
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStimuli-responsive temporary adhesives: enabling debonding on demand through strategic molecular design
Nicholas D.
Blelloch
,
Hana J.
Yarbrough
 and
Katherine A.
Mirica
*
and
Katherine A.
Mirica
*
Burke Laboratory, Department of Chemistry, Dartmouth College, Hanover, New Hampshire 03755, USA. E-mail: katherine.a.mirica@dartmouth.edu; Web: http://www.miricagroup.com
First published on 22nd October 2021
Abstract
Stimuli-responsive temporary adhesives constitute a rapidly developing class of materials defined by the modulation of adhesion upon exposure to an external stimulus or stimuli. Engineering these materials to shift between two characteristic properties, strong adhesion and facile debonding, can be achieved through design strategies that target molecular functionalities. This perspective reviews the recent design and development of these materials, with a focus on the different stimuli that may initiate debonding. These stimuli include UV light, thermal energy, chemical triggers, and other potential triggers, such as mechanical force, sublimation, electromagnetism. The conclusion discusses the fundamental value of systematic investigations of the structure–property relationships within these materials and opportunities for unlocking novel functionalities in future versions of adhesives.
 Katherine A. Mirica, Hana J. Yarbrough and Nicholas D. Blelloch | Nicholas D. Blelloch received his BA in Chemistry from Middlebury College in 2016. He recently (2021) earned his PhD in Chemistry from Dartmouth College under the guidance of Prof. Katherine A. Mirica. His interests involve research at the interface of fundamental chemistry and materials science and enjoying New England's outdoor adventures. Hana J. Yarbrough earned her BSc degree chemistry in 2020 from St. Mary's College of Maryland. She is a second-year Ph.D. student at Dartmouth College working with Prof. Katherine Mirica. Her research interests include adhesion science, polymeric materials, and materials engineering. Katherine A. Mirica is an Associate Professor of Chemistry at Dartmouth College. She obtained a B.S. in Chemistry from Boston College in 2004, where she worked in the laboratory of Lawrence T. Scott. She earned her Ph.D. in Chemistry under the guidance of George M. Whitesides at Harvard University in 2011 and completed her postdoctoral training with Timothy M. Swager at the Massachusetts Institute of Technology in 2015. She launched her independent career at Dartmouth College in 2015. Her current research interests span the study of structure–function relationships of molecularly precise materials, self-assembly, gas sensors, and adhesion science. |
Introduction
The importance of stimuli-responsive temporary adhesives
The development and adaptation of adhesive materials, particularly stimuli-responsive temporary adhesives, has significantly contributed to the advancement of human civilization and economic growth.1,2 From the Paleolithic use of tar-like substances for tool manufacturing to modern adhesive applications in medicine,3–7 electronics,8–11 construction,12 and consumer products,13 adhesives permeate much of our daily life.14 For example, over 50 billion Post-It notes—those ubiquitous brightly-colored notes that stick to surfaces, but are easily removed and rearranged—are sold each year worldwide.15,16Stimuli-responsive adhesives represent a class of materials that undergo a physical and/or chemical change in response to an applied stimulus or stimuli. These alterations are intended to strengthen or weaken the adhesive performance. Overcoming fundamental knowledge gaps regarding the structure–property relationships in temporary adhesives—a pursuit motivated by modern technological challenges that demand new function—will enhance the broad utility of these materials. Stimuli-responsive adhesives that release adherends on demand have utility in art conservation and restoration,17 biomedical engineering,3 microelectronics manufacturing,10,18 and office supplies.19 For example, this type of adhesive is extremely promising for biomedical applications, where fragile surfaces, such as organs or healing wounds, must be protected during removal.20–23 In contrast, stimuli-responsive materials that demonstrate permanent adhesion after a stimulating event are especially useful in dentistry,6,7 wound dressings,5,23,24 and aerospace engineering.25,26
Unique characteristics of stimuli-responsive temporary adhesives
A stimuli-responsive adhesive is a material designed to promote the joining of two surfaces that exhibits a change in its mechanical and/or adhesive properties after being exposed to an external stimulus or stimuli. While stimuli-responsive permanent adhesives, also known as structural adhesives, exist,27–30 this perspective is concerned with stimuli-responsive temporary adhesives, a class of materials that exhibit the conflicting properties of bonding, resistance to mechanical stress and strain, and on-demand debonding.29,31–42,162 All temporary adhesives are stimuli-responsive; once two surfaces are bonded, a temporary adhesive requires some form of energy input to debond. This energy input is delivered by an external stimulus or stimuli.Stimuli-responsive temporary adhesives possess several unique characteristics compared with permanent adhesives. First, these adhesives may be removed without risking damage to fragile surfaces by utilizing a strategic stimulus that triggers debonding selectively and specifically.27,29,43,44 Second, through strategic molecular design, these materials exhibit stimuli-specific reactivity.38,45,46 Potential stimuli include light, heat, various chemicals, pressure, mechanical force, magnetism, or electricity. The strategic selection of an appropriate stimulus or stimuli means that temporary adhesives can be used in a wide variety of applications without risk of unintended debonding. Finally, the diversity of temporary adhesive systems presents numerous advantages, such as conformal contact to the surface, dissipation of stress and strain over a large bonded area, dampening of vibrations, improved resistance to fatigue and electrochemical corrosion, and easy application to delicate surfaces.14,47–51
Fundamentals of bonding and debonding
Adhesion originates from a combination of physical and chemical mechanisms. A general definition of adhesion has been provided by Wu:“Adhesion refers to the state in which two dissimilar bodies are held together by intimate interfacial contact such that mechanical force or work can be transferred across the interface. The interfacial forces holding the two phases together may arise from van der Waals forces, chemical bonding, or electrostatic interaction. Mechanical strength of the system is determined not only by the interfacial forces, but also by the mechanical properties of the interfacial zone and the two bulk phases.”52
From this definition, it follows that an adhesive is a material that promotes adhesion between two dissimilar bodies. Additionally, all functional adhesives must also promote cohesion, which is analogous to adhesion, except that it refers to transfer of mechanical force or work through a uniform bulk.
As suggested by Wu, the origins of adhesion (and cohesion) have traditionally been divided into different classical schools of thought: mechanical theory, electrostatic theory, diffusion theory, adsorption theory, and chemical bonding theory.27,29,30,44 Mechanical adhesion relies on the physical interlocking of adhesive and adherend.29,53 The electrostatic theory originates from the attraction between opposite electric charges that create electrostatic forces at the interface of the bond.54–60 Alternatively, diffusion theory, the most applicable for polymeric systems, states that the molecules in the adhesive and adherend can diffuse into the adjoining material, creating an adhesive interaction.29 Adsorption theory states that, once two surfaces are brought into contact, attractive forces will act between them, resulting in adhesion.27,29,30,61 Finally, the chemical bonding theory centers on the concept of creating covalent chemical bonds between the adherend-adhesive interface.29
Adsorption theory (also referred to as wettability theory) and chemical bonding theory are the most relevant to stimuli-responsive temporary adhesives because they are concerned with molecular-level interactions. When two molecular species are brought into proximity (i.e., sub-nm-scale distances), varied interactions form between them (Table 1). These interactions, the type and strength of which depend on the chemical identity of the two species, enable the successful transfer of force or work.27,30,61,62 Consequently, both permanent and temporary adhesives are strategically designed to promote the formation of strong interactions at the adhesive–adherend interface.
| Type of interaction | Typical strength (kJ mol−1) | Distance dependence |
|---|---|---|
| Covalent | 200–1000 | r −1 |
| Coulombic | 170–1500 | r −1 |
| Hydrogen bond | 5–150 | r −2 |
| Halogen bond | 5–150 | r −2 |
| Cation–π | 5–150 | r −2 |
| Ion–dipole | 10–50 | r −2 |
| Dipole–dipole | 5–20 | r −3 |
| Quadrupole–quadrupole | 4–30 | r −3 |
| Dipole-induced dipole | 1–5 | r −6 |
| London dispersion | 1–10 | r −6 |
Stimuli-responsive adhesives are unique in that the interactions present at the interface and within the bulk of these materials can be strategically controlled. In these materials, introduction of an external stimulus weakens or breaks the intermolecular interactions responsible for adhesion, resulting in on-demand debonding. The exact mechanism that promotes stimulus-induced debonding depends on the molecular design of an adhesive. For example, a stimulus may initiate a chemical change within the adhesive that weakens the cohesive strength of the material (e.g., stimuli-induced decrease in the cross-linking density within a polymer network).64–67 Alternatively, a stimulus may affect adhesive interactions (e.g., preferential formation of adhesive–stimuli interactions relative to adhesive–adherend interactions).68–71Table 2 summarizes a range of examples for specific release mechanisms.
| Trigger | Key structural feature for debonding | Release mechanism | Maximum adhesive strength | Strength after initial debonding | Highlights | Selected applications | Reference |
|---|---|---|---|---|---|---|---|
| a Strength during peel test. b Strength after rebonding and cyclic testing. c No single responsive group responsible for debonding. d Reported as adhesive force, adhesive strength not reported. | |||||||
| UV light |

|
Photoisomerization | 5.0 MPa | 0.003 MPa | Sugar alcohol scaffold | Coatings, dental medicine | 66 |
| UV light |
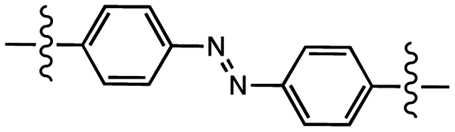
|
Photoisomerization | 0.571 MPa | Not reported | Reversible adhesion via reverse photoisomerization (455 nm) | Soft nanomaterials | 86 |
| UV light |
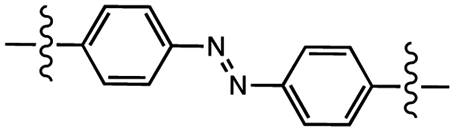
|
Photoisomerization | 1.7 MPa | 0.1 MPa | Healing of polymer film via photoisomerization cycling | Transfer printing | 92 |
| UV light |

|
Increasing degree of cross-linking | 279.1 N m−1a | 2.1 N m−1a | Rigidification of polymer network with UV-active cross-linkers | Microelectronics manufacturing | 67 |
| UV light and heat (100 °C) |

|
Depolymerization via backbone cleavage | 6.9 MPa | Not reported | Irreversible depolymerization resulting in volatile monomers | Materials recycling | 87 |
| UV light (365 nm) |

|
Photoisomerization | 1.6 MPa | 0.2 MPa | Liquification due to flexible UV-active joint | Optical switching, image storage | 96 |
| UV light (320–390 nm) |

|
Dynamic covalent bond exchange | ∼6.5 MPa | No significant changeb | Phase transition of semi-crystalline polymer results in debonding and shape memory | Aircraft parts | 99 |
| Heat (70 °C and 150 °C) | 5.3 MPa | ||||||
| Heat (150 °C) |

|
Reverse Diels–Aider reaction | 8.0 MPa | 2.0 MPa (rapid cooling)b | Reversible adhesion of cross-linked polymer depends on cooling rate | Materials recycling, thermoset adhesives | 106 |
| 5.9 MPa (slow cooling)b | |||||||
| Heat (60 °C) |

|
Depolymerization via backbone cleavage; disrupting intermolecular interactions | 3.0–5.8 MPa | Not reported | Sugar–acid supramolecular polymer | Biodegradable biocompatible materials | 107 |
| Heat |

|
Disrupting intermolecular interactions | 0.1 MPa | 0.05 MPa | Semi-crystalline shape memoty polymer | Microelectronics manufacturing | 108 |
| Heat (<200 °C) |

|
Gas formation and volumetric expansion | 68–88 MPa | 27–67 MPa | Decarboxylation increases porosity into polymer matrix | Dental cements, microelectronics manufacturing | 109 |
| Heat (160 °C) |

|
Dynamic covalent bond exchange | 1.06 MPa | No significant changeb | Self-immolative polymer with dynamic covalent bond cross-linkers | Thermoset adhesives | 65 |
| Fluoride ions |

|
Depolymerization via backbone cleavage | Irreversible debonding | ||||
| Fluoride ions |

|
Depolymerization via end cap cleavage | 2.86–11.41 MPa | 1.82–7.25 MPa | semi-crystalline self-immolative polymer | Materials recycling thermoset adhesives | 148 |
| Fluoride ions |

|
Breaking cross-linkers | 0.51 MPa | 0.05 MPa | Polymer with self-immolative cross-linkers | Tissue adhesion, wound dressings | 110 |
| Metal ions |

|
Metal ion-mediated molecular recognition | 0.001 MPa (Cu2+) | Not reported | Supramolecular hydrogel adhesive | Wet substrates | 68 |
| Metal ions |

|
Metal ion-mediated molecular recognition | Not reported | Not reported | Mussel-inspired self-assembled monolayer | Metal oxide surface coatings | 155 |
| Water |

|
Solubilizing the polymer network | 0.013–1.88 MPa | No significant changeb | Self-healing supramolecular polymer | Tissue adhesion, wound dressings | 71 |
| Heat |

|
Disrupting intermolecular interactions | |||||
| Reduced pressure and heat (90% of melting) | NAc | Sublimation | 0.22–0.33 MPa | No significant changeb | Polycyclic aromatic hydrocarbon | Microelectronics manufacturing | 32 |
| Reduced pressure and heat | NAc | Sublimation | 1.24 MPa | Not reportedb | Polycyclic aromatic hydrocarbon | Microelectronics manufacturing | 31,162 |
| Directional force | NAc | Self-peeling due to curvature | 0.055 Nd | No significant changeb | Hydrogel layered with mushroom structured arrays and copolymer adhesive | Wet/dry substrates, mobile robots | 227 |
Scope and focus of this perspective
The goal of this perspective is to emphasize recent fundamental and applied advances in the molecular design of stimuli-responsive temporary adhesives capable of debonding on demand. We categorize our discussion of the latest strategies for designing stimuli-responsive temporary adhesives according to four types of stimuli initiating debonding, which also govern the release mechanism: UV light, thermal energy, chemical triggers, and other approaches (mechanical force, sublimation, and electromagnetism) (Table 2). Throughout this perspective, we aim to: (i) detail how the complexities of adhesion phenomena that span length scales are incorporated into the rational design of moieties that respond to stimuli by debonding substrates; (ii) discuss the advantages and disadvantages to triggering debonding with particular stimuli in applications with unique challenges, such as microelectronic manufacturing or wound dressings; and (iii) highlight the emergence of stimuli-responsive temporary adhesives as multifunctional materials afforded by key structure–property relationships. We conclude by underscoring the opportunities to elucidate fundamental structure–property relationships and a perspective to unlocking the potential functionalities of this class of materials.This article provides a perspective over the field and, consequently, is not comprehensive. Specifically, we do not provide a thorough discussion of the fundamentals of adhesion,27,44,72 or of permanent adhesives.29,30,73,74 The specific examples highlighted in this perspective article do not constitute an exhaustive list of all stimuli-responsive temporary adhesive systems, or even all stimuli used in these systems.14,38,42,75–81 Interested readers are directed to the aforementioned comprehensive reviews on these topics.14,38,42,75–81
UV-triggered release from adhesion
Ultraviolet (UV) light is an ideal stimulus for initiating debonding of temporary adhesives because of its ease of application and real-time spatiotemporal control of exposure, energy, and stoichiometry.59,82,83 The three most employed UV-induced debonding mechanisms are (i) photoliquification and photoisomerization,66,84–86 (ii) the fabrication of UV-curable cross-linkers,67 and (iii) selective depolymerization of adhesive systems containing photobase generators.87 For strategically designed adhesives, light induces various changes in the material that diminish the intermolecular interactions responsible for bonding. These changes may be reversible, enabling the adhesive to rebond substrates after initial exposure to light.88–91 Alternatively, UV-sensitive irreversible temporary adhesives can promote permanent debonding by increasing the polymer side-chain cross-linking density, creating interpenetrating networks, or by employing photodegradable linkers.42 Although the central focus of this section is to discuss triggering debonding, we note that UV light may also be used to enhance adhesive strength.69,70 A report from Bardeen and coworkers revealed that UV light can initiate molecular and polymeric movements, which ultimately improves adhesion by increasing interactions at the adhesive–substrate interface.69Adhesive liquification via photoisomerization
Eliciting debonding of a polymeric adhesive with UV light can be accomplished with a solid-to-liquid phase transition, a mechanism known as photoliquification or photomelting.92–94 A UV-sensitive polymeric adhesive must be strategically designed with groups that respond by altering the bulk configuration of the polymer chains, such as packing, volume, or flexibility. One of the most well-known photoswitchable compounds, azobenzene, undergoes reversible photoisomerization between the cis- and trans-state, which also corresponds to distinct solid and liquid phases.64,95The photoisomerization of azobenzene is limited, if not entirely inhibited, in highly crystalline systems with insufficient volume to allow for geometrical rearrangement. For this reason, Akiyama et al. published an early report of azobenzene photoliquification from a highly ordered solid. The authors demonstrated that a sugar alcohol scaffold functionalized with azobenzene-containing moieties underwent reversible, isothermal photoinduced crystal-to-liquid phase transitions (Fig. 1a).66 Using quartz adherends, the crystalline adhesive supported shear strengths up to 500 N cm−2. Upon photoliquification (λmax = 365 nm, 20 mW cm−2), single-lap joints could only support 0.3 N cm−2 in shear. Bond strength was fully restored upon irradiation with visible light (λmax = 510 nm, ∼20 mW cm−2). The demonstration of reversible bonding, in addition to the other reversible processes of this polymer, highlights the power of controlling bulk properties of a well-ordered solid through a photoisomerization mechanism.
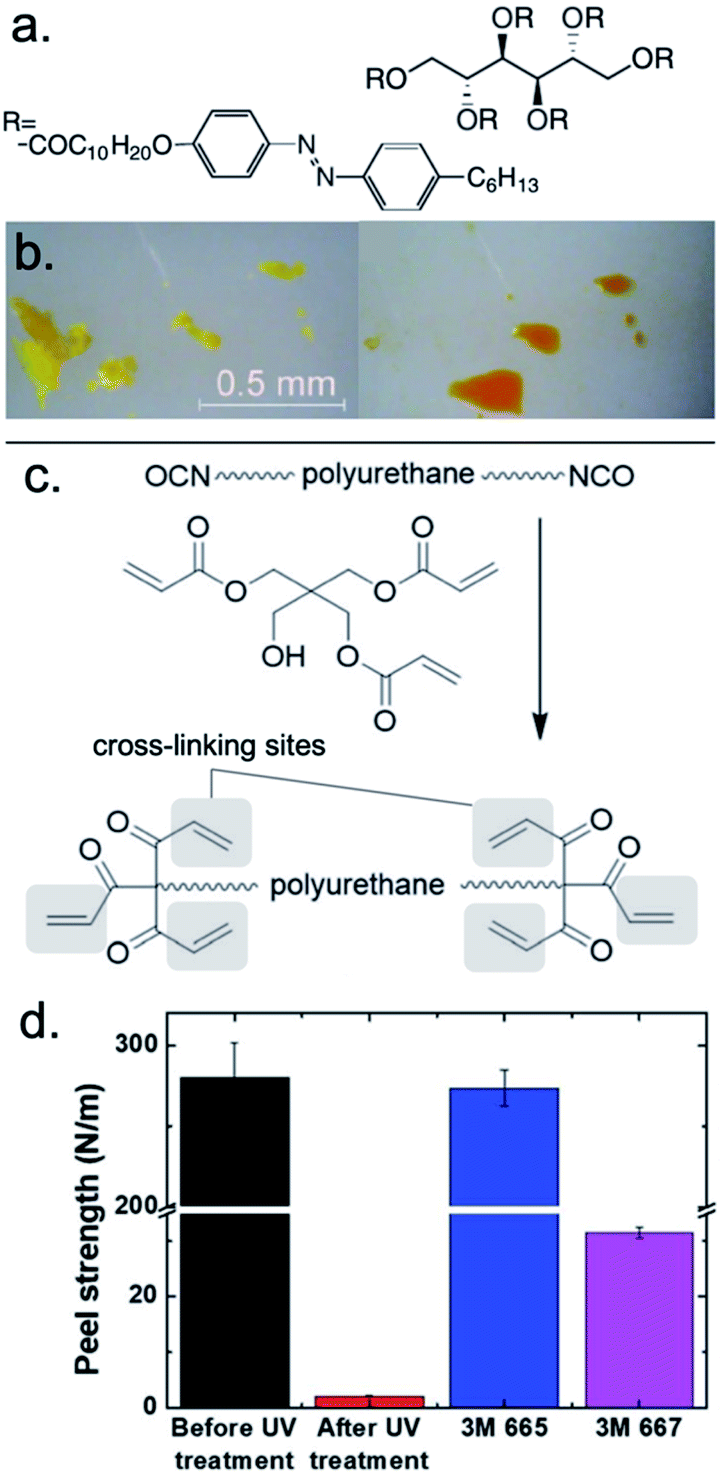 | ||
| Fig. 1 UV-triggered release from adhesion (a) sugar alcohol scaffold functionalized with azobenzene-containing moieties (b) reversible, isothermal photoinduced crystal-to-liquid phase transitions after UV irradiation for 3 min (λ = 365 nm, 40 mW cm−2). Adapted with permission from ref. 66. Copyright 2012 Wiley-VCH.66 (c) Synthetic scheme of polyurethane acrylate, which produces the PSA when mixed with an acrylic polymer, epoxy cross-linker, and Irgacure 184. (d) Peel strengths of the PSA before and after UV irradiation compared to 3M Scotch 665 and 667. Adapted from ref. 67 with permission of The Royal Society of Chemistry.67 | ||
Photoliquification occurs when the ordered structure of a material is perturbed. These perturbations reduce the packing capability of flexible polymer chains. Siloxane oligomers functionalized with aromatic rings are known to produce well-ordered crystalline domains. Ordering of responsive groups across a range of length scales promotes a material to be multifunctional and multiresponsive. Using this phenomenon as inspiration, Meijer and coworkers designed liquid crystals based on telechelic dimethylsiloxane oligomers with azobenzene end groups.86 Upon synthesis, the materials presented a lamellar microstructure in which amorphous dimethylsiloxane oligomers separated crystalline azobenzene monolayers. In shear experiments, the yellow wax resisted forces up to 571 kPa, depending on the length of the oligomers. On-demand debonding was achieved within seconds, with 365 nm light inducing a trans-to-cis isomerization of azobenzene units to produce a red oil. The reverse photoisomerization (cis-to-trans) occurred within 1–2 days under ambient conditions, or nearly instantaneously under blue light irradiation (λmax = 455 nm, 200 mW cm−2). Authors demonstrated that the high degree of molecular ordering due to crystalline azobenzene domains can be drastically reduced by photoisomerization, ultimately leading to debonding of the liquid material.
Specific applications may require that the temporary polymeric adhesives undergo structural changes after the initial bonding, while maintaining a strong bond. In such cases, the material should have a relatively low glass transition temperature (Tg) for optimal processing and high Tg when stiffness is desired. Wu and coworkers utilized RAFT-synthesized acrylate- and methacrylate-based homopolymers containing azobenzene side groups attached via flexible spacers.92 In the azobenzene trans-configuration, the polymers exhibited glass transition temperatures (Tg) above room temperature, but in the cis-configuration, the Tg values dropped below 0 °C. Up to five-time cyclability of the photoisomerization and, consequently, of adhesion and debonding, was confirmed by UV-Vis absorption spectroscopy promoted cycling the photoisomerization, and healing damage to the polymer films. The trans-to-cis isomerization also decreased resistance to shear from 1.7 MPa to 0.1 MPa. In this work, authors reported one of the first polymers that can reversibly transition between hard and soft states throughout the whole material or at a precise location.
As an example of non-azobenzene photoisomerization for debonding, Saito et al. developed a light-melt liquid crystal adhesive with a cyclooctatetraene reactive moiety and rigid anthracene “wings”.96 Initially, the material assumed a V-shaped columnar-stacking arrangement, which promoted strong aromatic stacking interactions between anthracene moieties in adjacent molecules. In this liquid crystalline phase, the adhesive was able to bond glass adherends with shear strengths of ∼1.6 MPa. Heating the adhesive to 70–135 °C eliminated the columnar stacking, but did not reduce adhesion; however, when coupled with UV-irradiation (365 nm), the anthracene moieties dimerized, reducing adhesion to ∼0.2 MPa. The length of time required for debonding was dependent on the intensity of light. Heating the debonded material above 160 °C, followed by cooling, restored the liquid crystal phase and the adhesive capabilities.
Fabrication of UV-curable cross-linkers
The adhesive properties of polymeric systems often vary as a function of their cross-linking density, which can be controlled photochemically.42 Kim et al. recently proposed the use of an irreversible pressure-sensitive adhesive (PSA) that can rapidly debond substrates upon UV curing during the fabrication of epidermal electronic devices (Fig. 1c).67 These ultrathin and/or flexible devices must be adhered to carrier substrates during their fabrication process, and a stimuli-responsive adhesive is required for their on-demand release after fabrication. The authors formulated an acrylic adhesive comprising butyl acrylate, 2-ethylhexyl acrylate, acrylic acid, ethyl acrylate, and 2-hydroxyethyl acrylate that was capable of bonding substrates with a peel strength of 279.9 N m−1. The formulation also included a pentaerythritol triacrylate (PETA)-terminated cross-linking oligomer. UV irradiation (3000 mJ cm−2) induced cross-linking between the acrylate moieties on the polymer and oligomer, which decreased the peel strength to 2.1 N m−1 (Fig. 1d). Cross-linking in this and similar polymer adhesives increased the storage density of the material, which violated the Dahlquist criterion of tack, and resulted in loss of adhesive capabilities.42Selective depolymerization via photobase generator
Finally, UV light can be used to completely degrade a photoresponsive temporary adhesive. With this approach, macromolecular adhesives may lose their adhesive capabilities upon depolymerization. For example, Sasaki et al. utilized a photodepolymerizable cross-linked poly(olefin sulfone) as a self-immolative temporary adhesive (Fig. 2).87 By mixing a cross-linkable poly(olefin sulfone), a cross-linking reagent, and a photobase generator, the researchers formulated a novel thermosetting adhesive. Upon bonding quartz surfaces, the adhesive resisted tensile stresses of 6.9 ± 0.2 N mm−2. Neither heating, nor UV irradiation (254 nm, 3.0 J cm−2) alone resulted in a significant decrease in the adhesive performance. However, a combination of UV irradiation and heating to 100 °C almost instantaneously resulted in a decrease in tensile strength. Within 15 min, tensile strength was reduced ∼95%. Within 1 h, tensile strength was nearly eliminated.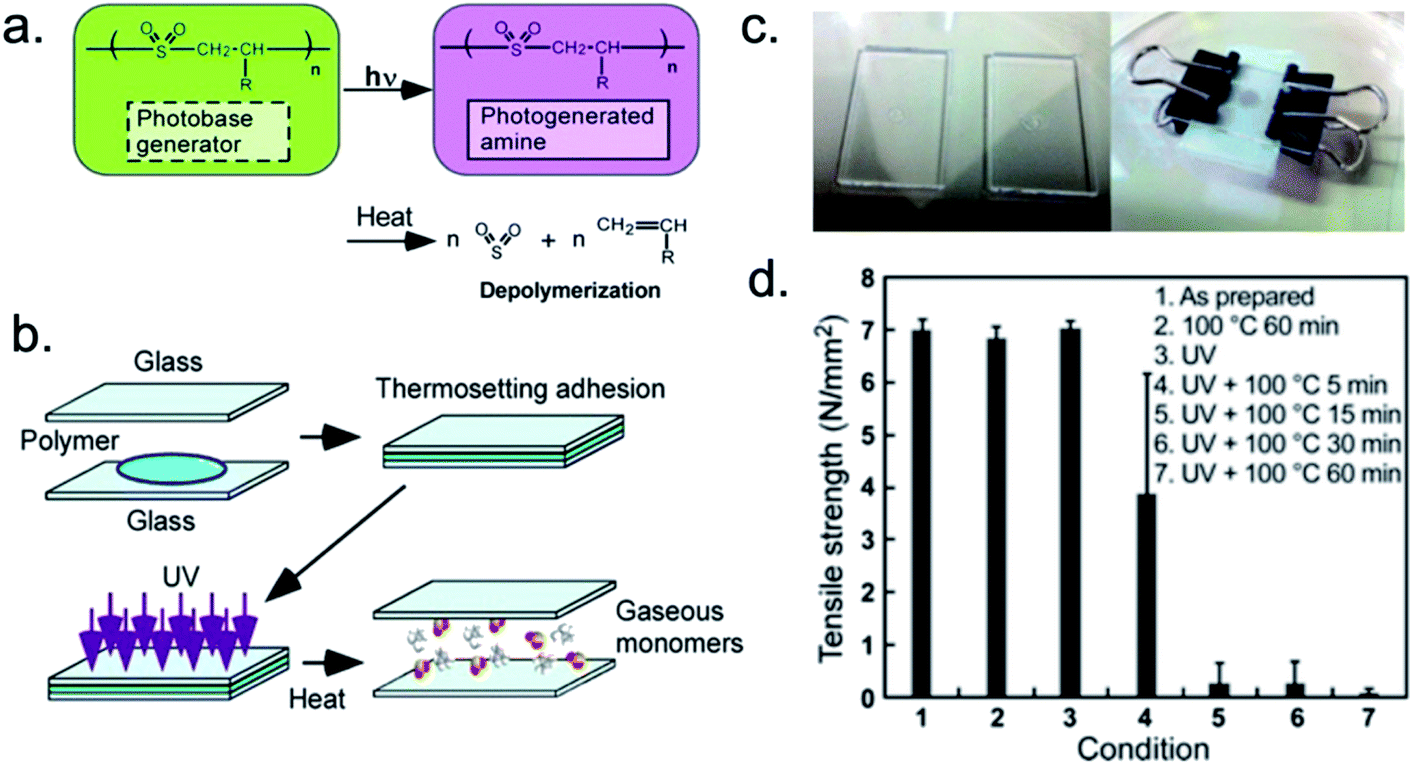 | ||
| Fig. 2 Photoinduced depolymerization of poly(olefin sulfone)s containing photobase generators and a sequence showing a photodetachable thermosetting adhesive. (a) Simultaneous exposure to UV light and heat induces depolymerization and produces a photogenerated amine. (b) Sequence of events resulting in debonding. (c) Photographs of glass slides with adhesive (left) and use of binder clips to apply pressure to glass slide assembly during melt-bonding (right). (d) Tensile strength of adhesive under various conditions. Adapted with permission of ref. 87. Copyright 2016 American Chemical Society.87 | ||
Debonding substrates with thermal energy
Implementing heat as a debonding stimulus has two significant advantages compared to UV light. First, limited penetration depth often limits the effect of UV light, which is of particular concern in dense polymeric systems or within the human body. Second, thermal energy can be more uniformly applied to adhesive materials and stimuli-responsive groups compared to light-controlled systems. For these reasons, the molecular design of heat-sensitive temporary adhesives is distinct from UV-sensitive temporary adhesives and requires a separate discussion. Current efforts to release substrates with heat involve the strategic selection of responsive groups, whether polymer backbones, cross-linkers, or functional groups, that debond through an existing or novel mechanism. This section of the perspective covers five well-established temperature-dependent debonding mechanisms of temporary adhesives: (i) alteration of dynamic covalent bonds; (ii) Diels–Alder chemistry; (iii) disruption of intermolecular interactions; (iv) gas formation; and (v) volumetric expansion.38Dynamic covalent bond exchange
Dynamic covalent bonds can provide structural support to an adhesive, but are also readily and reversibly cleaved with heat or UV light. Embedding polymeric systems with ester, disulfide, or imine bonds, among others, is a common design tactic to reversibly modify the viscoelastic properties of the material.97,98 Applying heat to an adhesive polymeric network with dynamic covalent bonds promotes bond exchange, ultimately leading to debonding.65,75 For example, Michal et al. detailed a cross-linked dynamic polymeric network capable of tiered adhesion and shape-memory.99 The authors prepared polymer thin films of (dithio)telechelic oligomers, tetrathiol cross-linkers, and cellulose nanocrystals (CNCs), then heated the films to 150 °C to initiate the formation of the disulfide dynamic network (Fig. 3a). Thiol oxidation of the oligomers and tetra-functionalized cross-linkers resulted in the dynamic polymer networks. By varying the percentage of CNCs, the crystallinity and storage moduli of the polymer networks were controlled. Two reductions in the shear moduli around 60–70 °C and 150 °C indicated dynamic bond exchange events that significantly affected the viscoelastic properties (Fig. 3c). After heating to 80 °C and cooling to room temperature, the polymer films adhered the glass substrates with a minimal to moderate increase in adhesion strength when compared to non-heated films (Fig. 3b and d). The authors attributed these observations to an improvement of surface wetting after melting the crystalline CNC domains. The adhesive strength between the adhesive and glass substrates increased after heating the polymer films to 150 °C followed by subsequent cooling to room temperature (Fig. 3b and d). Restructuring of the disulfide bonds and melting the CNC crystalline domains justified the improved adhesive strength because of improved interfacial contact and stronger interactions. Semi-crystalline dynamic polymer adhesives are particularly well suited for applications where it is desirable to instantaneously alter the shape of adhered surfaces, as is the case with aircraft part manufacturing. | ||
| Fig. 3 Cross-linked dynamic polymeric network capable of tiered adhesion and shape-memory. (a) Synthesis of dynamic polymeric networks 2a–c from oligomers and tetrathiol cross-linkers. (b) Tiered adhesion achieved by heating dynamic polymeric network to 80 °C or 150 °C. (c) Reduction in shear storage moduli for 2a–c and non-dynamic network 2d upon heating. (d) Lap shear stress of glass slides bonded with 2a–d and 0.136 MPa contact pressure during cooling. Adapted with permission from ref. 99. Copyright 2016 American Chemical Society.99 | ||
Dismantling cross-linkers via reverse Diels–Alder reactions
The thermal reversibility of Diels–Alder chemistry is an alternative strategy for heat-controlled temporary polymeric adhesives, as the forward and reverse reactions can be cycled with high fidelity and little to no side products.100,101 Installing diene and dienophile pairs into a polymeric system can create functional groups whose structure and function respond to changes in thermal energy. The monomers or polymer end groups may be functionalized as the diene or dienophile to modulate adhesion through Diels–Alder reactions.102–105Das et al. developed a random copolymer from hexyl methacrylate (HMA) and 2-hydroxyethyl methacrylate (HEMA), where the HEMA side groups were functionalized with furane to produce an adhesive (FMP) (Fig. 4a).106 The two key features of this copolymer comprised the HMA chains, which facilitated structural flexibility, and the furane side groups, which can react with 1,1′-(methylenedi-4,1-phenylene)bismale-imide (BMI) to form Diels–Alder cross-linkers (Fig. 4b). The FMP materials showed a wide range of shear strength (0.20 ± 0.04 MPa to 4.4 ± 0.4 MPa) and cohesive failure for aluminum substrates, which suggested that cross-linking with BMI could increase the lap shear strength. A series of adhesives was synthesized by varying the degree of BMI cross-linking within the FMP (Fig. 4b). This systematic study correlated an increase in adhesive strength and adhesive failure of aluminum substrates bonded with the cross-linked FMP to the degree of cross-linking. The observed effect of cross-linking on adhesion existed until a distinct point, where unreacted BMI began to lower the adhesive strength. Adhesion was temporarily diminished by heating the FMP material to 150 °C, then reinstated once the material cooled to 80 °C, which was the temperature required for the Diels–Alder reaction to occur (Fig. 4c). The authors found that the cooling rate determined the capability of the cooled cross-linked FMP material to rebond (Fig. 4c). The rapidly cooled cross-linked FMP sample demonstrated a lap shear strength (2.0 ± 0.4 MPa) that was more like the non-cross-linked FMP sample (1.1 ± 0.1 MPa) than to the FMP sample prior to heating and slow cooling (7.9 ± 1.1 MPa). The lower observed strength upon rapid cooling was likely due to the inability of the polymer to reform the Diels–Alder cross-linkers before reaching room temperature. This study demonstrated how controlling the debonding response and mechanism can be used to discover novel properties of stimuli-responsive polymeric adhesives. Additionally, the authors offered this polymeric adhesive as a more sustainable approach to recycling thermosetting adhesives, which are characteristically difficult to remove and repurpose.
 | ||
| Fig. 4 Temporary reversible adhesive composed of a random copolymer cross-linked with a Diels–Alder dienophile. (a) Synthetic scheme for a random copolymer with diene-functionalized side chains (FMP). (b) Diels–Alder cross-linkers formed by heating FMP and a dienophile (BMI). (c) Heating bonded aluminum substrates initiates debonding and slow cooling enables rebonding. Adapted from ref. 106 with permission of The Royal Society of Chemistry.106 | ||
Forming and deconstructing networks of intermolecular interactions
A recent report from Wu et al. demonstrated how the molecular and structural features of supramolecular polymeric adhesives can be manipulated to increase adhesion or cohesion and promote debonding.107 The authors synthesized supramolecular polymeric adhesives from naturally occurring acids and sugars (Fig. 5a). The functional groups of these acids and sugars directed formation of extensive 3-D hydrogen bond networks, which the authors highlighted as a driving force for polymerization and strong cohesion. The acid–sugar adhesives displayed high viscosities until heated to 60 °C, at which point the viscosities were significantly diminished and debonding was achieved. This observation was attributed to thermal energy dismantling the hydrogen bond network and reversing the polymerization reaction. Disruption of the hydrogen bond network due to heat illustrated how altering a bulk property, viscosity in the present study, can promote the reduction of cohesive interactions. All acid–sugar polymers in this study demonstrated adhesion on steel, glass, poly(methyl methacrylate) (PMMA), and poly(tetrafluoroethylene) (PTFE); the strongest adhesion occurred with steel substrates (3.0–5.42 MPa) and the weakest occurred with PTFE (0.22–0.38 MPa). The relative degree of hydrogen bonding between the acid–sugar polymers and the substrates justified the higher adhesive strengths (Fig. 5b). Like the cohesive interactions, the application of heat resulted in loss of adhesive interactions because the interfacial hydrogen bonds were no longer present. After cooling, the heated adhesives reformed the hydrogen bond network, restoring all cohesive and adhesive interactions. Based on these results, the authors suggested these acid–sugar adhesives should be further explored as non-toxic, biodegradable alternatives to traditional petroleum-based adhesives. | ||
| Fig. 5 Reversible supramolecular polymeric adhesives formed from naturally occurring acids and sugars. (a) Chemical structures of the acid and sugar monomers. (b) Interfacial interactions between acid–sugar adhesives and two types of substrates. This figure has been published in S. Wu, C. Cai, F. Li, Z. Tan and S. Dong, Supramolecular Adhesive Materials from Natural Acids and Sugars with Tough and Organic Solvent-Resistant Adhesion, CCS Chem., 2020, 1690–1700 is available online at DOI: 10.31635/ccschem.020.202000318.107 | ||
Zhao and coworkers designed a shape memory reversible dry adhesive based on the crystalline transition of polycaprolactone (Fig. 6).108 Shape memory stamps that exhibit reversible adhesion have been developed for applications in transfer printing. The authors synthesized a butyl acrylate/polycaprolactone diacrylate (BA/PCLDA) copolymer by radical initiated copolymerization. The PCLDA component provided crystallinity to the samples. Mechanical properties (e.g., moduli) were modulated at room temperature, or above the melting point by tuning the BA and PCLDA content, respectively. At low temperatures, the material was in a crystalline state that exhibited a high Young's modulus and at higher temperatures transitioned into a rubbery state that exhibited a low Young's modulus. Upon loading (adhering) in the rubbery state, the adhesive could be released with the application of 2 ± 1 N cm−2 of force. However, if the heated stamp was cooled such that it transitioned to a crystalline state while loaded, it required 13 ± 2 N cm−2 of force for removal. In microelectronics manufacturing, adhesives need to be capable of strongly bonding microdevices to a solid mount but are often limited in the ability to remove the fragile devices without damage. These soft reversible adhesives offer the strong bonding character of traditional epoxy resins and the low modulus of a shape-memory polymer to maximize efficiency of microdevice manufacturing.
 | ||
| Fig. 6 (a) Illustration of the transfer process enabled by the adhesive with shape memory microstructure. (b) Chemical structures of PCLDA and BA. (c) Adhesion strength of non-structured samples with different BA contents utilizing different protocols. Reprinted by permission from Springer Nature: Springer Nature CJPS ref. 108. Copyright 2018.108 | ||
Gas formation and volumetric expansion
Highly cross-linked polymeric adhesives are desirable for their high stability, stiffness, and adhesive strengths. Consequently, these characteristics also define the limitations of these polymers when they need to be reworked or debonded after the initial application. One option is to install responsive groups that allow the material to undergo plastic deformation at low temperatures and debond via gas formation or joint expansion at slightly higher temperatures.38 Gorsche et al. reported a highly cross-linked polymer adhesive with a glass transition temperature below the temperature that initiates a decarboxylation reaction. This strategy creates a porous network that leads to mechanical failure of the adhered joint.14,38,109 The utility of this type of adhesive and debonding mechanism is particularly apparent in dental cements, where the adhesive is required to strongly bond surfaces, but may need to be structurally malleable without completely debonding.Considering heat as a debonding stimulus encapsulates the challenge of designing a stimuli-responsive temporary adhesive that readily responds when triggered and discriminates against ambient temperatures in everyday use. Furthermore, this debonding stimulus has led to other properties, such as shape memory99,108 or malleability,109 being layered with adhesive properties, resulting in multi-functional adhesive materials.
Modulating adhesion with chemical triggers
Nature began the iterative design process for materials long before humans embarked on a similar endeavor. Consequently, current research in stimuli-responsive temporary adhesives leverages the ever-evolving principles of biological systems, function, simplicity, and dissipation, to unlock the next-generation of materials.110 This section of the perspective discusses temporary adhesives that debond in response to chemical triggers, such as fluoride ions, metal ions, water, pH, and enzymes through similar processes as signaling events in biological systems.Self-immolation of polymer backbones and cross-linkers
A single chemical triggering event initiating a dramatic change in the properties of a material is the cornerstone of self-immolative polymers (SIPs).111–113 This class of polymers undergoes an autocatalytic depolymerization upon exposure to an external stimulus. Linear SIPs must be synthesized with specific terminating groups, which creates end caps that respond to targeted stimuli. During the polymerization reaction, these end caps trap the polymer in a metastable state, as the enthalpy associated with bond formation along the backbone does not counteract the entropic cost of polymerization at higher temperatures.114,115 The molecular design of SIPs most commonly centers around the type of polymer backbone and the end cap identities.77,116 The endless combinations of backbones and end caps lends well to a vast range of applications including, but not limited to, sensing,117–122 drug delivery,123–126 lithography,127–138 electronics,139–143 and smart composites.144–146 Maximizing the potential of SIPs in these applications relies on a radical difference in the properties of the polymer and the monomer states. The use of SIPs as stimuli-responsive temporary adhesives constitutes an emerging application for this class of polymers and chemically triggered debonding on demand.For applications where adhesives need to be cleanly removed from delicate surfaces, such as wound dressings or microelectronics, significant effort has been allocated to achieving the balance of strong adhesion when desirable and elimination of adhesion when no longer necessary. Phillips and coworkers were among the first to propose and develop a SIP system as a temporary adhesive, noting the amplified responses in response to a small quantity of the trigger that is characteristic of these polymeric systems.147 A poly(benzyl ether) macro-cross-linked poly(norbornene) matrix fused glass surfaces with resistance to shear stresses up to 0.51 ± 0.10 MPa (Fig. 7a). Exposure of the polymer to the fluoride ion triggered the cleavage of the reactive end cap, tert-butyldimethylsilyl (TBS) group, resulting in the destabilization of the crosslinking units (Fig. 7b). The cross-linked polymer then underwent rapid depolymerization of the crosslinking units through sequential quinone methide elimination reactions, reducing the resistance to shear to 0.05 ± 0.02 MPa. By strategically selecting the length of the cross-linkers, the authors altered the time required for depolymerization, and consequently, for debonding. As initially shown by Phillips and coworkers, conceptualizing SIPs as temporary adhesives incorporates a chemical trigger that specifically targets the end cap to dismantle the structural ordering from the molecular to the bulk material scale.
 | ||
| Fig. 7 (a) Schematic representation of the macro-cross-linked adhesive and its stimuli-induced de-cross-linking by depolymerization. (b) A specific polymer and the stimulus (fluoride) used in this work. (c) Response of a control polymer 1 (loaded with 0.3% of a TBS cross-linker) and reactive polymer 8 (loaded with 40% of another TBS cross-linker) to increasing fluoride concentrations. (d) Fluoride-initiated debonding of polypropylene on a glass slide in response to solvent with fluoride ions. Reprinted with permission from ref. 147. Copyright 2015 Wiley-VCH.147 | ||
Aiming to increase the robustness of SIP temporary adhesives, Greenland and coworkers imparted symbiotic relationships between responsive groups that resulted in reworkable fluoride-responsive polyurethane adhesives.148 This adhesive system contained fluoride-responsive depolymerizable units and polyester chains connected through urethane linkers. By varying the chemical identity of the polyol and the diisocyanate linker, the researchers were able to synthesize and characterize four amorphous polymers and one crystalline polymer (Fig. 8a). Mechanical and adhesive properties were engineered into the materials by incorporating hydrogen bonding moieties into the polymer structures. In mechanical testing, the crystalline adhesive exhibited a tensile modulus of 2035 MPa, two orders of magnitude larger than the amorphous adhesives. The dramatic increase in the tensile modulus suggested that the intermolecular interactions that result in a well-ordered crystal (e.g., hydrogen bonds) were much stronger than those present within an amorphous sample. Similarly, the amorphous polymers exhibited a significant (∼75%) reduction in their ultimate tensile strength (UTS) upon exposure to fluoride, whereas the crystalline polymer experienced only a 9% reduction in UTS (Fig. 8c). This finding suggested that the hydrogen-bonding components in the amorphous polymer were lost during fluoride-induced degradation, whereas the crystalline nature of the other sample maintained those strong intermolecular interactions even as the polymer backbone was degraded (Fig. 8b). By distinguishing the functionalities responsible for structural malleability and debonding, the authors demonstrated specific and tunable control over the bulk properties of the polyurethane materials.
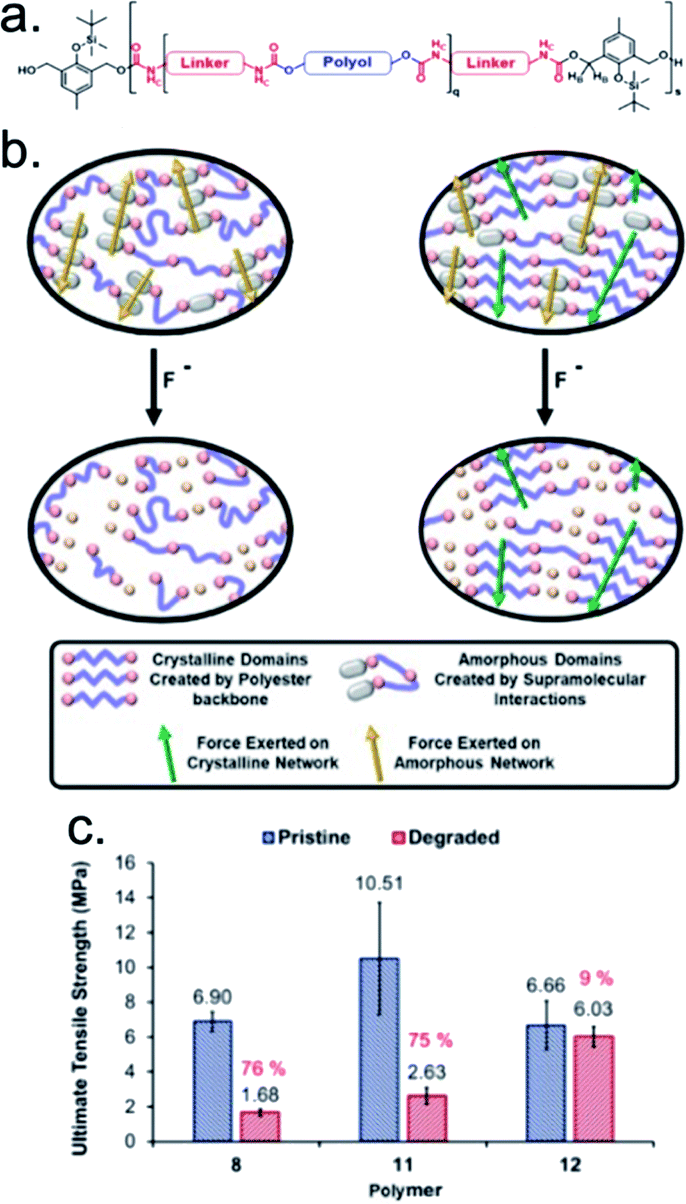 | ||
| Fig. 8 (a) General structure of the polymer containing the degradable unit with varying polyols and diisocyanate linkers. (b) Schematic showing the proposed differences in the transfer of force between amorphous polymers 8–11 and the crystalline polymer 12 before and after degradation. (c) The ultimate tensile strength of polymer 8, 11, and 12, calculated from the stress–strain curves. Errors are calculated from the standard deviation (n = 5). Adapted with permission from ref. 148. Copyright 2019 Elsevier.148 | ||
More recently, Kim and coworkers expanded on the use of fluoride to trigger self-immolative on-demand adhesion to improve the processability and end-of-life degradation of highly cross-linked polymer networks.65 The authors synthesized a dual cross-linked functional thermoset polymeric adhesive that displayed reversible bonding capabilities and total degradation in response to fluoride (Fig. 9a). Strategic selection of two cross-linked polymers imparted specific reactivity to the thermoset. One polymer contained 1,2,3-triazole units, which can undergo reversible alkylation reactions, and the other contained fluoride-reactive TBS moieties, which imparted the self-immolative behavior. During synthesis, the copolymer composition was varied by adjusting the ratio of the two monomers, known as the feed ratio. Once synthesized, the thermoset adhered glass slides and rebonded the slides upon heating to 140 °C (Fig. 9b). Three successful cycles of bonding and debonding showed reversibility (Fig. 9d). When adhered samples were tested to failure, they exhibited an average shear strength of 1.06 ± 0.14 MPa before delamination of the glass adherends (Fig. 9c). Exposure to the fluoride at the end of use completely degraded and destroyed adhesion of the self-immolative polymer backbone of the thermoset (Fig. 9e). This study highlights a strategy for designing multifunctional, multiresponsive temporary adhesives with properties that were previously inaccessible to exhibit simultaneously.
 | ||
| Fig. 9 Dual cross-linked thermoset with reversible adhesion. (a) Molecular design of functional thermoset adhesive with dynamic and self-immolative cross-linkers. (b) Photograph of bonded glass assemblies (bonding with 75% feed molar ratio) holding a 1 kg pink dumbbell. (c) Heating the polymeric adhesives activates lap shear strengths as a function of the feed molar ratio. (d) Lap shear strength after cyclic debonding and rebonding at 140 °C. (e) Percent reduction in shear strength depends on time and the incorporation percentage of the self-immolative cross-linkers. Adapted with permission from ref. 65. Copyright 2020 American Chemical Society.65 | ||
Metal ion-mediated molecular recognition
The structural network of an adhesive can be manipulated via the dynamic nature of coordination chemistry by introducing competition for metal–ligand bonding. These dynamic complexes have been identified as a driving force for supramolecular assembly and molecular recognition events due to the directionality and strength of the coordination bonds.149This section highlights two responsive functionalities, catechol moieties and host–guest cross-linking interactions, that modulate adhesion and facilitate debonding on demand through competition for coordination to a metal ion. By dwelling on the slippery surfaces of marine landscapes, mussels evolved adhesive threads to strongly, yet temporarily, attach themselves to wet substrates in the presence of currents and waves.150,151 Mussel-inspired adhesives exhibit several potential advantages over other adhesive design strategies: (i) catechol-based dynamic bonds that are able to break and reform during energy dissipation; (ii) the variety of intermolecular interactions that can promote in adhesion on wet surfaces; and (iii) utility in applications where antioxidative properties and biocompatibility are crucial, such as bioelectronics and tissue adhesion.152–154
Hawker and coworkers utilized the reversible complexation of catechol moieties with metal ions to form removable and recoverable self-assembled monolayers (SAMs) on metal oxide surfaces.155 In this study, catechol-functionalized tri(ethylene oxide) monolayers were adhered to metal (i.e., Ti, Al, Fe) oxide surfaces. As the strength of catechol–metal binding is heavily dependent on the identity of the metal,156–158 debonding was achieved by the addition of a Fe(III) metal ion solution. The Fe(III) ion atoms acted as competitors, against the metal oxide surface, for which the catechol moieties preferentially coordinated. While the authors did not quantify the adhesive strength of the SAM, they confirmed its removal upon washing with a solution of FeCl3 with angle-dependent X-ray absorption near edge structure (NEXAFS) spectroscopy. Furthermore, static water contact angle measurements confirmed that the SAM formation/removal cycling was possible up to the tested four-cycle limit. This temporary adhesive uses mussel-inspired design strategies to incorporate the competition for metal atom coordination as a viable debonding mechanism.
Another strategy, reported by Nakamura et al., is the removal of metal atoms to shift preferential coordination from the interfacial interaction of substrates to one between polymer chains within one material.68 The authors reported a hydrogel material functionalized with host moieties, β-cyclodextrin (βCD) rings, and guest moieties, 2,2′-bipyridyl (BIPY) units, that can respond by binding to metal ions (Fig. 10a). A second hydrogel featured another guest moiety, N-tert-butylacrylamide (N-tBuAAm), with which the βCD rings from the βCD–BIPY hydrogel can coordinate when the BIPY unit complexes with a metal ion (Fig. 10b). Metal ions modulated the surface interactions between the βCD–BIPY hydrogel and the N-tBuAAm hydrogel and resulted in adhesive interactions (Fig. 10c). Immersing the βCD–BIPY hydrogels in aqueous CuCl2 solutions produced the strongest adhesive interactions (1000 Pa ± 200 Pa). The addition of a metal chelating agent, ethylenediaminetetraacetic acid tetrasodium salt (EDTA·4Na), promoted reversible debonding (Fig. 10d). Metal ions that did not modulate adhesion, such as Mg2+, Mn2+, and Fe2+, failed to form complexes with BIPY units (Fig. 10d). In addition to modulating adhesion with molecular recognition, this hydrogel adhesive from Nakamura et al. benefits from host–guest interactions and supramolecular assembly.
 | ||
| Fig. 10 Metal-ion mediated interfacial adhesion based on host–guest interactions between two hydrogels. (a) Structures of βCD–BIPY hydrogel and N-tBuAAm hydrogel. (b) Metal ion modulates adhesion by forming new host–guest interaction between βCD–BIPY hydrogel and N-tBuAAm hydrogel. Adapted from ref. 68. Copyright 2014 Springer Nature (http://creativecommons.org/licenses/by/4.0/).68 | ||
Hydrogen bond competition
Covalent bonds and intermolecular interactions serve as the basis for the structural support of the polymeric network in a temporary adhesive material. If a small molecule trigger were to act as a competitive inhibitor of those key structural interactions, the adhesive would experience a significant modification to the bulk properties, which may in turn enable debonding of substrates.A recent study from Gao et al. reported the design and synthesis of a supramolecular polymeric assembly with metallocyclic cross-linkers that showed emissive, self-healing, and adhesive properties.71 Metallacycles were formed through metal–ligand complexation of a bifunctional platinum species and a bipyridyl triphenyl amine derivative; this complexation created a fluorescent building block that can install two polymer chains about 180° apart. End groups of tetra(poly(ethylene glycol)) (PEG) star polymers were equipped with alkyne groups. Then, alkyne-functionalized star polymers were cross-linked with the metallacycles via an amino-yne click reaction (Fig. 11a). The intricate supramolecular network contained three functionalities that imparted a unique blend of properties: (i) reversible Pt–N bonds that promoted self-healing (Fig. 11c); (ii) PEG chains that resulted in an abundance of interfacial hydrogen bonds and enhanced solubility; and (iii) bipyridyl triphenyl amine units that impart fluorescent properties. Glass slides that were melt-bonded with the supramolecular network (n = 111) exhibited an adhesive strength of 1.88 MPa, where the unmodified PEG had a strength of 0.62 MPa (n = 111) (Fig. 11b). Authors noted that this supramolecular adhesive was capable of bonding glass, plastic, leaves, metal, foam, and rubber; this observation was attributed to the extensive interfacial hydrogen bond network due to the PEG chains. Release from adhesion was achieved by exposing the bonded assemblies to water that solubilized the supramolecular adhesive. This result was attributed to water forming hydrogen bonds with the PEG chains, thereby competitively inhibiting the interfacial interactions to the substrate. The debonding was shown to be completely reversible following the removal of water, with no significant change in the adhesive strength after the initial dehydration event (Fig. 11d). This reported stimuli-responsive temporary adhesive joins the growing efforts to combine multiple desirable properties, thereby increasing complexity and organization, within a single material.
 | ||
| Fig. 11 Metal-ion mediated interfacial adhesion based on host–guest interactions between two hydrogels. (a) Structures of βCD–BIPY hydrogel and N-tBuAAm hydrogel. (b) Addition of a metal ion creates a reversible host–guest interaction to the BIPY unit. (c) Metal ion modulates adhesion by forming new host–guest interaction between βCD–BIPY hydrogel and N-tBuAAm hydrogel. (d) Adhesive interactions not observed after EDTA chelation or the addition of a metal ion that cannot complex with BIPY. Reprinted from ref. 71 with permission from Elsevier.71 | ||
Selective control of polymers with pH
Acids and bases are commonly used to control the properties of responsive polymers, and consequently, it is not surprising that pH is often utilized to promote on-demand debonding of temporary adhesives. For example, Cohen and Rubner reported a pH-sensitive Layer-by-Layer (LbL) multilayered hierarchical structure that promotes strategic separation of individual layers.60 In this work, discrete layers of poly(vinyl alcohol) (PVA), poly(acrylic acid) (PAA), and poly(methacrylic acid) (PMA) were connected through hydrogen-bonding interactions. However, the differences in pH stability limit interlayer diffusion of the polymer chains. When the assembled structures were subjected in solutions of increasing pH, the polyacids became increasingly ionized. Past a critical pH value, which varied depending on the chemical moieties present at the interface, film disassembly resulted in delamination. The authors showed that they could selectively choose where debonding occurred by altering the order in which each film was deposited.In another example, Lee and coworkers described a catechol-containing adhesive that utilized pH to control oxidation state and adhesive properties of a catechol-based smart adhesive.159 In its fully reduced state, a copolymer of dopamine methacrylamide (DMA) and 3-acrylamido phenylboronic acid (AAPBA) successfully adhered to borosilicate glass. Depending on the adhesive formulation, the authors measured adhesive strengths up to Fmax = −16 ± 0.60 mN and Wadh = 2000 ± 25 mJ m−2 at pH = 9. However, upon oxidation, the catechol moieties assumed their quinone forms, drastically reducing adhesive strength. At pH = 3, the materials exhibited adhesive strengths of only Fmax = −2.4 ± 1.1 mN and Wadh = 180 ± 87 mJ m−2. Returning to pH = 9 resulted in a 90 and 76% recovery of Fmax and Wadh, respectively.
Biocompatible degradation via enzymes
Adhesive materials for use within or on the human body come with a unique set of requirements and challenges: (i) ability to join wet surfaces; (ii) clean removal without damage to tissue; (iii) biocompatibility of constituents; and (iv) mechanical robustness.3,20–22,160 One strategy to overcome all of these obstacles is to create a polymeric network with cross-linkers that can be broken down by biocompatible chemical stimuli, such as enzymes.20–23 Mooney and coworkers reported a class of temporary adhesive materials with strategically selected polymeric hydrogels and cross-linkers that created a sturdy polymeric network with approximately 90% water.20 Authors constructed the hydrogels with a bridging polymer, chitosan, as an adhesive interface and with either an ionic or covalent network, to form the tough gel. The adhesive strength of these materials ranged between 0.01–0.08 MPa. This system showed a strong dependence on identity and weight percentage of the incorporated covalent cross-linker. Hydrogels with cleavable ester bonds in the covalent network exhibited degradation when implanted into mice, albeit at different rates, which is ascribed to the hydrolysis of the ester bonds. In similar in vivo experiments, hydrogels with gelatin methacrylate (GelMA) and hyaluronic acid methacrylate (HAMA) showed either no significant degradation or slower degradation rates, respectively. This observation was attributed to the lack of collagenase, which degrades gelatin, and low concentration of hyaluronidases, which degrade hyaluronic acid (HA), at the site of implantation. Additionally, authors reasoned that the HAMA hydrogel showed delayed degradation due to the low molecular weight (10 kDa) of HA, where the hyaluronidases have a high specificity for higher molecular weight HA. Authors demonstrated debonding on demand of skin surfaces with a triggering solution, that include lysozyme to cleave the glycoside bond subunits of the chitosan adhesive interface. About ten minutes after applying the trigger solution, the adhesive was removed from skin without leaving residue or causing damage. This study was the first to report control over degradation rates of bioadhesives through molecular engineering.Chemical signals govern biological communications.161 Because these systems have progressed to maximize efficiency and optimize function as a means of survival, the evolution of structure–property–function relationships provides inspiration for designing innovative materials to meet the world's growing needs.110 Next-generation stimuli-responsive temporary adhesive materials should borrow insights from biological communication processes to enable multiresponsive and multifunctional via distinct responsive groups.
Other approaches to triggering release from adhesion
Photo-, thermally-, and chemically responsive materials constitute the majority of temporary adhesives; however, other stimuli, including pressure, mechanical force, magnetism, and electrical fields, can also be employed to initiate on demand debonding of substrates. These non-traditional stimuli adhesives are immensely valuable in applications that require robust bonding in harsh conditions, such as high temperatures, frequent solvent exposure, or direct sunlight.Sublimation-induced release from adhesion
Sublimable adhesives are a general class of compounds that can release bonded substrates through the direct solid-to-gas phase transition of the material without the application of solvent or external mechanical force.31,32,162 While these materials respond to external pressure, they do not rely on pressure to bond the adhesive with a surface. Instead, a reduction in the ambient pressure, often coupled with an increase in temperature, initiate sublimation of the adhesive materials and, consequently, release of adherends.163 While specific debonding conditions depend on the exact chemical nature of the adhesive utilized, general conditions involve exposing a bonded sample to vacuum of >100 mTorr while heating it to 90% of its melting temperature. Sublimable adhesives possess numerous unique characteristics, including their ease of application, tailored ability to withstand mechanical stress and strain, recyclability, and tunable on-demand debonding.Sublimable adhesives can promote bonding through a process in which a small amount of molten material is sandwiched between two adherends, and then allowed to cool and fuse into a polycrystalline film (Fig. 12a). This application procedure enables individual molecules to self-assemble at the adhesive–adherend interface, maximizing favorable intermolecular interactions, resulting in preferential orientation of the adhesive at the interface that promotes adhesion relative to non-preferentially oriented films. Applying a liquid-phase adhesive also promotes conformal contact between adhesive and adherend, which reinforces adhesion.
 | ||
| Fig. 12 Crystal engineering of molecular solids as sublimable adhesives. (a) Small molecule adhesives are capable of bonding surfaces through a melt-bonding process and releasing them on demand through sublimation. (b) Selected adhesives characterized included iodine (IOD), (+)-camphor (CAM), hexachlorobenzene (HCB), octacyclic sulfur (OCS), 2,3-dihydroxynaphthalene (DH-NAP), octafluoronaphthalene (OFN), naphthalene (NAP), and anthracene (ANT). (c) Like all other molecular solids tested, camphor promotes strong bonding by assuming a preferential orientation relative to the surface upon melt-bonding that enables the formation of strong intermolecular interactions at the adhesive adherend interface. (d) Strategic selection of small molecule adhesive enables turning of the resulting adhesive properties, with quantified shear strengths up to 1.24 ± 0.15 MPa. Inset: a 50 lbs. (22.7 kg) dumbbell suspended from a camphor-adherend lap joint comprising two glass microscope slides (adhesive mass = 20 ± 3 mg, film thickness = 51 ± 6 μm). Adapted with permission from ref. 31, 32 and 162. Copyright 2020 American Chemical Society.31,32,162 | ||
Our laboratory was the first to quantitatively characterize sublimable adhesives.31,32,162 For a proof-of-concept demonstration, we selected a series of polycyclic aromatic hydrocarbons (PAHs)—naphthalene, anthracene, pyrene, and multiple naphthalene derivatives—because their crystal structures, intermolecular interactions, and enthalpies of phase transitions were well characterized. Mechanical testing confirmed that all PAHs studied (Fig. 12b) were capable of bonding glass substrates with shear strengths comparable to double-sided scotch tape (DST). Furthermore, we determined that it was possible to tailor the macroscopic properties of these adhesives by systematically altering the surface chemistry and roughness of the adherends.
We then expanded the scope of our investigations to study a broad range of molecular solids as sublimable adhesives, including both organic and inorganic molecular solids. We found that all materials tested successfully bonded glass adherends (Fig. 12d). In particular, (+)-camphor, which exhibited a shear strength of 1240 ± 150 kPa, was the strongest sublimable adhesive reported. The principles and methods of crystal engineering can be leveraged toward tailoring the mechanical performance of molecular solids as temporary adhesives.164–177
Sublimable adhesives have the potential to find numerous applications where photo-, thermal-, or chemically-responsive adhesives are unsuitable, such as construction,12 robotics,178 and microelectronics manufacturing.9,179 Additionally, the ability to quantitatively study the structure–property relationships between molecular-level phenomena and macroscopic properties has fundamental value to the field of crystal engineering.
Gecko-inspired adhesives are force-sensitive
While most stimuli-responsive temporary adhesives are designed to withstand force, the sticky pads on gecko feet and gecko-inspired adhesives are a clear example of materials that strategically debond upon the application of mechanical force. The gecko has evolved the ability to scale vertical walls, as well as to adhere to surfaces underwater and in space.180–187 Consequently, it has been a longstanding inspiration for bioinspired synthetic adhesives.Gecko feet and gecko-inspired adhesives exhibit anisotropic attachment, the ability to withstand large forces applied in one direction while releasing in response to small forces applied in a different direction.188 Adhesion is achieved primarily through van der Waals interactions at the adhesive–adherend interface;189,190 although capillary forces are also present at the interface, they are not believed to represent a dominant contribution to adhesion.191–195 The bottom of a gecko foot contains a hierarchical structure that terminate in arrays of setae and spatular tips, ∼0.2 μm long and ≥0.2 μm in diameter.195,196 The combined adhesive force between each spatular tip contribute to a total shear strength of 225 ± 50 kPa for a single gecko digit (i.e., toe).189–195,197 Similarly, gecko-inspired adhesives consist of micron-scale adhesive fibrils, often molded out of silicon rubber (e.g., poly(dimethyl siloxane) (PDMS) and poly(vinyl siloxane) (PVS)), that mimic a natural gecko foot.180,182,188,198–215
Geckos can control the adhesive state of their feet by controlling the directionality of applied forces. Close observation reveals that geckos “roll” their toes onto a surface to engage adhesion in a standard peeling mechanism.216–218 The setae deform such that they form acute angles with the surface, enhancing van der Waals interactions and adhesion. For release, the toes are rolled off the surface, causing the setae to form larger angles to the surface, weakening van der Waals interactions, and decreasing adhesion.
Numerous gecko-inspired synthetic adhesives employ a force-sensitive peeling mechanism to achieve debonding.219–226 While many of these materials rely on light or heat to induce the peeling behavior, it is the driving force of that peeling motion that causes debonding. In a representative example, Zhou and coworkers utilized a poly(acrylamide-isopropyl acrylamide-acrylic acid) hydrogel coupled with a PDMS micropillar array to achieve force-sensitive bonding and debonding (Fig. 13).227 A poly(dopamine methacrylamide-co-methoxyethyl acrylate) coating enabled bonding in both wet and dry environments. Heat was used to shrink the hydrogel and induce curving. The resulting peeling behavior was the driving force for debonding.
 | ||
| Fig. 13 Gecko-inspired hydrogel with mussel-inspired copolymer coating and mushroom-structured arrays that undergoes debonding through a self-peeling mechanism. Reprinted with permission from ref. 227. Copyright 2021 American Chemical Society.227 | ||
Gecko adhesion is defined by seven key characteristics: (i) anisotropic attachment; (ii) high pull-off force-to-preload ratio; (iii) low detachment force; (iv) material independence due to van der Waals adhesion; (v) self-cleaning; (vi) anti-self-matting, and (vii) nonstick default state.188 Depending on their design, gecko-inspired adhesives may exhibit any combination of these characteristics; however, no synthetic material has successfully demonstrated all seven. Force-sensitive materials that exhibit anisotropic attachment offer a unique opportunity for scientists and engineers to study the origin of adhesion and probe the geometric considerations that enable strong attachment.
Magnetically induced molecular movements
Stimuli that require direct access to the adhesive material to provoke debonding, such as UV light or chemical triggers, are not suitable for applications that bond impermeable or opaque substrates.228 To overcome this obstacle, adhesives may be responsive to magnetic fields with ferromagnetic or conductive additives, such as iron-based powders, aluminum, cobalt, or nickel.38 Applying a magnetic field elicits molecular motions, rotations, or vibrations, that transfers thermal energy to the adhesives, which can lead to debonding with the previously discussed mechanisms.38,228,229 In one example from Greenland and coworkers, a composite adhesive with Fe3O4 particles (8 wt%) bonded wood, aluminum, glass, and polyvinyl chloride (PVC) substrates with a tensile strength of 1.35 MPa.228 Authors hypothesized that an oscillating magnetic field (OMF) would heat the material due to hysteresis loss, which would lead to debonding by dismantling the intermolecular interaction network. An OMF was applied to this adhesive, leading to complete debonding from 30 seconds to five minutes, depending on the wt% loading of the Fe3O4 particles.Delamination with electrochemistry
Electrochemical debonding methods are another answer to the obstacle of chemically impermeable or optically opaque substrates; these methods may be especially useful in bonding metal surfaces. Current work with electrochemical stimuli seeks to elucidate the mechanisms responsible for delamination. Proposed mechanisms include: (i) electrochemical reactions at the interface, (ii) phase separation, (iii) conduction of ions at metal–resin interface, and (iv) dissolution of metal oxide coating.230–232 Catechol-containing materials are redox active, and can be switched between strong adhesion (i.e., catechol) and weak adhesion (i.e., quinone) states upon the application of an electric current. Lee and coworkers reported the wet adhesion of a titanium electrode and a catechol-modified surface.233 A platinum electrode placed in the buffered solution completed the electric circuit. Initially, adhered samples were able withstand a tensile load of 70 mN after 10 mN preloading. However, increasing the circuit voltage from 0 to 9 V resulted in a 96 and 100% decrease in the work and strength of adhesion, respectively.Summary and future outlook
Continued investigations into fundamental structure–property relationships of stimuli-responsive temporary adhesives pave the way for next-generation technologies.29,31,32,162,234–246 These materials exhibit the conflicting, yet simultaneous, properties of bonding, resistance to mechanical stress and strain, and on-demand debonding, delineating them from traditional permanent adhesives. Molecular design strategies enable the intentional perturbation of intermolecular interactions within these adhesives or at the interfaces they form with substrates through the strategic application of an external stimulus or stimuli. The weakening or breaking of these adhesive and cohesive interactions results in on demand debonding of adherends.This perspective systematically outlined the various molecular design strategies utilized in stimuli-responsive temporary adhesives, as well as demonstrated the benefits of these materials over other fastening systems. Common stimuli include UV light, thermal energy, various chemicals, pressure, mechanical force, magnetism, and electric fields. By studying the response of different adhesive systems to these stimuli, researchers can investigate the physical and chemical interactions that form across interfaces and formulate hypotheses for regulating those interactions. Additionally, they can examine the fundamental nature of interfacial interactions, the origins of adhesion, and structure–property relationships between molecular-level design and macroscopic mechanical and adhesive properties. Ultimately, future stimuli-responsive temporary adhesives are poised to be utilized in novel technologies and applications that are incompatible with current adhesives, enabling the development of materials with new and valuable properties.
Looking toward the future, there is opportunity for rapid technological advancement in at least three areas: (i) the development of characterization techniques for stimuli-responsive temporary adhesives; (ii) the development of chemically responsive temporary adhesives; and (iii) the development of crystalline stimuli-responsive molecular solids. As technology advances, new techniques will be necessary to characterize stimuli-responsive temporary adhesives that exhibit novel properties. For example, our laboratory developed methods to quantify the adhesive and cohesive strengths of polycrystalline molecular solids.31,32,162 Valuable work has already been focused on quantifying, for example, the ability of materials to store and dissipate mechanical energy or transfer it across interfaces,247–264 the deformability of thin films of solution-processable small molecules,265 or the surface interactions between two surfaces in contact.266 Similarly, researchers have begun studying the impact of chemical and physical defects in determining mechanical properties.267–275 These efforts should be continued and expanded upon. Future inspiration for adhesive characterization techniques can be adapted from standard industry test methods (e.g., ASTM or TAPPI procedures) commonly applied in other fields of materials characterization.276,277
As research uncovers the fundamental structure–property relationships that govern the molecular behavior of materials, there is great opportunity for further development of chemically-responsive temporary adhesives. Recent work has uncovered several viable debonding stimuli, including thermal energy, UV light, chemical species, pressure, mechanical force, magnetism, and electrical fields; while there are advantages to these stimuli, the chemical world has unrivaled diversity and, consequently, immense potential for future work. Chemically responsive temporary adhesives have numerous benefits including: (i) they can be utilized in environments that may contain strong UV light and/or high temperatures; (ii) they can be multi-responsive, where one material exhibits a unique mechanical and adhesive response to two or more stimuli; and (iii) they can be orthogonal, where sequential bonding and debonding can be achieved using different stimuli. Prior studies on self-immolative polymers are an excellent example of the variety in potential molecular design strategies in the context of adhesion science.116,147 Expanding research efforts to develop materials that respond to novel chemical stimuli will uncover the fundamental chemical basis of strong adhesive interactions and promote as-yet-undiscovered applications.
Finally, in specific regard to crystalline temporary adhesives, the design of next-generation stimuli-responsive materials will emerge from future progress towards answering two open questions. First, what fundamental structure–property relationships govern how the mechanical properties of crystals arise from specific intermolecular interactions?278,279 Elucidating these fundamental relationships will expand the design and synthetic strategies employed to promote and control intermolecular and interfacial interactions.280–283 Second, how can the adhesive and mechanical properties of a material be predicted from its molecular or crystal structure?284–286 If the adhesive and mechanical properties could be predicted from a crystal structure, then designing a material with architectural features or orientations that promote adhesion and debonding would be possible. Expanding the fundamental understanding of the structure–property relationships that govern adhesion will expand the availability of molecular design strategies that can be utilized to promote and control stimuli-responsive temporary adhesives.
Author contributions
NDB: conceptualization, project administration, writing, reviewing, and editing; HJY: conceptualization, writing, reviewing, and editing; KAM: supervision, organization, writing, reviewing, and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the United States Army Research Office (contract number W911NF1920089), the 3M Non-Tenured Faculty Award, the Sloan Research Fellowship, and the Camille Dreyfus Teacher-Scholar Award. KAM acknowledges support from the start-up funds provided by Dartmouth College.References
- P. P. A. Mazza, F. Martini, B. Sala, M. Magi, M. P. Colombini, G. Giachi, F. Landucci, C. Lemorini, F. Modugno and E. Ribechini, J. Archaeol. Sci., 2006, 33, 1310–1318 CrossRef.
- P. R. B. Kozowyk, M. Soressi, D. Pomstra and G. H. J. Langejans, Sci. Rep., 2017, 7, 8033 CrossRef CAS.
- V. Bhagat and M. L. Becker, Biomacromolecules, 2017, 18, 3009–3039 CrossRef CAS.
- C. E. Brubaker and P. B. Messersmith, Langmuir, 2012, 28, 2200–2205 CrossRef CAS PubMed.
- M. Li, Z. Zhang, Y. Liang, J. He and B. Guo, ACS Appl. Mater. Interfaces, 2020, 12, 35856–35872 CrossRef CAS PubMed.
- S. Matsunaga, A. Tamura, M. Fushimi, H. Santa, Y. Arisaka, T. Nikaido, J. Tagami and N. Yui, ACS Appl. Polym. Mater., 2020, 2(12), 5756–5766 CrossRef CAS.
- A. M. Schenzel, Karlsruher Institut für Technologie, Advanced Debonding on Demand Systems for Dental Adhesives, Doctoral Dissertation, Karlsruher Institut für Technologie, 2017.
- Handbook of 3D Integration, Volumes 1 and 2: Technology and Applications of 3D Integrated Circuits, ed. P. Garrou, C. Bower and P. Ramm, Wiley-VCH, Singapore, 1st edn, 2012 Search PubMed.
- P. Tanskanen, Acta Mater., 2013, 61, 1001–1011 CrossRef CAS.
- R. Puligadda, S. Pillalamarri, W. Hong, C. Brubaker, M. Wimplinger and S. Pargfrieder, Mater. Res. Soc. Symp. Proc., 2007, 970, 239–249 CAS.
- K. M. Lee, O. Phillips, A. Engler, P. A. Kohl and B. P. Rand, ACS Appl. Mater. Interfaces, 2018, 10, 28062–28068 CrossRef CAS PubMed.
- H. Ishikawa, K. Seto, S. Shimotuma, N. Kishi and C. Sato, Int. J. Adhes. Adhes., 2005, 25, 193–199 CrossRef CAS.
- Emerging Trends in Mobile Robotics: Proceedings of the 13th International Conference on Climbing and Walking Robots and the Support Technologies for Mobile Machines, ed. H. Mochiyama, M. O. Tokhl, H. Fujimoto and G. S. Virk, World Scientific Publishing Co. Inc., Singapore, 2010 Search PubMed.
- A. Hutchinson, Y. Liu and Y. Lu, J. Adhes., 2017, 93, 737–755 CrossRef CAS.
- N. Glass and T. Hume, The 'halleluja moment' behind the invention of the Post-it note, 2013, https://www.cnn.com/2013/04/04/tech/post-it-note-history Search PubMed.
- History Timeline Post-it® Notes, 2021, https://www.post-it.com/3M/en_US/post-it/contact-us/about-us Search PubMed.
- E. Carretti, M. Bonini, L. Dei, B. H. Berrie, L. V. Angelova, P. Baglioni and R. G. Weiss, Acc. Chem. Res., 2010, 43, 751–760 CrossRef CAS PubMed.
- J.-T. Dong, W.-K. Zou, F. Chen and Q. Zhao, Chin. J. Polym. Sci., 2018, 36, 953–959 CrossRef CAS.
- B. Halford, STICKY NOTES: Serendipitous chemical discovery and a bright idea led to a new product that is ubiquitous, 2004, https://pubsapp.acs.org/cen/whatstuff/stuff/8214sci3.html Search PubMed.
- B. R. Freedman, O. Uzun, N. M. M. Luna, A. Rock, C. Clifford, E. Stoler, G. Östlund-Sholars, C. Johnson and D. J. Mooney, Adv. Mater., 2021, 33(17), 2008553 CrossRef CAS PubMed.
- L. Zhou, C. Dai, L. Fan, Y. Jiang, C. Liu, Z. Zhou, P. Guan, Y. Tian, J. Xing, X. Li, Y. Luo, P. Yu, C. Ning and G. Tan, Adv. Funct. Mater., 2021, 31(14), 2007457 CrossRef CAS.
- X. Chen, H. Yuk, J. Wu, C. S. Nabzdyk and X. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 15497–15503 CrossRef CAS PubMed.
- H. Yuk, C. E. Varela, C. S. Nabzdyk, X. Mao, R. F. Padera, E. T. Roche and X. Zhao, Nature, 2019, 575, 169–174 CrossRef CAS PubMed.
- C. Ghobril and M. W. Grinstaff, Chem. Soc. Rev., 2015, 44, 1820–1835 RSC.
- NMAB ad hoc Committee on Structural Adhesives for Aerospace Use, Aerospace Structural Adhesives, 1974 Search PubMed.
- J. A. Reuter, The Correlation of Flat and Tubular Scarf Joints, Massachusetts Institute of Technology, 1983 Search PubMed.
- D. E. Packham, in Handbook of Adhesion Technology, Springer International Publishing, Cham, 2017, p. 31 Search PubMed.
- M. J. Troughton, Handbook of plastics joining: a practical guide, William Andrew, 2008 Search PubMed.
- S. Ebnesajjad and A. H. Landrock, Adhesives Technology Handbook, Elsevier, San Diego, California, USA, 3rd edn, 2009 Search PubMed.
- A. Pizzi and K. L. Mittal, Handbook of Adhesive Technology, CRC Press, 3rd edn, 2018 Search PubMed.
- N. D. Blelloch, H. T. Mitchell, C. C. Tymm, D. W. van Citters and K. A. Mirica, Chem. Mater., 2020, 32(32), 9882–9896 CrossRef CAS.
- H. T. Mitchell, M. K. Smith, N. D. Blelloch, D. W. Van Citters and K. A. Mirica, Chem. Mater., 2017, 29, 2788–2793 CrossRef CAS.
- D. J. Gardner, in Handbook of Adhesive Technology, ed. A. Pizzi and K. L. Mittal, CRC Press, 3rd edn, 2018, p. 18 Search PubMed.
- D. E. Packham, in Handbook of Adhesion Technology, Springer International Publishing, Cham, 2017, p. 31 Search PubMed.
- K. L. Mittal, Adhesive Joints: Formation, Characteristics, and Testing, Springer US, 1984 Search PubMed.
- R. D. Adams, Adhesive Bonding: Science, Technology and Applications, CRC Press, 2005 Search PubMed.
- G. Gierenz and W. Karmann, Adhesives and Adhesive Tapes, Wiley-VCH, 2001 Search PubMed.
- C. Bandl, W. Kern and S. Schlögl, Int. J. Adhes. Adhes., 2020, 99, 102585 CrossRef CAS.
- G. Jeevi, S. K. Nayak and M. Abdul Kader, J. Adhes. Sci. Technol., 2019, 1–24 Search PubMed.
- L. H. Lee, Fundamentals of Adhesion, Springer US, 1991 Search PubMed.
- A. Kinoch, Adhesion and Adhesives: Science and Technology, Springer Netherlands, 1987 Search PubMed.
- D. K. Hohl and C. Weder, Adv. Opt. Mater., 2019, 7, 1900230 CrossRef.
- A. Pizzi and K. L. Mittal, Handbook of Adhesive Technology, CRC Press, 3rd edn, 2018 Search PubMed.
- D. J. Gardner, in Handbook of Adhesive Technology, ed. A. Pizzi and K. L. Mittal, CRC Press, 3rd edn, 2018, p. 18 Search PubMed.
- K. M. Herbert, S. Schrettl, S. J. Rowan and C. Weder, Macromolecules, 2017, 50, 8845–8870 CrossRef CAS.
- C. Heinzmann, C. Weder and L. M. De Espinosa, Chem. Soc. Rev., 2016, 45, 342–358 RSC.
- C. Creton and E. Papon, MRS Bull., 2003, 28, 419–421 CrossRef CAS.
- R. A. Pethrick, Proc. Inst. Mech. Eng., Part L, 2015, 229, 349–379 CAS.
- D. W. Oplinger, Mechanical Fastening and Adhesive Bonding, in Handbook of Composites, ed. S. T. Peters, Springer US, 2002 Search PubMed.
- L. Liu, Z. Liu, Y. Ren, X. Zou, W. Peng, W. Li, Y. Wu, S. Zheng, X. Wang and F. Yan, Angew. Chem., Int. Ed., 2021, 60, 8948–8959 CrossRef CAS PubMed.
- H. Kim, H. Witt, T. A. Oswald and M. Tarantola, ACS Appl. Mater. Interfaces, 2020, 12, 33516–33529 CrossRef CAS PubMed.
- S. Wu, Polymer Interface and Adhesion, M. Dekker, 1982 Search PubMed.
- K. W. Allen, Int. J. Adhes. Adhes., 1993, 13, 67–72 CrossRef CAS.
- B. v. Derjaguin and V. P. Smilga, J. Appl. Phys., 1967, 38, 4609–4616 CrossRef.
- K. L. Mittal, Physicochemical Aspects of Polymer Surfaces, Springer US, 1983 Search PubMed.
- Surface Contamination: Genesis, Detection, and Control, ed. K. L. Mittal, Springer US, 1979 Search PubMed.
- W. Possart, Int. J. Adhes. Adhes., 1988, 8, 77–83 CrossRef CAS.
- A. D. Roberts, J. Phys. D: Appl. Phys., 1977, 10, 1801–1819 CrossRef CAS.
- X. Hu, E. McIntosh, M. G. Simon, C. Staii and S. W. Thomas, Adv. Mater., 2016, 28, 715–721 CrossRef CAS PubMed.
- H. Lee, C. Sample, R. E. Cohen and M. F. Rubner, ACS Macro Lett., 2013, 2, 924–925 CrossRef CAS.
- A. Kinoch, Adhesion and Adhesives: Science and Technology, Springer Netherlands, 1987 Search PubMed.
- L. H. Lee, Fundamentals of Adhesion, Springer US, 1991 Search PubMed.
- D. A. McQuarrie and J. D. Simon, Physical Chemistry: A Molecular Approach, University Science Books, Herdon, VA, 7th edn, 1997 Search PubMed.
- Y. Zhou, M. Chen, Q. Ban, Z. Zhang, S. Shuang, K. Koynov, H. J. Butt, J. Kong and S. Wu, ACS Macro Lett., 2019, 8, 968–972 CrossRef CAS.
- Y. Oh, J. Park, J.-J. Park, S. Jeong and H. Kim, Chem. Mater., 2020, 32(15), 6384–6391 CrossRef CAS.
- H. Akiyama and M. Yoshida, Adv. Mater., 2012, 24, 2353–2356 CrossRef CAS PubMed.
- S. W. Kim, Y. H. Ju, S. Han, J. S. Kim, H. J. Lee, C. J. Han, C. R. Lee, S. B. Jung, Y. Kim and J. W. Kim, J. Mater. Chem. A, 2019, 7, 22588–22595 RSC.
- T. Nakamura, Y. Takashima, A. Hashidzume, H. Yamaguchi and A. Harada, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- S. H. Mostafavi, F. Tong, T. W. Dugger, D. Kisailus and C. J. Bardeen, Macromolecules, 2018, 51, 2388–2394 CrossRef CAS.
- A. M. Asadirad, S. Boutault, Z. Erno and N. R. Branda, J. Am. Chem. Soc., 2014, 136, 3024–3027 CrossRef CAS PubMed.
- K. Gao, Z. Zhang, L. Ma, L. Chen, X. Chen, Y. Zhang and M. Zhang, Giant, 2020, 4, 100034 CrossRef.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, Burlington, Massachusetts, USA, 2011 Search PubMed.
- E. M. Petrie, in Handbook of Plastics and Elastomers, ed. C. A. Harper, McGraw-Hill, New York, NY, 2002 Search PubMed.
- M. J. Troughton, Handbook of Plastics Joining: A Practical Guide, William Andrew, 2008 Search PubMed.
- S. Kaiser, S. V. Radl, J. Manhart, S. Ayalur-Karunakaran, T. Griesser, A. Moser, C. Ganser, C. Teichert, W. Kern and S. Schlögl, Soft Matter, 2018, 14, 2547–2559 RSC.
- Q. Guo, J. Chen, J. Wang, H. Zeng and J. Yu, Nanoscale, 2020, 12, 1307–1324 RSC.
- R. E. Yardley, A. R. Kenaree and E. R. Gillies, Macromolecules, 2019, 52, 6342–6360 CrossRef CAS.
- M. K. Corpinot and D. K. Bučar, Cryst. Growth Des., 2019, 19, 1426–1453 CrossRef CAS.
- O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef CAS.
- R. A. Pethrick, Proc. Inst. Mech. Eng., Part L, 2015, 229, 349–379 CAS.
- C. Heinzmann, C. Weder and L. M. De Espinosa, Chem. Soc. Rev., 2016, 45, 342–358 RSC.
- X. Hu, J. Shi and S. W. Thomas, Polym. Chem., 2015, 6, 4966–4971 RSC.
- A. Gandioso, M. Cano, A. Massaguer and V. Marchán, J. Org. Chem., 2016, 81, 11556–11564 CrossRef CAS PubMed.
- Z. Wu, C. Ji, X. Zhao, Y. Han, K. Müllen, K. Pan and M. Yin, J. Am. Chem. Soc., 2019, 141, 7385–7390 CrossRef CAS PubMed.
- S. Saito, S. Nobusue, E. Tsuzaka, C. Yuan, C. Mori, M. Hara, T. Seki, C. Camacho, S. Irle and S. Yamaguchi, Nat. Commun., 2016, 7, 12094 CrossRef CAS PubMed.
- R. H. Zha, G. Vantomme, J. A. Berrocal, R. Gosens, B. De Waal, S. Meskers and E. W. Meijer, Adv. Funct. Mater., 2018, 28, 1–8 CrossRef.
- T. Sasaki, S. Hashimoto, N. Nogami, Y. Sugiyama, M. Mori, Y. Naka and K. v. Le, ACS Appl. Mater. Interfaces, 2016, 8, 5580–5585 CrossRef CAS PubMed.
- M.-M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS PubMed.
- J. Zhang, Q. Zou and H. Tian, Adv. Mater., 2013, 25, 378–399 CrossRef CAS PubMed.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- M. Irie, Chem. Rev., 2000, 100, 1683 CrossRef CAS PubMed.
- H. Zhou, C. Xue, P. Weis, Y. Suzuki, S. Huang, K. Koynov, G. K. Auernhammer, R. Berger, H.-J. Butt and S. Wu, Nat. Chem., 2017, 9, 145–151 CrossRef CAS PubMed.
- Y. Norikane, Y. Hirai and M. Yoshida, Chem. Commun., 2011, 47, 1770–1772 RSC.
- M. Hoshino, E. Uchida, Y. Norikane, R. Azumi, S. Nozawa, A. Tomita, T. Sato, S. Adachi and S. Koshihara, J. Am. Chem. Soc., 2014, 136, 9158–9164 CrossRef CAS PubMed.
- Z. Wu, C. Ji, X. Zhao, Y. Han, K. Müllen, K. Pan and M. Yin, J. Am. Chem. Soc., 2019, 141, 7385–7390 CrossRef CAS PubMed.
- S. Saito, S. Nobusue, E. Tsuzaka, C. Yuan, C. Mori, M. Hara, T. Seki, C. Camacho, S. Irle and S. Yamaguchi, Nat. Commun., 2016, 7, 12094 CrossRef CAS PubMed.
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS PubMed.
- L. Li, X. Chen and J. M. Torkelson, ACS Appl. Polym. Mater., 2020, 2, 4658–4665 CrossRef CAS.
- B. T. Michal, E. J. Spencer and S. J. Rowan, ACS Appl. Mater. Interfaces, 2016, 8, 11041–11049 CrossRef CAS PubMed.
- P. J. Boul, P. Reutenauer and J. M. Lehn, Org. Lett., 2005, 7, 15–18 CrossRef CAS PubMed.
- A. J. Inglis, L. Nebhani, O. Altintas, F. G. Schmidt and C. Barner-Kowollik, Macromolecules, 2010, 43, 5515–5520 CrossRef CAS.
- J. H. Aubert, J. Adhes., 2003, 79, 609–616 CrossRef CAS.
- D. H. Turkenburg, H. van Bracht, B. Funke, M. Schmider, D. Janke and H. R. Fischer, J. Appl. Polym. Sci., 2017, 134, 1–11 CrossRef.
- K. Luo, T. Xie and J. Rzayev, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4992–4997 CrossRef CAS.
- A. J. Inglis, L. Nebhani, O. Altintas, F. G. Schmidt and C. Barner-Kowollik, Macromolecules, 2010, 43, 5515–5520 CrossRef CAS.
- S. Das, S. Samitsu, Y. Nakamura, Y. Yamauchi, D. Payra, K. Kato and M. Naito, Polym. Chem., 2018, 9, 5559–5565 RSC.
- S. Wu, C. Cai, F. Li, Z. Tan and S. Dong, CCS Chem., 2020, 1690–1700 Search PubMed.
- J.-T. Dong, W.-K. Zou, F. Chen and Q. Zhao, Chin. J. Polym. Sci., 2018, 36, 953–959 CrossRef CAS.
- C. Gorsche, C. Schnoell, T. Koch, N. Moszner and R. Liska, Macromolecules, 2018, 51, 660–669 CrossRef CAS.
- G. M. Whitesides, Interface Focus, 2015, 5(4) DOI:10.1098/rsfs.2015.0031.
- H. Ito, W. P. England and M. Ueda, J. Photopolym. Sci. Technol., 1990, 3, 219–233 CrossRef CAS.
- P. Scrimin and L. J. Prins, Chem. Soc. Rev., 2011, 40, 4488 RSC.
- A. Sagi, R. Weinstain, N. Karton and D. Shabat, J. Am. Chem. Soc., 2008, 130, 5434–5435 CrossRef CAS PubMed.
- F. S. Danton and K. J. Ivin, Nature, 1948, 162, 705–707 CrossRef.
- R. B. Grubbs and R. H. Grubbs, Macromolecules, 2017, 50, 6979–6997 CrossRef CAS.
- G. I. Peterson, M. B. Larsen and A. J. Boydston, Macromolecules, 2012, 45, 7317–7328 CrossRef CAS.
- A. Sagi, R. Weinstain, N. Karton and D. Shabat, J. Am. Chem. Soc., 2008, 130, 5434–5435 CrossRef CAS PubMed.
- R. Weinstain, A. Sagi, N. Karton and D. Shabat, Chem.–Eur. J., 2008, 14, 6857–6861 CrossRef CAS PubMed.
- S. Gnaim and D. Shabat, J. Am. Chem. Soc., 2017, 139, 10002–10008 CrossRef CAS PubMed.
- G. G. Lewis, J. S. Robbins and S. T. Phillips, Macromolecules, 2013, 46, 5177–5183 CrossRef CAS.
- G. G. Lewis, J. S. Robbins and S. T. Phillips, Chem. Commun., 2014, 50, 5352–5354 RSC.
- P. Scrimin and L. J. Prins, Chem. Soc. Rev., 2011, 40, 4488 RSC.
- M. T. Gambles, B. Fan, A. Borecki and E. R. Gillies, ACS Omega, 2018, 3, 5002–5011 CrossRef CAS PubMed.
- K. Kumar and A. P. Goodwin, ACS Macro Lett., 2015, 4, 907–911 CrossRef CAS PubMed.
- B. Fan, R. E. Yardley, J. F. Trant, A. Borecki and E. R. Gillies, Polym. Chem., 2018, 9, 2601–2610 RSC.
- K. Kumar, E. J. Castaño, A. R. Weidner, A. Yildirim and A. P. Goodwin, ACS Macro Lett., 2016, 5, 636–640 CrossRef CAS PubMed.
- H. Ito and C. G. Willson, Polym. Eng. Sci., 1983, 23, 1012–1018 CrossRef CAS.
- H. Ito, W. P. England and M. Ueda, J. Photopolym. Sci. Technol., 1990, 3, 219–233 CrossRef CAS.
- H. Ito, M. Ueda and R. Schwalm, J. Vac. Sci. Technol., B: Microelectron. Process. Phenom., 1988, 6, 2259–2263 CrossRef CAS.
- H. Ito and R. Schwalm, J. Electrochem. Soc., 1989, 136, 241–245 CrossRef CAS.
- J.-F. de Marneffe, B. T. Chan, M. Spieser, G. Vereecke, S. Naumov, D. Vanhaeren, H. Wolf and A. W. Knoll, ACS Nano, 2018, 12, 11152–11160 CrossRef PubMed.
- L. L. Cheong, P. Paul, F. Holzner, M. Despont, D. J. Coady, J. L. Hedrick, R. Allen, A. W. Knoll and U. Duerig, Nano Lett., 2013, 13, 4485–4491 CrossRef CAS PubMed.
- O. Coulembier, A. Knoll, D. Pires, B. Gotsmann, U. Duerig, J. Frommer, R. D. Miller, P. Dubois and J. L. Hedrick, Macromolecules, 2010, 43, 572–574 CrossRef CAS.
- A. W. Knoll, D. Pires, O. Coulembier, P. Dubois, J. L. Hedrick, J. Frommer and U. Duerig, Adv. Mater., 2010, 22, 3361–3365 CrossRef CAS PubMed.
- M. J. Skaug, C. Schwemmer, S. Fringes, C. D. Rawlings and A. W. Knoll, Science, 2018, 359, 1505–1508 CrossRef CAS PubMed.
- M. J. Bowden and E. A. Chandross, J. Electrochem. Soc., 1975, 122, 1370–1374 CrossRef CAS.
- Y. Jiang and J. M. J. Frechet, Macromolecules, 1991, 24, 3528–3532 CrossRef CAS.
- O. P. Lee, H. Lopez Hernandez and J. S. Moore, ACS Macro Lett., 2015, 4, 665–668 CrossRef CAS.
- H. L. Hernandez, S.-K. Kang, O. P. Lee, S.-W. Hwang, J. A. Kaitz, B. Inci, C. W. Park, S. Chung, N. R. Sottos, J. S. Moore, J. A. Rogers and S. R. White, Adv. Mater., 2014, 26, 7637–7642 CrossRef CAS PubMed.
- C. W. Park, S.-K. Kang, H. L. Hernandez, J. A. Kaitz, D. S. Wie, J. Shin, O. P. Lee, N. R. Sottos, J. S. Moore, J. A. Rogers and S. R. White, Adv. Mater., 2015, 27, 3783–3788 CrossRef CAS PubMed.
- K. M. Lee, O. Phillips, A. Engler, P. A. Kohl and B. P. Rand, ACS Appl. Mater. Interfaces, 2018, 10, 28062–28068 CrossRef CAS PubMed.
- J. Jiang, O. Phillips, A. Engler, M. H. Vong and P. A. Kohl, Polym. Adv. Technol., 2019, 30, 1198–1204 CrossRef CAS.
- O. Phillips, A. Engler, J. M. Schwartz, J. Jiang, C. Tobin, Y. A. Guta and P. A. Kohl, J. Appl. Polym. Sci., 2019, 136, 47141 CrossRef.
- E. M. Lloyd, H. Lopez Hernandez, A. M. Feinberg, M. Yourdkhani, E. K. Zen, E. B. Mejia, N. R. Sottos, J. S. Moore, S. R. White, E. C. Feinberg, M. Yourdkhani, E. K. Zen, E. B. Mejia, N. R. Sottos, J. S. Moore and S. R. White, Chem. Mater., 2019, 31, 398–406 CrossRef CAS.
- J. M. Lobez and T. M. Swager, Angew. Chem., Int. Ed., 2010, 49, 95–98 CrossRef CAS PubMed.
- J. M. Lobez and T. M. Swager, Macromolecules, 2010, 43, 10422–10426 CrossRef CAS.
- H. Kim, H. Mohapatra and S. T. Phillips, Angew. Chem., Int. Ed., 2015, 54, 13063–13067 CrossRef CAS PubMed.
- T. S. Babra, M. Wood, J. S. Godleman, S. Salimi, C. Warriner, N. Bazin, C. R. Siviour, I. W. Hamley, W. Hayes and B. W. Greenland, Eur. Polym. J., 2019, 119, 260–271 CrossRef CAS.
- T. Nakamura, Y. Kaneko, E. Nishibori and T. Nabeshima, Nat. Commun., 2017, 8, 1–6 CrossRef PubMed.
- C. Zhang, B. Wu, Y. Zhou, F. Zhou, W. Liu and Z. Wang, Chem. Soc. Rev., 2020, 49, 3605–3637 RSC.
- G. P. Maier and A. Butler, JBIC, J. Biol. Inorg. Chem., 2017, 22, 739–749 CrossRef CAS PubMed.
- W. Wei, J. Yu, C. Broomell, J. N. Israelachvili and J. H. Waite, J. Am. Chem. Soc., 2013, 135, 377–383 CrossRef CAS PubMed.
- D. S. Hwang, H. Zeng, Q. Lu, J. Israelachvili and J. H. Waite, Soft Matter, 2012, 8, 5640–5648 RSC.
- B. K. Ahn, S. Das, R. Linstadt, Y. Kaufman, N. R. Martinez-Rodriguez, R. Mirshafian, E. Kesselman, Y. Talmon, B. H. Lipshutz, J. N. Israelachvili and J. H. Waite, Nat. Commun., 2015, 6, 8663 CrossRef CAS PubMed.
- T. Kang, D. X. Oh, J. Heo, H.-K. Lee, S. Choy, C. J. Hawker and D. S. Hwang, ACS Appl. Mater. Interfaces, 2015, 7, 24656–24662 CrossRef CAS PubMed.
- K. S. Rajan, S. Mainer and J. M. Davis, Bioinorg. Chem., 1978, 9, 187–203 CrossRef CAS.
- R. K. Boggess and R. B. Martin, J. Am. Chem. Soc., 1975, 97, 3076–3081 CrossRef CAS PubMed.
- B. Grgas-Kužnar, Vl. Simeon and O. A. Weber, J. Inorg. Nucl. Chem., 1974, 36, 2151–2154 CrossRef.
- A. R. Narkar, B. Barker, M. Clisch, J. Jiang and B. P. Lee, Chem. Mater., 2016, 28, 5432–5439 CrossRef CAS PubMed.
- H. Yuk, C. E. Varela, C. S. Nabzdyk, X. Mao, R. F. Padera, E. T. Roche and X. Zhao, Nature, 2019, 575, 169–174 CrossRef CAS PubMed.
- R. Frick, L. Bich and A. Moreno, Acta Biotheor., 2019, 67, 103–128 CrossRef PubMed.
- N. D. Blelloch, H. T. Mitchell, L. C. Greenburg, D. W. van Citters and K. A. Mirica, Chem. Mater., 2021 DOI:10.1021/acs.cgd.1c00593.
- D. A. McQuarrie and J. D. Simon, Physical Chemistry: A Molecular Approach, University Science Books, Herndon, VA, 1st edn, 1997 Search PubMed.
- D. Braga and F. Grepioni, Acc. Chem. Res., 2000, 33, 601–608 CrossRef CAS PubMed.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS PubMed.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- C. M. Reddy, M. T. Kirchner, R. C. Gundakaram, K. A. Padmanabhan and G. R. Desiraju, Chem.–Eur. J., 2006, 12, 2222–2234 CrossRef CAS PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- S. L. Cockroft, J. Perkins, C. Zonta, H. Adams, S. E. Spey, C. M. R. Low, J. G. Vinter, K. R. Lawson, C. J. Urch and C. A. Hunter, Org. Biomol. Chem., 2007, 5, 1062 RSC.
- M. Swart, M. Solà and F. M. Bickelhaupt, J. Comput. Chem., 2011, 32, 1117–1127 CrossRef CAS PubMed.
- A. Huller, Faraday Discuss. Chem. Soc., 1980, 69, 66–74 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- S. L. Cockroft, C. A. Hunter, K. R. Lawson, J. Perkins and C. J. Urch, J. Am. Chem. Soc., 2005, 127, 8594–8595 CrossRef CAS PubMed.
- C. Sutton, C. Risko and J.-L. Brédas, Chem. Mater., 2016, 28, 3–16 CrossRef CAS.
- T. Kang, X. Banquy, J. Heo, C. Lim, N. A. Lynd, P. Lundberg, D. X. Oh, H.-K. Lee, Y.-K. Hong, D. S. Hwang, J. H. Waite, J. N. Israelachvili and C. J. Hawker, ACS Nano, 2016, 10, 930–937 CrossRef CAS PubMed.
- D. Kitagawa, K. Kawasaki, R. Tanaka and S. Kobatake, Chem. Mater., 2017, 29, 7524–7532 CrossRef CAS.
- H. Mochiyama, M. O. Tokhi, H. Fujimoto and G. S. Virk, Emerging Trends in Mobile Robotics: Proceedings of the 13th International Conference on Climbing and Walking Robots and the Support Technologies for Mobile Machines, World Scientific Publishing Co. Inc., Singapore, 2010 Search PubMed.
- Handbook of 3D Integration, Volumes 1 and 2: Technology and Applications of 3D Integrated Circuits, ed. P. Garrou, C. Bower and P. Ramm, Wiley-VCH, Singapore, 1st edn, 2012 Search PubMed.
- Y. Mengüç and M. Sitti, in Polymer Adhesion, Friction, and Lubrication, ed. H. Zeng, John Wiley & Sons, Inc., 1st edn, 2013, p. 723 Search PubMed.
- A. Jagota and C.-Y. Hui, Mater. Sci. Eng., R, 2011, 72, 253–292 Search PubMed.
- L. F. Boesel, C. Greiner, E. Arzt and A. del Campo, Adv. Mater., 2010, 22, 2125–2137 CrossRef CAS PubMed.
- D. Sameoto and C. Menon, Smart Mater. Struct., 2010, 19, 103001 CrossRef.
- A. Majumder, A. Sharma and A. Ghatak, in Microfluidics and Microfabrication, Springer US, Boston, MA, 2010, pp. 283–307 Search PubMed.
- M. K. Kwak, C. Pang, H.-E. Jeong, H.-N. Kim, H. Yoon, H.-S. Jung and K.-Y. Suh, Adv. Funct. Mater., 2011, 21, 3606–3616 CrossRef CAS.
- K. Autumn, Am. Sci., 2006, 94, 124 CrossRef.
- M. Kamperman, E. Kroner, A. del Campo, R. M. McMeeking and E. Arzt, Adv. Eng. Mater., 2010, 12, 335–348 CrossRef CAS.
- K. Autumn, MRS Bull., 2007, 32, 473–478 CrossRef.
- E. Arzt, S. Gorb and R. Spolenak, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10603–10606 CrossRef CAS PubMed.
- K. Autumn, Y. A. Liang, S. T. Hsieh, W. Zesch, W. P. W. P. Chan, T. W. Kenny, R. Fearing and R. J. Full, Nature, 2000, 405, 681–685 CrossRef CAS PubMed.
- G. Huber, S. N. Gorb, R. Spolenak and E. Arzt, Biol. Lett., 2005, 1, 2–4 CrossRef PubMed.
- W. Sun, P. Neuzil, T. S. Kustandi, S. Oh and V. D. Samper, Biophys. J., 2005, 89, L14–L17 CrossRef CAS PubMed.
- P. H. Niewiarowski, S. Lopez, L. Ge, E. Hagan and A. Dhinojwala, PLoS One, 2008, 3, e2192 CrossRef PubMed.
- M. S. Prowse, M. Wilkinson, J. B. Puthoff, G. Mayer and K. Autumn, Acta Biomater., 2011, 7, 733–738 CrossRef PubMed.
- G. Huber, H. Mantz, R. Spolenak, K. Mecke, K. Jacobs, S. N. Gorb and E. Arzt, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16293–16296 CrossRef CAS PubMed.
- W. R. Hansen and K. Autumn, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 385–389 CrossRef CAS PubMed.
- K. Jin, Y. Tian, J. S. Erickson, J. Puthoff, K. Autumn and N. S. Pesika, Langmuir, 2012, 28, 5737–5742 CrossRef CAS PubMed.
- S. Kim and M. Sitti, Appl. Phys. Lett., 2006, 89, 261911 CrossRef.
- H. Gao and H. Yao, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7851–7856 CrossRef CAS PubMed.
- A. del Campo, C. Greiner and E. Arzt, Langmuir, 2007, 23, 10235–10243 CrossRef CAS PubMed.
- S. Kim, E. Cheung and M. Sitti, Langmuir, 2009, 25, 7196–7199 CrossRef CAS PubMed.
- A. V. Spuskanyuk, R. M. McMeeking, V. S. Deshpande and E. Arzt, Acta Biomater., 2008, 4, 1669–1676 CrossRef CAS PubMed.
- M. P. Murphy, B. Aksak and M. Sitti, J. Adhes. Sci. Technol., 2007, 21, 1281–1296 CrossRef CAS.
- B. Aksak, M. P. Murphy and M. Sitti, Langmuir, 2007, 23, 3322–3332 CrossRef CAS PubMed.
- A. J. Crosby, M. Hageman and A. Duncan, Langmuir, 2005, 21, 11738–11743 CrossRef CAS PubMed.
- M. Lamblet, E. Verneuil, T. Vilmin, A. Buguin, P. Silberzan and L. Léger, Langmuir, 2007, 23, 6966–6974 CrossRef CAS PubMed.
- H. E. Jeong, J.-K. Lee, H. N. Kim, S. H. Moon and K. Y. Suh, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5639–5644 CrossRef CAS PubMed.
- C. Greiner, A. del Campo and E. Arzt, Langmuir, 2007, 23, 3495–3502 CrossRef CAS PubMed.
- E. Cheung and M. Sitti, J. Adhes., 2011, 87, 547–557 CrossRef CAS.
- P. Glass, H. Chung, N. R. Washburn and M. Sitti, Langmuir, 2010, 26, 17357–17362 CrossRef CAS PubMed.
- J. Davies, S. Haq, T. Hawke and J. P. Sargent, Int. J. Adhes. Adhes., 2009, 29, 380–390 CrossRef CAS.
- D. Soto, G. Hill, A. Parness, N. Esparza, M. Cutkosky and T. Kenny, Appl. Phys. Lett., 2010, 97, 053701 CrossRef.
- A. Parness, D. Soto, N. Esparza, N. Gravish, M. Wilkinson, K. Autumn and M. Cutkosky, J. R. Soc., Interface, 2009, 6, 1223–1232 CrossRef CAS PubMed.
- M. P. Murphy, S. Kim and M. Sitti, ACS Appl. Mater. Interfaces, 2009, 1, 849–855 CrossRef CAS PubMed.
- N. J. Glassmaker, T. Himeno, C.-Y. Hui and J. Kim, J. R. Soc., Interface, 2004, 1, 23–33 CrossRef CAS PubMed.
- Y. Tian, N. Pesika, H. Zeng, K. Rosenberg, B. Zhao, P. McGuiggan, K. Autumn and J. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19320–19325 CrossRef CAS PubMed.
- S. Hu, S. Lopez, P. H. Niewiarowski and Z. Xia, J. R. Soc., Interface, 2012, 9, 2781–2790 CrossRef PubMed.
- Y. Tian, J. Wan, N. Pesika and M. Zhou, Sci. Rep., 2013, 3, 1382 CrossRef CAS PubMed.
- T. Xie and X. Xiao, Chem. Mater., 2008, 20, 2866–2868 CrossRef CAS.
- S. Baik, G. W. Hwang, S. Jang, S. Jeong, K. H. Kim, T.-H. Yang and C. Pang, ACS Appl. Mater. Interfaces, 2021, 13, 6930–6940 CrossRef CAS PubMed.
- E. Kizilkan, J. Strueben, A. Staubitz and S. N. Gorb, Sci. Robot., 2017, 2, eaak9454 CrossRef PubMed.
- E. Kizilkan and S. N. Gorb, Adv. Mater., 2018, 30, 1704696 CrossRef PubMed.
- H. Shahsavan, S. M. Salili, A. Jákli and B. Zhao, Adv. Mater., 2017, 29, 1604021 CrossRef PubMed.
- H. Tian, H. Liu, J. Shao, S. Li, X. Li and X. Chen, Soft Matter, 2020, 16, 5599–5608 RSC.
- S. Song and M. Sitti, Adv. Mater., 2014, 26, 4901–4906 CrossRef CAS PubMed.
- J. Shintake, V. Cacucciolo, D. Floreano and H. Shea, Adv. Mater., 2018, 30, 1707035 CrossRef PubMed.
- Y. Zhang, S. Ma, B. Li, B. Yu, H. Lee, M. Cai, S. N. Gorb, F. Zhou and W. Liu, Chem. Mater., 2021, 33, 2785–2795 CrossRef CAS.
- S. Salimi, T. S. Babra, G. S. Dines, S. W. Baskerville, W. Hayes and B. W. Greenland, Eur. Polym. J., 2019, 121, 109264 CrossRef.
- A. G. Gillies, J. Kwak and R. S. Fearing, Adv. Funct. Mater., 2013, 23, 3256–3261 CrossRef CAS.
- S. Leijonmarck, A. Cornell, C.-O. Danielsson and G. Lindbergh, J. Electrochem. Soc., 2011, 158, 109–114 CrossRef.
- S. Leijonmarck, A. Cornell, C. O. Danielsson, T. Åkermark, B. D. Brandner and G. Lindbergh, Int. J. Adhes. Adhes., 2012, 32, 39–45 CrossRef CAS.
- J. Jeong, J. Adhes. Interface, 2018, 19, 84–94 Search PubMed.
- M. S. Akram Bhuiyan, J. D. Roland, B. Liu, M. Reaume, Z. Zhang, J. D. Kelley and B. P. Lee, J. Am. Chem. Soc., 2020, 142, 4631–4638 CrossRef CAS PubMed.
- G. N. Alexandrovich, L. E. Alexandrovna, H. D. Jur’evich, B. I. Alexandrovna and P. A. Vladimirovna, Res. J. Pharm., Biol. Chem. Sci., 2018, 9(2), 723–730 CAS.
- K. Takeuchi, M. Fujino and T. Suga, IEEE Trans. Compon., Packag., Manuf. Technol., 2017, 7, 1713–1720 CAS.
- J. M. Boyne, E. J. Millan and I. Webster, Int. J. Adhes. Adhes., 2001, 21, 49–53 CrossRef CAS.
- K. Ebe, H. Seno and K. Horigome, J. Appl. Polym. Sci., 2003, 90, 436–441 CrossRef CAS.
- K. Horigome, K. Ebe and S. Kuroda, J. Appl. Polym. Sci., 2004, 93, 2889–2895 CrossRef CAS.
- S. R. Trenor, T. E. Long and B. J. Love, J. Adhes., 2005, 81, 213–229 CrossRef CAS.
- N. Saiki, O. Yamazaki and K. Ebe, J. Appl. Polym. Sci., 2008, 108, 1178–1183 CrossRef CAS.
- H. Ishikawa, K. Seto, S. Shimotuma, N. Kishi and C. Sato, Int. J. Adhes. Adhes., 2005, 25, 193–199 CrossRef CAS.
- P. Tanskanen, Acta Mater., 2013, 61, 1001–1011 CrossRef CAS.
- G. Jeevi, S. K. Nayak and M. Abdul Kader, J. Adhes. Sci. Technol., 2019, 1–24 Search PubMed.
- M. A. Schwartz, J. Cell Sci., 2008, 121, 1771 CrossRef CAS PubMed.
- V. P. Patil, J. D. Sandt, M. Kolle and J. Dunkel, Science, 2020, 367, 71–75 CrossRef CAS PubMed.
- B. Polster, Nature, 2002, 420, 476 CrossRef CAS PubMed.
- D. Rodriquez, S. Savagatrup, E. Valle, C. M. Proctor, C. McDowell, G. C. Bazan, T.-Q. Q. Nguyen and D. J. Lipomi, ACS Appl. Mater. Interfaces, 2016, 8, 11649–11657 CrossRef CAS PubMed.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- S. Savagatrup, D. Rodriquez, A. D. Printz, A. B. Sieval, J. C. Hummelen and D. J. Lipomi, Chem. Mater., 2015, 27, 3902–3911 CrossRef CAS.
- S. R. Dupont, M. Oliver, F. C. Krebs and R. H. Dauskardt, Sol. Energy Mater. Sol. Cells, 2012, 97, 171–175 CrossRef CAS.
- C. Bruner, N. C. Miller, M. D. McGehee and R. H. Dauskardt, Adv. Funct. Mater., 2013, 23, 2863–2871 CrossRef CAS.
- S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Macromolecules, 2014, 47, 1981–1992 CrossRef CAS.
- O. Awartani, B. I. Lemanski, H. W. Ro, L. J. Richter, D. M. DeLongchamp and B. T. O'Connor, Adv. Energy Mater., 2013, 3, 399–406 CrossRef CAS.
- T. Kim, J.-H. Kim, T. E. Kang, C. Lee, H. Kang, M. Shin, C. Wang, B. Ma, U. Jeong, T.-S. Kim and B. J. Kim, Nat. Commun., 2015, 6, 8547 CrossRef CAS.
- H.-C. Wu, S. J. Benight, A. Chortos, W.-Y. Lee, J. Mei, J. W. F. To, C. Lu, M. He, J. B.-H. Tok, W.-C. Chen and Z. Bao, Chem. Mater., 2014, 26, 4544–4551 CrossRef CAS.
- J. Liang, L. Li, D. Chen, T. Hajagos, Z. Ren, S.-Y. Chou, W. Hu and Q. Pei, Nat. Commun., 2015, 6, 7647 CrossRef CAS PubMed.
- A. D. Printz, S. Savagatrup, D. Rodriquez and D. J. Lipomi, Sol. Energy Mater. Sol. Cells, 2015, 134, 64–72 CrossRef CAS.
- A. D. Printz, A. V Zaretski, S. Savagatrup, A. S.-C. Chiang and D. J. Lipomi, ACS Appl. Mater. Interfaces, 2015, 7, 23257–23264 CrossRef CAS.
- S. Savagatrup, A. S. Makaram, D. J. Burke and D. J. Lipomi, Adv. Funct. Mater., 2014, 24, 1169–1181 CrossRef CAS.
- S. Savagatrup, A. D. Printz, T. F. O'Connor, A. V. Zaretski, D. Rodriquez, E. J. Sawyer, K. M. Rajan, R. I. Acosta, S. E. Root and D. J. Lipomi, Energy Environ. Sci., 2015, 8, 55–80 RSC.
- E. Boujo and M. Sellier, Phys. Rev. Fluid., 2019, 4, 64802 CrossRef.
- M. A. Fardin, Rheol. Bull., 2014, 83(16–17), 30 Search PubMed.
- C. Clanet, F. Hersen and L. Bocquet, Nature, 2004, 427, 29 CrossRef CAS PubMed.
- L. Gong, L. Xiang, J. Zhang, J. Chen and H. Zeng, Langmuir, 2019, 35, 15914–15936 CrossRef CAS PubMed.
- D. Rodriquez, S. Savagatrup, E. Valle, C. M. Proctor, C. McDowell, G. C. Bazan, T. Q. Nguyen and D. J. Lipomi, ACS Appl. Mater. Interfaces, 2016, 8, 11649–11657 CrossRef CAS PubMed.
- J. Israelachvili, Intermolecular and Surface Forces, Elsevier Inc., 3rd edn, 2011 Search PubMed.
- X. Zhong, A. G. Shtukenberg, M. Liu, I. A. Olson, M. Weck, M. D. Ward and B. Kahr, Cryst. Growth Des., 2019, 19, 6649–6655 CrossRef CAS.
- I. A. Olson, A. G. Shtukenberg, B. Kahr and M. D. Ward, Rep. Prog. Phys., 2018, 81, 96501 CrossRef PubMed.
- X. Zhong, A. G. Shtukenberg, T. Hueckel, B. Kahr and M. D. Ward, Cryst. Growth Des., 2018, 18, 318–323 CrossRef CAS.
- I. A. Olson, A. G. Shtukenberg, G. Hakobyan, A. L. Rohl, P. Raiteri, M. D. Ward and B. Kahr, J. Phys. Chem. Lett., 2016, 7, 3112–3117 CrossRef CAS PubMed.
- F. Dong, M. Liu, V. Grebe, M. D. Ward and M. Weck, Chem. Mater., 2020, 32, 6898–6905 CrossRef CAS.
- A. G. Shtukenberg, M. D. Ward and B. Kahr, Chem. Rev., 2017, 117, 14042–14090 CrossRef CAS PubMed.
- L. N. Poloni, Z. Zhu, N. Garcia-Vázquez, A. C. Yu, D. M. Connors, L. Hu, A. Sahota, M. D. Ward and A. G. Shtukenberg, Cryst. Growth Des., 2017, 17, 2767–2781 CrossRef CAS PubMed.
- M. H. Lee, A. Sahota, M. D. Ward and D. S. Goldfarb, Curr. Rheumatol. Rep., 2015, 17, 33 CrossRef PubMed.
- Q. Saleem, R. D. Wildman, J. M. Huntley and M. B. Whitworth, Meas. Sci. Technol., 2003, 14, 2027–2033 CrossRef CAS.
- ASTM International, Standards & Publications, 2021, https://www.astm.org/Standard/standards-and-publications.html.
- TAPPI, TAPPI Standards, Technical Information Papers (TIPS) and Useful Methods, 2021, https://www.tappi.org/publications-standards/standards-methods/standardsonline Search PubMed.
- M. K. Mishra, U. Ramamurty and G. R. Desiraju, Curr. Opin. Solid State Mater. Sci., 2016, 20, 361–370 CrossRef CAS.
- A. K. Nangia and G. R. Desiraju, Angew. Chem., Int. Ed., 2019, 58, 4100–4107 CrossRef CAS PubMed.
- V. Videnova-Adrabińska, J. Mol. Struct., 1996, 374, 199–222 CrossRef.
- C. B. Aakeröy and A. M. Beatty, Aust. J. Chem., 2001, 54, 409–421 CrossRef.
- G. R. Desiraju, Chem. Commun., 1997, 1475–1482 RSC.
- A. Nangia and G. R. Desiraju, in Design of Organic Solids, ed. E. Weber, Springer-Verlag Berlin Heidelberg, 1998, pp. 57–95 Search PubMed.
- A. Parkin, G. Barr, W. Dong, C. J. Gilmore, D. Jayatilaka, J. J. McKinnon, M. A. Spackman and C. C. Wilson, CrystEngComm, 2007, 9, 648 RSC.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC.
- G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565–573 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |