
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrofluoromethylation of alkenes with fluoroiodomethane and beyond†
Sandrine M.
Hell‡
 ,
Claudio F.
Meyer‡
,
Sebastiano
Ortalli‡
,
Jeroen B. I.
Sap
,
Xuanxiao
Chen
and
Véronique
Gouverneur
*
,
Claudio F.
Meyer‡
,
Sebastiano
Ortalli‡
,
Jeroen B. I.
Sap
,
Xuanxiao
Chen
and
Véronique
Gouverneur
*
University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: veronique.gouverneur@chem.ox.ac.uk
First published on 11th August 2021
Abstract
A process for the direct hydrofluoromethylation of alkenes is reported for the first time. This straighforward silyl radical-mediated reaction utilises CH2FI as a non-ozone depleting reagent, traditionally used in electrophilic, nucleophilic and carbene-type chemistry, but not as a CH2F radical source. By circumventing the challenges associated with the high reduction potential of CH2FI being closer to CH3I than CF3I, and harnessing instead the favourable bond dissociation energy of the C–I bond, we demonstrate that feedstock electron-deficient alkenes are converted into products resulting from net hydrofluoromethylation with the intervention of (Me3Si)3SiH under blue LED activation. This deceptively simple yet powerful methodology was extended to a range of (halo)methyl radical precursors including ICH2I, ICH2Br, ICH2Cl, and CHBr2F, as well as CH3I itself; this latter reagent therefore enables direct hydromethylation. This versatile chemistry was applied to 18F-, 13C-, and D-labelled reagents as well as complex biologically relevant alkenes, providing facile access to more than fifty products for applications in medicinal chemistry and positron emission tomography.
Introduction
The introduction of fluoroalkyl groups has garnered significant interest in medicinal chemistry, enabling the modulation of biological and physicochemical properties of lead candidates for drug discovery.1–3 Whilst the fields of radical trifluoromethylation and difluoromethylation have been extensively explored,4–10 the fluoromethyl radical has received far less attention.11–13 This is unexpected as the fluoromethyl group features frequently in pharmaceutical drugs, more often to improve metabolic stability by serving as a bioisosteric replacement of functional groups responsible for poor performance.14,15 In recent years, several reagents for the generation of the CH2F radical have been developed.16–20 Often, efficient activation of these reagents requires harsh reaction conditions, such as elevated temperatures, strong oxidants, or strong reductants. Furthermore, many of these reagents are either expensive, highly toxic or non-commercial, requiring multistep syntheses for their preparation. As part of our growing interest in developing “minimalistic” procedures for the late-stage hydrofluoroalkylation of alkene-containing biologically active molecules,21–23 we sought to develop an operationally simple method for the direct hydrofluoromethylation of alkenes, as an attractive strategy for the introduction of this motif to C(sp3)-enriched backbones (Scheme 1).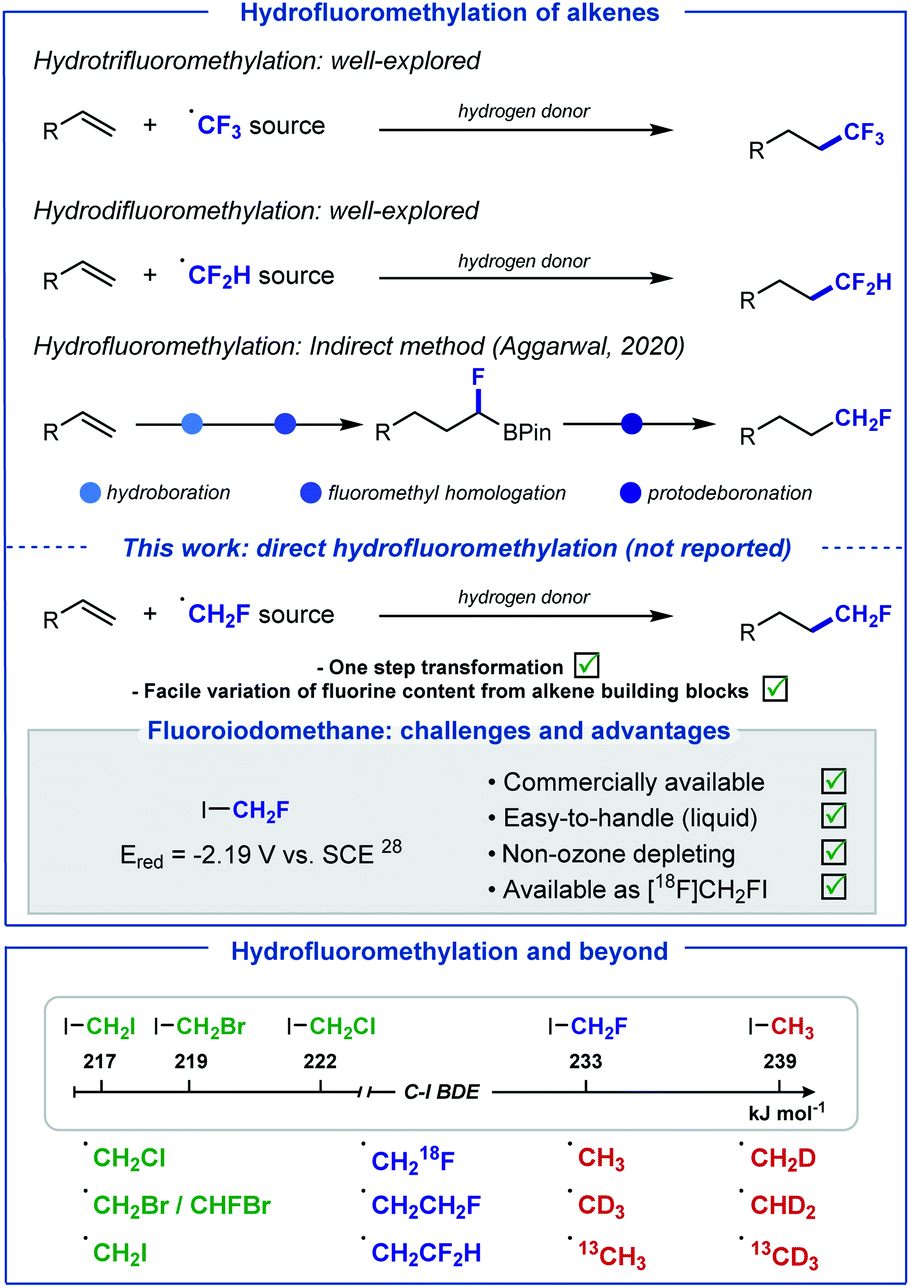 | ||
| Scheme 1 Hydro(per)fluoromethylation of alkenes. This work: direct silyl radical-mediated hydrofluoromethylation of electron-deficient alkenes and extension to numerous hydro(halo)methylation reactions. | ||
In 2020, an indirect method for the hydrofluoromethylation of alkenes was developed by Aggarwal and co-workers;13 this elegant multi-step procedure starts with the conversion of alkenes into boronic esters, subsequent treatment at low temperature (−78 °C) with in situ formed fluoroiodomethyl lithium to generate fluoroboronic esters, and a final protodeboronation. Our aim was to develop a one-step method that avoids operational complexity and over-engineering, ideally using fluoroiodomethane which is a non-ozone depleting, easy to handle and inexpensive commercial CH2F radical precursor. We noted that fluoroiodomethane has found applications as an electrophilic or nucleophilic fluoromethylation reagent as well as in cross-coupling reactions,24–27 but has not been explored in the context of radical chemistry.
The high reduction potential of CH2FI (Ered = −2.19 V vs. saturated calomel electrode (SCE) in MeCN),28 much closer to MeI (Ered = −2.39 V vs. SCE in MeCN)28 than CF3I (Ered = −1.22 V vs. SCE in MeCN),29 encouraged the implementation of an activation pathway exploiting instead the favourable bond dissociation energy (BDE) of C–I (BDE (FH2C–I) = 233 kJ mol−1) versus C–F (BDE (IH2C–F) = 460 kJ mol−1).30 Since the pioneering work of Chatgilialoglu,31 tris(trimethylsilyl)silane (TTMSS) has found ample applications as a powerful tool for mild radical generation via the activation of alkyl halides.32–36 In addition, TTMSS has valuably complemented Giese-type reactions, a commonly exploited platform for late-stage functionalisation, by providing a suitable alternative to traditional toxic tin-based reagents.37 Consequently, we envisioned that the supersilyl radical (TMS)3Si˙ would be well suited to release ˙CH2F from CH2FI. Subsequent Giese-type addition of ˙CH2F to the electron-deficient alkene would generate a carbon-centered radical intermediate. Hydrogen-atom transfer (HAT) between this electrophilic species and hydridic (TMS)3SiH would afford the desired hydrofluoromethylated product, and (TMS)3Si˙ entering chain propagation. Initiation for this process would be triggered by photolytic C–I cleavage of FH2C–I.38 This method offers the prospect of being applicable to a range of other halo-containing alkyl radicals, provided that competitive hydrogen atom abstraction with (TMS)3SiH does not occur prior to Giese addition. Herein, we report the realisation of this strategy with a wide range of haloiodomethanes for the direct hydrohalomethylation of electron-deficient alkenes including biologically relevant molecules. The method was extended to 18F-hydrofluoromethylation and hydromethylation with iodomethane along with five of its D and 13C isotopomers.
Results and discussion
Preliminary experiments were conducted with N-phenyl acrylamide (1a) (Table 1).39 Various combinations of silanes and solvents revealed that the desired hydrofluoromethylated product (2a) was obtained in 71% with (TMS)3SiH in MeCN at room temperature under blue light irradiation for 16 h (entry 1).40 The addition of fac-Ir(ppy)3 (0.5 mol%) did not lead to significant improvement (entry 2). The simpler protocol was therefore retained for further investigations. Control experiments indicate that the reaction was not effective in absence of light (entry 3), and unsuccessful in absence of silane or in presence of the radical scavenger TEMPO (entries 4 and 5). No deuterium incorporation was observed in the product when the reaction was performed in CD3CN.40 These data corroborate our proposed radical chain propagation mechanism, initiated by blue-light homolysis of the CH2F–I bond.38 Giese addition of the fluoromethyl radical to an electron-deficient alkene furnishes an electrophilic carbon-centered radical intermediate, capable of undergoing HAT with (TMS)3SiH. The resulting silyl radical enables chain propagation by abstracting iodine from CH2FI to afford (TMS)3SiI along with ˙CH2F.40|
|
||
|---|---|---|
| Entry | Deviations from standard conditions | Yield (%) |
| a Reaction conditions: CH2FI (0.2 mmol), 1a (0.1 mmol), (TMS)3SiH (0.12 mmol), solvent (0.6 mL) under blue light (λmax = 450 nm) irradiation for 16 h. Yields of isolated products. b Addition of 4.0 equiv. of TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl). TEMPO-CH2F was observed in quantitative 19F NMR analysis of the crude reaction mixture using α,α,α-trifluorotoluene as an internal standard. fac-Ir(ppy)3 = tris[2-phenylpyridinato-C2,N]iridium(III). | ||
| 1 | None | 71 |
| 2 | With fac-Ir(ppy)3 (0.5 mol%) | 75 |
| 3 | No light | Traces |
| 4 | No silane | 0 |
| 5 | With TEMPO (4.0 equiv.)b | 0 |
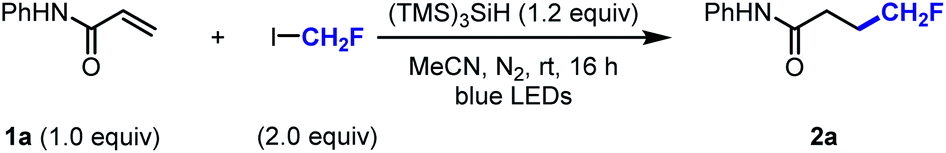
With the optimised reaction conditions in hand, we sought to explore the scope of this hydrofluoromethylation protocol (Scheme 2A). Various functional groups, such as methoxy, nitrile, halide, ketone, ether, amide, ester, aniline, and sulfone were tolerated. The addition of fac-Ir(ppy)3 (0.5 mol%) led to higher yields for selected substrates.40N-Aryl acrylamides bearing electron-withdrawing and electron-donating groups afforded the desired products in moderate to excellent yields (2a–d). The hydrofluoromethylation of N-heteroaryl acrylamides, such as pyridyl and benzothiazyl was also successful (2e, 2f). Alkenes substituted with sulfones and esters were competent substrates generating 2g and 2h in moderate yield. As deuteration can improve metabolic stability,41 we investigated the hydrofluoromethylation of a deuterated alkene (1i) that was successfully converted into [D3]2i. The gem-disubstituted alkene 1j provided 2j in 64% yield. Pleasingly, the internal alkene 1k was reactive under our reaction conditions and afforded fluoromethylcyclobutane 2k in moderate yield. This result is significant as 1,2-disubstituted fluoroalkyl cyclobutanes currently require multiple steps for their preparation.42 A non-cyclic trisubstituted alkene afforded the product in 57% yield (2l). Styrene derivatives such as 1m and 1n afforded the desired products in synthetically useful yields (2m, 2n). Our protocol is amenable to scale-up as demonstrated by the 10 mmol scale hydrofluoromethylation of N-benzylmaleimide affording 2o in 88% yield. The synthesis of fluorinated pyrrolidine 2p, amine 2q, alcohol 2r and carboxylic acid 2s was performed in two steps, offering a pathway to diversify the range of products within reach from CH2FI. The late-stage hydrofluoromethylation of complex biologically active molecules was considered next. The anti-cancer drug ibrutinib as well as estrone, tyrosine and ethacrynic acid derivatives afforded the desired hydrofluoromethylated products in good yields (2t–w). The tolerance of functional groups was investigated with a robustness screening.40 These experimental data provide an overview of the many heteroarenes (e.g. pyridazine, 1,3,5-triazine, indole, benzothiazole or oxazole) that are tolerated under the optimised reaction conditions.
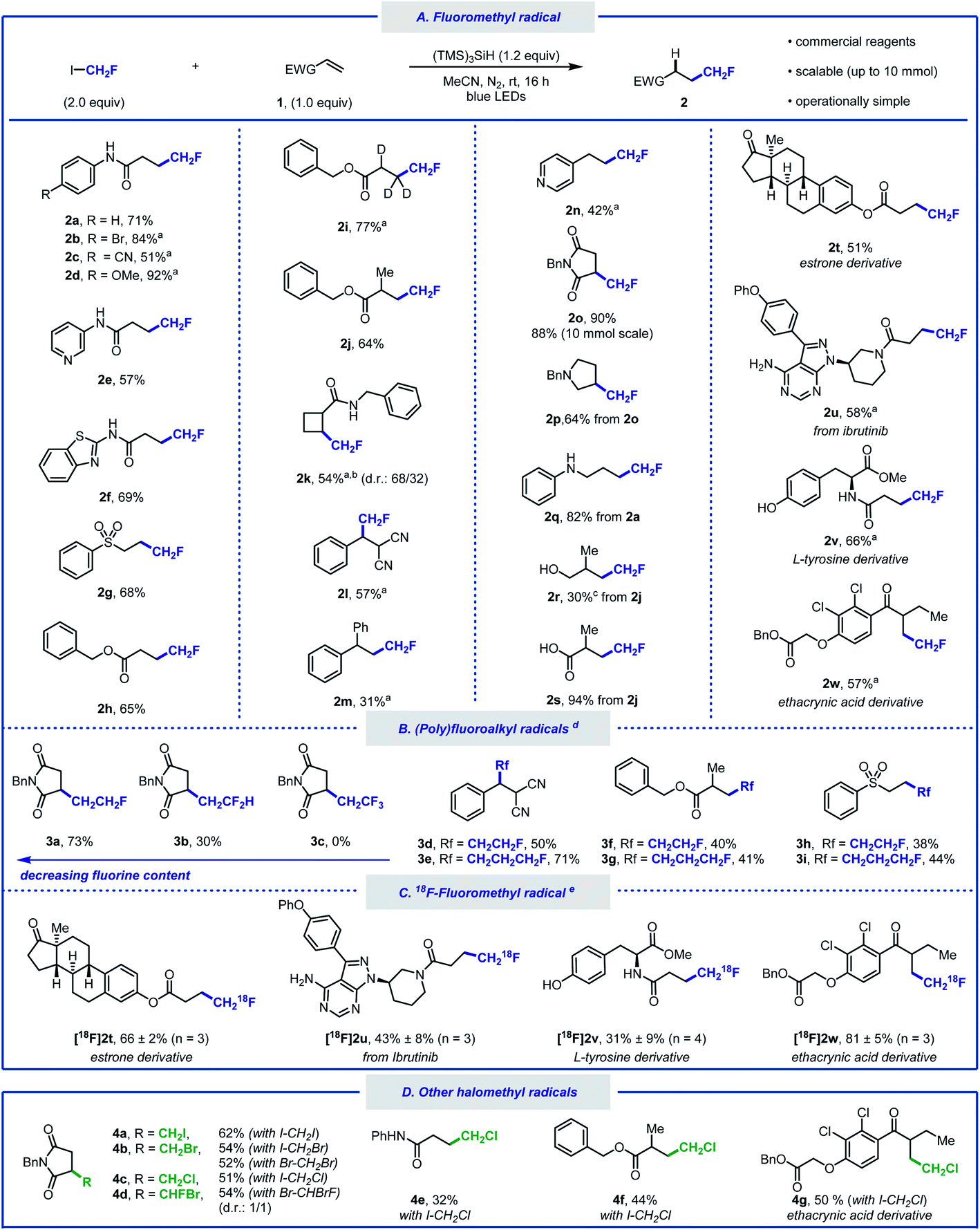 | ||
| Scheme 2 Substrate scope of the hydro(halo)methylation of electron-deficient alkenes. Reaction conditions: alkene (0.5 mmol), (TMS)3SiH (0.6 mmol), radical precursor (1.0 mmol), MeCN (3.0 mL), N2 atmosphere, blue light (λmax = 450 nm) irradiation for 16 h, room temperature. (a) Reaction performed with fac-Ir(ppy)3 (0.5 mol%). (b) Product isolated as a mixture of diastereomers. (c) Yield determined by quantitative 1H NMR using triphenylmethane as an internal standard. (d) Reaction performed using (poly)fluoroalkyl iodide. (e) 18F-Hydrofluoromethylation performed with [18F]CH2FI. Reaction conditions: [18F]CH2FI (10 MBq), alkene (0.1 mmol), (TMS)3SiH (0.12 mmol), fac-Ir(ppy)3 (0.5 mol%), DMF (0.6 mL), blue light (λmax = 450 nm) irradiation for 20 min, room temperature. | ||
Whilst additives containing nucleophilic functional groups such as alcohols and anilines were tolerated, side reactivity arising from nucleophilic substitution was observed.40 Competitive alkylation was suppressed when using 1.0 equivalent of CH2FI, albeit at the expense of reduced yield for the hydrofluoromethylated product. Aliphatic amines were tolerated but yields did not exceed 30%.40 The hydrofluoromethylation of alkenes not bearing electron-withdrawing groups was possible albeit significantly less efficient.40 With a protocol relying on the favourable C–I bond dissociation energy and considering the importance of homologation in medicinal chemistry,43 we considered the generation of products from a series of homologated fluoroiodoalkanes (Scheme 2B).44,45 Hydrofluoroalkylation of alkenes 1g, 1j and 1l provided effortlessly the homologous series of products 3d–i. Specifically, the fluoroethyl radical was efficiently generated applying similar silyl radical activation, and 3a was isolated in good yield. The introduction of the fluoroethyl radical was successfully performed on linear terminal, gem-disubstituted, and trisubstituted alkenes (3d, 3f, 3h). The method was further extended to fluoroiodopropane as shown with the synthesis of 3e, 3g, and 3i. Precursors featuring additional fluorine atoms were less suitable with the difluoroethylated product 3b isolated in 30%, and no product observed when attempting to prepare the hydrotrifluoroethylated product 3c. Increased fluorine content enhances radical electrophilicity, thereby encouraging undesired H-atom abstraction from (TMS)3SiH.40
Given the success of our protocol, we further investigated the applicability of our method for the generation of [18F]CH2F radical from [18F]CH2FI (Scheme 2C).46–48 Compounds labelled with the radioisotope F-18 are important for applications in Positron Emission Tomography (PET).49–53 The synthesis of [18F]CH2FI in high molar activity (Am) is well-established and has been automated.54–57 To date, this labelled reagent is mainly employed for the electrophilic 18F-fluoromethylation of phenols.58,59 We now demonstrate that [18F]CH2FI is well suited for [18F]CH2F radical chemistry. Specifically, Ibrutinib, an estrone, a tyrosine, and an ethacrynic acid derivative underwent 18F-hydrofluoromethylation in radiochemical yields up to 81% ([18F]2t–w). This reaction was best performed for 20 minutes at ambient temperature in the presence of fac-Ir(ppy)3 under blue-light irradiation. This method offers an alternative to nucleophilic 18F-fluorination with [18F]fluoride for precursors that are either unstable, require complex multiple steps synthesis, or lead predominantly to elimination products. Haloiodomethanes other than fluoroiodomethane were also considered as they would allow for the one-step introduction of reactive halomethyl groups to alkenes (Scheme 2D). Controlled activation of reagents such as ICH2X (X = Cl, Br, I) would enable their use for example as ˙CH2+ synthon. To date, only few examples for the generation and use of halomethyl radicals have been reported.60–64 When diiodomethane was employed under the standard reaction conditions, N-benzylmaleimide underwent hydroiodomethylation in 62% yield (4a). Similarly, hydrobromomethylation (from dibromomethane or bromoiodomethane), hydrochloromethylation (from chloroiodomethane), and hydrobromofluoromethylation (from dibromofluoromethane) provided the corresponding halomethyl alkanes in moderate yields (4b–4d).23,65 Other alkenes afforded the hydrochloromethylated products in moderate yields (4e–4g). Although full conversion of starting material was observed for these reactions, purification via silica gel chromatography led to elimination, which is reflected in the lower yield for these compounds upon isolation.
Competition experiments were performed to calibrate the reactivity of fluoroiodomethane versus other alkyl iodides (Scheme 3). When equimolar amounts of iodomethane and fluoroiodomethane were subjected to the standard reaction conditions, product resulting from fluoromethyl radical addition was obtained in 74% yield (2n), along with 25% of the hydromethylated product 5a. When the reaction was carried out with equimolar amounts of iodoethane, products 2n and 6 were formed in close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Additional competition experiments showed that the iso-propyl and tert-butyl adducts (7, 8) were formed preferentially over the hydrofluoromethylated product. The reactivity of these alkyl iodides therefore decreases in the following order: tBuI > iPrI > CH2FI ∼ EtI > MeI.
:1 ratio. Additional competition experiments showed that the iso-propyl and tert-butyl adducts (7, 8) were formed preferentially over the hydrofluoromethylated product. The reactivity of these alkyl iodides therefore decreases in the following order: tBuI > iPrI > CH2FI ∼ EtI > MeI.
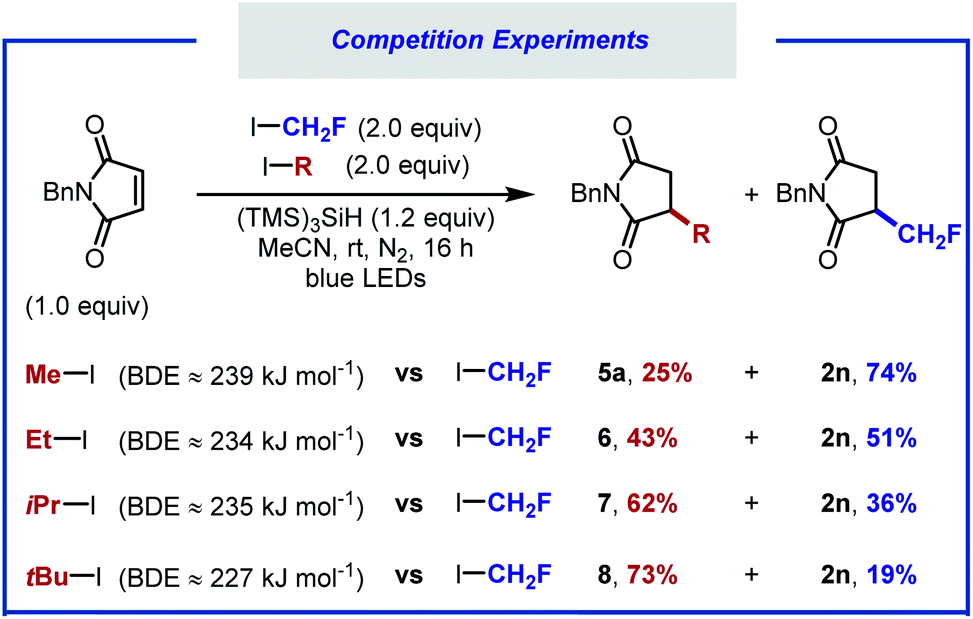 | ||
| Scheme 3 Competition experiments with equimolar amounts of different alkyl iodides. | ||
A notable outcome of this study was the observation that net methane addition across the double bond took place with iodomethane. Currently, protocols for the generation of the methyl radical from iodomethane (BDECH3–I = 239 kJ mol−1, Ered = −2.39 V vs. SCE in MeCN) remain underdeveloped.34,66 In recent years, the methyl radical has been generated from numerous precursors.67–71 The formation of the methyl radical often requires harsh reaction conditions, limiting the applicability of these protocols. Furthermore, the use of the methyl radical towards application to isotopic labelling is far from trivial. Iodomethane, on the other hand, can provide effortless access to a variety of useful isotopologues that would otherwise be beyond reach. The straightforwardness of our protocol prompted us to optimise the hydromethylation of alkenes using iodomethane as methyl radical precursor (Scheme 4). We noted significant gas release when applying our reaction conditions, attributed to methane resulting from competitive HAT between the methyl radical and MeCN (BDENCCH2–H = 389 kJ mol; BDECH3–H = 439 kJ mol−1).69 A screen of solvents, reactants stoichiometry and photocatalysts allowed for hydromethylation to occur in up to 93% yield (5a).40 Under the optimised reaction conditions consisting of 4.0 equivalents of MeI, 3.0 equivalents of (TMS)3SiH and 1,2-difluorobenzene as solvent, in combination with photocatalyst MesAcrBF4 (0.5 mol%), the hydromethylation of various alkenes took place in good to excellent yield (5b–f). Considering that bioactive compounds containing stable heavy isotopes are useful for example as mass spectroscopy standards,41,72 the hydromethylation of an ethacrynic acid derivative was performed with CH3I, CH2DI, CHD2I, CD3I, 13CH3I, and 13CD3I. All six isotopologues (5h–5m) were obtained in moderate yield.73–75
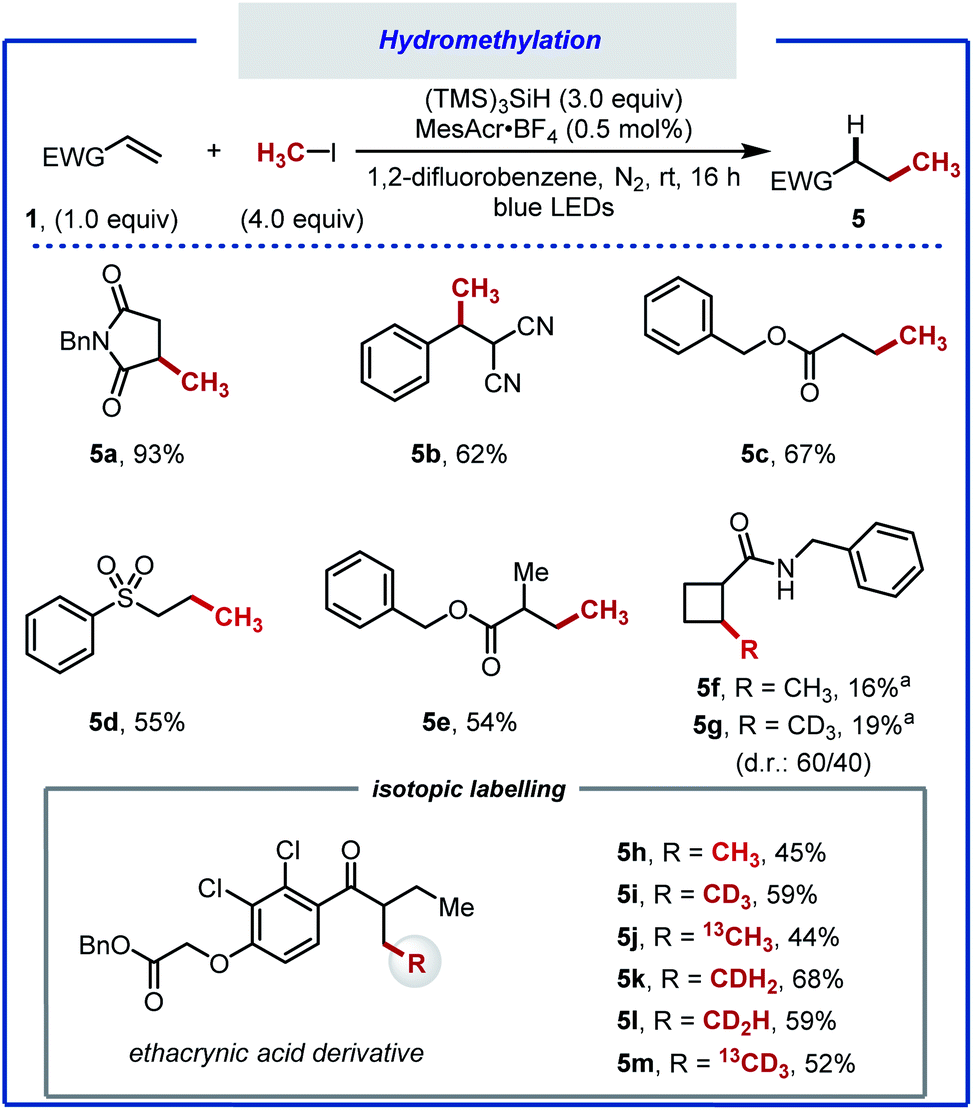 | ||
| Scheme 4 Hydromethylation of electron-deficient alkenes. Reaction conditions: alkene (0.5 mmol), (TMS)3SiH (1.5 mmol), iodomethane (2.0 mmol), 1,2-difluorobenzene (3.0 mL), N2 atmosphere, blue light (λmax = 450 nm) irradiation for 16 h, room temperature. (a) Product isolated as a mixture of diastereomers. | ||
Conclusions
In conclusion, the first direct hydrofluoromethylation of a broad range of electron-deficient alkenes has been developed using fluoroiodomethane. Mechanistically, the process harnesses known principles; so its core value is rooted in its immediate synthetic power. With the current global necessity “to do more with less”, this minimalistic and mild chemical method stands out as it is operationally simple with the supersilyl radical precursor (TMS)3SiH being the only chemical required in addition to the reaction partners. The mild reaction conditions are compatible with complex biologically active molecules such as Ibrutinib. The methodology was successfully adapted for the 18F-labelling of complex alkenes, and offers a new C–CH218F disconnection strategy for radiotracer development. The method was extended to additional fluoroiodoalkanes enabling facile product homologation, as well as multiple (halo)methyl radicals including the methyl radical itself and five of its D and 13C isotopomers.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
S. M. H., C. F. M., S. O. and J. B. I. S. performed the experiments and analysed the results. C. F. M. and X. C. performed the cyclic voltammetry experiments. S. M. H., C. F. M., S. O., J. B. I. S. and V. G. designed the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Prof. Benjamin Davis and Prof. Jason J. Davis for insightful comments on the manuscript, and useful discussions. This project has received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 721902 (S. M. H. and C. F. M.). J. B. I. S. acknowledges financial support from an EPSRC Doctoral Prize (EP/T517811/1).Notes and references
- Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, J. Med. Chem., 2017, 60, 797–804 CrossRef CAS PubMed.
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- D. A. Nagib and D. W. C. Macmillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed.
- A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950–8958 CrossRef CAS PubMed.
- T. Koike and M. Akita, Top. Catal., 2014, 57, 967–974 CrossRef CAS.
- C. Le, T. Q. Chen, T. Liang, P. Zhang and D. W. C. Macmillan, Science, 2018, 360, 1010–1014 CrossRef CAS PubMed.
- T. Koike and M. Akita, Org. Biomol. Chem., 2019, 17, 5413–5419 RSC.
- J. B. I. Sap, C. F. Meyer, N. J. W. Straathof, N. Iwumene, C. W. am Ende, A. A. Trabanco and V. Gouverneur, Chem. Soc. Rev., 2021, 50, 8214–8247 RSC.
- X. J. Tang, Z. Zhang and W. R. Dolbier, Chem.–Eur. J., 2015, 21, 18961–18965 CrossRef CAS PubMed.
- Y. Li, C. Ni, J. Liu, L. Zhang, J. Zheng, L. Zhu and J. Hu, Org. Lett., 2006, 8, 1693–1696 CrossRef CAS PubMed.
- G. K. S. Prakash, S. Chacko, S. Alconcel, T. Stewart, T. Mathew and G. A. Olah, Angew. Chem., Int. Ed., 2007, 46, 4933–4936 CrossRef CAS PubMed.
- V. Fasano, N. Winter, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2020, 59, 8502–8506 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- B. M. Johnson, Y. Z. Shu, X. Zhuo and N. A. Meanwell, J. Med. Chem., 2020, 63, 6315–6386 CrossRef CAS PubMed.
- Y. Fujiwara, J. A. Dixon, F. O’hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herlé, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95–99 CrossRef CAS PubMed.
- X. J. Tang and W. R. Dolbier, Angew. Chem., Int. Ed., 2015, 54, 4246–4249 CrossRef CAS PubMed.
- L. An, Y. L. Xiao, Q. Q. Min and X. Zhang, Angew. Chem., Int. Ed., 2015, 54, 9079–9083 CrossRef CAS PubMed.
- N. Noto, T. Koike and M. Akita, ACS Catal., 2019, 9, 4382–4387 CrossRef CAS.
- J. Sheng, H.-Q. Ni, S.-X. Ni, Y. He, R. Cui, G.-X. Liao, K.-J. Bian, B.-B. Wu and X.-S. Wang, Angew. Chem., Int. Ed., 2021, 60, 15020–15027 CrossRef CAS PubMed.
- S. Mizuta, S. Verhoog, K. M. Engle, T. Khotavivattana, M. O'Duill, K. Wheelhouse, G. Rassias, M. Médebielle and V. Gouverneur, J. Am. Chem. Soc., 2013, 135, 2505–2508 CrossRef CAS PubMed.
- C. F. Meyer, S. M. Hell, A. Misale, A. A. Trabanco and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 8829–8833 CrossRef CAS PubMed.
- C. F. Meyer, S. M. Hell, J. B. I. Sap, A. Misale, A. Peschiulli, D. Oehlrich, A. A. Trabanco and V. Gouverneur, Tetrahedron, 2019, 75, 130679 CrossRef.
- J. Hu, B. Gao, L. Li, C. Ni and J. Hu, Org. Lett., 2015, 17, 3086–3089 CrossRef CAS PubMed.
- G. Parisi, M. Colella, S. Monticelli, G. Romanazzi, W. Holzer, T. Langer, L. Degennaro, V. Pace and R. Luisi, J. Am. Chem. Soc., 2017, 139, 13648–13651 CrossRef CAS PubMed.
- N. Y. Wu, X. H. Xu and F. L. Qing, ACS Catal., 2019, 9, 5726–5731 CrossRef CAS.
- R. Senatore, M. Malik, M. Spreitzer, W. Holzer and V. Pace, Org. Lett., 2020, 22, 1345–1349 CrossRef CAS PubMed.
- Cyclic voltammetry experiments of CH2FI and CH3I are described in the ESI.†.
- S. M. Bonesi and R. Erra-Balsells, J. Chem. Soc., Perkin Trans. 2, 2000, 2, 1583–1595 RSC.
- Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, 2002 Search PubMed.
- C. Chatgilialoglu, C. Ferreri, Y. Landais and V. I. Timokhin, Chem. Rev., 2018, 118, 6516–6572 CrossRef CAS PubMed.
- J. J. Devery, J. D. Nguyen, C. Dai and C. R. J. Stephenson, ACS Catal., 2016, 6, 5962–5967 CrossRef CAS.
- P. Zhang, C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087 CrossRef CAS PubMed.
- R. Kumar, N. J. Flodén, W. G. Whitehurst and M. J. Gaunt, Nature, 2020, 581, 415–420 CrossRef CAS PubMed.
- S. M. Hell, C. F. Meyer, A. Misale, J. B. I. Sap, K. E. Christensen, M. C. Willis, A. A. Trabanco and V. Gouverneur, Angew. Chem., Int. Ed., 2020, 59, 11620–11626 CrossRef CAS PubMed.
- S. M. Hell, C. F. Meyer, G. Laudadio, A. Misale, M. C. Willis, T. Noël, A. A. Trabanco and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 720–725 CrossRef CAS PubMed.
- C. Chatgilialoglu and J. Lalevée, Molecules, 2012, 17, 527–555 CrossRef CAS PubMed.
- Initiation leading to alkyl radical from alkyl iodide via light mediated C–I bond homolyses is documented. For a recent example, see: Y. Cheng, C. Mück-Lichtenfeld and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 16832–16836 CrossRef CAS PubMed.
- Current methods available to access γ-fluoro carbonyl derivatives such as 2a are multistep sequences starting with γ-hydroxybutyric acid (or equivalent), a controlled substance with neurotoxic properties.
- For further details, see ESI.†.
- S. Kaur and S. Gupta, Global Journal of Pharmacy & Pharmaceutical Science, 2017, 1, 555566 Search PubMed.
- A. Skreminskiy, D. M. Volochnyuk and O. Oleksandr, Eur. J. Org. Chem., 2021, 2021, 87–95 CrossRef.
- L. M. Lima, M. A. Alves and D. N. Do Amaral, Curr. Top. Med. Chem., 2019, 19, 1734–1750 CrossRef CAS PubMed.
- L. M. Kreis, S. Krautwald, N. Pfeiffer, R. E. Martin and E. M. Carreira, Org. Lett., 2013, 15, 1634–1637 CrossRef CAS PubMed.
- B. He, Q. Pan, Y. Guo, Q. Y. Chen and C. Liu, Org. Lett., 2020, 22, 6552–6556 CrossRef CAS PubMed.
- L. Trump, A. Lemos, B. Lallemand, P. Pasau, J. Mercier, C. Lemaire, A. Luxen and C. Genicot, Angew. Chem., Int. Ed., 2019, 58, 13149–13154 CrossRef CAS PubMed.
- C. W. Kee, O. Tack, F. Guibbal, T. C. Wilson, P. G. Isenegger, M. Imiołek, S. Verhoog, M. Tilby, G. Boscutti, S. Ashworth, J. Chupin, R. Kashani, A. W. J. Poh, J. K. Sosabowski, S. Macholl, C. Plisson, B. Cornelissen, M. C. Willis, J. Passchier, B. G. Davis and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 1180–1185 CrossRef CAS PubMed.
- H. Doi, M. Goto and M. Suzuki, Bull. Chem. Soc. Jpn., 2012, 85, 1233–1238 CrossRef CAS.
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed.
- S. L. Pimlott and A. Sutherland, Chem. Soc. Rev., 2011, 40, 149–162 RSC; G. C. Van de Bittner, E. L. Ricq and J. M. A. Hooker, Acc. Chem. Res., 2014, 47, 3127–3134 CrossRef CAS PubMed.
- M. S. Placzek, W. Zhao, H. Y. Wey, T. M. Morin and J. M. Hooker, Semin. Nucl. Med., 2016, 46, 20–27 CrossRef PubMed.
- M. G. Campbell, J. Mercier, C. Genicot, V. Gouverneur, J. M. Hooker and T. Ritter, Nat. Chem., 2017, 9, 1–3 CrossRef CAS PubMed.
- D. Van der Born, A. Pees, A. J. Poot, R. V. A. Orru, A. D. Windhorst and D. J. Vugts, Chem. Soc. Rev., 2017, 46, 4709–4773 RSC.
- L. Zheng and M. S. Berridge, Appl. Radiat. Isot., 2000, 52, 55–61 CrossRef CAS.
- J. Bergman, O. Eskola, P. Lehikoinen and O. Solin, Appl. Radiat. Isot., 2001, 54, 927–933 CrossRef CAS PubMed; M. R. Zhang, M. Ogawa, K. Furutsuka, Y. Yoshida and K. Suzuki, J. Fluorine Chem., 2004, 125, 1879–1886 CrossRef.
- M. R. Zhang, J. Maeda, M. Ogawa, J. Noguchi, T. Ito, Y. Yoshida, T. Okauchi, S. Obayashi, T. Suhara and K. Suzuki, J. Med. Chem., 2004, 47, 2228–2235 CrossRef CAS PubMed.
- F. T. Chin, C. L. Morse, H. U. Shetty and V. W. Pike, J. Labelled Compd. Radiopharm., 2006, 49, 17–31 CrossRef CAS.
- M. R. Zhang, J. Maeda, K. Furutsuka, Y. Yoshida, M. Ogawa, T. Suhara and K. Suzuki, Bioorg. Med. Chem. Lett., 2003, 13, 201–204 CrossRef CAS PubMed.
- J. Zessin, O. Eskola, P. Brust, J. Bergman, J. Steinbach, P. Lehikoinen, O. Solin and B. Johannsen, Nucl. Med. Biol., 2001, 28, 857–863 CrossRef CAS PubMed.
- S. Bailleux, P. Dréan, Z. Zelinger, S. Civiš, H. Ozeki and S. Saito, J. Chem. Phys., 2005, 122, 134302–134306 CrossRef PubMed.
- A. M. Del Hoyo and M. G. Suero, Eur. J. Org. Chem., 2017, 15, 2122–2125 CrossRef.
- A. M. Del Hoyo, A. G. Herraiz and M. G. Suero, Angew. Chem., Int. Ed., 2017, 56, 1610–1613 CrossRef CAS PubMed.
- J. Dong, X. Wang, Z. Wang, H. Song, Y. Liu and Q. Wang, Chem. Commun., 2019, 55, 11707–11710 RSC.
- Q. Zhu and D. G. Nocera, J. Am. Chem. Soc., 2020, 142, 17913–17918 CrossRef CAS PubMed.
- F. Chen, X. H. Xu and F. L. Qing, Org. Lett., 2021, 23, 2364–2369 CrossRef CAS PubMed.
- L. Hu, X. Liu and X. Liao, Angew. Chem., Int. Ed., 2016, 55, 9743–9747 CrossRef CAS PubMed.
- X. Bao, T. Yokoe, T. M. Ha, Q. Wang and J. Zhu, Nat. Commun., 2018, 9, 1–8 CrossRef PubMed.
- A. Hu, J. J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed.
- G. Laudadio, Y. Deng, K. Wal, W. Van Der, D. Ravelli, M. Nunõ, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92–96 CrossRef CAS PubMed.
- S. K. Kariofillis, B. J. Shields, M. A. Tekle-Smith, M. J. Zacuto and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 7683–7689 CrossRef CAS PubMed.
- R. W. Pipal, K. T. Stout, P. Z. Musacchio, S. Ren, T. J. A. Graham, S. Verhoog, L. Gantert, T. G. Lohith, A. Schmitz, H. S. Lee, D. Hesk, E. D. Hostetler, I. W. Davies and D. W. C. MacMillan, Nature, 2021, 589, 542–554 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744–7765 CrossRef CAS PubMed.
- K. Kloiber, R. Spitzer, M. Tollinger, R. Konrat and C. Kreutz, Nucleic Acids Res., 2011, 39, 4340–4351 CrossRef CAS PubMed.
- G. Nestor, T. Anderson, S. Oscarson and A. M. Gronenborn, J. Am. Chem. Soc., 2017, 139, 6210–6216 CrossRef CAS PubMed.
- F. A. Gallagher, R. Woitek, M. A. McLean, A. B. Gill, R. M. Garcia, E. Provenzano, F. Riemer, J. Kaggie, A. Chhabra, S. Ursprung, J. T. Grist, C. J. Daniels, F. Zaccagna, M. C. Laurent, M. Locke, S. Hilborne, A. Frary, T. Torheim, C. Boursnell, A. Schiller, I. Patterson, R. Slough, B. Carmo, J. Kane, H. Biggs, E. Harrison, S. S. Deen, A. Patterson, T. Lanz, Z. Kingsbury, M. Ross, B. Basu, R. Baird, D. J. Lomas, E. Sala, J. Wason, O. M. Rueda, S. F. Chin, I. B. Wilkinson, M. J. Graves, J. E. Abraham, F. J. Gilbert, C. Caldas and K. M. Brindle, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2092–2098 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03421a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |