706363 CrossRef PubMed.
- S. Chandrabose, K. Chen, A. J. Barker, J. J. Sutton, S. K. K. Prasad, J. Zhu, J. Zhou, K. C. Gordon, Z. Xie, X. Zhan and J. M. Hodgkiss, J. Am. Chem. Soc., 2019, 141, 6922–6929 CrossRef CAS PubMed.
- L. Ma, S. Zhang, H. Yao, Y. Xu, J. Wang, Y. Zu and J. Hou, ACS Appl. Mater. Interfaces, 2020, 12, 18777–18784 CrossRef CAS PubMed.
- M. Azzouzi, T. Kirchartz and J. Nelson, Trends Chem., 2019, 1, 49–62 CrossRef CAS.
- V. Coropceanu, X.-K. Chen, T. Wang, Z. Zheng and J.-L. Brédas, Nat. Rev. Mater., 2019, 4, 689–707 CrossRef.
- X. Liu, B. P. Rand and S. R. Forrest, Trends Chem., 2019, 1, 815–829 CrossRef CAS.
- S. M. Menke, N. A. Ran, G. C. Bazan and R. H. Friend, Joule, 2018, 2, 25–35 CrossRef CAS.
- K. Vandewal, K. Tvingstedt, A. Gadisa, O. Inganas and J. V. Manca, Nat. Mater., 2009, 8, 904–909 CrossRef CAS PubMed.
- J. Benduhn, K. Tvingstedt, F. Piersimoni, S. Ullbrich, Y. Fan, M. Tropiano, K. A. McGarry, O. Zeika, M. K. Riede, C. J. Douglas, S. Barlow, S. R. Marder, D. Neher, D. Spoltore and K. Vandewal, Nat. Energy, 2017, 2, 17053 CrossRef CAS.
- D. C. Coffey, B. W. Larson, A. W. Hains, J. B. Whitaker, N. Kopidakis, O. V. Boltalina, S. H. Strauss and G. Rumbles, J. Phys. Chem. C, 2012, 116, 8916–8923 CrossRef CAS.
- X. Liu, Y. Li, K. Ding and S. Forrest, Phys. Rev. Appl., 2019, 11, 024060 CrossRef CAS.
- R. A. Marcus, J. Chem. Phys., 1965, 43, 679–701 CrossRef CAS.
- C. P. Hsu, Phys. Chem. Chem. Phys., 2020, 22, 21630–21641 RSC.
- T. Liu and A. Troisi, J. Phys. Chem. C, 2011, 115, 2406–2415 CrossRef CAS.
- L. Zhu, Y. Yi and Z. Wei, J. Phys. Chem. C, 2018, 122, 22309–22316 CrossRef CAS.
- A. Classen, C. L. Chochos, L. Lüer, V. G. Gregoriou, J. Wortmann, A. Osvet, K. Forberich, I. McCulloch, T. Heumüller and C. J. Brabec, Nat. Energy, 2020, 5, 711–719 CrossRef CAS.
- C. Sun, S. Qin, R. Wang, S. Chen, F. Pan, B. Qiu, Z. Shang, L. Meng, C. Zhang, M. Xiao, C. Yang and Y. Li, J. Am. Chem. Soc., 2020, 142, 1465–1474 CrossRef CAS PubMed.
- F. D. Eisner, M. Azzouzi, Z. Fei, X. Hou, T. D. Anthopoulos, T. J. S. Dennis, M. Heeney and J. Nelson, J. Am. Chem. Soc., 2019, 141, 6362–6374 CrossRef CAS PubMed.
- D. Bartesaghi, C. Perez Idel, J. Kniepert, S. Roland, M. Turbiez, D. Neher and L. J. Koster, Nat. Commun., 2015, 6, 7083 CrossRef CAS PubMed.
- X. Du, T. Heumueller, W. Gruber, O. Almora, A. Classen, J. Qu, F. He, T. Unruh, N. Li and C. J. Brabec, Adv. Mater., 2020, 32, 1908305 CrossRef CAS PubMed.
- F. Gajdos, H. Oberhofer, M. Dupuis and J. Blumberger, J. Phys. Chem. Lett., 2014, 5, 2765–2766 CrossRef CAS PubMed.
- F. Hamada and A. Saeki, ChemSusChem, 2021, 14, 1–8 CrossRef PubMed.
- C. Liu, K. Huang, W.-T. Park, M. Li, T. Yang, X. Liu, L. Liang, T. Minari and Y.-Y. Noh, Mater. Horiz., 2017, 4, 608–618 RSC.
- G. Kupgan, X.-K. Chen and J.-L. Brédas, ACS Mater. Lett., 2019, 1, 350–353 CrossRef CAS.
- S. Himmelberger and A. Salleo, MRS Commun., 2015, 5, 383–395 CrossRef CAS.
- M. Kim, S. U. Ryu, S. A. Park, K. Choi, T. Kim, D. Chung and T. Park, Adv. Funct. Mater., 2020, 30, 1904545 CrossRef CAS.
- J. Wang, J. Zhang, Y. Xiao, T. Xiao, R. Zhu, C. Yan, Y. Fu, G. Lu, X. Lu, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2018, 140, 9140–9147 CrossRef CAS PubMed.
- S. Dai, J. Zhou, S. Chandrabose, Y. Shi, G. Han, K. Chen, J. Xin, K. Liu, Z. Chen, Z. Xie, W. Ma, Y. Yi, L. Jiang, J. M. Hodgkiss and X. Zhan, Adv. Mater., 2020, 32, 2000645 CrossRef CAS PubMed.
- H. Lai, H. Chen, J. Zhou, J. Qu, P. Chao, T. Liu, X. Chang, N. Zheng, Z. Xie and F. He, iScience, 2019, 17, 302–314 CrossRef CAS PubMed.
- G. Han, T. Hu and Y. Yi, Adv. Mater., 2020, 32, 2000975 CrossRef CAS PubMed.
- X. Du, Y. Yuan, L. Zhou, H. Lin, C. Zheng, J. Luo, Z. Chen, S. Tao and L. S. Liao, Adv. Funct. Mater., 2020, 30, 1909837 CrossRef CAS.
- B. T. Luppi, D. Majak, M. Gupta, E. Rivard and K. Shankar, J. Mater. Chem. A, 2019, 7, 2445–2463 RSC.
- M. K. Etherington, J. Wang, P. C. Y. Chow and N. C. Greenham, Appl. Phys. Lett., 2014, 104, 063304 CrossRef.
- D. Veldman, S. C. J. Meskers and R. A. J. Janssen, Adv. Funct. Mater., 2009, 19, 1939–1948 CrossRef CAS.
- C. W. Schlenker, K. S. Chen, H. L. Yip, C. Z. Li, L. R. Bradshaw, S. T. Ochsenbein, F. Ding, X. S. Li, D. R. Gamelin, A. K.-Y. Jen and D. S. Ginger, J. Am. Chem. Soc., 2012, 134, 19661–19668 CrossRef CAS PubMed.
- A. Rao, P. C. Chow, S. Gelinas, C. W. Schlenker, C. Z. Li, H. L. Yip, A. K.-Y. Jen, D. S. Ginger and R. H. Friend, Nature, 2013, 500, 435–439 CrossRef CAS PubMed.
- X. K. Chen, T. Wang and J. L. Brédas, Adv. Energy Mater., 2017, 7, 1602713 CrossRef.
- Y. Jin, Y. Zhang, Y. Liu, J. Xue, W. Li, J. Qiao and F. Zhang, Adv. Mater., 2019, 31, 1900690 CrossRef PubMed.
- S. Kappaun, C. Slugovc and E. J. List, Int. J. Mol. Sci., 2008, 9, 1527–1547 CrossRef CAS PubMed.
- S. Goswami, J. L. Hernandez, M. K. Gish, J. Wang, B. Kim, A. P. Laudari, S. Guha, J. M. Papanikolas, J. R. Reynolds and K. S. Schanze, Chem. Mater., 2017, 29, 8449–8461 CrossRef CAS.
- L. Yang, W. Gu, L. Lv, Y. Chen, Y. Yang, P. Ye, J. Wu, L. Hong, A. Peng and H. Huang, Angew. Chem., Int. Ed., 2018, 57, 1096–1102 CrossRef CAS PubMed.
- S. Liu, H. Zhang, Y. Li, J. Liu, L. Du, M. Chen, R. T. K. Kwok, J. W. Y. Lam, D. L. Phillips and B. Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 15189–15193 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- M. J. Leitl, V. A. Krylova, P. I. Djurovich, M. E. Thompson and H. Yersin, J. Am. Chem. Soc., 2014, 136, 16032–16038 CrossRef CAS PubMed.
- L. Qin, X. Liu, X. Zhang, J. Yu, L. Yang, F. Zhao, M. Huang, K. Wang, X. Wu, Y. Li, H. Chen, K. Wang, J. Xia, X. Lu, F. Gao, Y. Yi and H. Huang, Angew. Chem., Int. Ed., 2020, 59, 15043–15049 CrossRef CAS PubMed.
- D. Kim, J. Phys. Chem. C, 2015, 119, 12690–12697 CrossRef CAS.
- J. Xue, Q. Liang, R. Wang, J. Hou, W. Li, Q. Peng, Z. Shuai and J. Qiao, Adv. Mater., 2019, 31, 1808242 CrossRef PubMed.
- B. Watts, W. J. Belcher, L. Thomsen, H. Ade and P. C. Dastoor, Macromolecules, 2009, 42, 8392–8397 CrossRef CAS.
- N. D. Treat, M. A. Brady, G. Smith, M. F. Toney, E. J. Kramer, C. J. Hawker and M. L. Chabinyc, Adv. Energy Mater., 2011, 1, 82–89 CrossRef CAS.
- L. Ye, B. A. Collins, X. Jiao, J. Zhao, H. Yan and H. Ade, Adv. Energy Mater., 2018, 8, 1703058 CrossRef.
- L. Ye, H. Hu, M. Ghasemi, T. Wang, B. A. Collins, J. H. Kim, K. Jiang, J. H. Carpenter, H. Li, Z. Li, T. McAfee, J. Zhao, X. Chen, J. L. Y. Lai, T. Ma, J. L. Bredas, H. Yan and H. Ade, Nat. Mater., 2018, 17, 253–260 CrossRef CAS PubMed.
- F. Zhao, C. Wang and X. Zhan, Adv. Energy Mater., 2018, 8, 1703147 CrossRef.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, 1953 Search PubMed.
- P. Knychała, K. Timachova, M. Banaszak and N. P. Balsara, Macromolecules, 2017, 50, 3051–3065 CrossRef.
- T. Nishi and T. Wang, Macromolecules, 1975, 8, 909–915 CrossRef CAS.
- C. Clarke, A. Eisenberg, J. La Scala, M. Rafailovich, J. Sokolov, Z. Li, S. Qu, D. Nguyen, S. Schwarz and Y. Strzhemechny, Macromolecules, 1997, 30, 4184–4188 CrossRef CAS.
- J. E. Mark, Physical Properties of Polymers Handbook, Springer, 2007 Search PubMed.
- T. Lindvig, M. L. Michelsen and G. M. Kontogeorgis, Fluid Phase Equilib., 2002, 203, 247–260 CrossRef CAS.
- M. Gao, Z. Liang, Y. Geng and L. Ye, Chem. Commun., 2020, 56, 12463–12478 RSC.
- L. Ye, S. Zhang, W. Ma, B. Fan, X. Guo, Y. Huang, H. Ade and J. Hou, Adv. Mater., 2012, 24, 6335–6341 CrossRef CAS PubMed.
- Z. Li, K. Jiang, G. Yang, J. Y. Lai, T. Ma, J. Zhao, W. Ma and H. Yan, Nat. Commun., 2016, 7, 13094 CrossRef CAS PubMed.
- F. Zhao, S. Dai, Y. Wu, Q. Zhang, J. Wang, L. Jiang, Q. Ling, Z. Wei, W. Ma, W. You, C. Wang and X. Zhan, Adv. Mater., 2017, 29, 1700144 CrossRef PubMed.
- T. Ma, K. Jiang, S. Chen, H. Hu, H. Lin, Z. Li, J. Zhao, Y. Liu, Y.-M. Chang, C.-C. Hsiao and H. Yan, Adv. Energy Mater., 2015, 5, 1501282 CrossRef.
- H. Hu, K. Jiang, G. Yang, J. Liu, Z. Li, H. Lin, Y. Liu, J. Zhao, J. Zhang, F. Huang, Y. Qu, W. Ma and H. Yan, J. Am. Chem. Soc., 2015, 137, 14149–14157 CrossRef CAS PubMed.
- L. Ye, W. Zhao, S. Li, S. Mukherjee, J. H. Carpenter, O. Awartani, X. Jiao, J. Hou and H. Ade, Adv. Energy Mater., 2017, 7, 1602000 CrossRef.
- S. Li, L. Ye, W. Zhao, H. Yan, B. Yang, D. Liu, W. Li, H. Ade and J. Hou, J. Am. Chem. Soc., 2018, 140, 7159–7167 CrossRef CAS PubMed.
- J. Wu, G. Li, J. Fang, X. Guo, L. Zhu, B. Guo, Y. Wang, G. Zhang, L. Arunagiri, F. Liu, H. Yan, M. Zhang and Y. Li, Nat. Commun., 2020, 11, 4612 CrossRef CAS PubMed.
- C. Yang, S. Zhang, J. Ren, M. Gao, P. Bi, L. Ye and J. Hou, Energy Environ. Sci., 2020, 13, 2864–2869 RSC.
- J. H. Carpenter, A. Hunt and H. Ade, J. Electron Spectrosc. Relat. Phenom., 2015, 200, 2–14 CrossRef CAS.
- S. A. Gevorgyan, M. V. Madsen, B. Roth, M. Corazza, M. Hösel, R. R. Søndergaard, M. Jørgensen and F. C. Krebs, Adv. Energy Mater., 2016, 6, 1501208 CrossRef.
- T. M. Clarke, C. Lungenschmied, J. Peet, N. Drolet, K. Sunahara, A. Furube and A. J. Mozer, Adv. Energy Mater., 2013, 3, 1473–1483 CrossRef CAS.
- W. R. Mateker and M. D. McGehee, Adv. Mater., 2017, 29, 1603940 CrossRef PubMed.
- N. Li, J. D. Perea, T. Kassar, M. Richter, T. Heumueller, G. J. Matt, Y. Hou, N. S. Guldal, H. Chen, S. Chen, S. Langner, M. Berlinghof, T. Unruh and C. J. Brabec, Nat. Commun., 2017, 8, 14541 CrossRef CAS PubMed.
- X. Du, T. Heumueller, W. Gruber, A. Classen, T. Unruh, N. Li and C. J. Brabec, Joule, 2019, 3, 215–226 CrossRef CAS.
- S. Li, L. Ye, W. Zhao, X. Liu, J. Zhu, H. Ade and J. Hou, Adv. Mater., 2017, 29, 1704051 CrossRef PubMed.
- S. Li, L. Ye, W. Zhao, S. Zhang, H. Ade and J. Hou, Adv. Energy Mater., 2017, 7, 1700183 CrossRef.
- Q. Yue, W. Liu and X. Zhu, J. Am. Chem. Soc., 2020, 142, 11613–11628 CrossRef CAS PubMed.
- L. Ma, S. Zhang, J. Wang, Y. Xu and J. Hou, Chem. Commun., 2020, 56, 14337–14352 RSC.
- C. J. Brabec, A. Distler, X. Du, H. J. Egelhaaf, J. Hauch, T. Heumueller and N. Li, Adv. Energy Mater., 2020, 10, 2001864 CrossRef CAS.
- K. Kranthiraja and A. Saeki, Adv. Funct. Mater., 2021, 31, 2011168 CrossRef CAS.
- W. Sun, Y. Zheng, K. Yang, Q. Zhang, A. A. Shah, Z. Wu, Y. Sun, L. Feng, D. Chen and Z. Xiao, Sci. Adv., 2019, 5, eaay4275 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |