
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDissipative operation of pH-responsive DNA-based nanodevices†
Davide
Mariottini‡
 a,
Daniele
Del Giudice‡
b,
Gianfranco
Ercolani
a,
Stefano
Di Stefano
*b and
Francesco
Ricci
*a
a,
Daniele
Del Giudice‡
b,
Gianfranco
Ercolani
a,
Stefano
Di Stefano
*b and
Francesco
Ricci
*a
aDipartimento di Scienze e Tecnologie Chimiche, Università di Roma Tor Vergata, Via della Ricerca Scientifica, 00133 Roma, Italy. E-mail: francesco.ricci@uniroma2.it
bDipartimento di Chimica, Università di Roma La Sapienza, ISB-CNR Sede Secondaria di Roma-Meccanismi di Reazione, P.le A. Moro 5, 00185 Roma, Italy. E-mail: stefano.distefano@uniroma1.it
First published on 19th July 2021
Abstract
We demonstrate here the use of 2-(4-chlorophenyl)-2-cyanopropanoic acid (CPA) and nitroacetic acid (NAA) as convenient chemical fuels to drive the dissipative operation of DNA-based nanodevices. Addition of either of the fuel acids to a water solution initially causes a rapid transient pH decrease, which is then followed by a slower pH increase. We have employed such low-to-high pH cycles to control in a dissipative way the operation of two model DNA-based nanodevices: a DNA nanoswitch undergoing time-programmable open–close–open cycles of motion, and a DNA-based receptor able to release-uptake a DNA cargo strand. The kinetics of the transient operation of both systems can be easily modulated by varying the concentration of the acid fuel added to the solution and both acid fuels show an efficient reversibility which further supports their versatility.
Introduction
One of the main features of chemical systems underlying living networks is the need to be continuously supplied with chemical or photophysical fuels to remain in a particular functional state. In the case of proteins, commonly cited examples are myosin, which allows muscle contraction,1 dynein and kinesin, responsible for moving cargo inside the cell,2 or ATP synthase that produces ATP, the typical biofuel, exploiting a proton gradient across membranes which drives the required unidirectional rotational motion.3 In most cases, turn on/off of such functional states involves molecular motions or assembly/disassembly of chemical species. For the above reasons, consuming a fuel to remain in a functional state is an essential character for an artificial system that aspires to have life-like properties. The functional state has to be maintained in a dissipative manner, that is until fuel exhaustion.4 Recently, artificial dissipative systems have been described in the fields of assembly/disassembly of aggregates,5 host–guest chemistry,6 and fully abiotic7 molecular machines. In this context, because of the high predictability, reversibility of the involved interactions, and programmability, synthetic DNA has emerged as a powerful material to engineer non-equilibrium machines, devices,8 and nanostructures with a transient behavior.9Among dissipative synthetic systems, those based on pH variations are particularly attractive due to the (at least apparent) ease of operation and the resemblance with naturally-occurring mechanisms. In fact, since biochemistry has evolved in water, pH variations are probably the most commonly used tool in the chemical control of biosystems.10 A number of artificial dissipative systems based on pH variations have appeared in the literature, ranging from time programmable aggregation/disaggregation of molecular assemblies11 to time-controlled motions of molecular machines.7 Different time-controlled DNA-based systems with dissipative pH changes have been also reported to date mostly based on conformational change DNA-based nanodevices involving cytosine-rich i-motif domains.12–14 Despite the elegant nature of these systems, however, i-motif domains remain challenging to engineer into functional devices and the rational modulation of their pKa is difficult to achieve. The pH dissipative cycles employed in these examples involve either the hydrolysis of β-butyrolactone,12 which allows high-to-low pH cycles, the use of a thiosulfate/sulfite oscillator,13 which requires continuous fuel supply, or, finally, the use of a complex, biocatalytic enzymatic reaction network.14
We and others have, during the recent years, demonstrated that triplex-forming DNA sequences employing Hoogsteen interactions are extremely versatile and their pH-responsiveness can be easily modulated15,16 and integrated not only into conformational change DNA switches17 but also into cargo-loading DNA receptors18,19 and reaction networks.20 It is also known that activated carboxylic acids such as 2-cyano-2-phenylpropanoic21 acid and its derivatives,22 trichloroacetic acid,23 and nitroacetic acid24 have been employed as versatile and convenient chemical fuels for controlling over time dissipative pH cycles of motions of molecular machines, both switches and motors,21–23 host–guest interactions24 and complex supramolecular assembly,25 both in aqueous and organic solvents. In water solution, these carboxylic acid fuels are converted by hydroxide ion into the corresponding carboxylates that slowly release CO2 to be transformed into the strongly basic carbanions R− (Fig. 1a). This remarkable sequence of reactions allows a nimble temporal control of the solution pH, which is simply realized by adding tiny amounts of a concentrated stock solution of fuel to a basic solution.
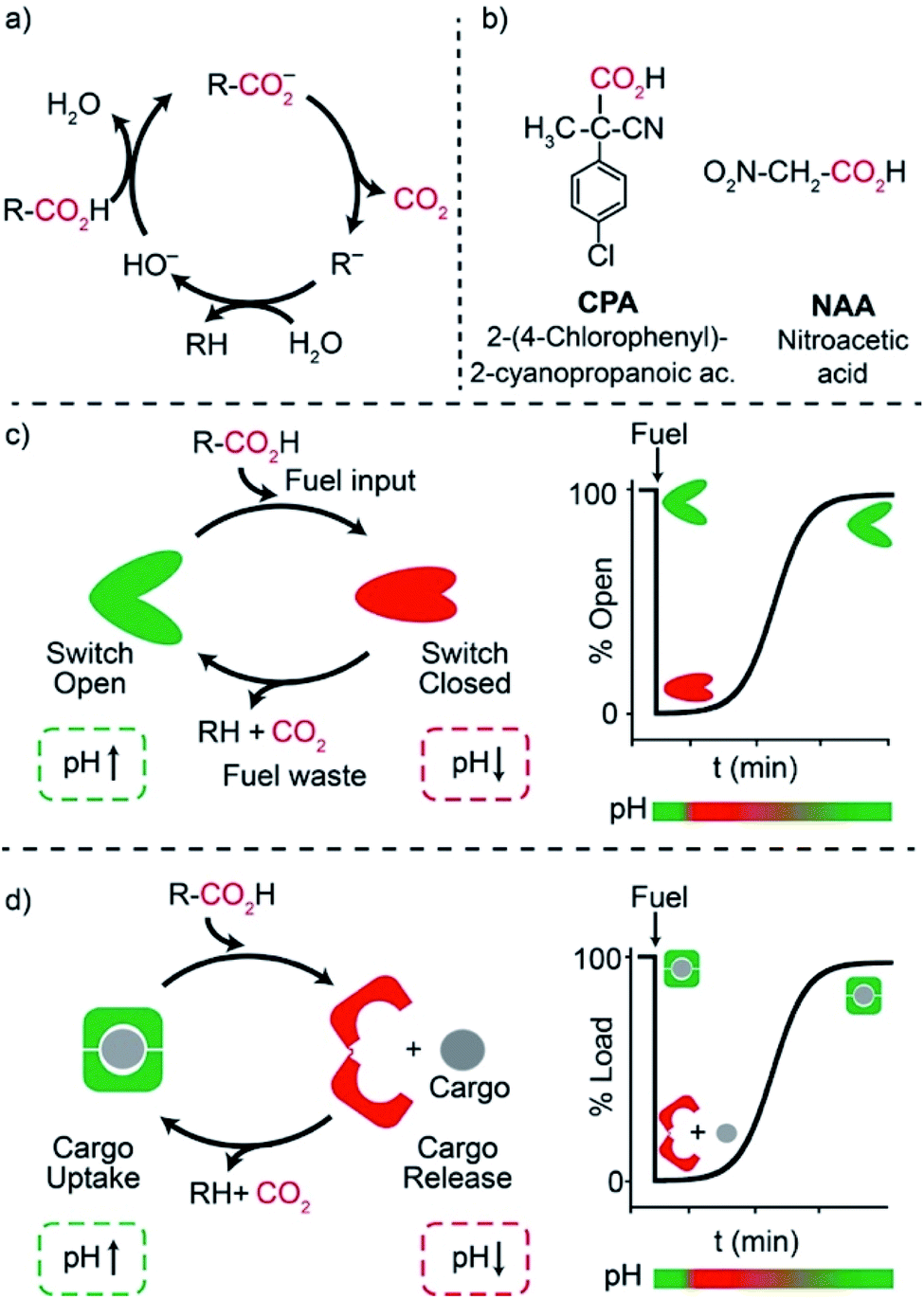 | ||
| Fig. 1 (a) Activated carboxylic acids allow the time-controlled pH change of a solution. (b) Chemical structures of the two activated carboxylic acids used in this work. (c) Dissipative control of a pH-responsive conformational change DNA nanoswitch and (d) of a pH-responsive cargo-loading DNA-based receptor. | ||
Results and discussion
Motivated by the above arguments, we report here the use of two different activated carboxylic acids, namely 2-(4-chlorophenyl)-2-cyanopropanoic acid (CPA) and nitroacetic acid (NAA) (Fig. 1b), as chemical fuels to drive the dissipative operation of two pH-responsive DNA devices based on triplex-forming Hoogsteen interactions: a conformational change DNA nanoswitch (Fig. 1c) and a cargo-loading DNA nanoscale receptor (Fig. 1d). Despite the same decarboxylation mechanism, the two fuels have somewhat different properties which can make one preferable over the other in specific applications. In particular, NAA decarboxylates faster than CPA, is more soluble and allows greater pH jumps, but, on the other hand, it is less stable and its solutions must be prepared just before use.In a first series of experiments, CPA was used as a chemical fuel to trigger the conformational change of a pH-responsive acid nanoswitch that can form an intramolecular triplex structure through Hoogsteen interactions between a hairpin duplex domain and a single-strand triplex-forming domain (see Fig. 2a). The nanoswitch16 is endowed with a pH-independent fluorescent probe and a quencher that allows following its open (duplex) or closed (triplex) state. pH and fluorescence time-course monitoring were carried out by adding the fuel acid to a weakly basic solution (pH = 7.5) containing a fixed concentration of the pH nanoswitch (25 nM) (Fig. 2b and c). Under these conditions the nanoswitch (pKa = 6.1; Fig. ESI 1†)16 is initially in an open conformation as supported by the high fluorescent signal recorded (Fig. 2c, t = 0). Upon addition of the fuel, the pH immediately drops from 7.5 to 5.0 inducing the closing of the nanoswitch. From this point onwards, decarboxylation of deprotonated fuel acid takes place, and the pH slowly increases to the final value of 6.4 (Fig. 2b).§ Consistently, fluorescence emission increases again due to the triplex to duplex transition. Since fluorescence emissions measured just before the addition of fuel, immediately after the addition of fuel, and at the end of the experiment, are 4.20, 0.20, and 3.20 a. u., respectively, it is apparent that, after 2 hours, 75% of the nanoswitch molecules have experienced a duplex → triplex → duplex cycle at the expense of CPA. Plotting the fluorescence data against the corresponding pH data leads to the binding profile in Fig. 2d with a pKa for the duplex–triplex transition which is in good accordance with the reported pKa of the switch.16 The time required for the nanoswitch to return to its initial duplex state can be modulated by varying the concentration of added fuel (Fig. 2e). In fact, it was previously shown that the pH dive and subsequent pH rise to the final, definite value increase on increasing the amount of the added carboxylic acid fuel undergoing decarboxylation.24 More specifically, we observe half-life times (defined as the time needed to return 50% of the switch to the initial open conformation) ranging from 30 to 127 min by varying the concentration of the added acid fuel from 125 to 300 μM (Fig. 2f). A control experiment was also carried out adding acetic acid instead of CPA to the DNA-based switch. Since acetic acid does not decarboxylate under the given conditions, after the initial duplex-to-triplex transition, no recovery of the fluorescence signal is observed, as expected (Fig. 2e).
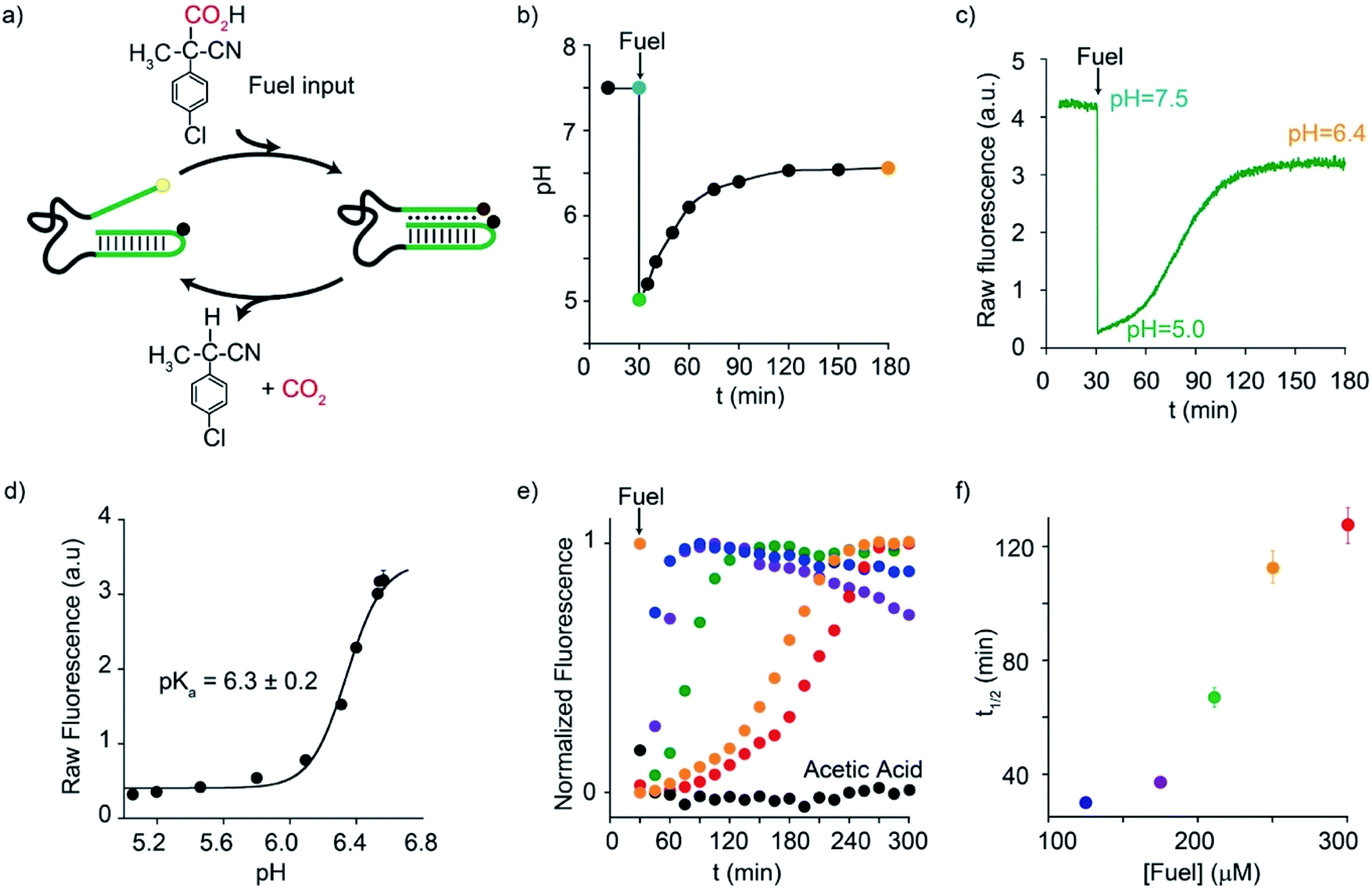 | ||
| Fig. 2 Dissipative cycles of motion of a DNA-based nanoswitch guided by triplex-to-duplex transitions by means of fuel CPA. (a) Cartoon representing the cycles of motion triggered by CPA. (b) pH-time monitoring of a 25.0 nM nanoswitch solution (initial pH = 7.5) after addition of the fuel. (c) Fluorescence-time monitoring of a 25.0 nM nanoswitch solution (initial pH = 7.5) after addition of the fuel. (d) Fluorescence-pH plot obtained by data points in Fig. 3b and c. (e) Fluorescence-time profiles obtained by addition of different concentrations of the acid fuel to a solution containing the nanoswitch (25 nM) at an initial pH of 7.5. (f) Half-life times of the triplex–duplex transition as a function of added fuel concentration. All reactions were carried out at 40 °C. | ||
In a second series of experiments, we used NAA as a chemical fuel to drive the controlled release-uptake of a cargo DNA strand from a pH-responsive DNA-based receptor. The latter is a stem-loop DNA sequence re-engineered with a pH-dependent domain located on the stem, far from the cargo recognition site (placed on the loop), that allows pH-dependent release-uptake of the cargo itself.19 At acidic pHs, the Hoogsteen interactions on the stem, in fact, stabilize the stem-loop structure thus hampering the ability of the receptor to bind the cargo DNA sequence (Fig. 3a, right). As the pH increases, the Hoogsteen interactions are destabilized and, as a result, the receptor can efficiently bind the cargo DNA (Fig. 3a, left). Release-uptake of the cargo strand from the receptor can be followed by a pH-independent optical pair on the DNA-receptor/cargo couple.
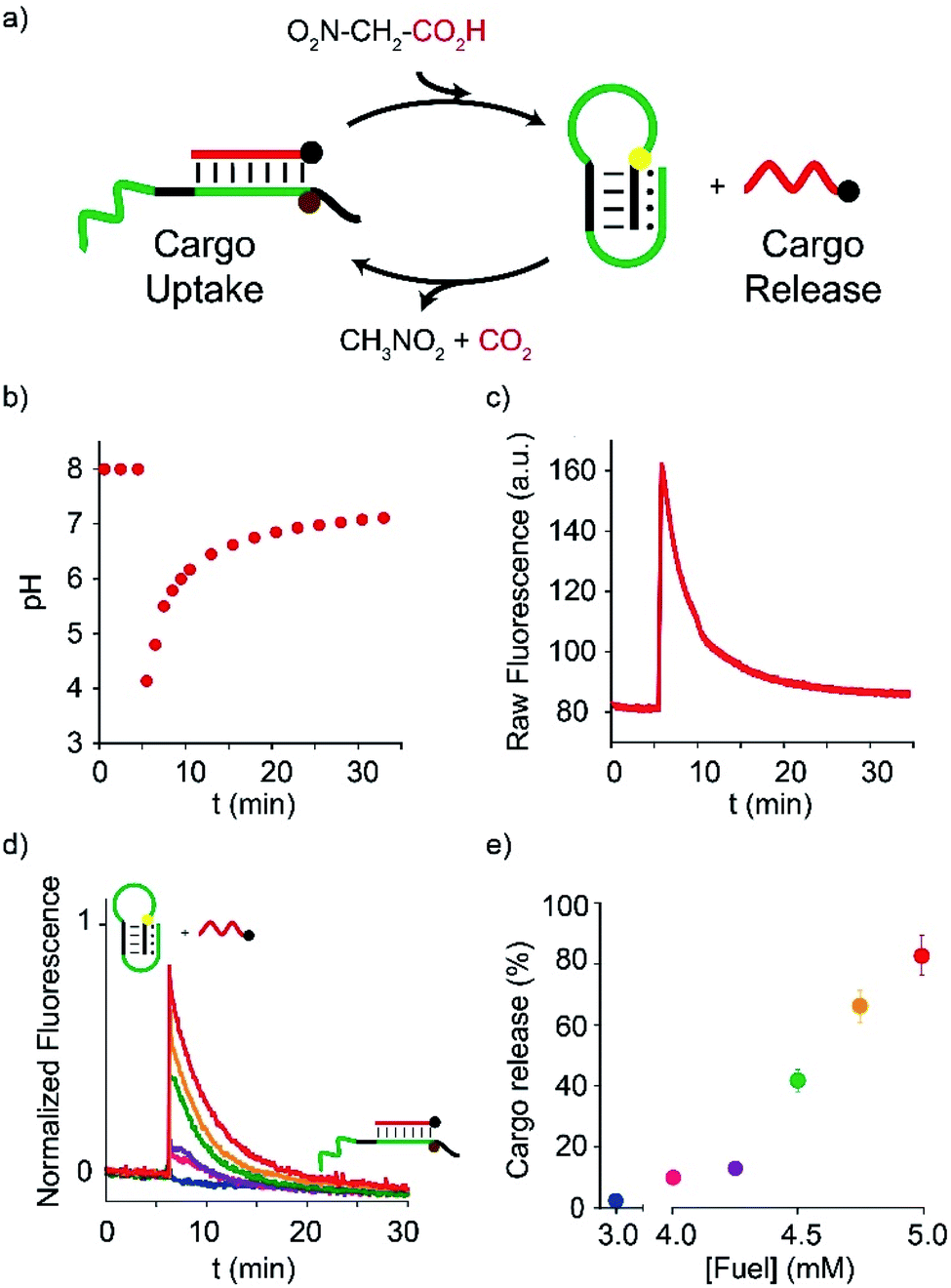 | ||
| Fig. 3 Dissipative release-uptake cycles of a DNA-cargo strand from a DNA-based receptor by fuel NAA. (a) Cartoon representing the unloading–loading cycles triggered by NAA. (b) pH-time monitoring of a 30.0 nM duplex·strand complex solution (initial pH = 8) after addition of the fuel. (c) Fluorescence-time monitoring of a 30.0 nM duplex·strand complex solution (initial pH = 8) after addition of the fuel. (d) Fluorescence-time profiles obtained after addition of increasing amounts of fuel to 30.0 nM duplex·strand complex (initial pH = 8). (e) Unloading percentage after addition of the given concentration of fuel (data obtained from Fig. 3d). All reactions were carried out at 25 °C. | ||
Also in this case, pH and fluorescence time-course experiments were recorded upon the addition of the acid fuel to a solution containing the receptor/cargo complex at a fixed pH (8.0). Immediately after the fuel addition (t = 5 min, Fig. 3b), the pH decreases from 8 to 4, and the fluorescence emission strongly increases (t = 5 min, Fig. 3c) suggesting that the cargo strand has been released from the receptor following the formation of the triplex structure on the stem. From this moment onwards, the deprotonated fuel starts to decarboxylate, the pH begins to rise again and the cargo is slowly re-loaded by the receptor (Fig. 3b§ and Fig. 3c). As a net result, a dissipative cycle of release-uptake has been realized by means of NAA. Furthermore, the amount of the DNA cargo transiently released from the receptor can be regulated by varying the amount of added fuel. More specifically, under the given conditions, addition of 4.0, 4.2, 4.5, 4.7, 5.0 mM causes the transient release of 10%, 15%, 50%, 70%, and 90% of the cargo strand, respectively (Fig. 3d and e). Successive release-uptake cycles can be achieved by subsequent additions of the fuel acid. In particular, three cycles have been obtained by three subsequent additions of NAA (Fig. ESI 2†). After each cycle, it is convenient to reset the pH to the initial value (i.e. pH = 8.0) by adding the required amount of NaOH, in order to obtain a better efficiency of the system.¶
Conclusions
In conclusion, we have demonstrated that the abiotic fuels CPA and NAA can be conveniently used to control the dissipative operation of pH-responsive DNA-based devices. In one case a conformational-change switch can be closed (triplex state) and then re-opened (duplex state) under the action of the pH variations (low → high pH cycles) driven by CPA, with the possibility to control at will the time required for the closing step (triplex → duplex transition). In the other case, NAA enables low → high pH cycles, which drive the autonomous release-uptake of a cargo DNA-strand to a DNA-based receptor. Remarkably, the amount of the cargo DNA transiently released in solution can be finely modulated by regulating the amount of the added fuel. It is noteworthy that the time programmable pH cycles driven by both fuels CPA and NAA (low → high pH cycles), are complementary to those reported by Walther and Heinen (high → low pH cycles),12 and, compared to the pH cycles based on the thiosulfate/sulfite oscillator,13 do not require a continuous fuel supply, moreover do not need the presence of enzymes that may limit the application range.14,26 As the number of responsive DNA-based devices is rapidly growing, the possibility to precisely control the energy dissipating pathway with different chemical fuels represents an essential step towards the design of DNA functional machines and materials with “life-like” behaviour.Experimental
Experimental details in ESI.†Author contributions
SDS, FR and GE conceived the general design. SDS, GE, DM, DDG and FR designed the experiments. DDG and DM performed the experiments. SDS, GE, DM, DDG and FR analyzed the data. SDS, GE and FR wrote the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Associazione Italiana per la Ricerca sul Cancro, AIRC (project n. 21965) (FR), by the European Research Council, ERC (Consolidator Grant project n. 819160) (FR) and by University of Roma La Sapienza (Progetti di Ricerca Grandi 2018, prot. n. RG1181641DCAAC4E) (SDS).Notes and references
- W. F. Harrington and M. E. Rodgers, Annu. Rev. Biochem., 1984, 53, 35–73 CrossRef CAS.
- N. Hirokawa, Science, 1998, 279, 519–526 CrossRef CAS PubMed.
- H. Noji, R. Yasuda, M. Yoshida and K. Kinosita Jr, Nature, 1997, 386, 299–302 CrossRef CAS.
- (a) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS; (b) K. Das, L. Gabrielli and L. J. Prins, Angew. Chem., Int. Ed., DOI:10.1002/anie.202100274; (c) Out-of-Equilibrium (Supra)molecular Systems and Materials, ed. N. Giuseppone and A. Walther, 2021, Wiley-VHC, Weinheim Search PubMed.
- Some selected examples are: (a) S. A. P. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519–5535 RSC; (b) F. della Sala, S. Neri, S. Maiti, J. L.-Y. Chen and L. J. Prins, Curr. Opin. Biotechnol., 2017, 46, 27–33 CrossRef CAS PubMed; (c) C. Pezzato, C. Cheng, J. F. Stoddart and R. D. Astumian, Chem. Soc. Rev., 2017, 46, 5491–5507 RSC; (d) A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC; (e) R. Merindol and A. Walther, Chem. Soc. Rev., 2017, 46, 5588–5619 RSC; (f) G. Ragazzon and L. J. Prins, Nat. Nanotechnol., 2018, 13, 882–889 CrossRef CAS PubMed; (g) R. D. Astumian, C. Pezzato, Y. Feng, Y. Qiu, P. R. McGonigal, C. Cheng and J. F. Stoddart, Mater. Chem. Front., 2020, 4, 1304–1314 RSC; (h) B. Rieß, R. K. Grötsch and J. Boekhoven, Chem, 2020, 6, 552–578 CrossRef; (i) M. Weißenfels, J. Gemen and R. Klajn, Chem, 2021, 7, 23–37 CrossRef; (j) L. S. Kariyawasam, M. M. Hossain and C. S. Hartley, Angew. Chem., Int. Ed., 2021, 60, 12648–12658 CrossRef CAS.
- C. S. Wood, C. Browne, D. M. Wood and J. R. Nitschke, ACS Cent. Sci., 2015, 1, 504–509 CrossRef CAS.
- C. Biagini and S. Di Stefano, Angew. Chem., Int. Ed., 2020, 59, 8344–8354 CrossRef PubMed.
- (a) B. Yurke, A. J. Turberfield, A. P. Mills Jr, F. C. Simmel and J. L. Neumann, Nature, 2000, 406, 605–608 CrossRef CAS PubMed; (b) T. Omabegho, R. Sha and N. C. Seeman, Science, 2009, 324, 67–71 CrossRef CAS; (c) J. Lloyd, C. H. Tran, K. Wadhwani, C. C. Samaniego, H. K. K. Subramanian and E. Franco, ACS Synth. Biol., 2018, 7, 30–37 CrossRef CAS PubMed; (d) E. Del Grosso, A. Amodio, G. Ragazzon, L. J. Prins and F. Ricci, Angew. Chem., Int. Ed., 2018, 57, 10489–10493 CrossRef CAS; (e) E. Del Grosso, G. Ragazzon, L. J. Prins and F. Ricci, Angew. Chem., Int. Ed., 2019, 17, 5582–5589 CrossRef PubMed; (f) E. Del Grosso, I. Ponzo, G. Ragazzon, L. J. Prins and F. Ricci, Angew. Chem., Int. Ed., 2020, 59, 21058–21063 CrossRef CAS; (g) L. Heinen and A. Walther, Sci. Adv., 2019, 5, eaaw0590 CrossRef CAS PubMed; (h) J. Deng, D. Bezold, H. J. Jessen and A. Walther, Angew. Chem., Int. Ed., 2020, 29, 12084–12092 CrossRef; (i) Z. Zhou, Y. Ouyang, J. Wang and I. Willner, J. Am. Chem. Soc., 2021, 143, 5071–5079 CrossRef CAS PubMed.
- (a) S. Agarwald and E. France, J. Am. Chem. Soc., 2019, 141, 7831–7841 CrossRef; (b) E. Del Grosso, L. J. Prins and F. Ricci, Angew. Chem., Int. Ed., 2020, 59, 13238–13245 CrossRef CAS; (c) J. Deng and A. Walther, J. Am. Chem. Soc., 2020, 142, 685–689 CrossRef PubMed; (d) S. Wang, L. Yue, V. Wulf, S. Lilienthal and I. Willner, J. Am. Chem. Soc., 2020, 142, 17480–17488 CrossRef CAS.
- A. Kurkdjian and J. Guern, Annu. Rev. Plant Physiol. Plant Mol. Biol., 1989, 40, 271–303 CrossRef CAS.
- (a) T. Heuser, A.-K. Steppert, C. Molano Lopez, B. Zhu and A. Walther, Nano Lett., 2015, 15, 2213–2219 CrossRef CAS PubMed; (b) E. Jee, T. Bánági Jr, A. F. Taylor and J. A. Pojman, Angew. Chem., Int. Ed., 2016, 55, 2127–2131 CrossRef CAS; (c) T. Heuser, E. Weyandt and A. Walther, Angew. Chem., Int. Ed., 2015, 54, 13258–13262 CrossRef CAS PubMed; (d) Y. Okamoto and T. R. Ward, Angew. Chem., Int. Ed., 2017, 56, 10156–10160 CrossRef CAS; (e) H. Che, B. C. Buddingh and J. C. M. van Hest, Angew. Chem., Int. Ed., 2017, 56, 12581–12585 CrossRef CAS PubMed; (f) H. E. Cingil, N. C. H. Meertens and I. K. Voets, Small, 2018, 14, 1802089 CrossRef; (g) P. Dowari, S. Das, B. Pramanik and D. Das, Chem. Commun., 2019, 55, 14119–14122 RSC; (h) S. Mondal, D. Podder, S. Kumar Nandi, S. Roy Chowdhury and D. Haldar, Soft Matter, 2020, 16, 10115–10121 RSC.
- L. Heinen and A. Walther, Chem. Sci., 2017, 8, 4100–4107 RSC.
- T. Liedl and F. C. Simmel, Nano Lett., 2005, 5, 1894–1898 CrossRef CAS PubMed.
- (a) L. Heinen, T. Heuser, A. Steinschulte and A. Walther, Nano Lett., 2017, 17, 4989–4995 CrossRef CAS PubMed; (b) X. Fan and A. Walther, Angew. Chem., Int. Ed., 2021, 60, 3619–3624 CrossRef CAS PubMed; (c) X. Fan and A. Walther, Angew. Chem., Int. Ed., 2021, 60, 11398–11405 CrossRef CAS PubMed.
- A. Idili, A. Vallée-Bélisle and F. Ricci, J. Am. Chem. Soc., 2014, 136, 5836–5839 CrossRef CAS.
- D. Mariottini, A. Idili, M. A. D. Nijenhuis, G. Ercolani and F. Ricci, J. Am. Chem. Soc., 2019, 141, 11367–11371 CrossRef CAS.
- A. Idili, K. W. Plaxco, A. Vallée-Bélisle and F. Ricci, ACS Nano, 2013, 7(12), 10863–10869 CrossRef CAS PubMed.
- D. Mariottini, A. Idili, A. Vallée-Bélisle, K. W. Plaxco and F. Ricci, Nano Lett., 2017, 17, 3225–3230 CrossRef CAS PubMed.
- A. Porchetta, A. Idili, A. Vallée-Bélisle and F. Ricci, Nano Lett., 2015, 15, 4467–4471 CrossRef CAS.
- J. Ren, Y. Hu, C.-H. Lu, W. Guo, M. A. García, F. Ricci and I. Willner, Chem. Sci., 2015, 6, 4190–4195 RSC.
- (a) J. A. Berrocal, C. Biagini, L. Mandolini and S. Di Stefano, Angew. Chem., Int. Ed., 2016, 55, 6997–7001 CrossRef CAS; (b) A. Ghosh, I. Paul, M. Adlung, C. Wickleder and M. Schmittel, Org. Lett., 2018, 20, 1046–1049 CrossRef CAS; (c) C. Biagini, F. Di Pietri, L. Mandolini, O. Lanzalunga and S. Di Stefano, Chem.–Eur. J., 2018, 24, 10122–10127 CrossRef CAS; (d) C. Biagini, G. Capocasa, V. Cataldi, D. Del Giudice, L. Mandolini and S. Di Stefano, Chem.–Eur. J., 2019, 25, 15205–15211 CrossRef CAS PubMed; (e) C. Biagini, G. Capocasa, D. Del Giudice, V. Cataldi, L. Mandolini and S. Di Stefano, Org. Biomol. Chem., 2020, 18, 3867–3873 RSC.
- (a) C. Biagini, S. Albano, R. Caruso, L. Mandolini, J. A. Berrocal and S. Di Stefano, Chem. Sci., 2018, 9, 181–188 RSC; (b) P. Franchi, C. Poderi, E. Mezzina, C. Biagini, S. Di Stefano and M. Lucarini, J. Org. Chem., 2019, 84, 9364–9368 CrossRef CAS; (c) D. Del Giudice, E. Spatola, R. Cacciapaglia, A. Casnati, L. Baldini, G. Ercolani and S. Di Stefano, Chem.–Eur. J., 2020, 26, 14954–14962 CrossRef CAS PubMed.
- (a) S. Erbas-Cakmak, S. D. P. Fielden, U. Karaca, D. A. Leigh, C. T. McTernan, D. J. Tetlow and M. R. Wilson, Science, 2017, 358, 340–343 CrossRef CAS PubMed; (b) C. Biagini, S. P. Fielden, D. A. Leigh, F. Schaufelberger, S. Di Stefano and D. Thomas, Angew. Chem., Int. Ed., 2019, 58, 9876–9880 CrossRef CAS.
- D. Del Giudice, E. Spatola, M. Valentini, C. Bombelli, G. Ercolani and S. Di Stefano, Chem. Sci., 2021, 12, 7460–7466 RSC.
- (a) A. Ghosh, I. Paul and M. Schmittel, J. Am. Chem. Soc., 2019, 141, 18954–18957 CrossRef CAS; (b) A. Ghosh, I. Paul and M. Schmittel, J. Am. Chem. Soc., 2021, 143, 5319–5323 CrossRef CAS PubMed.
- T. Heuser, E. Weyandt and A. Walther, Angew. Chem., Int. Ed., 2015, 54, 13258–13262 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03435a |
| ‡ These authors contributed equally. |
| § The fact that the final pH differs from the initial one (about one unit lower) depends on the multiple acid–base equilibria involving fuel by-products including CO2. More specifically, in the case of NAA the complex network of acid–base equilibria associated to the reaction has been analysed and discussed in detail in a previous work (ref. 24). An analogous behaviour is expected for CPA. As to the role of CO2, one may ask whether there are differences when the system is operated either in an open or closed reactor. Of course, in a closed system an increase in the partial pressure of CO2 is expected that translates into a higher concentration of CO2 in solution. However, we recorded only a slightly lower final pH value (ca. 0.3 units) when the experiment was carried out in a closed reactor. |
| ¶ If the pH is not reset to 8 after each release-uptake cycle by adding the required amount of NaOH, the system loses efficiency, probably due to the accumulation of reaction products that someway affect the decarboxylation process (see Fig. ESI 3† and ref. 24). |
| This journal is © The Royal Society of Chemistry 2021 |