
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstrate induced generation of transient self-assembled catalytic systems
Syed Pavel
Afrose
,
Chandranath
Ghosh
and
Dibyendu
Das
 *
*
Department of Chemical Sciences & Centre for Advanced Functional Materials, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur, West Bengal 741246, India. E-mail: dasd@iiserkol.ac.in
First published on 8th November 2021
Abstract
Living matter is sustained under non-equilibrium conditions via continuous expense of energy which is coordinated by complex organized events. Spatiotemporal control over exquisite functions arises from chemical complexity under non-equilibrium conditions. For instance, extant biology often uses substrate binding events to access temporally stable protein conformations which show acceleration of catalytic rates to subsequently degrade the substrate. Furthermore, thermodynamically activated but kinetically stable esters (GTP) induce the change of conformation of cytoskeleton proteins (microtubules) which leads to rapid polymerization and triggers an augmentation of catalytic rates to subsequently degrade the ester. Importantly, high-energy assemblies composed of non-activated building blocks (GDP-tubulin) are accessed utilizing the energy dissipated from the catalytic conversion of GTP to GDP from the assembled state. Notably, some experimental studies with simple self-assembled systems have elegantly mimicked the phenomena of substrate induced transient generation of catalytic conformations. Through this review, we endeavour to highlight those select studies which have used simple building blocks to demonstrate substrate induced self-assemblies that subsequently show rate acceleration to convert the substrate into waste. The concept of substrate induced self-assembly of building blocks and rate acceleration from the assembled state has the potential to play a predominant role in the preparation of non-equilibrium systems. The design strategies covered in this review can inspire the possibilities of accessing high energy self-assembled structures that are seen in living systems.
 Syed Pavel Afrose | Syed Pavel Afrose is currently pursuing his PhD in the Department of Chemistry, Indian Institute of Science Education and Research (IISER) Kolkata, India, under the supervision of Dr Dibyendu Das. He completed both his BSc and MSc degree from Visva Bharati University, India. His research interest is to design dynamic reaction networks showing advanced traits akin to living matter. |
 Chandranath Ghosh | Chandranath Ghosh is currently pursuing his PhD in the Department of Chemistry, Indian Institute of Science Education and Research (IISER) Kolkata, India, under the supervision of Dr Dibyendu Das. He completed his BSc from Midnapore College and MSc degree from Banaras Hindu University, India. His research interest is exploring the catalytic potential of amyloid based short peptide sequences under equilibrium and out of equilibrium conditions. |
 Dibyendu Das | Dibyendu Das leads an interdisciplinary group that focuses on the areas of systems chemistry and chemical evolution and developing functional materials based on peptide nanotechnology. He is an Associate Professor at the Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, India. He conducted his PhD work at IACS, Kolkata, India and Post-Doctoral research at Emory University, USA. |
1. Introduction
Living matter is an emergent collective ensemble of cooperative machineries that accesses functions with remarkable spatiotemporal control.1–3 The illustrations of such temporal functions are diverse, for instance, moving a payload, organization of the intracellular structure, cell division and so forth.1–4 The flux of chemical energy provided by high energy substrates drives such complex and highly synchronised processes in a far-from-equilibrium regime.5–10 Steps of energy consumption and dissipation are dynamically coupled via biocatalytic steps that generate the desired outputs and provide a regulatory mechanism of the metabolic networks. Extant enzymes thus play pivotal roles in facilitating such complex interrelated reactions required for the sustainment of the far-from-equilibrium state of living matter.1–4Contemporary biology uses the binding of molecular substrates to induce change of protein conformations which are catalytically proficient.11–13 For example, peptide-based substrates induce change of conformations in proteases like caspase-1 and the main protease (Mpro) of SARS-CoV where the binding drives the formation of the catalytically active dimers (Fig. 1a).14,15 Upon completion of the catalytic cycles, the active dimers dissociate back to the catalytically less proficient monomers. The functional regulation observed in such complex proteins is also found in the switching mechanism of the energy driven flagellar motor movements.16
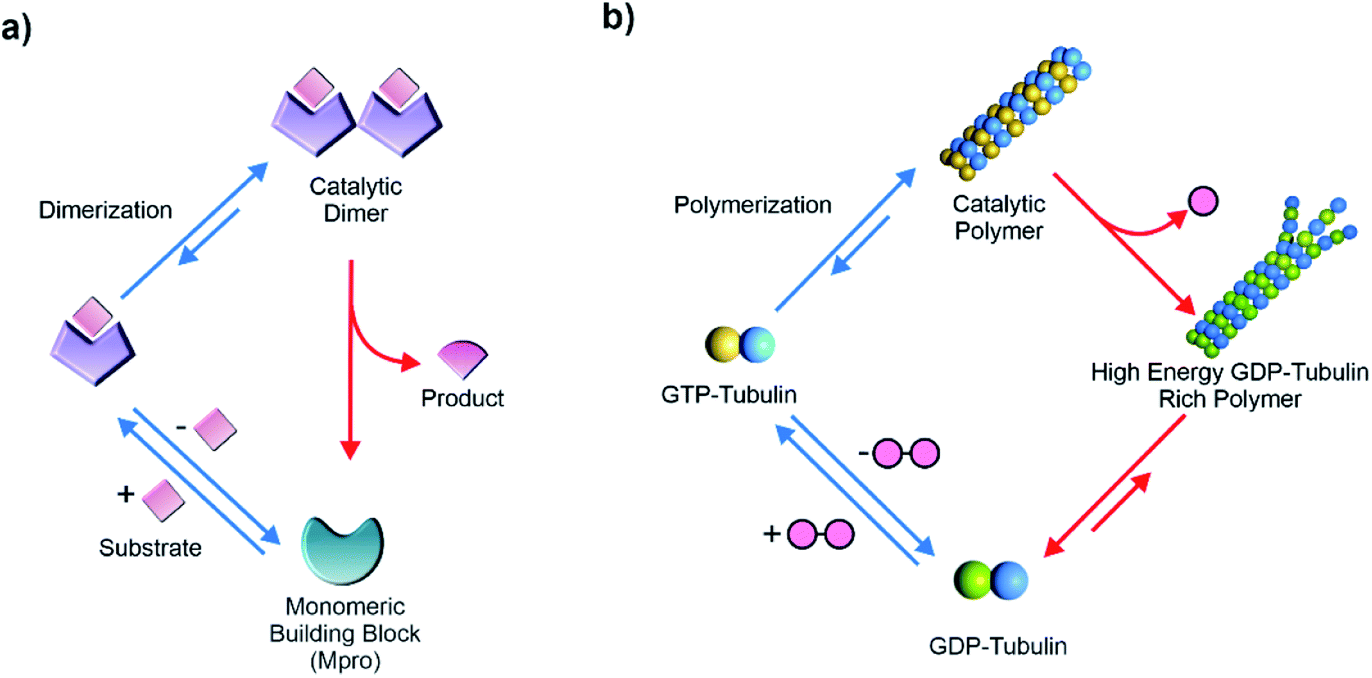 | ||
| Fig. 1 (a) Representative scheme showing the substrate induced conformation change and dimerization of catalytic SARS CoV main protease (Mpro). (b) GTP induced dynamic polymerization of catalytic cytoskeleton proteins. | ||
Interestingly, substrate driven generation of catalytically active non-equilibrium polymers is observed in the cytoskeleton filaments for performing mechanical work with acute spatiotemporal control. Such dynamic polymers are exploited across life forms as an evolutionary strategy for vital roles, from mitotic spindle formation, cellular integrity to muscle movement and so forth. The transient polymerization of the cytoskeleton proteins is regulated by the binding of thermodynamically activated substrates like nucleoside triphosphates.13,17 For instance, in the case of microtubules, GTP binding with tubulin building block induces a change of conformation and leads to formation of polymers which subsequently catalyse the rapid hydrolysis of GTP (Fig. 1b). Although the GTP-tubulin polymerization is thermodynamically driven, the GTP consumption releases energy which drives the formation of the non-equilibrium GDP-tubulin rich polymers (high-energy assemblies composed of non-activated building blocks).4,13,17 These high energy assemblies eventually collapse and tubulin dimers are regenerated, re-establishing the equilibrium. Acceleration of catalytic rates by the tubulin polymers plays a critical role in the energy release and storage in the non-equilibrium polymeric states. Notably, such transfer of energy to the non-equilibrium polymers features the installation of a kinetic asymmetry in its energy consumption pathways, first revealed from the recent simulations of Astumian and Prins et al.18–21 Kinetic asymmetry is generated due to the binding of GTP exclusively with building blocks (and not with the assemblies) and also due to the rate acceleration and energy dissipation specifically from the polymerized state. This kinetic preference is vital for accessing the non-equilibrium high energy assemblies (GDP-tubulin) and also for the directionality in the coupled processes of GTP-GDP conversion and microtubule polymerization. It was further noted that if experiments are carried out under stationary conditions (open system), the presence of kinetic asymmetry can allow access to the high energy self-assembled state as the most populated one.20 In a closed system, high energy states may also be transiently accessed in the absence of kinetic asymmetry. In such cases switching of environmental conditions due to batchwise addition of the substrate leads to the temporal alteration of energy landscape.20
The rate acceleration, feedback loops and time delays are crucial not only for the oscillations observed in microtubule length but also for the sustainment of various biochemical oscillators.1 Towards this end, the non-equilibrium polymers of biology have inspired some elegant attempts where acceleration of catalytic rates has been witnessed as a result of interaction with the substrate in purely chemical based systems. This review aims to highlight such contributions where a substrate induced self-assembled state has been accessed on batchwise addition of a substrate that showed an increase of catalytic prowess to subsequently degrade the substrate. As observed in the case of microtubules, many of the covered examples suggest the binding of the substrate primarily with the building blocks and show rate acceleration specifically from the assembled state. Although the presence of high energy assemblies or installation of kinetic asymmetry was neither demonstrated nor investigated in these examples, yet these substrate-induced catalytic systems can in principle play a predominant role in the preparation of non-equilibrium systems where high energy assemblies can be accessed.19,20 This review aims to engage these examples with an eye on the factors that could possibly have installed kinetic asymmetry in the system, i.e., the substrate preferentially interacting with the monomeric building block and the substrate to product conversion occurring specifically from the self-assembled catalytic states.
Examples of diverse approaches such as cooperative effects, metal–ligand interactions, host–guest supramolecular interactions, hydrophobic interactions and phase separation that have been aptly utilized to achieve the catalytic states have been discussed. We have intentionally kept the sections minimal to underpin the elements of substrate induced assembly from the context of morphological diversity. Due to the presence of in-depth reviews in the field of chemically fuelled non-equilibrium systems, we have not included studies which do not demonstrate substrate induced generation of catalytic systems.22–26
2. Substrate induced formation of micelles and vesicles
The proximity of catalytically active residues via self-assembly can result in cooperative effects which can be an interesting yet simple way of achieving acceleration of catalytic rates. In 2018, Prins, Chen and co-workers utilised anionic HPNPP, a model substrate used in the study of RNA phosphodiester hydrolysis, to induce the dissipative assembly of a surfactant (C16TACN, Fig. 2a) coordinated to Zn2+.27 HPNPP was found to drastically reduce the aggregation concentration of the surfactant resulting in the formation of vesicles. The increased proximity of the Zn2+ ions in the self-assembled vesicular state resulted in rate acceleration via cooperative effects, subsequently degrading the substrate HPNPP to the cyclic phosphate and p-nitrophenolate (Fig. 2a). Important to note here, just like in the case of microtubules, the substrate interacted with the surfactant (building block) and the degradation of the substrate apparently occurred primarily from the assembled state via cooperative effects. The presence of such a cooperative catalytic mechanism can be important to install kinetic asymmetry in the energy consumption pathways which can in principle help in the accessing of higher energy states.20,27 To investigate the accelerated rates from the assemblies, the concentration of the surfactant was varied which revealed a sudden increase in initial rates of hydrolysis as the critical assembly concentration (CAC) was reached. This suggested the role of the vesicular assemblies in degrading the substrate (Fig. 2b). Furthermore, changing the concentration of the metal ion yielded a sigmoidal profile of the initial rates, which implied that the cooperative effect of the ions was at play (Fig. 2c). Hydrolysis of the ester resulted in the formation of smaller assemblies, suggesting that the products were able to stabilize the assemblies to a certain extent. Nonetheless, the results demonstrated the substrate induced generation of catalytic vesicles where enhancement of rates was an important factor for the realization of the transient self-assembled state.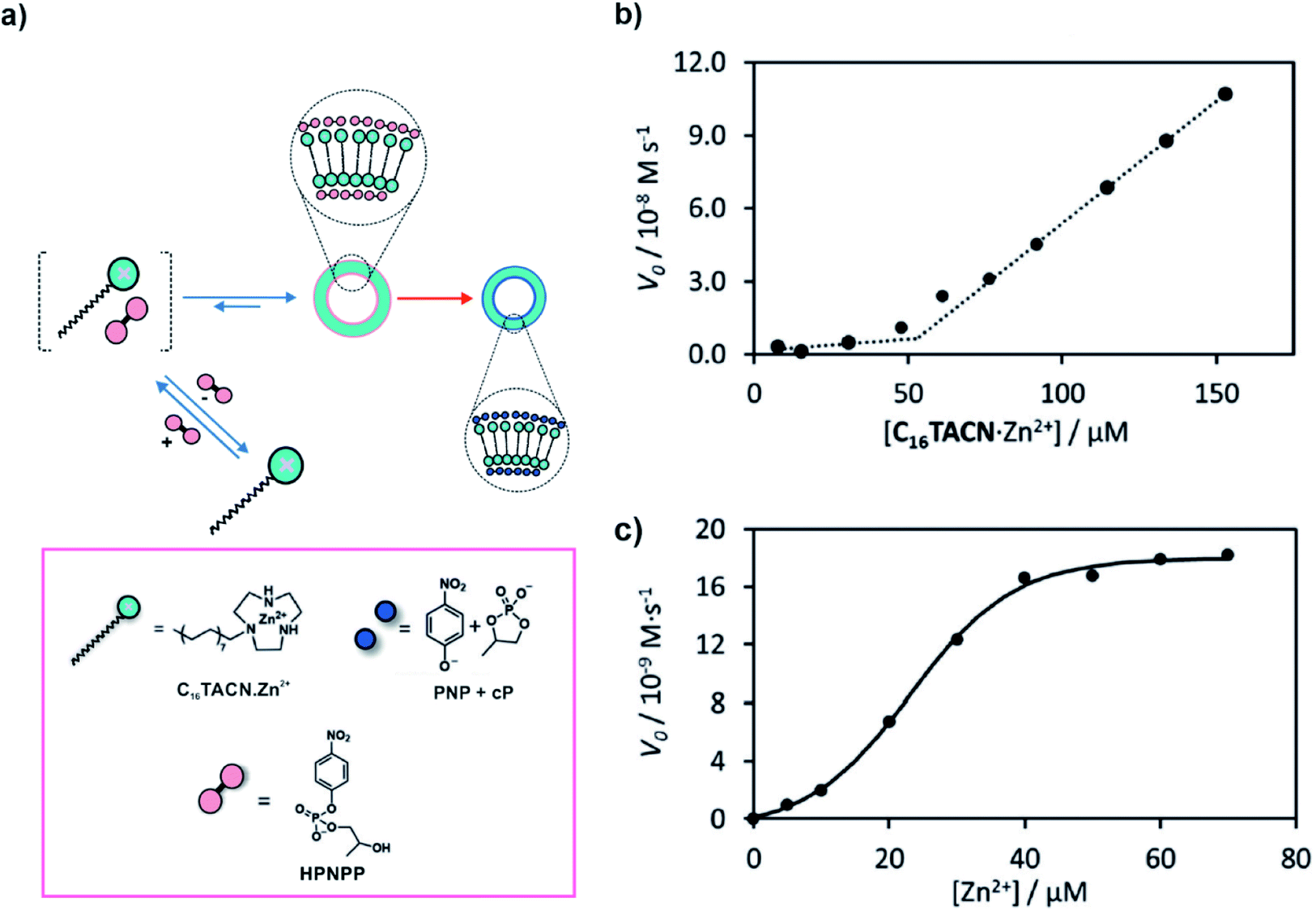 | ||
| Fig. 2 (a) The anionic substrate HPNPP induces the assembly of the surfactant C16TACN·Zn2+ to vesicles. Cooperative effects lead to enhancement of catalytic activity resulting in the cleavage of the substrate. (b) Variation of the initial speed of HPNPP hydrolysis ([HPNPP] = 62 μM) with increasing concentrations of C16TACN·Zn2+. The dotted lines are the linear fit to the first three and last three data points. (c) Variation of the initial speed of HPNPP hydrolysis with increasing concentrations of Zn2+ ([HEPES buffer] = 5 mM, [C16TACN] = 50 μM, [HPNPP] = 500 μM, 40 °C). The solid line represents the sigmoidal fit of the experimental data. Adapted from ref. 27 with permission from John Wiley and Sons. | ||
The catalytic metathesis reaction between phase separated hydrophobic and hydrophilic components has been utilised to access a kinetically stable amphiphile in an autocatalytic manner in the presence of a 2nd generation Grubbs catalyst. However, in contrast to what is observed in autocatalysis where the concentration is maximised and equilibrium ‘death’ phase is reached, Fletcher and co-workers observed transient assembly (Fig. 3a). Briefly, the authors devised an autopoietic system based on small molecules where an activated amphiphile was accessed.28 Phase separated alkenes (1 and 2) of contrasting polarity were used to metathesize the formation of amphiphile 3via a ruthenium-based Grubbs catalyst already present in the mixture (Fig. 3a). The resultant thermodynamically activated amphiphile acted like a building block to self-assemble into aggregates. The self-assembly had catalytic efficiency which facilitated the autocatalytic formation of 3. Interestingly, after a certain concentration of the thermodynamically activated amphiphile was accessed, the amphiphile was degraded through a second metathesis reaction, giving rise to the water-soluble thermodynamic product 4 (Fig. 3b, black curve). Notably, there was a time delay prior to the formation of the thermodynamic product 4 (Fig. 3b) and did not occur from the beginning as self-assembly of 3 was catalysing the formation of 4. In contrast to classical examples of autocatalytic reactions, the exponential phase of the autocatalytic reaction was observed away from the equilibrium. In this case the autocatalytic generation of the kinetically stable amphiphile resulted in self-assemblies which subsequently fostered the second metathesis reaction leading to the formation of the equilibrium product 4. When the mixture was seeded with 20 mol% of the transient species 3, the formation of 4 was observed from the beginning (Fig. 3c, black curve). The fact that the generation of 4 and the autonomous disassembly of the transient amphiphile occurred after a certain concentration was reached shows that the amphiphile needed to self-assemble in order to produce the equilibrium product (Fig. 3b and c). This is similar to the disassembly of microtubules as assembly of the GTP-tubulin is essential for catalysis and dissipation. Most importantly, it signifies that the design strategy can in principle control the dynamic instability of the autopoietic moiety.
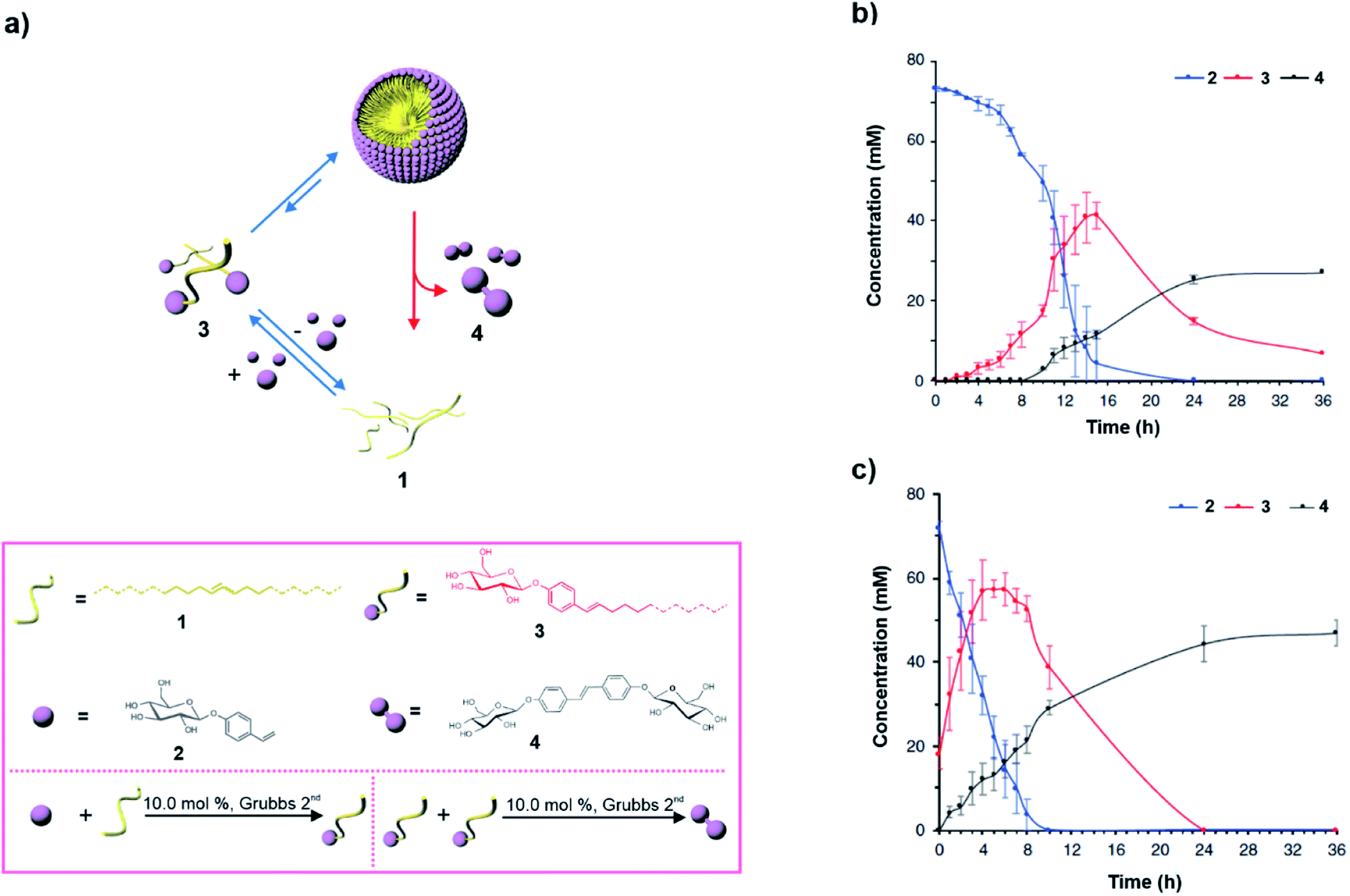 | ||
| Fig. 3 (a) In a biphasic mixture, compounds 1 and 2 are metathesized by a ruthenium catalyst present in the medium (not shown in the scheme for clarity) to create the thermodynamically activated amphiphile 3. After 3 accesses a micellar structure, a second metathesis reaction produces the thermodynamically stable product 4, and results in disassembly. Structures of the compounds and the reactions are shown in the box. (b) Change of the concentration of different species in the biphasic mixture with time. (c) Change of the concentration of different species in the biphasic mixture in the presence of 20 mol% of seed 3. Adapted from ref. 28 with permission from Nature Publishing House. | ||
3. Substrate induced formation of fibrillar networks
Cooperativity among catalytic sites has been proposed to be an effective way of installing kinetic asymmetry in chemical reaction cycles for the realization of dissipative self-assembly.27 In 2019, Das and co-workers demonstrated substrate induced generation of catalytic nanostructures and rate acceleration from the assembled state.29 Briefly, an amphiphilic molecule having a lipid chain coupled to histidine was used (C18H, Fig. 4a). In the presence of p-nitrophenol and a high energy coupling agent (EDC), C18H was partially converted to a p-nitro phenyl ester (C18H-NP, Fig. 4a) which readily co-assembled with the unreacted C18H. With time, the co-assembled system showed the emergence of a fibrillar network. The formation of such co-assembled fibres resulted in rate acceleration via cooperative effects of the proximal histidine residues. This consequently resulted in energy dissipation predominantly from the assembled fibrous states, resulting in autonomous disassembly. Time dependent UV-vis spectra showed formation of C18H-NP followed by the consumption of the formed ester as indicated by the absorbance changes (Fig. 4b). As controls, when a short chain analogue of C18H (acetyl histidine, C2H, Fig. 4b) incapable of forming assemblies or a non-catalytic analogue containing a phenylalanine instead of histidine (C18F) was used, gradual formation of the ester was observed (Fig. 4b). Cooperativity between catalytic histidine residues was investigated with the help of pH dependent activity studies. The rates at different pH from 5.5 to 7.5 exhibited a bell-shaped profile with the highest hydrolytic rate at a pH close to the pKa of histidine. At pH 7.9, base catalysed formation of the ester was observed instead (Fig. 4c). Moreover, the lifetime of the fibres was dependent on the stability of the esters as less hydrolysable analogues yielded longer lifetimes of the fibres. The accelerated catalysis of the substrate from the self-assembled states due to cooperative catalysis makes this system significant from the context of designing reaction networks involving kinetic asymmetry. This in turn can help accessing high energy states with unique properties. Although not attempted by the authors, it would be interesting to probe the system under stationary conditions to investigate the time dependent evolution of the system and any possible generation of high energy assemblies.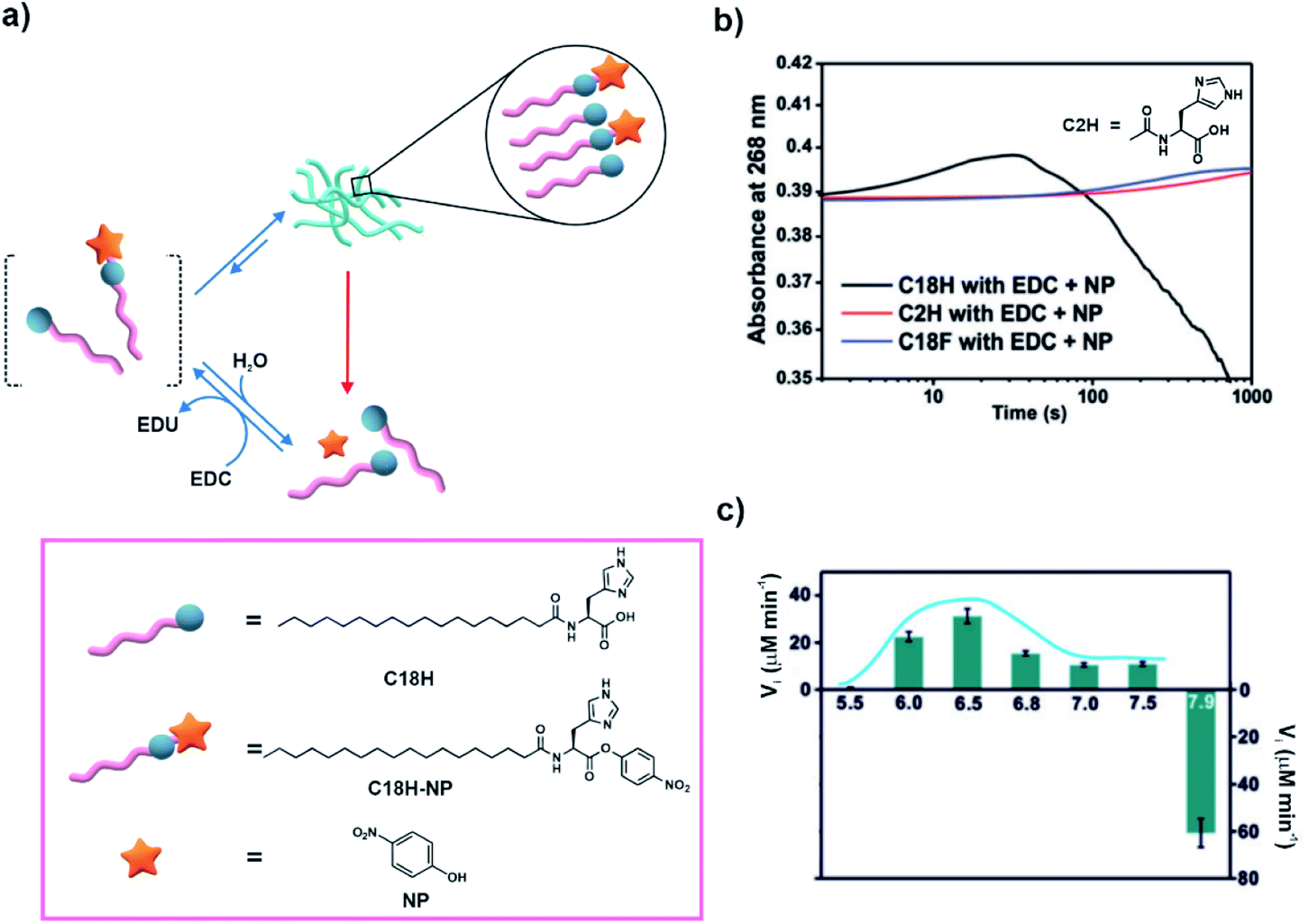 | ||
| Fig. 4 (a) Amphiphile C18H co-assembles with the in situ generated C18H-NP to form transient fibrous networks. Rate acceleration is observed in the assembled state due to cooperative effects and subsequently results in disassembly. Structures of the molecules are shown in the box. (b) Change of absorbance at 268 nm with time for different samples. (c) Rate of hydrolysis of C18H-NP with a variation of pH from 5.5 to 7.5 and rate of formation of the ester at pH 7.9. Adapted from ref. 29 with permission from John Wiley and Sons. | ||
As a follow-up study, Das and co-workers removed the need of using nitrophenol and a coupling agent to access the ester in the medium. Instead, they used the p-nitrophenyl ester of a phenylalanine based amphiphile (C18F-NP, Fig. 5a). C18F-NP acted as a thermodynamically active substrate to induce the co-assembly with C18H.30 When mixed together, C18F-NP induced the time-delayed generation of helical nanostructures as an emergent conformation as observed by electron microscopic techniques. Utilizing cooperative effects emanating from the proximally located histidines, the helical morphologies rapidly catalysed the hydrolysis of the ester C18F-NP from the assembled state and demonstrated a time-delayed sigmoidal rise in activity (Fig. 5b). The accelerated hydrolysis produced p-nitrophenol (NP) and the hydrolysed product C18F (Fig. 5a). Such enhanced degradation of the substrate C18F-NP by the helical fibers implied energy dissipation almost exclusively from the assembled state. Such a co-assembly approach leading to cooperativity among catalytic sites can prove to be useful for the realization of high energy assemblies. The importance of the substrate for inducing the transient helical morphologies that were required for enhanced hydrolysis was indicated by control experiments. When the substrate was added to an aged equilibrated solution of the building block, the helical morphology was not accessed and the system showed significantly slower rates of hydrolysis (Fig. 5b, SYS1 and SYS2 refer to substrate induced assembly and substrate added after equilibration of the building block respectively). Furthermore, when an ester lacking the lipid chain (p-nitrophenyl acetate, PNPA) was used, the authors observed a reversal of trends in hydrolytic rates (Fig. 5c). The above-mentioned reports mimic the ester induced self-assembly and the subsequent rate acceleration as seen in microtubules.
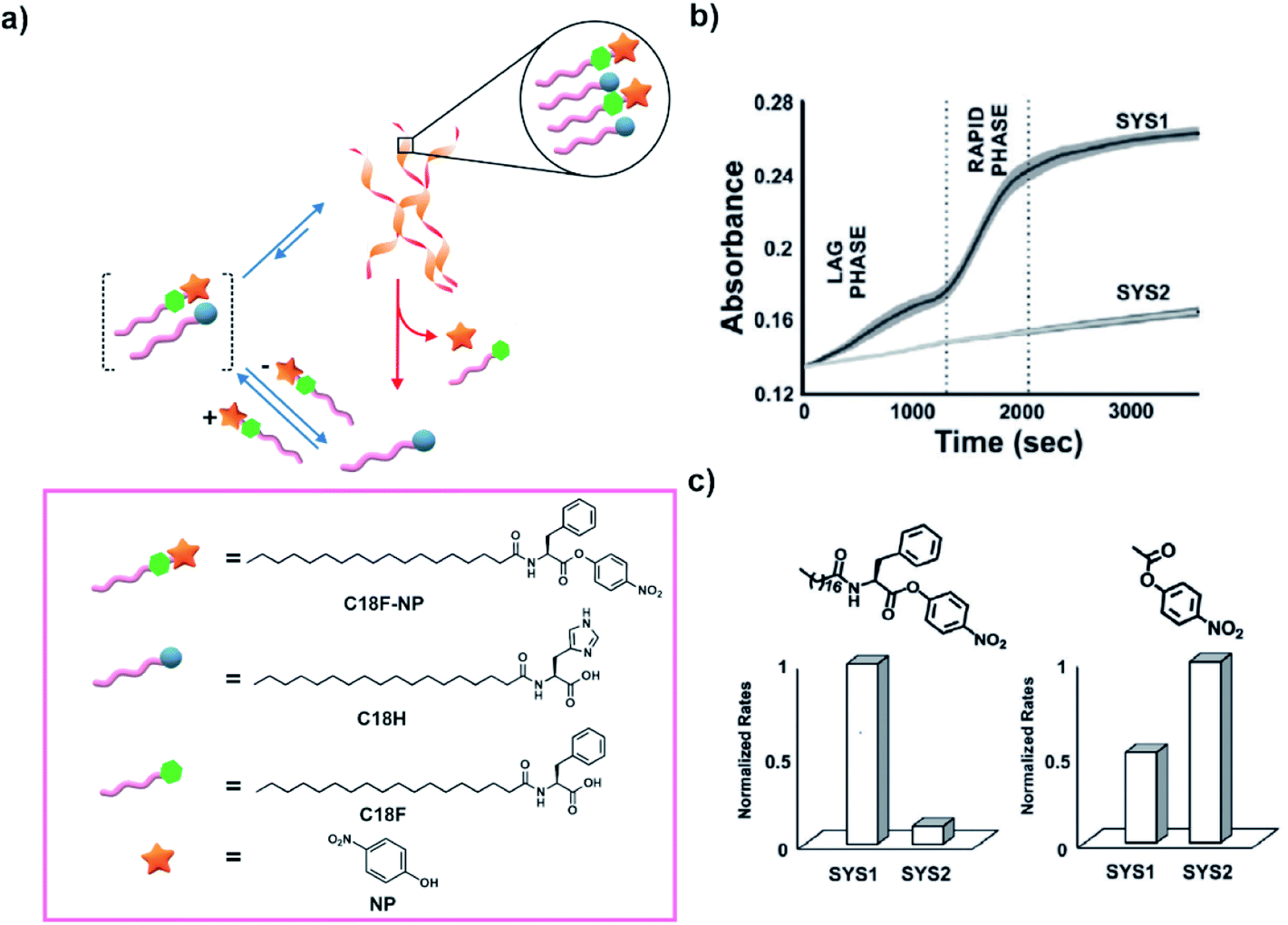 | ||
| Fig. 5 (a) High energy ester C18F-NP undergoes co-assembly with C18H to drive the transient generation of helical morphologies. Hydrolysis of the substrate C18F-NP is accelerated from the assembled state due to cooperative effects and results in disassembly. Structures of the molecules are shown in the box. (b) Time-course kinetics of the hydrolysis of C18F-NP for SYS1 and SYS2 (SYS1 and SYS2 refer to the substrate induced assembly and substrate added after equilibration of the building block respectively). (c) Bar diagram of normalized hydrolytic rates of C18F-NP (left) and PNPA (right) in SYS1 and SYS2. Adapted from ref. 30 with permission from John Wiley and Sons. | ||
4. Substrate induced formation of catalytic macrocycles
The complex behaviour of biochemical systems can be partly attributed to spatiotemporal control of enzyme concentrations through the underlying regulatory mechanisms. Despite the advancement in the development of synthetic catalysts, the control over catalyst concentration with time is rather less explored. In 2014, Otto and co-workers developed one of the early examples of substrate induced selection of a catalytic moiety in a dynamic chemical network (Fig. 6a). A small molecular guest was shown to alter host concentrations temporally.31 It was observed that an aqueous medium containing a dithiol building block on aerial oxidation gave rise to an equilibrated library consisting mainly of catenane type of molecules (11) and a trace amount of tetrameric macrocycles (12, Fig. 6a). Interestingly, in the presence of 10 (Fig. 6a), the transition state analogue (TSA) of a cyclic aza-Cope rearrangement, the composition of the system was altered, resulting in the amplification of the tetrameric macrocycles over the catenanes (Fig. 6a and b). Importantly, this TSA induced transient host which also happened to be the catalyst, subsequently converted TSA to the rearranged acyclic ketone 8 (Fig. 6a and c). On exhaustion of the TSA, the library returned to the equilibrium composition dominated by the catenane. This approach is appealing in terms of developing catalysts which work on demand, making it important in terms of fabricating non-equilibrium systems with life-like traits.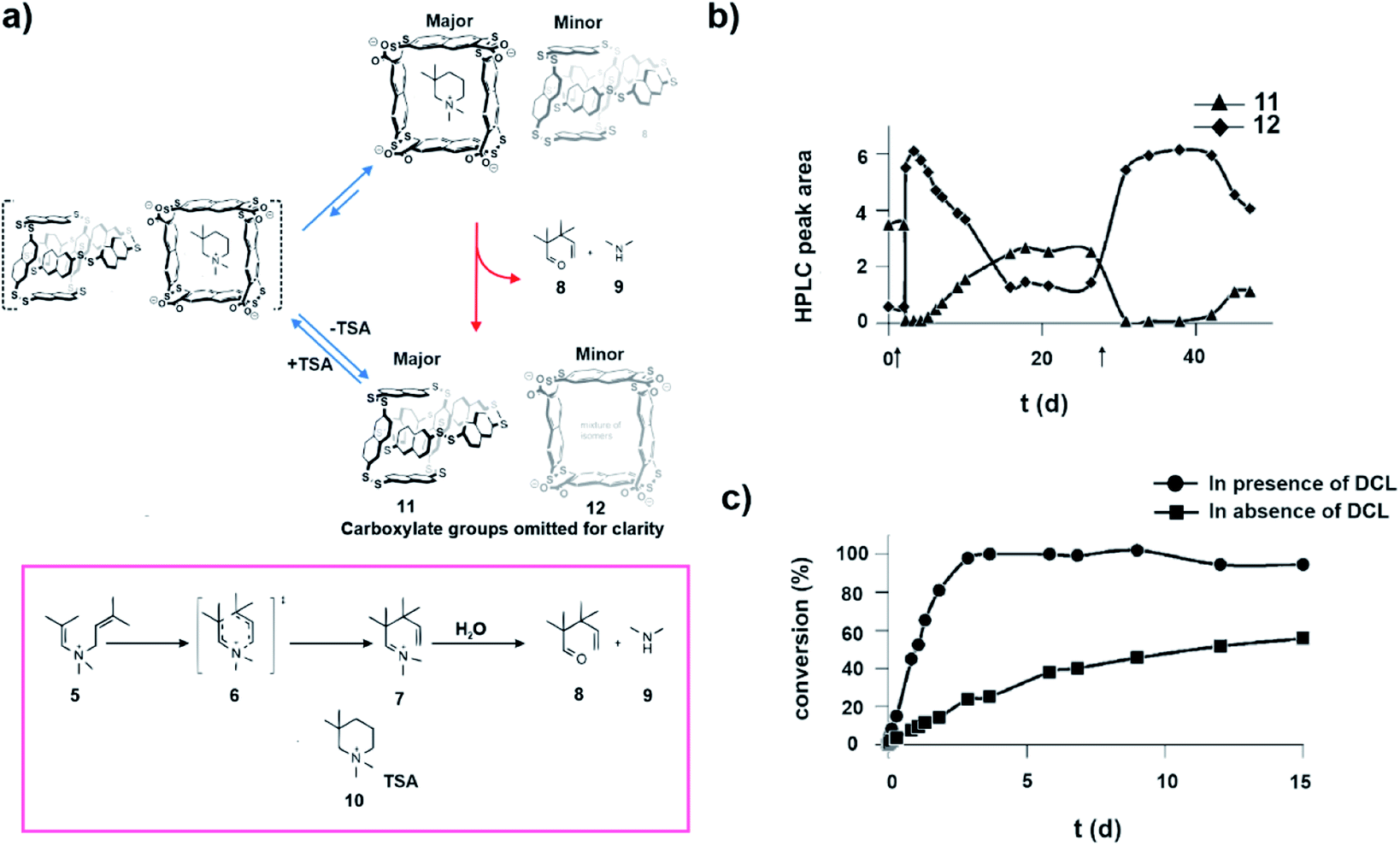 | ||
| Fig. 6 (a) Transition state analogue TSA drives the distribution of the library towards the transient macrocycle which subsequently catalyses the degradation of TSA via the aza-Cope reaction and restores the equilibrium. The aza-Cope reaction and structures of the involved species are shown. (b) Change in composition of the dynamic disulfide network over the course of the aza-Cope rearrangement, showing the rapid conversion of catenanes 11 into tetramers 12 upon addition of the substrate (denoted by the black arrows). (c) Appearance of product 9 as a function of time in the presence and absence of a dynamic combinatorial library (DCL) made from a dithiol building block. Adapted from ref. 31 with permission from John Wiley and Sons. | ||
In a recent study, the concept of substrate induced generation of the dimeric state of proteins inspired the development of a synthetic system involving transient catalytic macrocycles (Fig. 1 and 7a). Wang and co-workers showed that a substrate can induce the generation of active dimers of the chiral macromolecular catalyst, rendering it capable of carrying out decarboxylative Mannich reaction with high enantioselectivity.32 They found out that sulfate anions were able to promote the dimerization of the macromolecular catalyst M containing two chiral diamines and two thioureas. Upon dimerization, an extroversive conformation was adopted by the macrocycles which formed hydrogen bonding with the interaction sites. This sulfate induced generation of the macrocycles inspired the authors to investigate the capability of the system to facilitate decarboxylative Mannich reaction. For this purpose, a cyclic substrate having a sulfamate group (13) was used to induce a similar dimerization with the expectation that complementary hydrogen bonds will augment the electrophilicity of the imine-based substrate and facilitate a nucleophilic attack (Fig. 7a). Furthermore, the formation of the hydrogen bonding network will lead to stabilization of the negative charge on the substrate after the nucleophilic attack. Indeed, the combination of the two factors enabled a β-ketoacid (14) to attack 13 to furnish the product 15 in high yields. Kinetic analyses demonstrated a second order dependence of the initial reaction rate on the concentration of the macrocycle which supported the dimerization prior to catalysis (Fig. 7b and c). The utilization of substrate induced dimerization which can subsequently enhance the reactivity of the complex towards conversion of the same substrate is reminiscent of the substrate induced systems of biology.
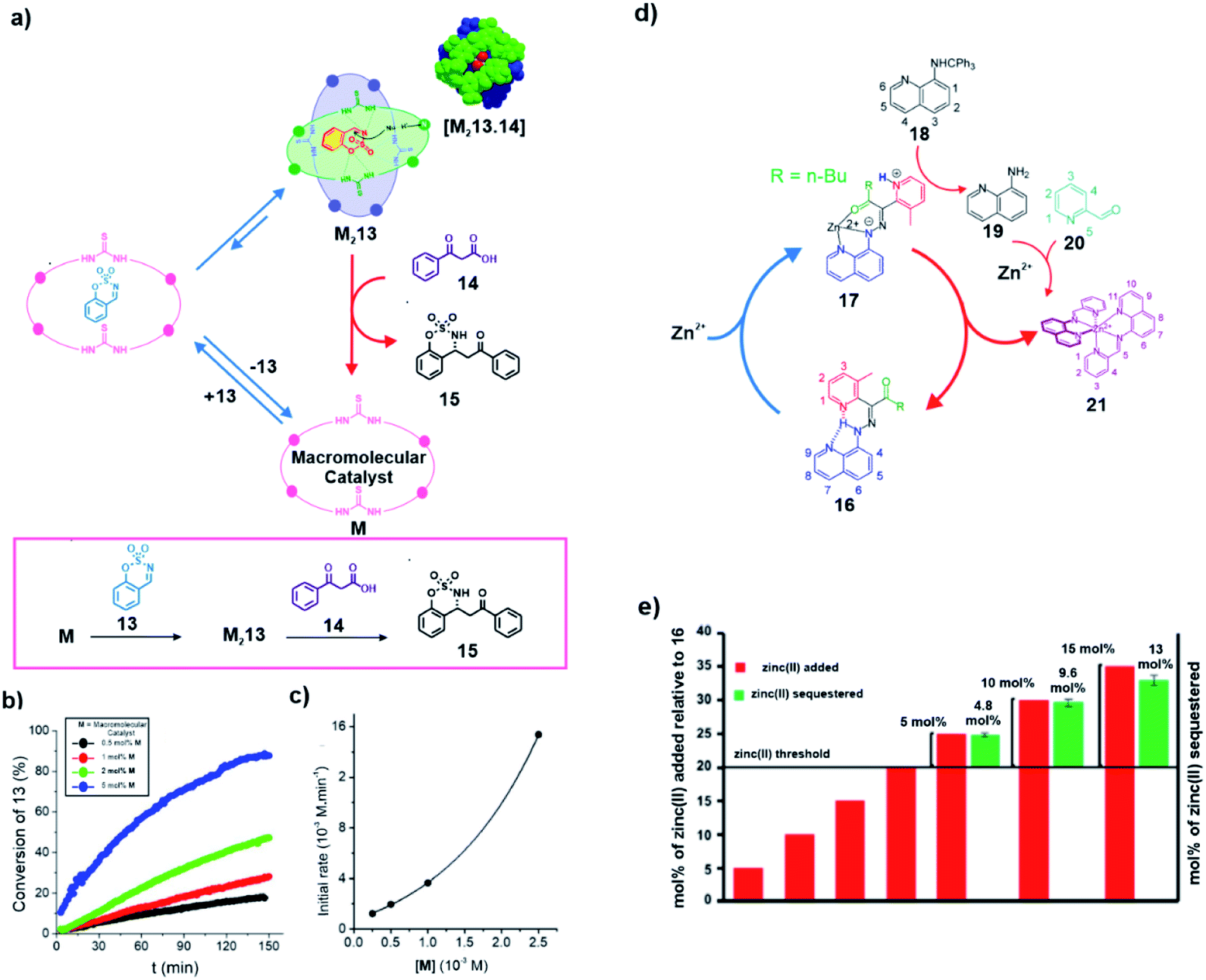 | ||
| Fig. 7 (a) Substrate induced generation of a macromolecular catalyst. The cyclic substrate 13 promotes the dimerization of the catalyst M to form an active ternary complex M213, capable of catalyzing a decarboxylative Mannich reaction. (b) Conversion of 13 over time with the varying mol% of the macromolecular catalyst. (c) Change of the initial reaction rate with varying concentration of the macromolecular catalyst. Adapted from ref. 32 with permission from John Wiley and Sons. (d) The presence of a threshold amount of Zn2+ (>20 mol%) induces the formation of catalytic complex 17 that initiates a cascade of events to yield 21 which sequestrates Zn2+ away from 17, leading to restoration of 16 and subsequent loss of catalytic ability. (e) Formation of 21 was achieved only after the addition of a certain threshold concentration of zinc(II) to a mixture of 16, 18 and 20. Adapted from ref. 33 with permission from the American Chemical Society. | ||
The development of a negative feedback loop using synthetic organic molecules is an important design strategy to mimic the complex biological systems. Unique to the examples reported in this review, Pramanik and Aprahamian demonstrated a metal ion induced complex signalling cascade using different hydrazine-based multicomponent switches (Fig. 7d). Coordination-coupled deprotonation (CCD) resulted in such metal ion induced generation of the catalytic complex 17 (Fig. 7d).33 Briefly, a threshold concentration of Zn2+ ions was required for the formation of the complex 17 on binding with the hydrazone switch 16. Complex 17 showed catalytic ability as it produced protons that assisted the transformation of 18 into 19. Compound 19 was able to condense with 20 (already present in the medium) to form 21. Interestingly, 21 acted as a capable ligand of Zn2+ subsequently stripping away the ion from 17, thus eliminating the chemical input that resulted in the generation of the catalyst. Finally, the generation of 16 stopped further release of protons and autonomously turned off the cascade. The fact that a certain concentration of metal ion was required to induce the formation of 21 was demonstrated from NMR based studies. It was observed that on addition of a certain concentration (25 mol%) of Zn2+, the yield of 21 matched well with the amount of excess metal ions added beyond a threshold of 20 mol% (Fig. 7e). This demonstrated that the chain of events acted as a negative feedback loop where the Zn2+ concentration was suppressed whenever it was reaching a certain threshold. It should be noted that although this work is not an example of substrate induced catalytic self-assemblies, yet, through this feedback driven system, a catalytic complex was accessed via a chemical input that was eventually removed via catalysis.
5. Substrate induced formation of functional membrane
In nature, non-equilibrium structures such as microtubules are formed in order to perform exquisite functions when needed. In 2014, Fyles and co-workers demonstrated transient generation of an amphiphile that was able to integrate within a bilayer membrane with the capability to demonstrate ion transport via a hydrophobic pore (Fig. 8a).34 The authors started with an equilibrated thiol functionalized amphiphile (22) which was incapable of transporting ions through a membrane (Fig. 8a). A highly active amine functionalized thioester (23) was able to induce the generation of the thioester 24via a thiol–thioester exchange. Owing to the unique design, the high energy molecule 24 installed a mechanism of self-destruction and underwent a slow intramolecular reaction where its carbonyl group was attacked by its amine to regenerate the inactive thiol 22 and the lactam 25. Transient accumulation of 24 was due to slower intramolecular cyclization (24 to 25) than the thioester exchange (22 to 24). Furthermore, control over the relative rates of formation and degradation of 24 was achieved by alteration of the length of the spacer between the amine and the carbonyl group of the thioester bond. Notably, in the presence of a pre-formed bilayer, the transiently formed 24 was capable of transporting ions across the membrane by forming a pore. The transport of ions was indicated by the changes in conductivity in the voltage-clamp experiments which concomitantly proved the temporal generation of the active species 24 (Fig. 8b and c). The temporal control of function in terms of the observation of transport via association with the bilayer was reminiscent of the dynamic functions of microtubules that have also been linked with transport of materials within the cell.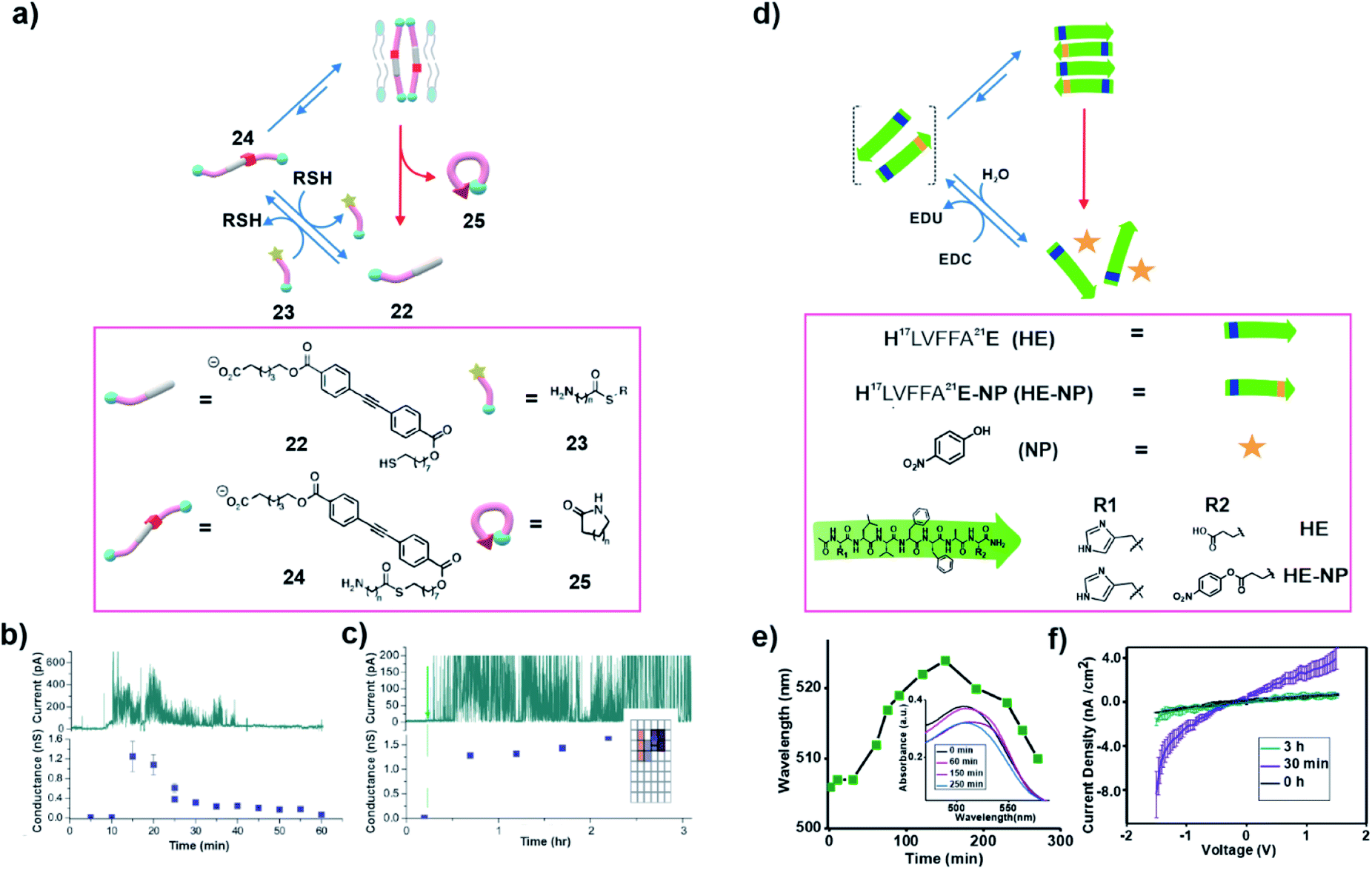 | ||
| Fig. 8 (a) An amine containing thioester 23 induces the conversion of 22 to 24 which is capable of transporting ions in the presence of a bilayer. Subsequently, 24 undergoes an intramolecular nucleophilic attack which produces the lactam 25 and regenerates the thiol building block. Transport activity of 24 in the absence (b) and presence (c) of 23. Adapted from ref. 34 with permission from the Royal Society of Chemistry. (d) Thermodynamically activated congener (HE-NP) induces the temporal polymerization of histidine functionalized cross β peptide (HE). (e) Time dependent UV-Vis study shows the temporal binding of Congo red. (f) Change in the current density with the modulation of the voltage (I–V plot) for the HE-NP system at different times. Adapted from ref. 37 with permission from John Wiley and Sons. | ||
Speaking of bilayers, hydrophobic mutations in amyloid sequences have been known to direct peptide bilayer assembly.35,36 Das and co-workers have recently demonstrated the transient polymerization of amyloid peptides due to hydrophobic collapse and accelerated catalysis from the self-assembled state (Fig. 8d).37 The core sequence of Aβ peptide (16KLVFFA22E) known to be associated with the neurodegenerative disease (e.g., Alzheimer's disease) was used. The lysine at the 16th position was mutated with histidine for its known hydrolytic competence. The mutated sequence H17LVFFA22E (HE) was thermodynamically activated by installing a high energy ester bond at the C terminal glutamic acid side chain by coupling with para-nitro phenol (Fig. 8d). This resulted in loss of the charge at the terminal and installed hydrophobicity in the sequence. The activated monomers (HLVFFAE-NP) readily co-assembled with the precursor HE strands, eventually forming fibrillar aggregates via hydrophobic collapse. The polymerization allowed the histidines to be placed in close proximity which led to an enhanced catalytic proficiency towards the hydrolysis of the activated esters via cooperative effects. The co-assembly of the activated monomer (HE-NP) with the building block (HE) and accelerated catalytic rates from the polymerized state is reminiscent of the GTP induced polymerization of microtubules. Although not sought for, the combination of these two facts might have installed kinetic asymmetry in the cycle. As mentioned above, it would be interesting to probe such systems under stationary conditions to investigate any possible generation of high energy assemblies. The temporal amyloid polymerization was corroborated by the transient binding of the Congo Red (CR) dye with the co-assembled HE and HE-NP system. The red shift of the absorbance maxima of CR upon binding to the assembly and blue shift after the disassembly of the system suggested the autonomous depolymerization (Fig. 8e). Notably, the temporal generation of the catalytic phases by self-propagation of short amyloid peptides exhibited transient enhancement of conductance due to the spatiotemporal paracrystalline packing of the β-sheets (Fig. 8f). A clear insight towards the generation of complex bioinspired devices was perceived via such dissipative self-assembly.
In a recent study, Lee and co-workers utilized two components of an aromatic substitution reaction to induce the formation of a vesicular state which catalysed the reaction, creating a product which subsequently resulted in disassembly (Fig. 9a).38 The transient assembly of the pyridine based amphiphilic compound 26 was induced by the two substrates, a hydrophobic thiol 27 and an aromatic halide 28 to form vesicles (Fig. 9a). Dynamic light scattering (DLS) study revealed formation of aggregates on addition of the substrates (Fig. 9b). The aggregation resulted in increased proximity of the substrates and thereby promoted an SNAr reaction between them (Fig. 9a and c). The generation of HCl as one of the products of the reaction caused the protonation of the pyridine unit, resulting in disassembly of the vesicles and subsequent release of the product. Interestingly, the overall cycle could be repeated through neutralization of the protonated 26 on addition of K2CO3 (step shown with the help of the black arrow in Fig. 9a).
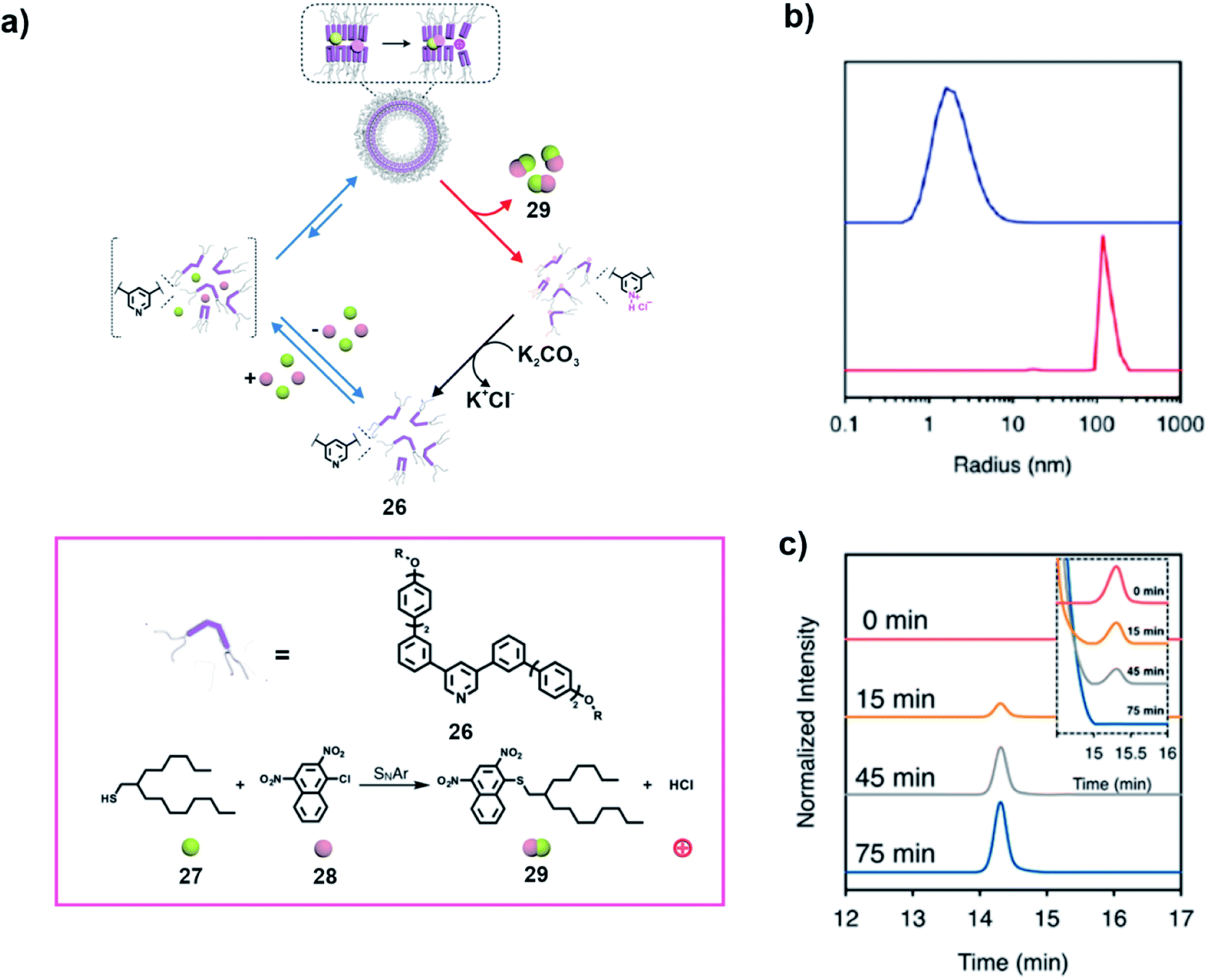 | ||
Fig. 9 (a) A thiol-based reagent 27 and an aryl halide 28 induce the self-assembly of a pyridine based amphiphile 26 to access a vesicular membrane which subsequently facilitates an SNAr reaction between 27 and 28. Generation of hydrochloric acid as a product leads to disassembly. Structures of the compounds and the reaction are shown in the box. (b) The blue line and red line depict the DLS profiles of 26 (75.3 μM) before and after addition of 27 (mole ratio 27/26 of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in aqueous solution. (c) HPLC chromatograms of the product generated in the reaction mixture (inset shows the decrease of 27) with time. Adapted from ref. 38 with permission from the American Chemical Society. :1) in aqueous solution. (c) HPLC chromatograms of the product generated in the reaction mixture (inset shows the decrease of 27) with time. Adapted from ref. 38 with permission from the American Chemical Society. | ||
6. Summary and outlook
In the current review, we have attempted to focus on the recent examples of synthetic systems where substrate induced generation of transient catalytic moieties have been demonstrated and an enhanced supramolecular catalysis has been witnessed. Substrates have been shown to bind to building blocks to induce the generation of a catalytically proficient complex that subsequently degrades the substrates to eventually reach a state consisting of the building blocks and waste. Notably, the design of most of the reviewed systems suggests that the substrate almost exclusively interacts with the building blocks while the subsequent reaction of substrate to product conversion occurs primarily from the induced catalytic state. It is important to note here that all the studies covered here use a closed system with substrate additions in batches and none demonstrate or seek the presence of a non-equilibrium high energy state composed of the unactivated building blocks.20 As seen in the case of GDP-tubulin higher energy polymers that are capable of doing mechanical work, the non-equilibrium states are accessed only when the energy released from the substrate to product conversions is exploited to shift the equilibrium. Recent simulations have suggested that the presence of a kinetic asymmetry in the energy dissipation pathways can help in the utilization of the released energy to populate the high energy non-equilibrium state under stationary conditions.18–20 Under such conditions, the detailed balance between forward and backward rates is broken when kinetic asymmetry leads to a preference in the directionality of the overall reaction cycle.20 Although none of the studies report or investigate the presence of kinetic asymmetry, the design principles of substrate induced catalytic assemblies can lead to the development of future synthetic systems involving high energy non-equilibrium assemblies with fascinating properties. Towards this end, recent discoveries show that supramolecular self-assembled systems can register a huge enhancement of catalytic rates upon assembly, utilizing features such as cooperative effects, multivalency, supramolecular binding pocket generation and so forth.39–48 These can contribute to the incorporation of autonomous reaction cycles which could play an important role in installing adaptivity to transient systems. Looking beyond, it is important to probe the ability of such transient reaction cycles to control functions as seen in non-equilibrium biological assemblies. Some initial reports are beginning to demonstrate the dynamic control of functions by the transient self-assembled systems.34,37,49–52Finally, strategies covered in the present review attempt to lay the foundation for future research, where it could be conceivable to engineer acutely adaptive and self-regulating functional systems. The designed metabolic networks of dynamically exchanging monomers that can access non-equilibrium states can help in the generation of materials and nanostructures ready for diverse fields, from materials science to biomedical areas and further consolidate the emerging field of systems chemistry.
Data availability
This is a review article and hence no experimental or computational data is associated with this article.Author contributions
S. P. A., C. G., and D. D. conceptualized the topics and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. D. is thankful to SJF Grant SB/SJF/2020-21/08, GOI for financial assistance. S. P. A. and C. G. thank UGC, India and SERB (EMR/2017/005126) for fellowships.References
- B. Novak and J. J. Tyson, Nat. Rev. Mol. Cell Biol., 2008, 9, 981–991 CrossRef CAS PubMed.
- D. Deamer and A. L. Weber, Cold Spring Harbor Perspect. Biol., 2010, 2, a004929 Search PubMed.
- I. Gasic and T. J. Mitchison, Curr. Opin. Cell Biol., 2019, 56, 80–87 CrossRef CAS PubMed.
- H. Hess and J. L. Ross, Chem. Soc. Rev., 2017, 46, 5570–5587 RSC.
- R. D. Astumian, Chem. Commun., 2018, 54, 427–444 RSC.
- R. Pascal, A. Pross and J. D. Sutherland, Open Biol., 2013, 3, 130156 CrossRef PubMed.
- A. Pross, J. Phys. Org. Chem., 2008, 21, 724–730 CrossRef CAS.
- J. L. England, Nat. Nanotechnol., 2015, 10, 919–923 CrossRef CAS PubMed.
- I. R. Epstein, Chem, 2019, 5, 1922–1923 CAS.
- C. Cheng, P. R. McGonigal, J. F. Stoddart and R. D. Astumian, ACS Nano, 2015, 9, 8672–8688 CrossRef CAS PubMed.
- D. Michel, Biochimie, 2016, 128–129, 48–54 CrossRef CAS PubMed.
- R. Gasper, S. Meyer, K. Gotthardt, M. Sirajuddin and A. Wittinghofer, Nat. Rev. Mol. Cell Biol., 2009, 10, 423–429 CrossRef CAS PubMed.
- S. Roychowdhury and M. M. Rasenick, Biochemistry, 1994, 33, 9800–9805 CrossRef CAS PubMed.
- D. Datta, C. L. McClendon, M. P. Jacobson and J. A. Wells, J. Biol. Chem., 2013, 288, 9971–9981 CrossRef CAS PubMed.
- S. Cheng, G. Chang and C. Chou, Biophys. J., 2010, 98, 1327–1336 CrossRef CAS PubMed.
- F. Wang, H. Shi, R. He, R. Wang, R. Zhang and J. Yuan, Nat. Phys., 2017, 13, 710–714 Search PubMed.
- M. Caplow and J. Shanks, J. Biol. Chem., 1990, 265, 8935–8941 CrossRef CAS.
- R. D. Astumian, Nat. Commun., 2019, 10, 3837 CrossRef PubMed.
- G. Ragazzon and L. J. Prins, Nat. Nanotechnol., 2018, 13, 882–889 CrossRef CAS PubMed.
- K. Das, L. Gabrielli and L. J. Prins, Angew. Chem., Int. Ed., 2021, 60, 20120–20143 CrossRef CAS PubMed.
- E. Penocchio, R. Rao and M. Esposito, Nat. Commun., 2019, 10, 3865 CrossRef PubMed.
- A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC.
- H. S. Azevedo, S. L. Perry, P. A. Korevaar and D. Das, Nat. Chem., 2020, 12, 793–794 CrossRef CAS PubMed.
- G. Ashkenasy, T. M. Hermans, S. Otto and A. F. Taylor, Chem. Soc. Rev., 2017, 46, 2543–2554 RSC.
- M. Weißenfels, J. Gemen and R. Klajn, Chem, 2021, 7, 23–37 Search PubMed.
- R. Merindol and A. Walther, Chem. Soc. Rev., 2017, 46, 5588–5619 RSC.
- P. Solís Muñana, G. Ragazzon, J. Dupont, C. Z.-J. Ren, L. J. Prins and J. L.-Y. Chen, Angew. Chem., Int. Ed., 2018, 57, 16469–16474 CrossRef PubMed.
- I. Colomer, S. M. Morrow and S. P. Fletcher, Nat. Commun., 2018, 9, 2239 CrossRef PubMed.
- S. Bal, K. Das, S. Ahmed and D. Das, Angew. Chem., Int. Ed., 2019, 58, 244–247 CrossRef CAS PubMed.
- S. P. Afrose, S. Bal, A. Chatterjee, K. Das and D. Das, Angew. Chem., Int. Ed., 2019, 58, 15783–15787 CrossRef CAS PubMed.
- H. Fanlo-Virgós, A. R. Alba, S. Hamieh, M. Colomb-Delsuc and S. Otto, Angew. Chem., Int. Ed., 2014, 53, 11346–11350 CrossRef PubMed.
- H. Guo, L.-W. Zhang, H. Zhou, W. Meng, Y.-F. Ao, D.-X. Wang and Q.-Q. Wang, Angew. Chem., Int. Ed., 2020, 59, 2623–2627 CrossRef CAS PubMed.
- S. Pramanik and I. Aprahamian, J. Am. Chem. Soc., 2016, 138, 15142–15145 CrossRef CAS PubMed.
- K. Dambenieks, P. H. Q. Vu and T. M. Fyles, Chem. Sci., 2014, 5, 3396–3403 RSC.
- F. U. Hartl, A. Bracher and M. H. Hartl, Nature, 2011, 475, 324–332 CrossRef CAS PubMed.
- E. Levy, M. D. Carman, I. J. Fernandez-Madrid, M. D. Power, I. Lieberburg, S. G. van Duinen, G. T. Bots, W. Luyendijk and B. Frangione, Science, 1990, 248, 1124–1126 CrossRef CAS PubMed.
- S. Bal, C. Ghosh, T. Ghosh, R. K. Vijayaraghavan and D. Das, Angew. Chem., Int. Ed., 2020, 59, 13506–13510 CrossRef CAS PubMed.
- H. Wang, Y. Wang, B. Shen, X. Liu and M. Lee, J. Am. Chem. Soc., 2019, 141, 4182–4185 CrossRef CAS PubMed.
- P. Pengo, S. Polizzi, L. Pasquato and P. Scrimin, J. Am. Chem. Soc., 2005, 127, 1616–1617 CrossRef CAS PubMed.
- C. Guarise, F. Manea, G. Zaupa, L. Pasquato, L. J. Prins and P. Scrimin, J. Pept. Sci., 2008, 14, 174–183 CrossRef CAS PubMed.
- S. Dhiman, R. Ghosh, S. Sarkar and S. J. George, Chem. Sci., 2020, 11, 12701–12709 RSC.
- S. Neri, S. Garcia Martin, C. Pezzato and L. J. Prins, J. Am. Chem. Soc., 2017, 139, 1794–1797 CrossRef CAS PubMed.
- C. Zhang, R. Shafi, A. Lampel, D. MacPherson, C. Pappas, V. Narang, T. Wang, C. Madarelli and R. V. Ulijn, Angew. Chem., Int. Ed., 2017, 56, 14511–14515 CrossRef CAS PubMed.
- M. P. van der Helm, C.-L. Wang, B. Fan, M. Macchione, E. Mendes and R. Eelkema, Angew. Chem., Int. Ed., 2020, 59, 20604–20611 CrossRef CAS PubMed.
- T. O. Omosun, M.-C. Hsieh, W. S. Childers, D. Das, A. K. Mehta, N. R. Anthony, T. Pan, M. A. Grover, K. M. Berland and D. G. Lynn, Nat. Chem., 2017, 9, 805–809 CrossRef CAS PubMed.
- J. Ottelé, A. S. Hussain, C. Mayer and S. Otto, Nat. Catal., 2020, 3, 547–553 CrossRef.
- J. Greenwald, W. Kwiatkowski and R. Riek, J. Mol. Biol., 2018, 430, 3735–3750 CrossRef CAS PubMed.
- P. Makam, S. S. R. K. C. Yamijala, K. Tao, L. J. W. Shimon, D. S. Eisenberg, M. R. Sawaya, B. M. Wong and E. Gazit, Nat. Catal., 2019, 2, 977–985 CrossRef CAS PubMed.
- G. Ashkenasy, ChemSystemsChem, 2019, 1, e1900008 CrossRef.
- M. Kumar, N. L. Ing, V. Narang, N. K. Wijerathne, A. I. Hochbaum and R. V. Ulijn, Nat. Chem., 2018, 10, 696–703 CrossRef CAS PubMed.
- S. Dhiman, K. Jalani and S. J. George, ACS Appl. Mater. Interfaces, 2020, 12, 5259–5264 CrossRef CAS PubMed.
- R. Chen, S. Neri and L. J. Prins, Nat. Nanotechnol., 2020, 15, 868–874 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |