
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel aggregation-induced enhanced emission aromatic molecule: 2-aminophenylboronic acid dimer†
Xiaopei
Li
ab,
Dongdong
Wang
a,
Yongjie
Zhang
a,
Wenqi
Lu
a,
Songqiu
Yang
 a,
Guangjin
Hou
a,
Zhenchao
Zhao
a,
Haijuan
Qin
c,
Yahui
Zhang
a,
Minmin
Li
a and
Guangyan
Qing
*a
a,
Guangjin
Hou
a,
Zhenchao
Zhao
a,
Haijuan
Qin
c,
Yahui
Zhang
a,
Minmin
Li
a and
Guangyan
Qing
*a
aCAS Key Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, P. R. China. E-mail: qinggy@dicp.ac.cn
bInstrumental Analysis Center, Dalian Polytechnic University, Dalian 116034, P. R. China
cResearch Centre of Modern Analytical Technology, Tianjin University of Science and Technology, Tianjin 300457, P. R. China
First published on 18th August 2021
Abstract
Aggregation-induced enhanced emission (AIEE) molecules have significant applications in optoelectronics, biomedical probes and chemical sensors, and large amounts of AIEE molecules have been reported since the concept of AIEE was proposed. Most aromatic AIEE molecules have complex structures consisting of multiple aromatic rings and/or polycyclic skeletons. In this study, we find that 2-aminophenylboronic acid (2-APBA) with a simple structure is highly emissive in the solid state. Further studies reveal that 2-APBA exists in a dimeric form, and the 2-APBA dimer is a novel AIEE molecule. The underlying AIEE mechanism is that the 2-APBA dimeric units aggregate through intermolecular interactions to produce highly ordered molecular packing without the presence of π–π stacking interactions that would lead to aggregation-caused quenching. Furthermore, the 2-APBA dimer aggregates could reversibly transform into its non-fluorescent monomer form driven by new kinds of dynamic covalent B–N and B–O bonds, illustrating its good potential in molecular recognition, nanogating, chemo/bio-sensing and controlled drug release.
Introduction
Aggregation-induced emission or aggregation-induced enhanced emission (AIEE) refers to a photophysical phenomenon in which molecular aggregates exhibit stronger emission than single molecules,1–8 and is a major discovery. It makes us realize that the natural process of aggregation, which was previously believed to have detrimental effects on emission,9 could lead to an enhanced emission. Because of its clear scientific implications, AIEE has attracted considerable research attention. Therefore, a wealth of AIEE mechanistic insights and various AIEE molecules have been developed, bringing forth plentiful innovative applications.10–16 For example, Zhao et al. developed novel aggregation-induced delayed fluorescence luminogens, which enable the development of highly efficient nondoped organic light-emitting diodes with negligible efficiency roll-off.17 Cai et al. synthesized a novel tetra-aromatic butadiene structure-based AIEE molecule that could be employed for real-time imaging of cancer cells upon freezing.18Aromatic molecules have been the focus of AIEE research due to their high emission efficiencies and high flexibilities.1 To hamper the intermolecular π–π stacking interactions that would lead to aggregation-caused quenching, aromatic AIEE molecules have been designed with common features: bulky molecular size and highly twisted spatial configuration.11,19–21 For example, hexaphenylsilole (HPS),22 in which the silole core is decorated with a total of six phenyl rings, and tetraphenylethene (TPE),23 in which the central olefin stator is surrounded by four peripheral aromatic rotors, are prototypical AIEE molecules. In the past two decades, many kinds of HPS and TPE derivatives have been synthesized. Until now, HPS and TPE are still important core parts in the design of luminescent materials with diverse functionalities.24,25 Behind the booming development of AIEE, however, there was little report on whether aromatic molecules without bulky molecular size and highly twisted spatial configuration could be AIEE active. The answer to this question will largely enrich the AIEE theory and provide new design ideas for AIEE molecules.
Aminophenylboronic acids (APBAs) are versatile molecules. They selectively and reversibly bind to 1,2- or 1,3-diols through boronic acid groups, and are vital recognition receptors for biomolecules with a cis-diol structure26,27 such as saccharides and glycoproteins. Therefore, APBAs have been widely used as building blocks for many saccharide and glycosylated biomolecule-involved applications,28–30 such as sensing, separation, targeted drug delivery and biological imaging. APBAs are also one of the best building blocks for the construction of organized architectures with dynamic covalent functionality.31,32 Furthermore, APBAs are crucial intermediates used for the introduction of aniline units into products through the Suzuki–Miyaura cross-coupling reaction.33
Notably, in this study, we determined that 2-APBA emitted strong fluorescence with a high absolute quantum yield (ΦF) of 81.3% in the solid state. Detailed analysis revealed that 2-APBA existed in a dimeric form, and the 2-APBA dimer was a highly crystalline AIEE molecule. What's more, the dynamic covalent B–N and B–O bonds were first identified from the disassembly/assembly of the 2-APBA dimer, reflecting the quenching and enhancement of fluorescence of the 2-APBA dimer. Finally, the potential applications of the 2-APBA dimer in CO2 detection and nanogating were illustrated.
Results and discussion
Influence of 2-APBA on the fluorescence of HBQ
A chance discovery, in which 10-hydroxybenzo[h]quinoline (HBQ) functioned as a fluorescent probe, in our research on the interaction between 2-APBA and N-acetylneuraminic acid (Neu5Ac, the most common species of sialic acid) encouraged us to conduct this study. HBQ is a typical excited-state intramolecular proton transfer (ESIPT) molecule;34,35 it exists as an enol in the ground state and undergoes an ESIPT process, resulting in keto tautomer-derived emission at 597 nm, when excited (Fig. 1A(I)).36 Aronoff et al. reported that phenylboronic acid (PBA) could interrupt the ESIPT of HBQ by reacting with HBQ. The PBA–HBQ complex emits strong fluorescence at 504 nm (Fig. 1A(II) and B).37 Subsequently, because of the competitive binding of PBA with Neu5Ac, HBQ is released from the PBA–HBQ complex on adding Neu5Ac to the PBA–HBQ solution, resulting in a gradual decrease in the intensity of the emission peak at 504 nm (Fig. 1A(III) and C). | ||
Fig. 1 (A) Luminescence behavior of HBQ under the influence of PBA, 2-APBA and Neu5Ac. (B and D) Fluorescence spectra and intensity changes (at 504 nm, inset) of HBQ (5.0 × 10−5 mol L−1) on addition of various equivalents of PBA (B) or 2-APBA (D) to the water–DMSO (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solution of HBQ. (C and E) Fluorescence spectra and intensity changes (at 504 nm, inset) of the HBQ–PBA mixture (C) or HBQ-2-APBA mixture (E) on addition of various equivalents of Neu5Ac to the water–DMSO (4:1, v/v) solution of HBQ–PBA or HBQ-2-APBA, λex = 367 nm. The concentrations of HBQ, PBA and 2-APBA were 5.0 × 10−5, 3.5 × 10−4 and 3.5 × 10−4 mol L−1, respectively. :1, v/v) solution of HBQ. (C and E) Fluorescence spectra and intensity changes (at 504 nm, inset) of the HBQ–PBA mixture (C) or HBQ-2-APBA mixture (E) on addition of various equivalents of Neu5Ac to the water–DMSO (4:1, v/v) solution of HBQ–PBA or HBQ-2-APBA, λex = 367 nm. The concentrations of HBQ, PBA and 2-APBA were 5.0 × 10−5, 3.5 × 10−4 and 3.5 × 10−4 mol L−1, respectively. | ||
However, when PBA was replaced by 2-APBA, distinct phenomena were observed. The emission peak of HBQ at 597 nm remained almost unchanged, and only a low emission peak appeared at 504 nm even when excess 2-APBA was added to the HBQ solution (Fig. 1D). This indicated that most of 2-APBA did not react with HBQ (Fig. 1A(IV)). Notably, upon the addition of Neu5Ac to the 2-APBA–HBQ solution, the emission peak at 504 nm increased rapidly to reach a maximum and then decreased with an increase in the Neu5Ac-to-2-APBA molar ratio (Fig. 1E). Similar results were observed when 3- and 4-APBA were tested (Fig. S1†). The above results prompted two interesting questions: why did boronic acid groups in 2-, 3- and 4-APBA not react with HBQ, and what roles did their amine groups play.
Structural characterization of 2-APBA
A B–N dative bond can reportedly form between an amine and a boronic acid group.38–40 Therefore, it was necessary to determine whether the B–N dative bonds existed in 2-, 3-, and 4-APBA, which precluded their reaction with HBQ. To answer this question, single crystals of 2-, 3-, and 4-APBA were grown. Only the single crystal of 2-APBA was obtained through slow evaporation of an acetone solution (Fig. S2†), and its structure was characterized through single crystal X-ray diffraction analysis (Tables S1 and S2†), which revealed no B–N dative bonds but B–N covalent bonds in 2-APBA. Through the formation of a boronic anhydride group and a B–N covalent bond, 2-APBA existed in a dimeric form (Fig. 2A).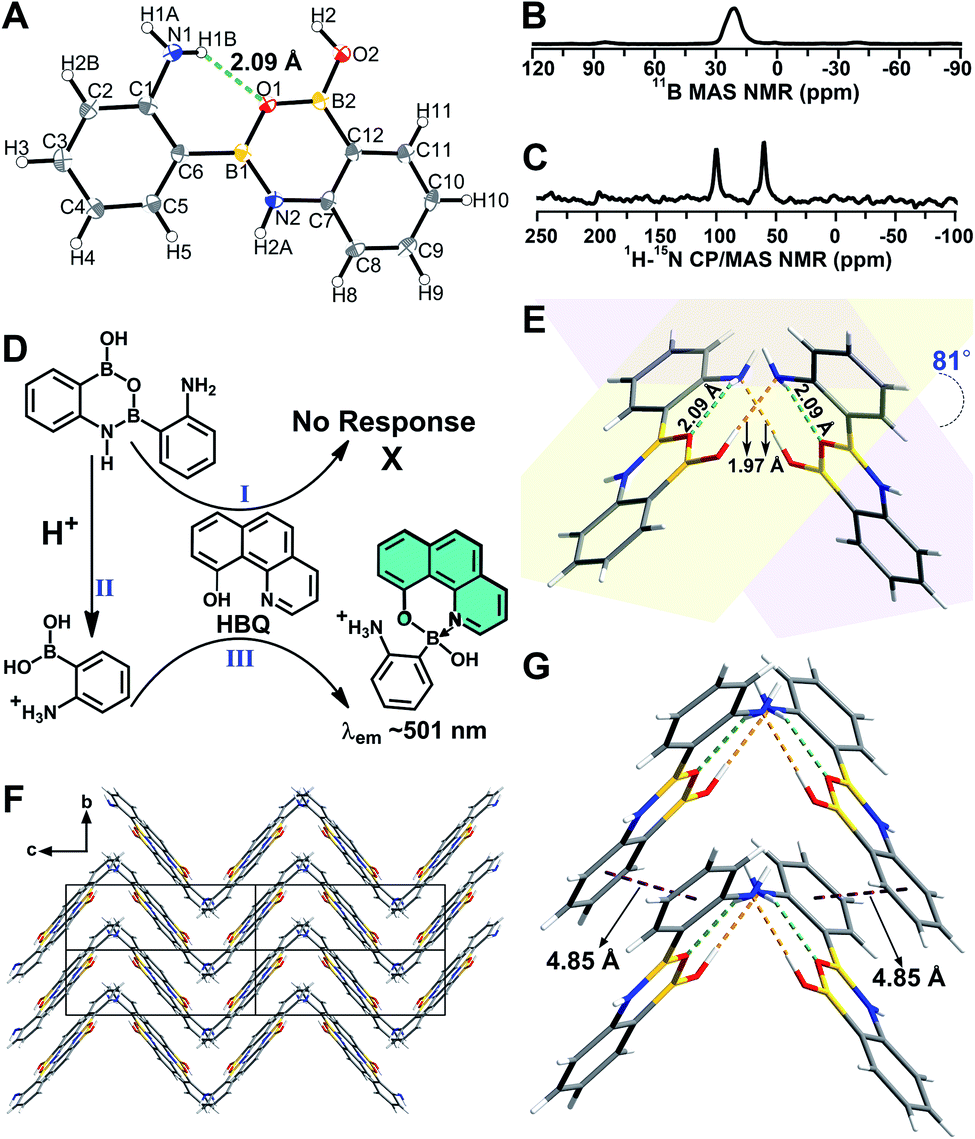 | ||
| Fig. 2 (A) Crystal structure of 2-APBA with thermal ellipsoids at 50% probability. The dashed green line represents an intramolecular N–H⋯O hydrogen bond. (B and C) 11B MAS (B) and 1H–15N CP-MAS (C) NMR spectra of the solid 2-APBA sample. (D) Explanation of the luminescence behavior of HBQ in response to 2-APBA and Neu5Ac. (E) Two dimers connected through intermolecular O–H⋯N hydrogen bonds (dashed orange lines). (F) Packing mode of 2-APBA viewed along the a axis (CCDC 2024024†). (G) Nearest distance between the phenyl rings (dashed plum lines) in the crystal structure. | ||
To verify that 2-APBA dimers were not produced during the growth of 2-APBA single crystals, the structure of the 2-APBA chemical used in the aforementioned experiments (Aladdin Reagent, Shanghai, Product no. A137630) was characterized meticulously. In the 11B magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectrum of a solid 2-APBA sample (Fig. 2B), only a strong peak derived from the three coordinated boron B[3]41,42 was observed, which was consistent with only B[3] species existing in the 2-APBA dimers. In the 1H–15N cross polarization (CP)-MAS NMR spectrum of the solid 2-APBA sample (Fig. 2C), two strong peaks were observed, confirming that two nitrogen (N) species existed in the sample. The peak at 60.6 ppm was the resonance signal of amine N,43,44 and the other peak at 100.2 ppm could be ascribed to the resonance signal of N in the B–N covalent bond. Furthermore, the dimeric structure of 2-APBA in DMSO-d6 solution was confirmed using 1H, 13C, 1H–1H COSY, 1H–1H NOESY, 1H–13C HSQC and 1H–13C HMBC NMR spectra (Fig. S3†). These results demonstrated that 2-APBA from Aladdin Reagent existed in the dimeric form, as did that from Alfa Aesar (Product no. L18069; Fig. S4†). By comparing the 1H NMR spectra of 2-APBA from Aladdin Reagent and Alfa Aesar with the one purified by HPLC (Fig. S4E†), it can be observed that 2-APBA from Aladdin Reagent had a higher purity than that from Alfa Aesar. Thus, unless otherwise noted, 2-APBA from Aladdin Reagent was used as received.
On the basis of the above results, the “abnormal” phenomena of 2-APBA (Fig. 1D and E) could be explained reasonably. Because the boronic acid groups were occupied in the 2-APBA dimers, they could not react with HBQ (Fig. 2D(I)). When Neu5Ac was added to the 2-APBA–HBQ solution, the 2-APBA dimers hydrolyzed into monomers with the aid of H+ ionized from Neu5Ac (Fig. 2D(II) and S5†). The 2-APBA monomers reacted with HBQ (Fig. 2D(III) and S6A†), resulting in an increase in the emission of HBQ at 504 nm with the increase in the molar ratio of Neu5Ac-to-2-APBA. However, excess Neu5Ac competed with HBQ to bind to 2-APBA monomers, resulting in a gradual decrease in the emission of HBQ at 504 nm after a maximum had been reached. For 3- and 4-APBA, boronic acid groups were involved in multiple intermolecular hydrogen bonds with amines, resulting in their “abnormal” phenomena (Fig. S6B–D†).
AIEE properties of the 2-APBA dimer
In the single crystal structure of 2-APBA, two dimers, whose dihedral angle was 81°, were edge-to-edge connected with each other through a pair of intermolecular O–H⋯N hydrogen bonds with a distance of 1.97 Å (Fig. 2E). Further ordered packing was realized through intermolecular N–H⋯π interactions and C–H⋯O hydrogen bonds (Fig. 2F, S7 and Table S3†). The nearest distance between the phenyl rings in the two adjacent layers was 4.85 Å (Fig. 2G), which suggested no π–π stacking interactions in the crystal structure of 2-APBA.45 The rigidified molecular conformation caused by intermolecular interactions and the absence of π–π stacking interaction revealed that the 2-APBA dimer might be AIEE active.19The following experimental data validated the aforementioned speculation. It was reported that the fluorescence of aromatic amines was strong in organic solvents but considerably quenched in water (e.g., ΦF = 0.19 and 0.010 for m-methylaniline in cyclohexane and H2O, respectively).46,47 The 2-APBA dimer was soluble in tetrahydrofuran (THF) and was molecularly dispersed at 50 μmol L−1. As shown in Fig. 3A and Table 1, the 2-APBA dimer emitted strong fluorescence in THF, with an absolute ΦF of 20.1%. In sharp contrast to the reported results,46,47 the fluorescence of the 2-APBA dimer was not quenched by water. With the increase of water content in THF–water mixtures, the fluorescence of the 2-APBA dimer enhanced gradually (Fig. 3A). The absolute ΦF of the 2-APBA dimer in water was 46.6% (Table 1), substantially larger than that of aniline and PBA in water (1.86% and 0.042%, respectively). The 2-APBA dimer was not readily soluble in water, and numerous particles existed in a 50 μmol L−1 aqueous solution of the 2-APBA dimer (Fig. S8 and Movie S1,† with the hydrodynamic diameter ranging from 40 to 400 nm). Thus, the fluorescence enhancement of the 2-APBA dimer in water might have originated from 2-APBA dimer aggregates. The formation of aggregates in water overshadowed the quenching effect of water on the emission of the 2-APBA dimer, leading to strong emission. As expected, the solid 2-APBA dimer sample emitted strong fluorescence (Fig. 3A, dashed purple line) with an absolute ΦF of 81.3%, markedly enhanced without the interference of water. These results indicated that aggregation enhanced the emission of the 2-APBA dimer, confirming that the 2-APBA dimer was AIEE active.
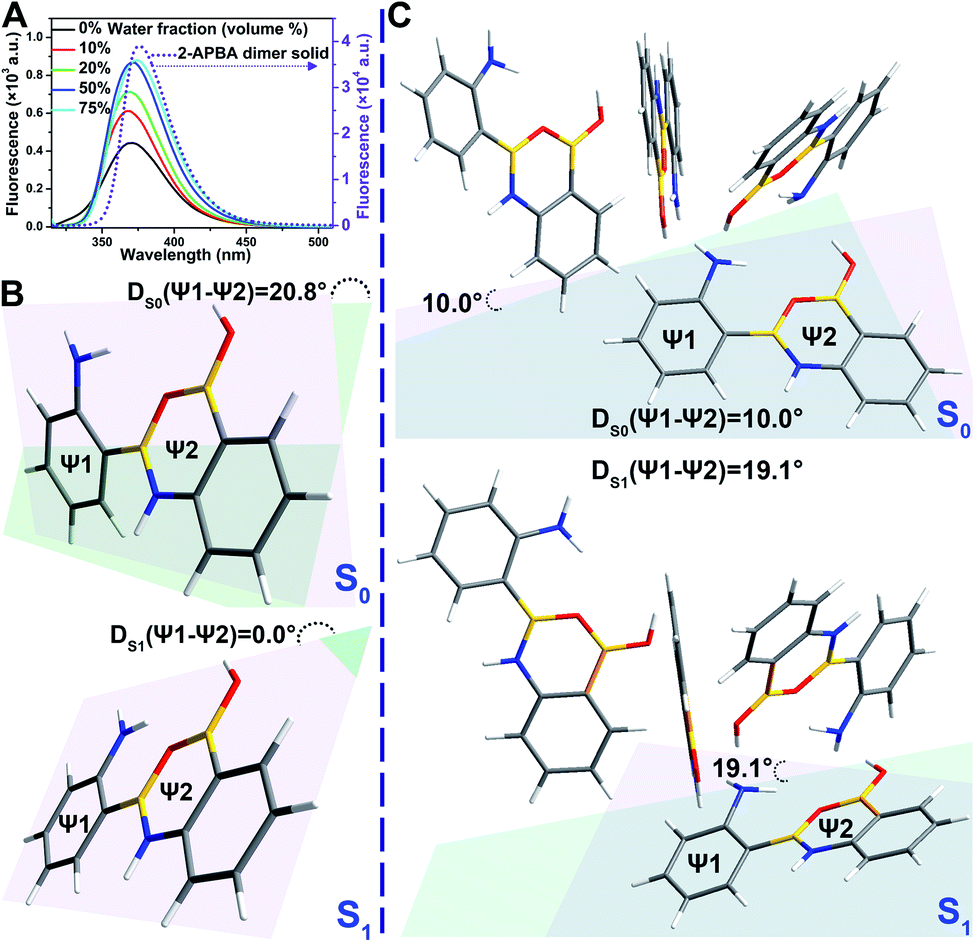 | ||
| Fig. 3 (A) Fluorescence spectra of the 2-APBA dimer (2.9 × 10−5 mol L−1) in THF–water mixtures with different amounts of water (volume%), λex = 300 nm. The amorphous 2-APBA dimer sample was used for preparing the solutions. The dashed purple line is the fluorescence spectrum of the solid 2-APBA dimer sample. (B and C) Optimized structures of the S0 and S1 states of one 2-APBA dimer (B) and four 2-APBA dimers (C). | ||
AIEE mechanism of the 2-APBA dimer
To investigate the AIEE mechanism of the 2-APBA dimer, time-resolved fluorescence measurements and theoretical calculations were performed. From the single molecule state (in THF) to the aggregate state (solid), the radiative decay rate (kF) of the 2-APBA dimer calculated from the absolute ΦF and fluorescence lifetime (τ, Table 1, eqn (S3) and (S4), and Fig. S9†) increased by a factor of 1.9; the total nonradiative decay rate (knr obtained from τ and eqn (S4)†) decreased by a factor of 9.2, resulting in a 17.4-fold increase in the ratio of kF to knr (kF/knr, Table 1). In addition, from the perspective of the lowest excited singlet state (S1) to the ground state (S0), time-dependent density functional theory calculations using the B3LYP function and TZVP basis set revealed that the variance of the dihedral angle between ψ1 and ψ2 in the dimer [DS1(ψ1 − ψ2) − DS0(ψ1 − ψ2) in Fig. 3B] was 20.8°, whereas it was only 9.1° [DS1(ψ1 − ψ2) − DS0(ψ1 − ψ2) in Fig. 3C] in the aggregated structure, which was simplified to four 2-APBA dimers connected by intermolecular interactions. The difference of the electric transition dipole moment from the S1 to the S0 state in the dimer (4.22) and in the aggregated structure (4.52) was small, implying that the aggregation of the 2-APBA dimer had a slight effect on the radiative decay. These results indicated that the intramolecular motion of the 2-APBA dimer was largely restricted in the aggregate state, which blocked the nonradiative decay pathways,48 leading to a sharp decrease in knr. Finally, the emission of the 2-APBA dimer enhanced considerably.To determine whether 3- and 4-APBA are also AIEE active, luminescence measurements were performed. In THF solution, the fluorescence intensity of the 2-APBA dimer was somewhat higher than that of 3- and 4-APBA (Fig. S10,† black lines). However, a remarkable difference was observed for the solid samples (Fig. S10,† red lines). The 2-APBA dimer emitted intense fluorescence. By comparison, the fluorescence of 3- and 4-APBA was weak. This phenomenon indicated that 3- and 4-APBA might not be AIEE active, which was further confirmed by the absolute ΦF. As shown in Fig. 4A, the absolute ΦF of the solid 3-APBA sample (22.7%) was close to that of 3-APBA in THF (21.2%) and water (20.1%). It was the same for 4-APBA (the absolute ΦF of 4-APBA in THF, water and the solid state was 5.3%, 5.8% and 5.6%, respectively). Besides, the knr of 3-APBA just decreased by a factor of 1.3, while the knr of 4-APBA increased by a factor of 4.1 from the single molecule state (in THF) to the aggregate state (solid) (Fig. S11, S12, Tables S4 and S5†). These data indicated that 3- and 4-APBA were not AIEE molecules.
 | ||
| Fig. 4 (A) Comparison of absolute ΦF of the 2-APBA dimer, and 3- and 4-APBA in THF, water and the solid state. (B) XRD patterns of the 2-APBA dimer, and 3- and 4-APBA. (C, H and J) SEM images of the samples on the silicon wafer prepared from the aqueous solution of the 2-APBA dimer (C), and 3- (H) and 4-APBA (J). (D, I and K) SEM images of the solid samples of the 2-APBA dimer (D), and 3- (I) and 4-APBA (K). (E and F) XRD pattern (E) and SEM image (F) of the amorphous 2-APBA dimer sample. (G) Fluorescence spectra of crystalline and amorphous 2-APBA dimer samples recorded under identical measurement conditions, λex = 300 nm. | ||
To understand why the 2-APBA dimer was AIEE active but 3- and 4-APBA were not, molecular packing in aggregates, which was reported to play a key role in the resultant luminescence properties,49,50 was investigated. In the single crystal structure of 2-APBA, the 2-APBA dimer molecules were ordered. Correspondingly, the 2-APBA dimer could self-assemble into well-ordered structures. The scanning electron microscopy (SEM) image of the 2-APBA dimer sample, which was prepared by dropping the 2-APBA dimer aqueous solution onto a clean silicon wafer, reveals that well-ordered aggregates mainly formed into rod-like crystals with an average length of approximately 5 μm (Fig. 4C and S13A and B†). It was worth noting that the 2-APBA dimer particles tended to aggregate on the edge of the silicon wafer during the evaporation of water (Fig. S13A†). Fig. 4C shows one aggregate on the edge of the silicon wafer, demonstrating that the sizes of the 2-APBA dimer aggregates on the silicon wafer were substantially larger than those recorded in water (Fig. S8†). The solid 2-APBA dimer sample was observed to have a regular cuboid shape (Fig. 4D), and its X-ray diffraction (XRD) pattern had several intense and sharp reflection peaks (Fig. 4B), further illustrating the highly ordered stacking of the 2-APBA dimer.
After melting the solid 2-APBA dimer sample under a nitrogen atmosphere and then rapidly quenching the molten 2-APBA dimer with liquid nitrogen, the sharp reflection peaks in the XRD pattern and ordered structures in the SEM image disappeared (Fig. 4E and F), which indicated that the ordered molecular packing of the 2-APBA dimer was destroyed, accompanied by the transformation from the crystalline state to the amorphous state. Correspondingly, the fluorescence intensity of the amorphous 2-APBA dimer sample was remarkably weaker than that of the crystalline 2-APBA dimer sample (Fig. 4G), further confirming the crucial role of crystallinity in the AIEE effect. For 3- and 4-APBA, no ordered structures were observed in the SEM images of 3- and 4-APBA (Fig. 4H–K and S13C and D†). The XRD pattern of 3-APBA exhibited less clear reflection peaks compared with that of the 2-APBA dimer, and the XRD pattern of 4-APBA contained only a weak and broad reflection peak (Fig. 4B and S14†). The detailed aggregation modes of 3- and 4-APBA are provided in Fig. S15–S27 and Tables S6 and S7.† Given these results, we concluded that the disordered molecular packing resulted in the non-AIEE properties of 3- and 4-APBA, whereas the ordered molecular packing without the presence of π–π stacking interactions (Fig. 2G) endowed the 2-APBA dimer with AIEE activity.
Dynamic covalent B–N and B–O bonds behind the AIEE effect of the 2-APBA dimer
Further application disclosed another attractive feature of 2-APBA that it could reversibly transform between its monomer form and dimer aggregates by alternately being treated with CO2 and N2, reflecting the dramatic AIEE fluorescence quenching and recovery. As shown in Fig. 5A, the fluorescence of the 2-APBA dimer in water decreased by 74% after bubbling CO2 for 2 s, and stabilized within 60 s. This process would be slower when CO2 was mixed with nitrogen (N2). Fig. 5B shows the time dependence of fluorescence intensity changes of the 2-APBA dimer solution during bubbling the CO2/N2 mixture. With increasing proportion of CO2, the fluorescence quenching speed accelerated gradually, and a linear relationship between −log10(I/I0) and the proportion of CO2 could be built in a range of 0–30% (Fig. S28†). The CO2-quenched fluorescence of the 2-APBA dimer could be explained to be the result of the dissolution of CO2 in water, which reduced the pH value of the solution (Fig. S29 and S30†). With the aid of H+, the 2-APBA dimer aggregates decomposed rapidly and finally hydrolyzed into the non-fluorescent 2-APBA monomers (Fig. 5E and S5†).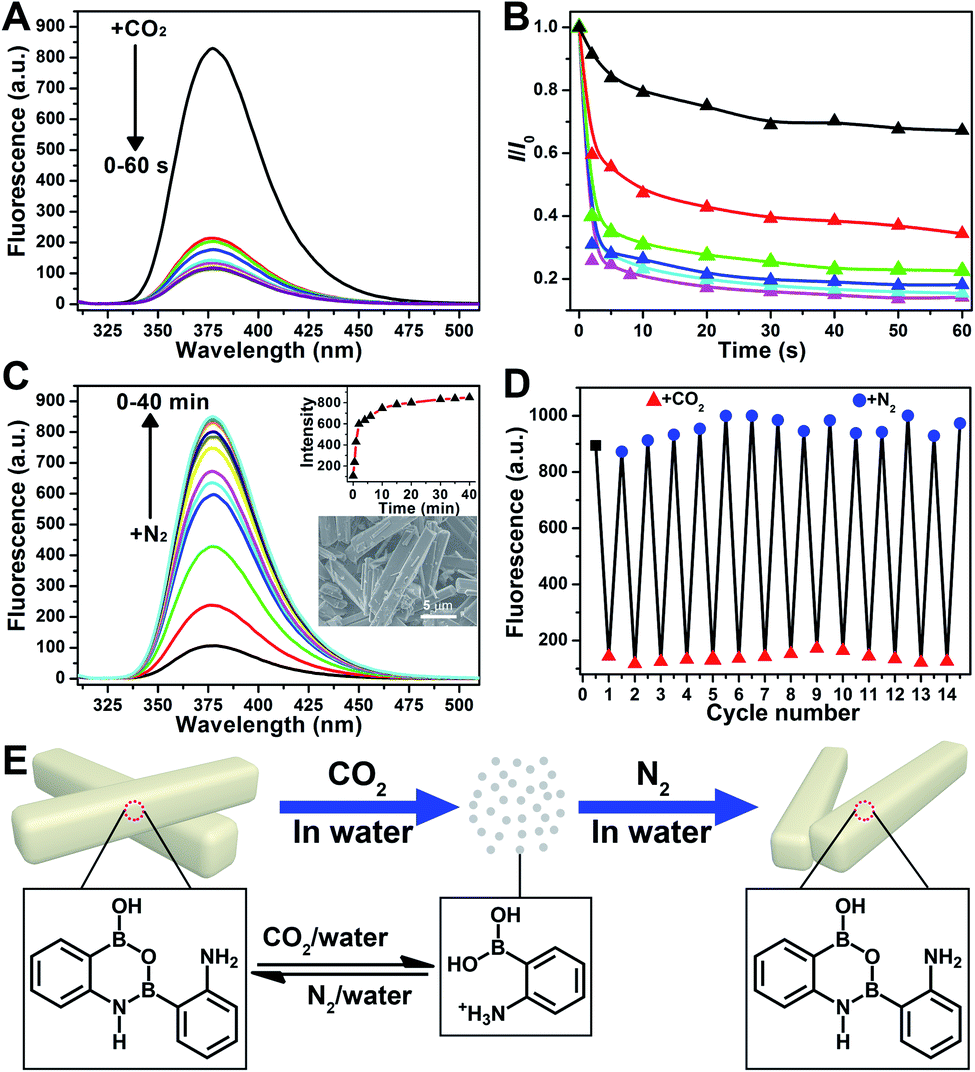 | ||
| Fig. 5 (A) Fluorescence spectra of the 2-APBA dimer in water with bubbling CO2 for different times (2.9 × 10−5 mol L−1). The gas flow rate is 60 mL min−1. λex = 300 nm. (B) Time dependence of the variation of the relative fluorescence intensities at 377.5 nm of the 2-APBA dimer (I/I0) in water with bubbling the CO2/N2 mixture (2.9 × 10−5 mol L−1). The proportions of CO2 in CO2/N2 mixtures are 10.0% (black), 25.0% (red), 33.3% (green), 50% (blue), 66.7% (cyan) and 100% (magenta), respectively. The total gas flow rate is 60 mL min−1. λex = 300 nm. (C) Fluorescence spectra of the 2-APBA dimer (2.9 × 10−5 mol L−1) and intensity changes (at 377.5 nm, inset in the upper-right corner) during bubbling N2 through an aqueous solution of the 2-APBA dimer saturated with CO2. The inset SEM image in the lower-right corner is of the 2-APBA dimer sample alternately treated with CO2 and N2. The gas flow rate is 60 mL min−1. λex = 300 nm. (D) Cycling experiment illustrating the variation of the fluorescence intensities of 2-APBA at 377.5 nm after alternately bubbling CO2 and N2 through an aqueous solution of the 2-APBA dimer. (E) Illustration of the reversible transformation of 2-APBA between dimer and monomer aggregates through alternate treatment with CO2 and N2. | ||
More interestingly, we found that the fluorescence of the 2-APBA dimer, after being quenched by CO2, increased to 79% of its original value after bubbling N2 for 6 min and returned to its original value after bubbling N2 for 40 min (Fig. 5C). Fourteen cycling experiments indicated that CO2-quenched fluorescence and the subsequent N2-induced fluorescence recovery were highly reversible (Fig. 5D). Here N2 was used to remove CO2 from the solution. When N2 was replaced by other inert gases, i.e., O2 and air, the highly reversible fluorescence quenching and recovery can also be observed (Fig. S31†). Detailed characterization data proved that 2-APBA returned back to its dimer form after removing CO2 from the solution (Fig. 5E and S32†). Under this condition, the resulting 2-APBA dimers maintained a strong self-assembly capacity, and numerous cuboid shape aggregates could be easily observed by SEM (inset of Fig. 5C), which contributed to the recovery of the fluorescence. The above results implied that the B–N and B–O bonds in 2-APBA dimers were dynamic covalent bonds, which could rapidly and reversibly break and form through the adjustment of the solution pH. To the best of our knowledge, this is the first example of a dynamic covalent system with both B–N and B–O bonds, which provides a new avenue for the construction of molecular architectures for advanced materials applications such as CO2 detection (Fig. S28†), nanogating, saccharide recognition and separation,51 chemo/bio-sensing,52 self-healing,53 supramolecular self-assembly54 and controlled drug release.55 In the following, a CO2-responsive nanogating system by using the 2-APBA dimer was presented.
In nature, CO2 is an important stimulus in modulating the activity of fluidic ion channels. For instance, CO2 controls the electrical signals of the odorant-gated ion channels in the olfactory sensory neurons of mosquitoes guiding them to navigate and locate the host.56 As another typical example, CO2 regulates the opening and closing of stomatal pores in plants' leaves by modulating the activity of the anion channels during the photosynthesis process.57 Inspired by nature, some delicate CO2-responsive nanogating examples have been reported; however, these systems required complicated fabrication technologies.56–58 The reversible breaking and reforming of the dynamic covalent B–N and B–O bonds in response to CO2 and N2, which led to the disassembly/assembly of the 2-APBA dimer, offered a simple strategy to fabricate a CO2-responsive nanogating system. A polyethylene terephthalate (PET) membrane with multiple conical nanochannels fabricated by the established procedures from our group (1 × 106 pores per cm2, the large and small opening were approximately 500 and 35 nm in diameter, respectively)59 was chosen as the nanochannel substrate, and 0.1 M KCl solution containing 2-APBA dimer aggregates (5.0 × 10−3 mol L−1) was chosen as the electrolyte. When bubbled with CO2, the 2-APBA dimer aggregates decomposed and hydrolyzed into the positively charged 2-APBA monomers (Fig. 5E) which interacted with the negatively charged carboxyl groups (–COO−) on the inner surface of the nanochannels, forming –COO−/2-APBA monomer ion pairs (Fig. 6A). Owing to the reduced surface charge of the nanochannels after the formation of ion pairs, an obvious decrease of the ion current was observed (Fig. 6B). After being bubbled with N2, the positively charged 2-APBA monomers dissociated from the ion pairs and returned back to the form of the 2-APBA dimer aggregates. Thus, the ion currents were readily recovered after N2 bubbling (Fig. 6B). Moreover, the switching characteristic was highly reversible (Fig. 6C). In sharp contrast, when there were no 2-APBA dimer aggregates in the electrolyte, no evident change of the ion current was observed upon alternate treatment with CO2 and N2 (Fig. S33†). The above results illustrated the potential application of the 2-APBA dimer in CO2 related nanogating and nanofluidic systems.60
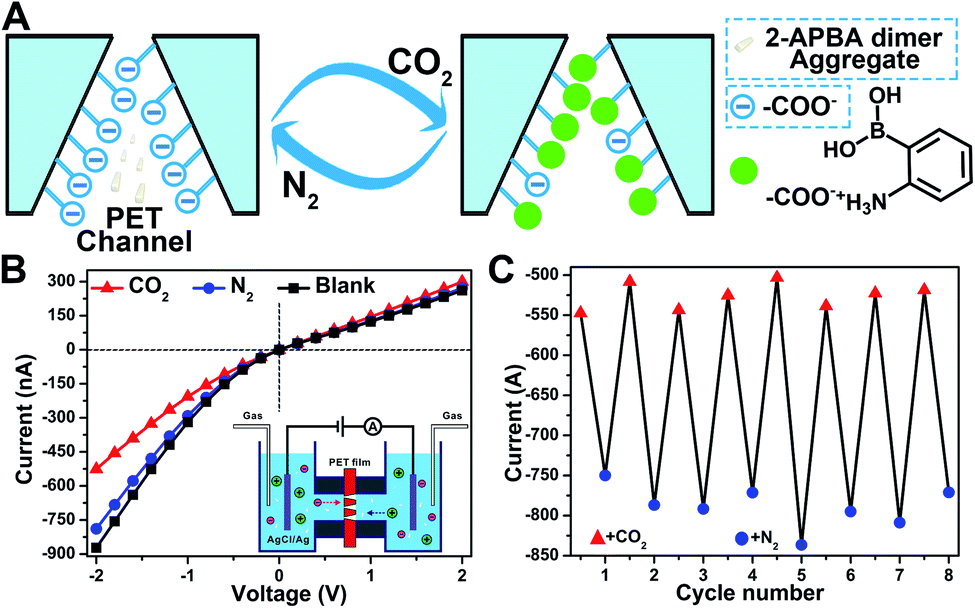 | ||
| Fig. 6 (A) Schematic of the design of CO2-responsive nanochannels by using the 2-APBA dimer. (B) Current–voltage (I–V) response recorded before and after CO2 and N2 bubbling through PET nanochannels in KCl solution containing 2-APBA dimer aggregates. I–V curves corresponding to CO2 and N2 bubbling are the average of the curves in the eight cycles in (C). (C) Transmembrane ionic current (at −2 V) switching of the PET conical nanochannels in 0.1 M KCl solution containing 2-APBA dimer aggregates upon alternate treatment with CO2 and N2. | ||
Conclusions
In summary, we demonstrated that the 2-APBA dimer, with a high ΦF of 81.3% in the solid state, is a novel AIEE molecule. Detailed analysis revealed that its remarkably high solid-state emission was attributed to ordered molecular packing without the presence of π–π stacking interactions. This study revealed that a simple molecule can also be AIEE active, shedding new light on AIEE effect. Moreover, a new kind of dynamic covalent bond (i.e., B–N and B–O bonds) was discovered from the ordered structure of the 2-APBA dimer. The convergence of B–N and B–O dynamic covalent chemistry and the AIEE effect provides a versatile platform for designing and synthesizing AIEE-active organoboron luminescent materials with excellent optoelectronic properties originating from the unique electronic nature and chemical characteristics of boron.61Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
X. L. and G. Q. conceived the study, designed the experiments and wrote the manuscript. X. L. performed most of the experiments. D. W. designed and performed the theoretical calculations. Y. Z. and W. L. designed the application experiments. S. Y. designed the time-resolved fluorescence experiments. G. H. designed the 11B MAS and 1H–15N CP-MAS NMR experiments. Z. Z. and H. Q. performed the NMR experiments. Y. Z. helped in growing the single crystals. M. L. helped in polishing the language.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21775116 and 21922411), DICP Innovation Funding (DICP-RC201801), and the LiaoNing Revitalization Talents Program (XLYC1802109).Notes and references
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed.
- F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 14192–14196 CrossRef PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS.
- J. Yang, M. Fang and Z. Li, Aggregate, 2020, 1, 6–18 CrossRef.
- G. C. Schmidt, Ann. Phys., 1896, 294, 103–130 CrossRef.
- G. C. Schmidt, Ann. Phys., 1921, 370, 247–256 CrossRef.
- J. Stark and P. Lipp, Z. Phys. Chem., 1913, 86, 36–41 Search PubMed.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- Kenry, B. Z. Tang and B. Liu, Chem, 2020, 6, 1195–1198 CAS.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS.
- G. Niu, R. Zhang, X. Shi, H. Park, S. Xie, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, TrAC, Trends Anal. Chem., 2020, 123, 115769 CrossRef CAS.
- J. Guan, R. Wei, A. Prlj, J. Peng, K.-H. Lin, J. Liu, H. Han, C. Corminboeuf, D. Zhao, Z. Yu and J. Zheng, Angew. Chem., Int. Ed., 2020, 59, 14903–14909 CrossRef CAS.
- H.-T. Feng, C. Liu, Q. Li, H. Zhang, J. W. Y. Lam and B. Z. Tang, ACS Mater. Lett., 2019, 1, 192–202 CrossRef CAS.
- J. Yang, Z. Chi, W. Zhu, B. Z. Tang and Z. Li, Sci. China Chem., 2019, 62, 1090–1098 CrossRef CAS.
- J. Guan, C. Shen, J. Peng and J. Zheng, J. Phys. Chem. Lett., 2021, 12, 4218–4226 CrossRef CAS PubMed.
- J. Huang, H. Nie, J. Zeng, Z. Zhuang, S. Gan, Y. Cai, J. Guo, S.-J. Su, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2017, 56, 12971–12976 CrossRef CAS.
- Z. He, P. Liu, S. Zhang, J. Yan, M. Wang, Z. Cai, J. Wang and Y. Dong, Angew. Chem., Int. Ed., 2019, 58, 3834–3837 CrossRef CAS PubMed.
- J. Li, J. Wang, H. Li, N. Song, D. Wang and B. Z. Tang, Chem. Soc. Rev., 2020, 49, 1144–1172 RSC.
- T. Wu, J. Huang and Y. Yan, Chem.–Asian J., 2019, 14, 730–750 CrossRef CAS.
- M. Gao and B. Z. Tang, Coord. Chem. Rev., 2020, 402, 213076 CrossRef CAS.
- Z. Zhao, B. He and B. Z. Tang, Chem. Sci., 2015, 6, 5347–5365 RSC.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726–23740 RSC.
- W. Wu, M. Shen, X. Liu, L. Shen, X. Ke and W. Li, Biosens. Bioelectron., 2020, 150, 111912 CrossRef CAS PubMed.
- Y. Li, Y. Dong, L. Cheng, C. Qin, H. Nian, H. Zhang, Y. Yu and L. Cao, J. Am. Chem. Soc., 2019, 141, 8412–8415 CrossRef CAS.
- M. Golabi, F. Kuralay, E. W. H. Jager, V. Beni and A. P. F. Turner, Biosens. Bioelectron., 2017, 93, 87–93 CrossRef CAS.
- S. E. Son, P. K. Gupta, W. Hur, H. Choi, H. B. Lee, Y. Park and G. H. Seong, Anal. Chim. Acta, 2020, 1134, 41–49 CrossRef CAS PubMed.
- D. Li, Y. Chen and Z. Liu, Chem. Soc. Rev., 2015, 44, 8097–8123 RSC.
- S. Munir, S. Hussain and S.-Y. Park, ACS Appl. Mater. Interfaces, 2019, 11, 37434–37441 CrossRef CAS.
- M. M. Elgohary, M. W. Helmy, E.-Z. A. Abdelfattah, D. M. Ragab, S. M. Mortada, J.-Y. Fang and A. O. Elzoghby, J. Controlled Release, 2018, 285, 230–243 CrossRef CAS.
- R. Nishiyabu, Y. Kubo, T. D. James and J. S. Fossey, Chem. Commun., 2011, 47, 1124–1150 RSC.
- R. Deng, M. J. Derry, C. J. Mable, Y. Ning and S. P. Armes, J. Am. Chem. Soc., 2017, 139, 7616–7623 CrossRef CAS.
- F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC.
- A. C. Sedgwick, L. Wu, H.-H. Han, S. D. Bull, X.-P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian and J. Yoon, Chem. Soc. Rev., 2018, 47, 8842–8880 RSC.
- W. Wang, M. Marshall, E. Collins, S. Marquez, C. Mu, K. H. Bowen and X. Zhang, Nat. Commun., 2019, 10, 1170 CrossRef PubMed.
- H. Marciniak, S. Hristova, V. Deneva, F. S. Kamounah, P. E. Hansen, S. Lochbrunner and L. Antonov, Phys. Chem. Chem. Phys., 2017, 19, 26621–26629 RSC.
- M. R. Aronoff, B. VanVeller and R. T. Raines, Org. Lett., 2013, 15, 5382–5385 CrossRef CAS.
- Y. Liu, Y. Lu and Z. Liu, Chem. Sci., 2012, 3, 1467–1471 RSC.
- Q. Yang, D. Huang, S. Jin, H. Zhou and P. Zhou, Analyst, 2013, 138, 4752–4755 RSC.
- L. Ren, Z. Liu, Y. Liu, P. Dou and H.-Y. Chen, Angew. Chem., Int. Ed., 2009, 48, 6704–6707 CrossRef CAS PubMed.
- F. Wang, R. Zhang, Q. Wu, T. Chen, P. Sun and A.-C. Shi, ACS Appl. Mater. Interfaces, 2014, 6, 21397–21407 CrossRef CAS PubMed.
- H. Noda, K. Motokura, Y. Wakabayashi, K. Sasaki, H. Tajiri, A. Miyaji, S. Yamaguchi and T. Baba, Chem.–Eur. J., 2016, 22, 5113–5117 CrossRef CAS.
- K. A. Thorn, J. C. Pennington, K. R. Kennedy, L. G. Cox, C. A. Hayes and B. E. Porter, Environ. Sci. Technol., 2008, 42, 2542–2550 CrossRef CAS.
- K. A. Thorn and K. R. Kennedy, Environ. Sci. Technol., 2002, 36, 3787–3796 CrossRef CAS PubMed.
- Y. Wang, G. Li, J. Zhang, Y. Jia, P. Pandey and S. Yang, Dyes Pigm., 2020, 174, 108025 CrossRef CAS.
- J. Oshima, S. Shiobara, H. Naoumi, S. Kaneko, T. Yoshihara, A. K. Mishra and S. Tobita, J. Phys. Chem. A, 2006, 110, 4629–4637 CrossRef CAS PubMed.
- S. Tobita, K. Ida and S. Shiobara, Res. Chem. Intermed., 2001, 27, 205–218 CrossRef CAS.
- J. Liu, L. Pan, Q. Peng and A. Qin, Molecules, 2017, 22, 1679 CrossRef.
- Q. Li and Z. Li, Acc. Chem. Res., 2020, 53, 962–973 CrossRef CAS PubMed.
- Q. Li and Z. Li, Sci. China Mater., 2020, 63, 177–184 CrossRef.
- S. D. Bull, M. G. Davidson, J. M. H. van den Elsen, J. S. Fossey, A. T. A. Jenkins, Y.-B. Jiang, Y. Kubo, F. Marken, K. Sakurai, J. Zhao and T. D. James, Acc. Chem. Res., 2013, 46, 312–326 CrossRef CAS.
- Y. Zhang, Y. Qi, S. Ulrich, M. Barboiu and O. Ramström, Mater. Chem. Front., 2020, 4, 489–506 RSC.
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS.
- K.-s. Kim, H. J. Cho, J. Lee, S. Ha, S. G. Song, S. Kim, W. S. Yun, S. K. Kim, J. Huh and C. Song, Macromolecules, 2018, 51, 8278–8285 CrossRef CAS.
- S. Ulrich, Acc. Chem. Res., 2019, 52, 510–519 CrossRef CAS PubMed.
- X. Shang, G. Xie, X.-Y. Kong, Z. Zhang, Y. Zhang, W. Tian, L. Wen and L. Jiang, Adv. Mater., 2017, 29, 1603884 CrossRef.
- Y. Xu, M. Zhang, T. Tian, Y. Shang, Z. Meng, J. Jiang, J. Zhai and Y. Wang, NPG Asia Mater., 2015, 7, e215 CrossRef CAS.
- Q. Zhai, H. Jiang, X. Zhang, J. Li and E. Wang, Talanta, 2016, 149, 280–284 CrossRef CAS.
- M. Li, Y. Xiong, W. Lu, X. Wang, Y. Liu, B. Na, H. Qin, M. Tang, H. Qin, M. Ye, X. Liang and G. Qing, J. Am. Chem. Soc., 2020, 142, 16324–16333 CrossRef CAS.
- H. Yang and G. Qing, Chem. Phys. Rev., 2021, 2, 021306 CrossRef.
- S. Mukherjee and P. Thilagar, J. Mater. Chem. C, 2016, 4, 2647–2662 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and measurements and supplementary figures and tables. CCDC 2024024. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03765j |
| This journal is © The Royal Society of Chemistry 2021 |