
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxa-spirocycles: synthesis, properties and applications†
Kateryna
Fominova
a,
Taras
Diachuk
a,
Dmitry
Granat
a,
Taras
Savchuk
a,
Vladyslav
Vilchynskyi
 a,
Oleksiy
Svitlychnyi
a,
Vladyslav
Meliantsev
a,
Igor
Kovalchuk
a,
Eduard
Litskan
a,
Vadym V.
Levterov
a,
Valentyn R.
Badlo
a,
Ruslan I.
Vaskevych
b,
Alla I.
Vaskevych
c,
Andrii V.
Bolbut
b,
Volodymyr V.
Semeno
a,
Rustam
Iminov
a,
Kostiantyn
Shvydenko
b,
Anastasiia S.
Kuznetsova
d,
Yurii V.
Dmytriv
ac,
Daniil
Vysochyn
a,
Vasyl
Ripenko
a,
Andrei A.
Tolmachev
a,
Olexandra
Pavlova
e,
Halyna
Kuznietsova
e,
Iryna
Pishel
e,
Petro
Borysko
e and
Pavel K.
Mykhailiuk
*a
a,
Oleksiy
Svitlychnyi
a,
Vladyslav
Meliantsev
a,
Igor
Kovalchuk
a,
Eduard
Litskan
a,
Vadym V.
Levterov
a,
Valentyn R.
Badlo
a,
Ruslan I.
Vaskevych
b,
Alla I.
Vaskevych
c,
Andrii V.
Bolbut
b,
Volodymyr V.
Semeno
a,
Rustam
Iminov
a,
Kostiantyn
Shvydenko
b,
Anastasiia S.
Kuznetsova
d,
Yurii V.
Dmytriv
ac,
Daniil
Vysochyn
a,
Vasyl
Ripenko
a,
Andrei A.
Tolmachev
a,
Olexandra
Pavlova
e,
Halyna
Kuznietsova
e,
Iryna
Pishel
e,
Petro
Borysko
e and
Pavel K.
Mykhailiuk
*a
aEnamine Ltd, Chervonotkatska 78, 02094 Kyiv, Ukraine. E-mail: Pavel.Mykhailiuk@gmail.com; Web: http://www.enamine.net Web: http://www.mykhailiukchem.org
bInstitute of Organic Chemistry, National Academy of Sciences of Ukraine, Murmanska St. 5, 02094 Kyiv, Ukraine
cNational Technical University of Ukraine, Igor Sikorsky Kiev Polytechnic Institute, Prosp. Peremohy 37, 03056 Kyiv, Ukraine
dNational Research Tomsk Polytechnic University, Lenin Ave. 30, 634050 Tomsk, Russia
eBienta, Chervonotkatska 78, 02094 Kyiv, Ukraine Web: http://www.bienta.net
First published on 27th July 2021
Abstract
A general approach to a new generation of spirocyclic molecules – oxa-spirocycles – was developed. The key synthetic step was iodocyclization. More than 150 oxa-spirocyclic compounds were prepared. Incorporation of an oxygen atom into the spirocyclic unit dramatically improved water solubility (by up to 40 times) and lowered lipophilicity. More potent oxa-spirocyclic analogues of antihypertensive drug terazosin were synthesized and studied in vivo.
Introduction
Saturated monocyclic units – cyclohexane, cyclopentane, piperidine, etc. – dominated in chemistry and in drug discovery for a long time.1 The situation started changing at the beginning of this century. In 2009, Lovering introduced the concept of “escape from flatland”,2 which already changed the way medicinal chemists think. Today, scientists tend to use small F(sp3)-rich molecules in their research.3,4 In 2010, saturated spirocycles were shown to possess improved physico-chemical characteristics over their common monocyclic counterparts.5 Since that time, spirocyclic molecules have been playing an important role in chemistry.6,7 In fact, more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 research manuscripts and 50000 patents on the topic have appeared during the last decade (Fig. 1).8
000 research manuscripts and 50000 patents on the topic have appeared during the last decade (Fig. 1).8
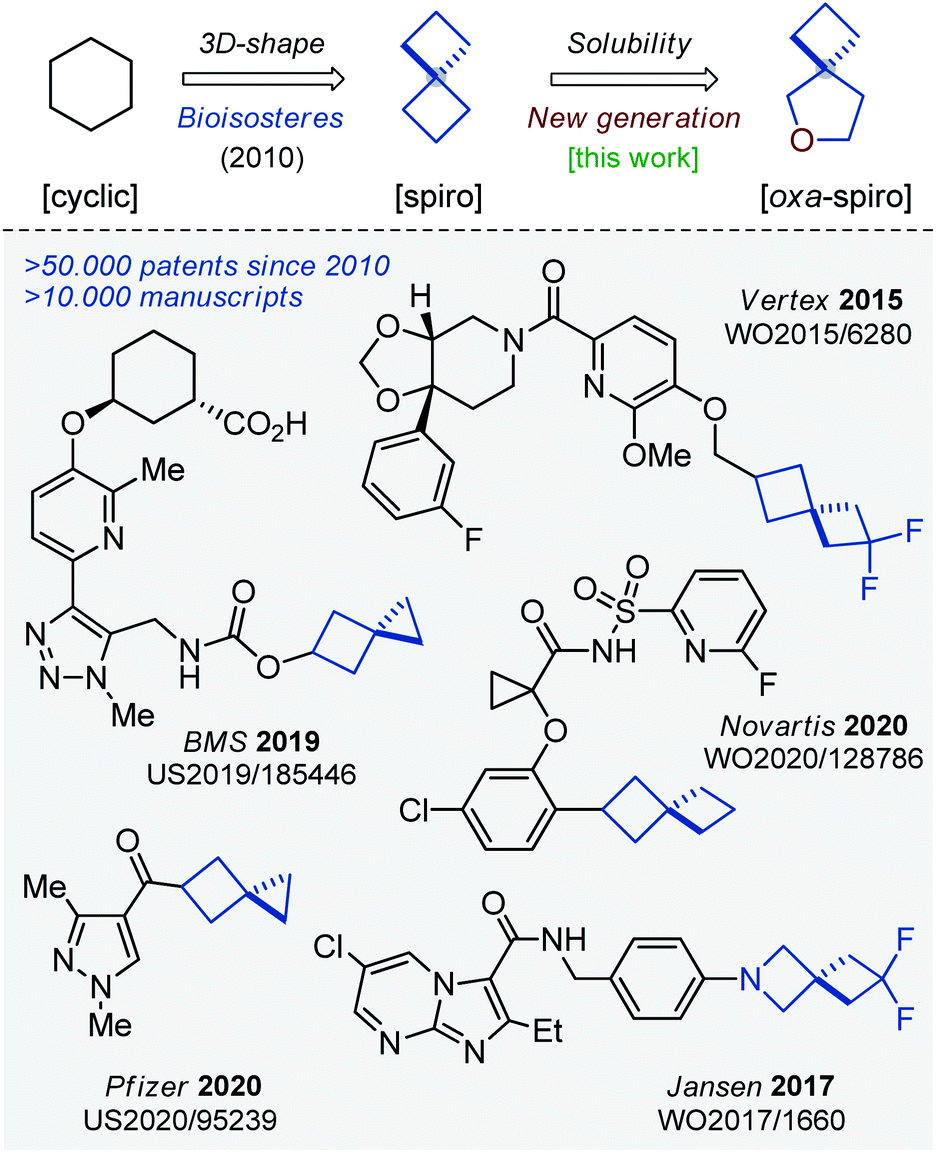 | ||
| Fig. 1 Spirocycles and their application in chemistry. | ||
Earlier, we reported on the preparation of oxa-bridged bicycles via iodocyclization of alkenyl alcohols.9 These compounds were designed as water-soluble analogues of popular bicyclo[1.1.1]pentanes. The work received positive feedback from both academy and industry, and therefore we decided to expand this tactic to a new generation of spirocycles – oxa-spirocycles (Fig. 1). Previously, oxa-spirocycles remained mostly in the shadow. For example, while three representative spirocyclic substituents (left, Fig. 2) are extremely popular in chemistry, the corresponding oxa-spirocyclic counterparts (right, Fig. 2) remain almost unknown.10
 | ||
| Fig. 2 Spirocycles and their unexplored oxa-counterparts. | ||
Of course, there was some interest in oxa-spirocycles before, but examples reported in the literature were rare and non-systematic.11 In 1985, Yoshida synthesized two substituted oxa-spirocycles via iodocyclization (Scheme 1).12 In 2008, Gouverneur studied the influence of the fluorine atom on the diastereoselectivity of the iodocyclization reaction. In this work, one example of the oxa-spirocyclic core is shown (Scheme 1).13 In 2011, Cernak and co-workers from Merck employed the Grubbs-metathesis reaction to synthesize oxa-spiropiperidines with reduced lipophilicity (Scheme 1).14 Later, Carreira used the same approach to prepare oxa-spiroazetidines.15 In 2013, Santini described the gold-catalyzed oxidative cyclization of propargyl alcohols into oxa-spirocyclic amines (Scheme 1).16 In the same year, Santini also developed an alternative approach to oxa-spirocyclic amines via oxidative intramolecular hydroxycyclization of alkenes.17 Subsequently, Cocotos realized an organocatalytic version of that method (Scheme 1).18 In 2018, Cossy developed a [Rh]-catalyzed cyclization of unsaturated alkoxyamines.19 All these reports described different topics and contained one to three examples of the desired oxa-spirocyclic molecules. Recently, Baik and Hong developed a [Pd]-mediated directed oxidative synthesis of sterically hindered oxa-spirocycles (Scheme 1).20 This work had an excellent scope, but needed an installation and the subsequent removal of a directing group.
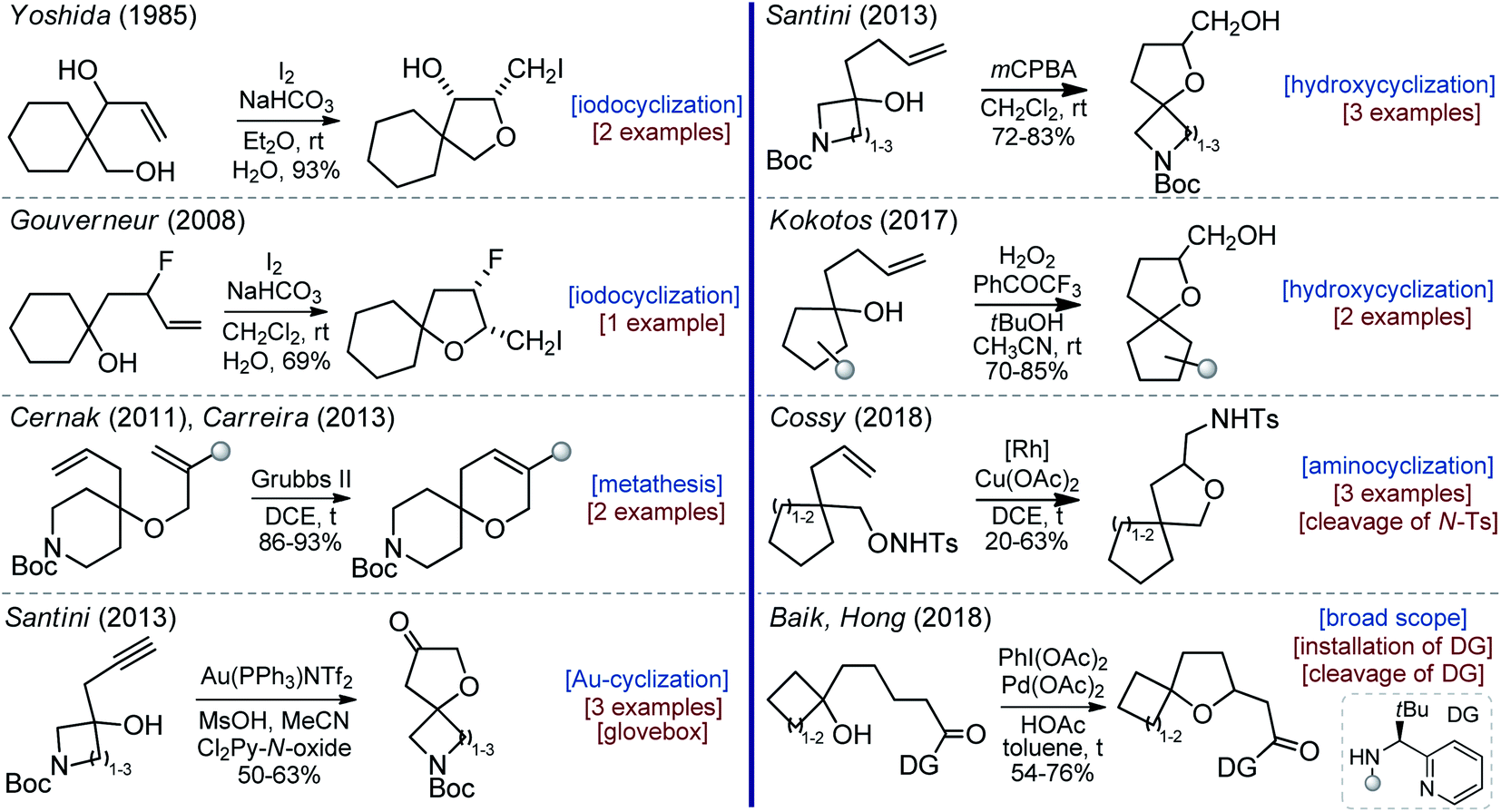 | ||
| Scheme 1 Literature precedents to oxa-spirocycles. | ||
Presumably, the absence of a general method to oxa-spirocycles is a primary reason why these molecules did not receive proper recognition from the scientific community.21 An ideal practical method should (a) employ inexpensive starting reagents; (b) not use protecting groups;22 and (c) provide oxa-spirocycles with a functional group that could be easily converted into a variety of other functional substituents: amines, alcohols, carboxylic acids, sulfonyl chlorides, etc. In this work, we present such an approach.
Results and discussion
Optimization
Based on our experience,9 and literature precedents,12,13 we studied iodocyclization of model alkene 4 (obtained by alkylation of ester 3 with LDA/allyl bromide; reduction with LiAlH4) into oxa-spirocyclic core 4a. We decided to specifically exploit iodocyclization, and not bromocyclization or hydroxycyclization, because the putative alcohols or bromides would be much less active in further modifications.Under the previously developed conditions (Table 1, entry 1) alkene 4 indeed predominantly afforded iodide 4a, although formation of side products was also observed. Separation of this mixture was problematic, especially on a gram scale. Performing the reaction in diethyl ether (entry 2) or dichloromethane (entry 3) still led to formation of a mixture of compounds. Finally, we screened various solvents and found that the transformation smoothly proceeded in acetonitrile to provide iodide 4a in almost quantitative yield (entry 7). Formation of side products was not observed in this experiment. Other inorganic bases also worked well, while pyridine and triethylamine gave moderate yields (entries 10–13). It is worth noting that N-iodosuccinimide could also be used although with a slightly lower efficiency (entry 14).
|
|
||
|---|---|---|
| Entry | Deviations from the literature | NMR yield 4aa,b (%) |
| a 2 mmol. b Yield determined by 1H NMR with CH2Br2 as an internal standard. c Isolated yield. NIS = N-iodosuccinimide, NIS = N-bromosuccinimide. | ||
| 1 | tBuOMe/H2O as a solvent | 71 |
| 2 | Et2O as a solvent | 67 |
| 3 | CH2Cl2 as a solvent | 65 |
| 4 | CHCl3 as a solvent | 69 |
| 5 | Dioxane as a solvent | 49 |
| 6 | THF as a solvent | 55 |
| 7 | CH3CN as a solvent | 96c (91) |
| 8 | DMF as a solvent | 41 |
| 9 | DMSO as a solvent | 34 |
| 10 | Na2CO3 as a base | 83 |
| 11 | KHCO3 as a base | 91 |
| 12 | NEt3 as a base | 57 |
| 13 | Py as a base | 42 |
| 14 | NIS instead of I2 | 85 |
| 15 | NBS instead of I2 | 83 (Br) |

Importantly, using the optimized conditions we could easily synthesize iodide 4a on a 26 g scale. The product was isolated from the reaction mixture by simple distillation and no additional purification was needed.
Scope
Having an optimized procedure in hand, we next studied its scope. Indeed, various five-membered oxa-spirocyclic iodides 5a–31a were easily prepared in 45–96% yield following the optimized protocol (Scheme 2). Among them were N-Boc-protected azetidines (8a, 16a, and 21a), pyrrolidines (10a) and piperidines (14a, 17a, and 29a). Labile ketal (7a) and ester groups (19a) were also compatible with the reaction conditions. Three (5a) to eight (31a)-membered cycles were incorporated into oxa-spirocyclic cores. Importantly, the popular oxetane ring23 was also successfully incorporated without decomposition (20a). Next, we decided to construct the oxetane ring via iodocyclization. The corresponding products 32a and 33a were obtained, although in low yields of 21–24% because the reaction was not selective. | ||
| Scheme 2 Scope of the iodocyclization step into oxa-spirocycles. Iodocyclization conditions: alkene (1 equiv.), NaHCO3 (3 equiv.), I2 (3 equiv.), CH3CN, rt, 1 h. aAlkene (1 equiv.), K2CO3 (4 equiv.), I2 (4 equiv.), CH3CN, rt, 48 h. Synthesis of amines, conditions: (i) iodide (1 equiv.), NaN3 (1.5 equiv.), DMSO, 85 °C. (ii) H2/Pd, MeOH, rt. b(ii) PPh3 (1.5 equiv.), H2O/THF, 50 °C. | ||
We also tried iodocyclization to construct the six-membered tetrahydropyrane ring. Under the standard conditions, however, incomplete conversions were observed. The reaction was slow, and some starting material remained. After optimization, we found that an excess of molecular iodine and a prolonged reaction time was needed. As a result, products 34a–42a were obtained in 45–64% yield (Scheme 2). Furthermore, the developed conditions were also used to synthesize dioxanes 43a and 44a in 58–63% yield and morpholines 45a–48a in 53–72% yield (Scheme 2).
Limitations
The developed protocol was not without limitations, however. While alcohols 32 and 33 provided the needed oxetanes 32a and 33a in low yields (Schemes 2 and 3), the corresponding azetidine-containing alcohol 49 unexpectedly afforded pyrrolidine 50 as a single stereoisomer in 92% yield (Scheme 3).24 The structure of the product was confirmed by X-ray analysis.25 Presumably, close proximity of a nitrogen atom and an iodonium moiety in the initially formed intermediate 51 led to an intramolecular nucleophilic attack providing another strained intermediate (52). Ring-opening of the azetidine ring in 52 with the iodide anion gave the observed pyrrolidine 50. | ||
| Scheme 3 Unexpected synthesis of pyrrolidine 50. | ||
Modifications
Several representative modifications of iodides 4a–48a were undertaken using standard chemical transformations to provide numerous oxa-spirocyclic derivatives. First, a simple reaction of iodides 4a–44a with sodium azide followed by reduction with either H2/Pd or PPh3 gave amines 4b–44b in 32–95% yield. Among them were not only monofunctional compounds, but also linkers with two functional groups: amino acids 19b and 39b and valuable diamines for medicinal chemistry 8b, 10b, 14b, 16b, 17b, 21b, 29b, 33b, 35b, 37b, 40b, 42b, and 44b (Scheme 2).Reaction of iodide 5a with potassium acetate in dimethylsulfoxide under heating, followed by hydrolysis of the ester group with sodium ethoxide, and the subsequent oxidation of the formed alcohol with PhI(OAc)2/TEMPO gave acid 5d (Scheme 4). Using that simple three-step strategy, seventeen other acids were synthesized. Especially worth noting are the unusual amino acids from Scheme 4 – that type of structure plays an important role in drug discovery programs.26
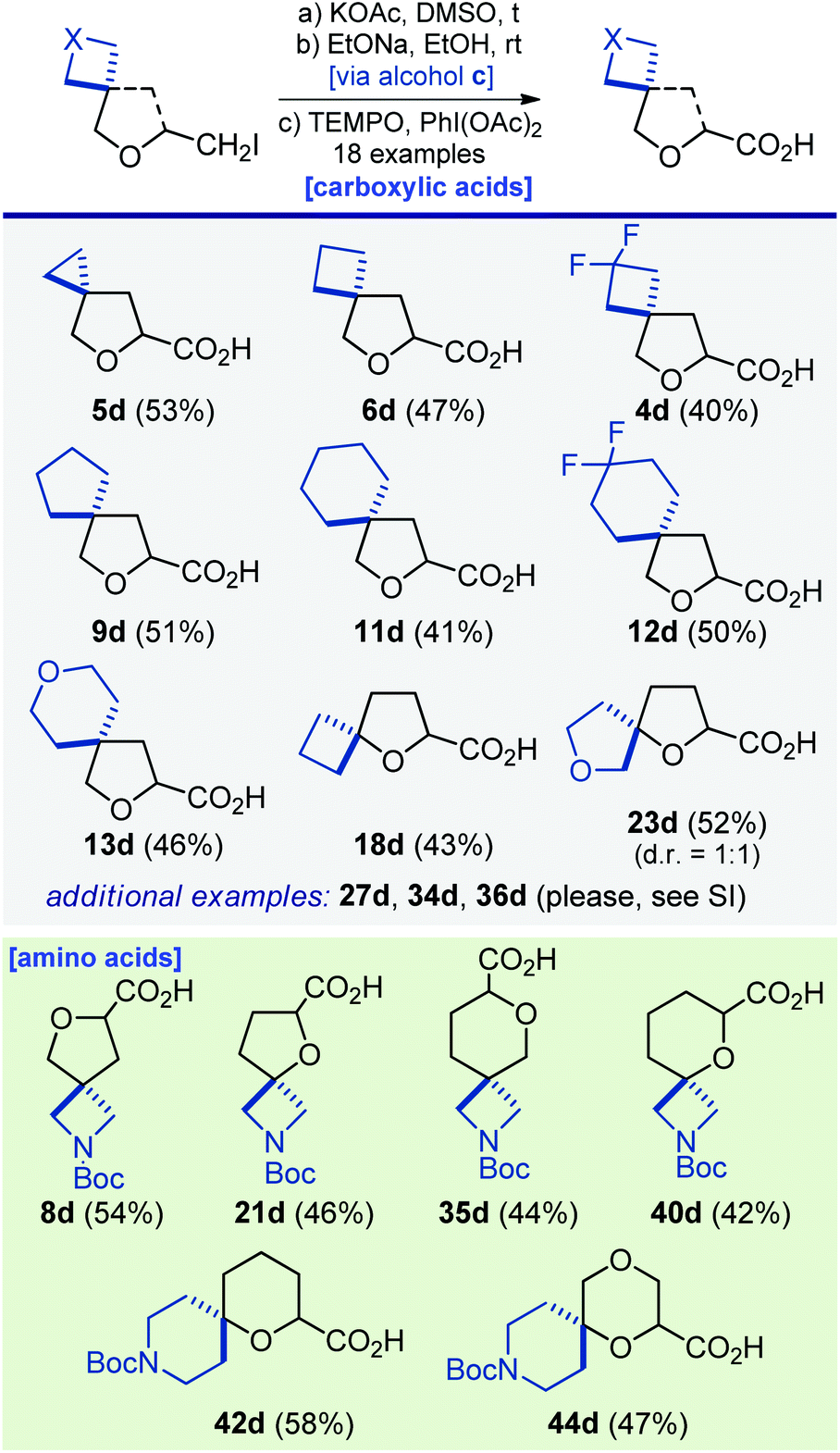 | ||
| Scheme 4 Synthesis of oxa-spirocyclic carboxylic acids and amino acids 4d–6d, 8d, 9d, 11d–13d, 18d, 21d, 23d, 27d, 34d–36d, 40d, 42d and 44d. | ||
Next, synthesis of some aliphatic sulfonyl chlorides – popular reagents for making bioactive sulfonamides27 – was undertaken. Reaction of iodide 16a with potassium thioacetate in dimethylsulfoxide at room temperature, followed by a direct oxidation of the formed intermediate with N-chlorosuccinimide in methanol gave aminosulfonyl chloride 16e (Scheme 5). Using this two-step strategy, sixteen other sulfonyl chlorides and aminosulfonyl chlorides – linkers – were easily obtained (Scheme 5).
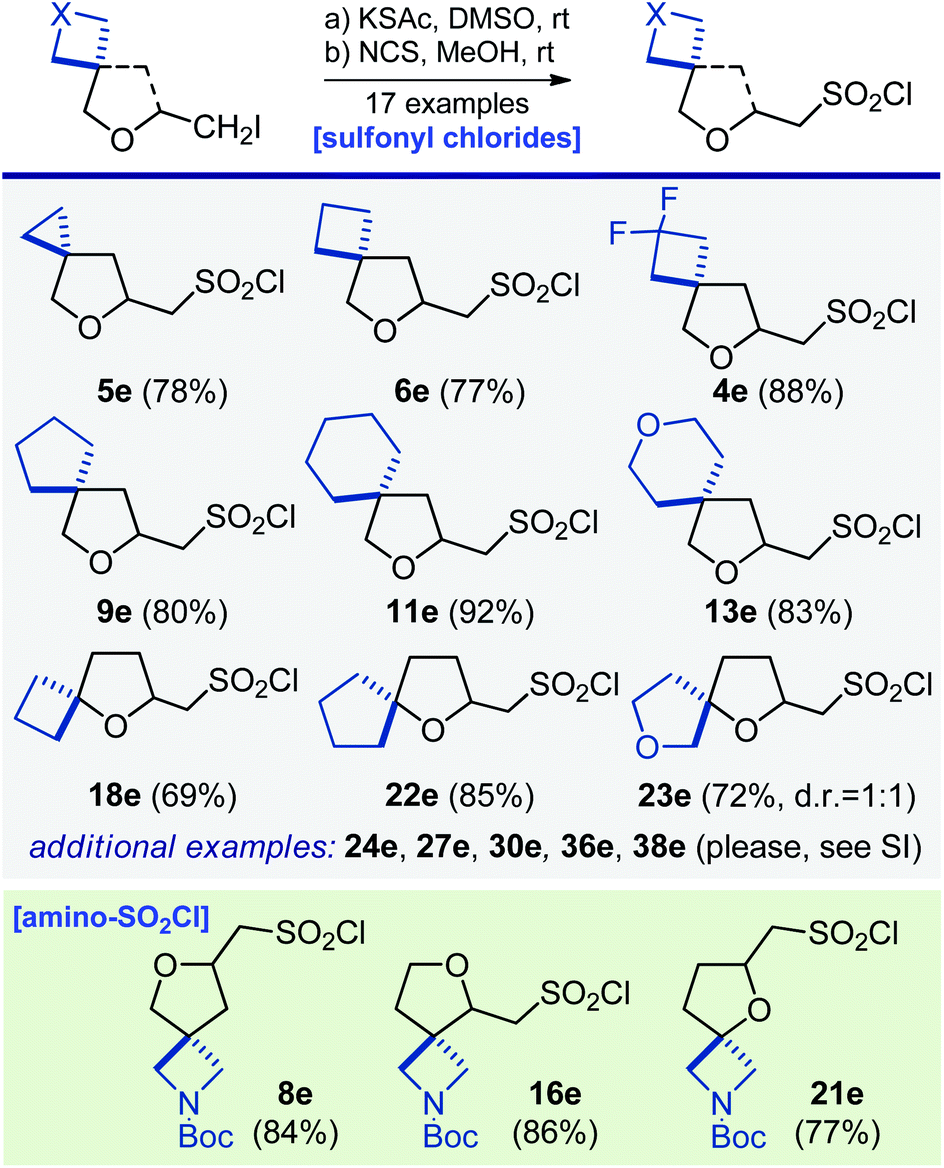 | ||
| Scheme 5 Synthesis of oxa-spirocyclic sulfonyl chlorides 4e–6e, 8e, 9e, 11e, 13e, 16e, 18e, 21e–24e, 27e, 30e, 36e, and 38e. | ||
Synthesis of several substituted acetic acids, homologues of carboxylic acids, was also performed. For example, reaction of iodide 40a with potassium cyanide, followed by alkali hydrolysis of the intermediate nitrile gave amino acid 40f (Scheme 6). Likewise, thirteen other mono- and bifunctional derivatives were obtained (Scheme 6).
 | ||
| Scheme 6 Synthesis of oxa-spirocyclic homoacids 5f, 6f, 8f, 9f, 13f, 18f, 21f, 29f, 34f–36f, 40f, and 44f. | ||
Recently, methylation was shown to have a profound impact on the activity of bioactive compounds – a “magic methyl” effect.28 On the other hand, in recent years substituted azetidines gained a lot of popularity in medicinal chemistry.29 In this context, we reduced the C–I bond in compounds 8a and 35a with hydrogen using palladium on charcoal and after acidic N-Boc cleavage obtained interesting methyl azetidines 8g and 35g in 85–88% yield (Scheme 7). Morpholine, in turn, is one of the most popular rings in drugs.1,30 Reduction of the C–I bond in iodides 45a–48a,31 followed by a Pd-catalyzed hydrogenative cleavage of the N-benzyl group afforded methyl-substituted morpholine-containing diamines 45g–48g in 68–85% yield (Scheme 7).
 | ||
| Scheme 7 Synthesis of methyl-azetidines 8g and 35g and methyl-morpholines 45g–48g. | ||
Reaction of iodide 6a with potassium cyanide in dimethylsulfoxide under heating, followed by reduction of the nitrile group with RANEY® nickel alloy gave amine 6h in 78% yield (Scheme 8) – a homologue of amine 6b (Scheme 2). Using the same tactic, amines 18h and 36h were also obtained. In addition, reduction of the C–I bond in compound 7a followed by acidic cleavage of the ketal moiety gave methyl ketone 7i in 63% yield (Scheme 8).
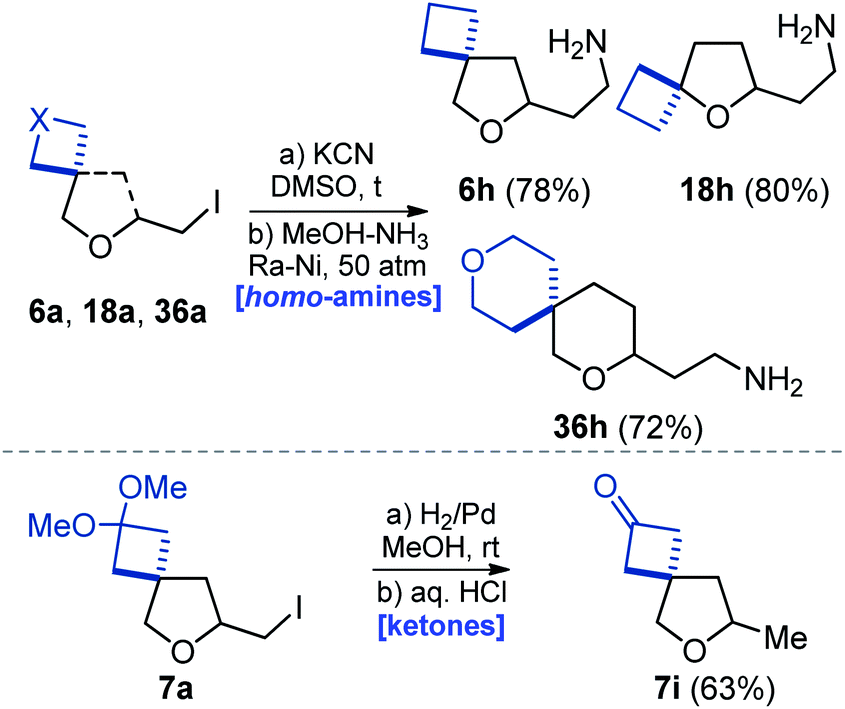 | ||
| Scheme 8 Synthesis of oxa-spirocyclic homoamines 6h, 18h, and 36h and ketone 7i. | ||
All modifications of oxa-spirocyclic iodides depicted in Schemes 4–8 represent (2e)-reactions. In this work, we also wanted to show that oxa-spirocyclic molecules are also compatible with radical (1e) modifications. Recently, Blackmond and Baran developed a practical [Cu]-catalyzed decarboxylative borylation of carboxylic acid derivatives.32 We used that radical transformation to convert oxa-spirocyclic acetic acids 14f, 18f, 21f, 34f, and 36f into the corresponding BPin-products (Scheme 9).33 In fact, three mono- (18j, 34j, and 36j) and one bifunctional (14j) organoboron compounds were obtained via the N-hydroxyphthalimide (NHPI) esters. Acid 21f, however, under identical conditions, gave reduced product 21j, presumably due to steric hindrance around the reaction center.
 | ||
| Scheme 9 Decarboxylative borylation of oxa-spirocyclic acids. | ||
In short summary, straightforward two to three step modifications of iodides 4a–48a using common (2e) and radical (1e) reactions allowed rapid synthesis of >150 novel or previously hardly accessible oxa-spirocyclic molecules. All products contained one or two appropriately protected functional groups – amino acids, diamines, aminosulfonyl chlorides, and amino boronates – suitable for direct use in medicinal chemistry programs. In terms of diversity and efficiency, this is the most useful method to access oxa-spirocyclic cores so far.
Application in organic synthesis
The synthetic approach to oxa-spirocycles described here not only provides entry into novel chemical space, but also significantly simplifies the preparation of known molecules. For example, in 2018, [Rh]-catalyzed cyclization of unsaturated alkoxyamines was developed (Scheme 1).19 In this project, the authors attempted [Rh]-catalyzed cyclization of substrate 55, but observed only “traces” of the needed N-tosyl intermediate 56 (Scheme 10). Alternatively, our approach allowed rapid preparation of the N-deprotected oxa-spiroamine 5b from the same starting alkene 54 in only three steps. Moreover, the synthesis was easily scaled up to 20 g.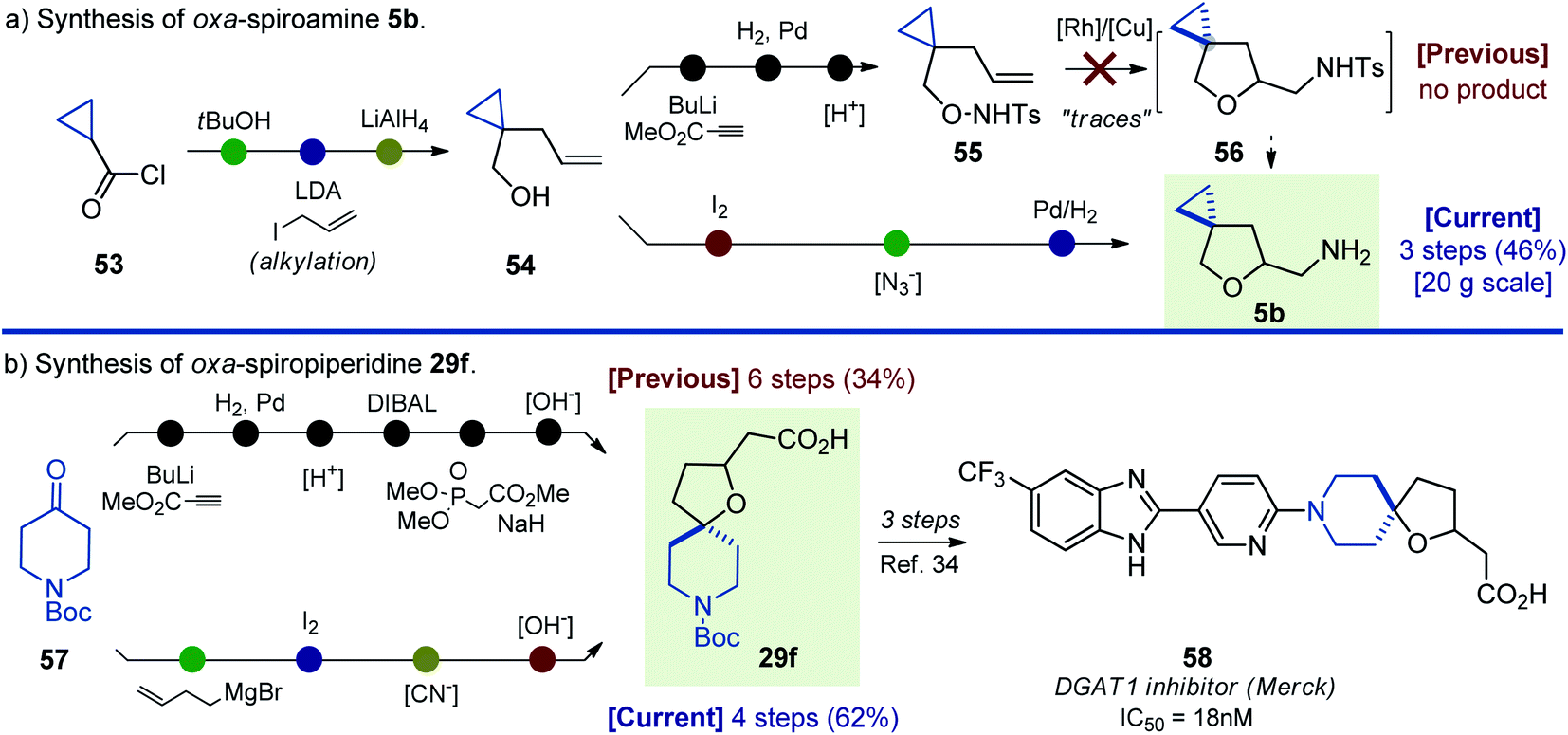 | ||
| Scheme 10 Synthesis of oxa-spiroamine 5b and oxa-spiropiperidine 29f: literature approaches vs. this work. | ||
Compound 58 was recently discovered as a potent DGAT1 inhibitor (Scheme 10).34 Synthesis of its key intermediate 29f was undertaken in six steps from the commercially available N-Boc piperidone 55 in 34% yield.14,35 In contrast, our approach allowed the preparation of oxa-spiropiperidine 29f in four steps from N-Boc piperidone in 62% yield.
Characterization
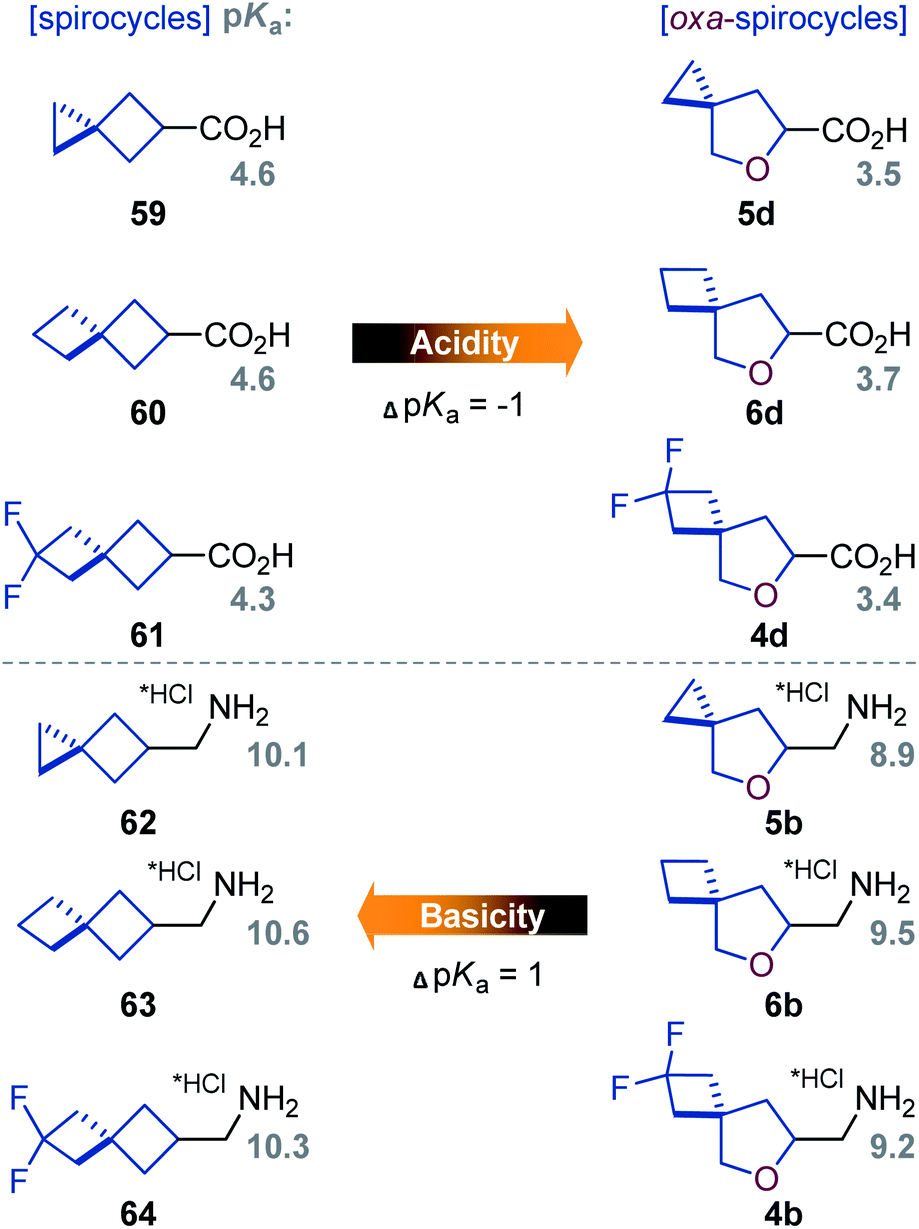 | ||
| Fig. 3 Experimental pKa values of acids 59–61 and 4d–6d, and conjugated amines 62·HCl–64·HCl and 4b·HCl–6b·HCl. | ||
To study the effect of incorporation of an oxygen atom on water solubility and lipophilicity of spirocyclic structures, we first synthesized model compounds 65–73 by standard amide coupling (Table 2).
D) and water solubility of model compounds 65–73
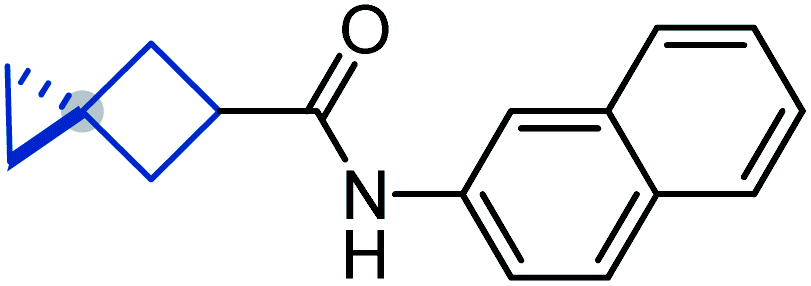


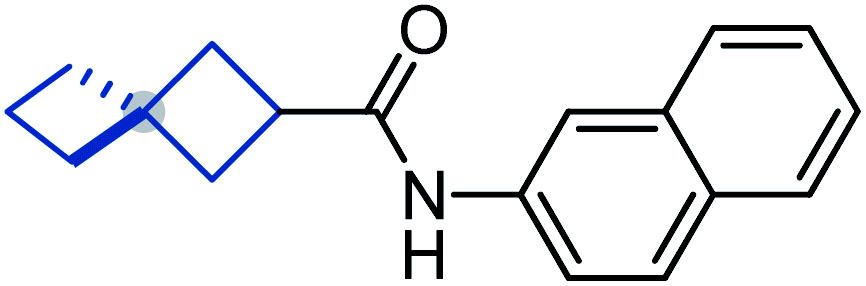
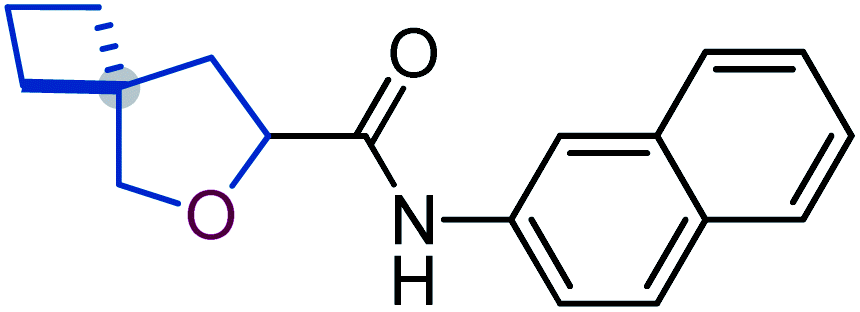


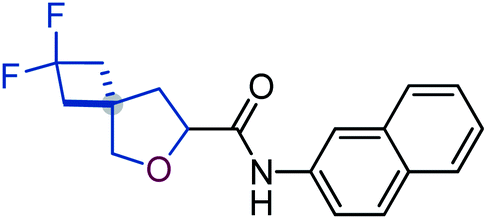
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) D7.4).
Incorporation of an oxygen atom into the spirocyclic unit also decreased lipophilicity. The lipophilicity index (logD) of oxa-spirocyclic models 67, 70, and 73 was ca. one order of magnitude lower than that of spirocyclic models 66, 69, and 72: 4.5 (66) vs. 3.6 (67); 4.9 (69) vs. 4.0 (70); 4.4 (72) vs. 3.6 (73).
D7.4).
Incorporation of an oxygen atom into the spirocyclic unit also decreased lipophilicity. The lipophilicity index (logD) of oxa-spirocyclic models 67, 70, and 73 was ca. one order of magnitude lower than that of spirocyclic models 66, 69, and 72: 4.5 (66) vs. 3.6 (67); 4.9 (69) vs. 4.0 (70); 4.4 (72) vs. 3.6 (73).
In brief, oxa-spirocyclic compounds have (a) dramatically higher solubility (up to 40 times), and (b) lower lipophilicity (ΔlogD = ca. 1) than common spirocycles.
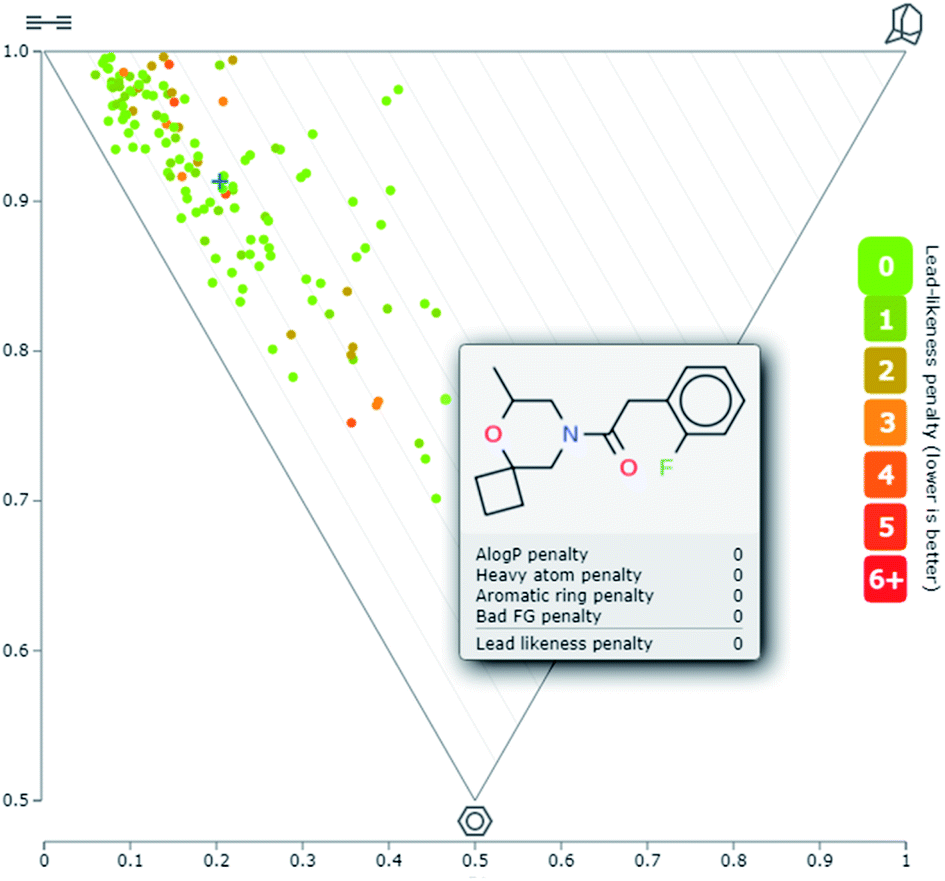
logP < 3 (Fig. 4). The mean lead-likeness index of all 130 compounds was 0.68. To assess the three-dimensionality of the library, the principal moments of inertia (PMI) plot was generated (Fig. 5). This plot confirmed that many of these lead-like compounds also showed significant shape diversity. The fraction of sp3-hybridised carbons, F(sp3), in the library was also analyzed, as it was previously shown to correlate with success in drug discovery projects.2 The average F(sp3) index was 0.79 which is significantly higher than that of a random molecule from the ZINC database (0.33).38
 | ||
| Fig. 4 Distribution of virtual molecules, logP (y)–MW (x), obtained by decoration of amines 4b, 32b, 43b, 8g, and 45g in LLAMA software. The chemical structure of a representative derivative of 4b is shown. | ||
 | ||
| Fig. 5 Principal moments of inertia (PMI) plot of virtual molecules, obtained by decoration of amines 4b, 32b, 43b, 8g, and 45g in LLAMA software. The chemical structure of a representative derivative of 45g is shown. | ||
 | ||
| Fig. 6 Antihypertensive drug terazosin (74) and oxa-spiro-substituted analogues 75, 76, 77, 78, and 79. | ||
Finally, we measured and compared the biological activity of terazosin (74) and all its analogues 75–79. Studies were conducted using 7.5 month-old spontaneously hypertensive (SHR) male rats with an average body weight of 329 ± 30 g and basal systolic blood pressure not less than 185 mm Hg.41 The compounds were dissolved in saline (74 and 76–79), or water containing 20% Captisol (75). Animals received 3 mg kg−1 compound in 5 ml kg−1 vehicle per os once. Five animals per group were assigned. Systolic and diastolic blood pressure (BP)42 were measured 15, 60, 120, 180 and 240 min after the dosing (Fig. 7 and 8). Statistical analysis was performed using two-way ANOVA with Tukey's multiple comparisons test.
 | ||
| Fig. 7 The dynamics of systolic blood pressure in SHR rats at different time-points after single PO administration of vehicle (saline or 20% Captisol) or test substances at a dose of 3 mg kg−1. Compounds were dissolved in saline (74 and 76–79) or 20% Captisol in water (75). Data are expressed as means ± SEM. *, **, *** – P < 0.05, 0.01, 0.001 compared with vehicle groups; # – P < 0.01 compared with terazosin treated group for the same time point. | ||
 | ||
| Fig. 8 The dynamics of diastolic blood pressure in SHR rats at different time-points after single PO administration of vehicle (20% Captisol) or test substances at a dose of 3 mg kg−1. Compounds were dissolved in saline (74 and 76–79) or 20% Captisol in water (75). Data are expressed as means ± SEM. *, ** – P < 0.05, 0.01 compared with vehicle groups for the same time point. | ||
Terazosin (74) and its oxa-spirocyclic analogues 75, 76, and 78 had similar biological profiles in vivo – all compounds demonstrated significant systolic BP decrease in comparison with corresponding vehicle groups after 15 min and 60 min of treatment (Fig. 7); while compounds 77 and 79 – only after 60 min (Fig. 7). For the 120 min time point, the BP lowering effect was statistically significant only for compound 78 in comparison with the vehicle. Compound 78 demonstrated prolonged effect even compared to terazosin (74), whereby terazosin had no statistically significant activity at 120 min. This effect may suggest that compound 78 modification may contribute to the prolongation of the drug action.
It should be noted that systolic BP decrease of the cyclopropane-containing analogue 75 after 15 min of treatment was significantly higher (Fig. 7) than that of the original drug terazosin (74).
Conclusions
During the recent decade, oxa-spirocycles undeservedly remained in the shadow compared to the more popular spirocyclic analogues (Fig. 2). The key reason was an absence of a general practical approach to them. In this work, we developed such an approach. Oxa-spirocycles were easily synthesized through the iodocyclization reaction. Using common (2e) and radical (1e) modifications, the obtained iodides 4a–48a were easily converted into >150 oxa-spirocyclic derivatives with appropriately protected functional groups, which could be directly used in medicinal chemistry projects. Incorporation of an oxygen atom into the spirocyclic unit was shown to dramatically increase its solubility (by up to 40 times: 66 vs. 67, Table 1) and lower lipophilicity. The developed protocol not only gave access to novel molecules, but also significantly simplified the synthesis of the known ones (Scheme 10). In addition, five oxa-spirocyclic analogues 75–79 of the antihypertensive drug terazosin (74) were prepared. Analogue 75 showed significantly higher potency in vivo than the original drug (Fig. 7 and 8).We believe that with this general simple approach to oxa-spirocyclic iodides and procedures for their modifications (Schemes 4–9), oxa-spirocycles will soon become very popular in chemistry.
Data availability
Supporting data for this article have been uploaded as part of the ESI material.†Author contributions
Conceptualization – PKM; investigation and methodology – KF, TD, DG, TS, VV, OS, VM, IK, EL, VVL, VRB, RIV, AIV, AVB, VVS, RI, KS, ASK, YVD, DV, VR, OP, HK; supervision – IP, PB, AAT, PKM; writing, original draft – PKM; writing, reviewing & editing – ASK, AAT, OP, HK, IP, PB, PKM.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Enamine Ltd for financial support. The authors are also grateful to Mr A. Zhemera and Dr D. Panov for the synthesis of model compounds 65–73. PM is grateful to Mrs I. Sadkova for the help with the preparation of the manuscript, and to Dr E. Rusanov (IOC) for X-ray analysis of compound 50. A part of this project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement No. 101000893 – BENOVELTY).Notes and references
- (a) A. R. D. Taylor, M. Maccoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef; (b) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed; (b) F. Lovering, RSC Med. Chem., 2013, 4, 515–519 RSC.
- For recent interesting examples on building 3D-shaped molecules, see: (a) M. S. Oderinde, E. Mao, A. Ramirez, J. Pawluczyk, C. Jorge, L. A. M. Cornelius, J. Kempson, M. Vetrichelvan, M. Pitchai, A. Gupta, A. Kumar Gupta, N. A. Meanwell, A. Mathur and T. G. M. Dhar, J. Am. Chem. Soc., 2020, 142(6), 3094–3103 CrossRef CAS; (b) A. Denisenko, P. Garbuz, S. Shishkina, N. M. Voloshchuk and P. Mykhailiuk, Angew. Chem., Int. Ed., 2020, 59, 20515–20521 CrossRef CAS; (c) T.-G. Chen, L. M. Barton, Y. Lin, J. Tsien, D. Kossler, I. Bastida, S. Asai, C. Bi, J. S. Chen, M. Shan, H. Fang, F. G. Fang, H.-W. Choi, L. Hawkins, T. Qin and P. S. Baran, Nature, 2018, 560, 350–354 CrossRef CAS PubMed; (d) B. A. Chalmers, H. Xing, S. Houston, C. Clark, S. Ghassabian, A. Kuo, B. Cao, A. Reitsma, C.-E. P. Murray, J. E. Stok, G. M. Boyle, C. J. Pierce, S. W. Littler, D. A. Winkler, P. V. Bernhardt, C. Pasay, J. J. De Voss, J. McCarthy, P. G. Parsons, G. H. Walter, M. T. Smith, H. M. Cooper, S. K. Nilsson, J. Tsanaktsidis, G. P. Savage and C. M. Williams, Angew. Chem., Int. Ed., 2016, 55, 3580–3585 CrossRef CAS; (e) R. Gianatassio, J. M. Lopchuk, J. Wang, C.-M. Pan, L. R. Malins, L. Prieto, T. A. Brandt, M. R. Collins, G. M. Gallego, N. W. Sach, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, Science, 2016, 351, 241–246 CrossRef CAS PubMed; (f) D. F. J. Caputo, C. Arroniz, A. B. Dürr, J. J. Mousseau, A. F. Stepan, S. J. Mansfield and E. A. Anderson, Chem. Sci., 2018, 9, 5295–5300 RSC; (g) M. L. J. Wong, J. J. Mousseau, S. J. Mansfield and E. A. Anderson, Org. Lett., 2019, 21, 2408–2411 CrossRef CAS; (h) J. Nugent, C. Arroniz, B. R. Shire, A. J. Sterling, H. D. Pickford, M. L. J. Wong, S. J. Mansfield, D. F. J. Caputo, B. Owen, J. J. Mousseau, F. Duarte and E. A. Anderson, ACS Catal., 2019, 9, 9568–9574 CrossRef CAS; (i) J. Nugent, B. R. Shire, D. F. J. Caputo, H. D. Pickford, F. Nightingale, I. T. T. Houlsby, J. J. Mousseau and E. A. Anderson, Angew. Chem., Int. Ed., 2020, 59, 11866–11870 CrossRef CAS PubMed; (j) J. M. Lopchuk, K. Fjelbye, Y. Kawamata, L. R. Malins, C.-M. Pan, R. Gianatassio, J. Wang, L. Prieto, J. Bradow, T. A. Brandt, M. R. Collins, J. Elleraas, J. Ewanicki, W. Farrell, O. O. Fadeyi, G. M. Gallego, J. J. Mousseau, R. Oliver, N. W. Sach, J. K. Smith, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 3209–3226 CrossRef CAS PubMed; (k) X. Ma, D. L. Sloman, Y. Han and D. J. Bennett, Org. Lett., 2019, 21, 7199–7203 CrossRef CAS PubMed; (l) J.-X. Zhao, Y. Chang, J. Elleraas, T. P. Montgomery, J. E. Spangler, S. K. Nair, M. D. Bel, G. M. Gallego, J. J. Mousseau, M. A. Perry, M. R. Collins, J. C. Vantourout and P. S. Baran, 1,2-Difunctionalized Bicyclo[1.1.1]pentanes: Long Sought After Mimetics for ortho/meta-Substituted Arenes, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2108881118 CrossRef CAS; (m) A. R. Gomez-Angel, J. R. Donald, J. D. Firth, C. De Fusco, R. I. Storer, D. J. Cox and P. O'Brien, Tetrahedron, 2021, 83, 131961 CrossRef CAS; (n) T. D. Downes, S. P. Jones, H. F. Klein, M. C. Wheldon, M. Atobe, P. S. Bond, J. D. Firth, N. S. Chan, L. Waddelove, R. E. Hubbard, D. C. Blakemore, C. De Fusco, S. D. Roughley, L. R. Vidler, M. A. Whatton, A. J. A. Woolford, G. L. Wrigley and P. O'Brien, Chem.–Eur. J., 2020, 26, 8969–8975 CrossRef CAS; (o) S. Rice, D. J. Cox, S. P. Marsden and A. Nelson, Chem. Commun., 2021, 57, 599–602 RSC; (p) S. Rice, D. J. Cox, S. P. Marsden and A. Nelson, Tetrahedron, 2019, 75, 130513 CrossRef CAS; (q) A. F. Trindade, E. L. Faulkner, A. G. Leach, A. Nelson and S. P. Marsden, Chem. Commun., 2020, 56, 8802–8805 RSC; (r) T. A. King, H. L. Stewart, K. T. Mortensen, A. J. P. North, H. F. Sore and D. R. Spring, Eur. J. Org. Chem., 2019, 2019, 5219–5229 CrossRef CAS; (s) T. J. Osberger, S. L. Kidd, T. A. King and D. R. Spring, Chem. Commun., 2020, 56, 7423–7426 RSC; (t) A. Sveiczer, A. J. P. North, N. Mateu, S. L. Kidd, H. F. Sore and D. R. Spring, Org. Lett., 2019, 21, 4600–4604 CrossRef CAS PubMed; (u) F. E. Held, A. A. Guryev, T. Fröhlich, F. Hampel, A. Kahnt, C. Hutterer, M. Steingruber, H. Bahsi, C. von Bojničić-Kninski, D. S. Mattes, T. C. Foertsch, A. Nesterov-Mueller, M. Marschall and S. B. Tsogoeva, Nat. Commun., 2017, 8, 15071 CrossRef; (v) F. E. Held, A. A. Guryev, T. Fröhlich, F. Hampel, A. Kahnt, C. Hutterer, M. Steingruber, H. Bahsi, C. von Bojničić-Kninski, D. S. Mattes, T. C. Foertsch, A. Nesterov-Mueller, M. Marschall and S. B. Tsogoeva, Nat. Commun., 2017, 8, 15071 CrossRef PubMed.
- (a) F. W. Goldberg, J. G. Kettle, T. Kogej, M. W. D. Perry and N. P. Tomkinson, Drug Discovery Today, 2015, 20, 11–17 CrossRef; (b) P. K. Mykhailiuk, Org. Biomol. Chem., 2019, 17, 2839–2849 RSC; (c) G. M. Locke, S. S. R. Bernhard and M. O. Senge, Chem.–Eur. J., 2019, 25, 4590–4647 CrossRef CAS PubMed.
- J. A. Burkhard, B. Wagner, H. Fischer, F. Schuler, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 3524–3527 CrossRef CAS.
- For recent examples, see: (a) M. Espinosa, H. Noda and M. Shibasaki, Org. Lett., 2019, 21, 9296–9299 CrossRef CAS PubMed; (b) N. J. Floden, N. J. Flodén, A. Trowbridge, D. Willcox, S. M. Walton, Y. Kim and M. J. Gaunt, J. Am. Chem. Soc., 2019, 141, 8426–8430 CrossRef CAS; (c) W.-Y. Siau and J. W. Bode, J. Am. Chem. Soc., 2014, 136, 17726–17729 CrossRef CAS PubMed; (d) G. Müller, T. Berkenbosch, J. C. J. Benningshof, D. Stumpfe and J. Bajorath, Chem.–Eur. J., 2017, 23, 703–710 CrossRef; (e) A. Sveiczer, A. J. P. North, N. Mateu, S. L. Kidd, H. F. Sore and D. R. Spring, Org. Lett., 2019, 21, 4600–4604 CrossRef CAS PubMed.
- Our contribution to the field: (a) A. A. Kirichok, I. Shton, M. Kliachyna, I. Pishel and P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2017, 56, 8865–8869 CrossRef CAS; (b) A. A. Kirichok, I. O. Shton, I. M. Pishel, S. A. Zozulya, P. O. Borysko, V. Kubyshkin, O. A. Zaporozhets, A. A. Tolmachev and P. K. Mykhailiuk, Chem.–Eur. J., 2018, 24, 5444–5449 CrossRef CAS PubMed; (c) B. A. Chalyk, A. A. Isakov, M. V. Butko, K. V. Hrebeniuk, O. V. Savych, O. V. Kucher, K. S. Gavrilenko, T. V. Druzhenko, V. S. Yarmolchuk, S. Zozulya and P. K. Mykhailiuk, Eur. J. Org. Chem., 2017, 2017, 4530–4542 CrossRef CAS; (d) B. Chalyk, M. Butko, O. Yanshyna, K. Gavrilenko, T. Druzhenko and P. K. Mykhailiuk, Chem.–Eur. J., 2017, 23, 16782–16786 CrossRef CAS; (e) K. Fominova, T. Diachuk, I. V. Sadkova and P. K. Mykhailiuk, Eur. J. Org. Chem., 2019, 2019, 3553–3559 CrossRef CAS.
- Recent reviews: (a) K. Undheim, Synthesis, 2015, 47, 2497–2522 CrossRef CAS; (b) E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257–8322 CrossRef CAS; (c) Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673–3682 CrossRef CAS PubMed.
- V. V. Levterov, Y. Panasyuk, V. O. Pivnytska and P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2020, 59, 7161–7167 CrossRef CAS.
- Only one compound from Fig. 2 with an oxa-spirocyclic core was reported before in a peer-reviewed manuscript: S. T. Tang, Y. Liu, X. Gao, P. Wang, P. Huang and A. Lei, J. Am. Chem. Soc., 2018, 140, 6006–6013 CrossRef CAS (compound 3q, Scheme 3)..
- (a) F. J. Sayago, M. Ángeles Pradera, C. Gasch and J. Fuentes, Tetrahedron, 2006, 62, 915–921 CrossRef CAS; (b) A. Roy, B. Achari and S. B. Mandal, Tetrahedron Lett., 2006, 47, 3875–3879 CrossRef CAS; (c) Y. Nassar and O. Piva, Org. Biomol. Chem., 2020, 18, 5811–5815 RSC.
- Y. Tamaru, S. Kawamura and Z. Yoshida, Tetrahedron Lett., 1985, 26, 2885–2888 CrossRef CAS.
- M. Tredwell, J. A. R. Luft, M. Schuler, K. Tenza, K. N. Houk and V. Gouverneur, Angew. Chem., Int. Ed., 2008, 47, 357–360 CrossRef CAS.
- T. Cernak, K. Dykstra, D. Levorse, A. Verras, J. Balkovec, R. Nargund and R. DeVita, Tetrahedron Lett., 2011, 52, 6457–6459 CrossRef CAS.
- D. B. Li, M. Rogers-Evans and E. M. Carreira, Org. Lett., 2013, 15, 4766–4769 CrossRef CAS.
- T. O. Painter, J. R. Bunn, F. J. Schoenen, J. T. Douglas, V. W. Day and C. Santini, J. Org. Chem., 2013, 78, 3720–3730 CrossRef CAS.
- S. Kumar, P. D. Thornton, T. O. Painter, P. Jain, J. Downard, J. T. Douglas and C. Santini, J. Org. Chem., 2013, 78, 6529–6539 CrossRef CAS.
- A. Theodorou and C. G. Kokotos, Green Chem., 2017, 19, 670–674 RSC.
- J. Escudero, V. Bellosta and J. Cossy, Angew. Chem., Int. Ed., 2018, 57, 574–578 CrossRef CAS.
- Y. Kim, S. T. Kim, D. Kang, T. I. Sohn, E. Jang, M. H. Baik and S. Hong, Chem. Sci., 2018, 9, 1473–1480 RSC.
- J. T. Kohrt, P. H. Dorff, M. Burns, C. Lee, S. V. O'Neil, R. J. Maguire, R. Kumar, M. Wagenaar, L. Price and M. S. Lall, Org. Process Res. Dev., 2021 DOI:10.1021/acs.oprd.1c00075 , asap..
- T. Gaich and P. S. Baran, J. Org. Chem., 2010, 75, 4657–4673 CrossRef CAS PubMed.
- J. A. Bull, R. A. Croft, O. A. Davis, R. Doran and K. F. Morgan, Chem. Rev., 2016, 116, 12150–12233 CrossRef CAS PubMed.
- For similar types of expansion of azetidines into pyrrolidines, see: (a) B. Drouillat, I. V. Dorogan, M. Kletskii, O. N. Burov and F. Couty, J. Org. Chem., 2016, 81, 6677–6685 CrossRef CAS PubMed; (b) J. Dolfen, E. B. Boydas, V. Van Speybroeck, S. Catak, K. Van Hecke and M. D'hooghe, J. Org. Chem., 2017, 82, 10092–10109 CrossRef CAS PubMed; (c) S. Dekeukeleire, M. D'hooghe and N. De Kimpe, J. Org. Chem., 2009, 74, 1644–1649 CrossRef CAS; (d) S. Dekeukeleire, M. D'hooghe, K. W. Törnroos and N. De Kimpe, J. Org. Chem., 2010, 75, 5934–5940 CrossRef CAS PubMed.
- The CCDC number of compound 50 is 2046173.
- M. A. T. Blaskovich, J. Med. Chem., 2016, 59, 10807–10836 CrossRef CAS.
- A. V. Bogolubsky, Y. S. Moroz, P. K. Mykhailiuk, S. E. Pipko, A. I. Konovets, I. V. Sadkova and A. Tolmachev, ACS Comb. Sci., 2014, 16, 192–197 CrossRef CAS PubMed.
- (a) E. J. Barreiro, A. E. Kümmerle and C. A. M. Fraga, Chem. Rev., 2011, 111, 5215–5246 CrossRef CAS PubMed; (b) H. Schönherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 CrossRef.
- For recent examples, see: (a) N. E. Behnke, K. Lovato, M. Yousufuddin and L. Kürti, Angew. Chem., Int. Ed., 2019, 58, 14219–14223 CrossRef CAS PubMed; (b) M. R. Becker, A. D. Richardson and C. S. Schindler, Nat. Commun., 2019, 10, 5095 CrossRef; (c) S. Stanković, H. Goossens, S. Catak, M. Tezcan, M. Waroquier, V. Van Speybroeck, M. D'hooghe and N. De Kimpe, J. Org. Chem., 2012, 77, 3181–3190 CrossRef; (d) S. Kenis, M. D'hooghe, G. Verniest, T. A. D. Thi, C. P. The, T. Van Nguyen and N. De Kimpe, J. Org. Chem., 2012, 77, 5982–5992 CrossRef CAS; (e) B. J. Wang and M. A. J. Duncton, J. Org. Chem., 2020, 85, 13317–13323 CrossRef CAS PubMed; (f) R. Gianatassio and D. Kadish, Org. Lett., 2019, 21, 2060–2063 CrossRef CAS PubMed; (g) A. Fawcett, A. Murtaza, C. H. U. Gregson and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 4573–4578 CrossRef CAS.
- A. Kumaria and R. K. Singh, Bioorg. Chem., 2020, 96, 103578 CrossRef.
- After reduction of the C–I bond in iodides 45a–48a, we washed the reaction mixture with aq. AgNO3 to completely remove the iodide-anion. Otherwise, the next step – Pd-catalyzed cleavage of the N–Bn bond – could be slow.
- J. Wang, M. Shang, H. Lundberg, K. S. Feu, S. J. Hecker, T. Qin, D. G. Blackmond and P. S. Baran, ACS Catal., 2018, 8, 9537–9542 CrossRef CAS.
- We tried three different protocols for decarboxylative borylation of redox-active ester 18f-NHPI. Two other protocols that we tried: (a) C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson, A. K. Chatterjee, M. Yan and P. S. Baran, Science, 2017, 356, 6342 Search PubMed; (b) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS . [Ni]- and [B2Cat2]-protocols did not provide the needed product, while the [Cu]-protocol (ref. 32) gave the needed BPin derivative 18j on a ca. 500 mg scale in 58% yield..
- T. Cernak, N. J. Gesmundo, K. Dykstra, Y. Yu, Z. Wu, Z. C. Shi, P. Vachal, D. Sperbeck, S. He, B. A. Murphy, L. Sonatore, S. Williams, M. Madeira, A. Verras, M. Reiter, C. H. Lee, J. Cuff, E. C. Sherer, J. Kuethe, S. Goble, N. Perrotto, S. Pinto, D. M. Shen, R. Nargund, J. Balkovec, R. J. DeVita and S. D. Dreher, J. Med. Chem., 2017, 60, 3594–3605 CrossRef CAS PubMed.
- O. Dirat, J. M. Elliott, I. T. Huscroft, R. A. Jelley, J. J. Kulagowski, P. A. Raubo, D. E. Shaw, F. Sternfeld, C. J. Swain. WO 2004/078750 A1, 2004.
- M. Morgenthaler, E. Schweizer, A. Hoffmann-Roeder, F. Benini, R. E. Martin, G. Jaeschke, B. Wagner, H. Fischer, S. Bendels, D. Zimmerli, J. Schneider, F. Diederich, M. Kansy and K. Muller, ChemMedChem, 2007, 2, 1100–1115 CrossRef CAS PubMed.
- I. Colomer, C. J. Empson, P. Craven, Z. Owen, R. G. Doveston, I. Churcher, S. P. Marsden and A. Nelson, Chem. Commun., 2016, 52, 7209–7212 RSC.
- J. J. Irwin, T. Sterling, M. M. Mysinger, E. S. Bolstad and R. G. Coleman, J. Chem. Inf. Model., 2012, 52, 1757–1768 CrossRef CAS.
- https://clincalc.com/DrugStats/Top300Drugs.aspx .
- https://clincalc.com/DrugStats/Drugs/Terazosin .
- The study design, animal selection, handling and treatment were in accordance with Bienta's Animal Care and Use Guidelines, and European Union directive 2010/63/EU.
- Blood pressure (BP) was measured by a tail-cuff method. We used a Coda non-invasive blood-pressure system (Kent Scientific Corporation, CT, USA).
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2046173. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03615g |
| This journal is © The Royal Society of Chemistry 2021 |