
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlbumin-targeting of an oxaliplatin-releasing platinum(IV) prodrug results in pronounced anticancer activity due to endocytotic drug uptake in vivo†
Hemma
Schueffl‡
 a,
Sarah
Theiner‡
b,
Gerrit
Hermann
b,
Josef
Mayr
c,
Philipp
Fronik
c,
Diana
Groza
a,
Sushilla
van Schonhooven
a,
Luis
Galvez
b,
Nadine S.
Sommerfeld
c,
Arno
Schintlmeister
d,
Siegfried
Reipert
e,
Michael
Wagner
d,
Robert M.
Mader
f,
Gunda
Koellensperger
b,
Bernhard K.
Keppler
cg,
Walter
Berger
af,
Christian R.
Kowol
*cf,
Anton
Legin
*c and
Petra
Heffeter
*af
a,
Sarah
Theiner‡
b,
Gerrit
Hermann
b,
Josef
Mayr
c,
Philipp
Fronik
c,
Diana
Groza
a,
Sushilla
van Schonhooven
a,
Luis
Galvez
b,
Nadine S.
Sommerfeld
c,
Arno
Schintlmeister
d,
Siegfried
Reipert
e,
Michael
Wagner
d,
Robert M.
Mader
f,
Gunda
Koellensperger
b,
Bernhard K.
Keppler
cg,
Walter
Berger
af,
Christian R.
Kowol
*cf,
Anton
Legin
*c and
Petra
Heffeter
*af
aInstitute of Cancer Research and Comprehensive Cancer Center, Medical University of Vienna, Borschkegasse 8a, A-1090 Vienna, Austria. E-mail: petra.heffeter@meduniwien.ac.at; Fax: +43-1-40160-957555; Tel: +43-1-40160-57594
bInstitute of Analytical Chemistry, Faculty of Chemistry, University of Vienna, Waehringer Str. 38, A-1090 Vienna, Austria
cInstitute of Inorganic Chemistry, Faculty of Chemistry, University of Vienna, Waehringer Str. 42, A-1090 Vienna, Austria. E-mail: anton.legin@univie.ac.at; christian.kowol@univie.ac.at; Fax: +43-1-4277-852601; Fax: +43-1-4277-9526; Tel: +43-1-4277-52610 Tel: +43-1-4277-52611
dCentre for Microbiology and Environmental Systems Science, Division of Microbial Ecology and Large-Instrument Facility for Environmental and Isotope Mass Spectrometry, University of Vienna, Djerassiplatz 1, A-1030 Vienna, Austria
eCore Facility Cell Imaging and Ultrastructure Research, University of Vienna, University Biology Building (UBB), Djerassiplatz 1, A-1030 Vienna, Austria
fDepartment of Medicine I and Comprehensive Cancer Center, Medical University of Vienna, Waehringer Guertel 18-20, 1090 Vienna, Austria
gResearch Cluster “Translational Cancer Therapy Research”, University of Vienna, Medical University of Vienna, Vienna, Austria
First published on 26th August 2021
Abstract
Oxaliplatin is a very potent platinum(II) drug which is frequently used in poly-chemotherapy schemes against advanced colorectal cancer. However, its benefit is limited by severe adverse effects as well as resistance development. Based on their higher tolerability, platinum(IV) prodrugs came into focus of interest. However, comparable to their platinum(II) counterparts they lack tumor specificity and are frequently prematurely activated in the blood circulation. With the aim to exploit the enhanced albumin consumption and accumulation in the malignant tissue, we have recently developed a new albumin-targeted prodrug, which supposed to release oxaliplatin in a highly tumor-specific manner. In more detail, we designed a platinum(IV) complex containing two maleimide moieties in the axial position (KP2156), which allows selective binding to the cysteine 34. In the present study, diverse cell biological and analytical tools such as laser ablation inductively-coupled plasma mass spectrometry (LA-ICP-MS), isotope labeling, and nano-scale secondary ion mass spectrometry (NanoSIMS) were employed to better understand the in vivo distribution and activation process of KP2156 (in comparison to free oxaliplatin and a non-albumin-binding succinimide analogue). KP2156 forms very stable albumin adducts in the bloodstream resulting in a superior pharmacological profile, such as distinctly prolonged terminal excretion half-life and enhanced effective platinum dose (measured by ICP-MS). The albumin-bound drug is accumulating in the malignant tissue, where it enters the cancer cells via clathrin- and caveolin-dependent endocytosis, and is activated by reduction to release oxaliplatin. This results in profound, long-lasting anticancer activity of KP2156 against CT26 colon cancer tumors in vivo based on cell cycle arrest and apoptotic cell death. Summarizing, albumin-binding of platinum(IV) complexes potently enhances the efficacy of oxaliplatin therapy and should be further developed towards clinical phase I trials.
Introduction
Colorectal cancer is the third most diagnosed malignancy and fourth leading cause of cancer-related deaths worldwide.1 A standard treatment against this type of cancer, especially in cases of advanced stages, is oxaliplatin combined in diverse treatment schemes (e.g. together with folinic acid and fluorouracil; FOLFOX).1 As this therapy is usually accompanied by (severe) side effects, there is significant interest in the development of new strategies to improve tolerability and efficacy of platinum-based anticancer therapy.2,3 During the last decades, platinum(IV) complexes came into focus of interest, as they are kinetically more inert than their platinum(II) counterparts and have a lower reactivity towards biomolecules.2,4,5 Noteworthy, these complexes are considered as prodrugs, which are activated by reduction.4–6 During this process the axial ligands are released and the corresponding cytotoxic square-planar platinum(II) analogues are formed. The best investigated platinum(IV) drug is satraplatin, which was tested in a clinical phase III trial against metastatic prostate cancer.2,4–6 However, despite significant impact on progression-free survival, the drug did not meet its primary endpoint of overall survival and, thus, failed regulatory approval.7,8 Noteworthy, there are indications that satraplatin lacks sufficient tumor specificity due to premature reduction/activation of the drug (e.g. in the red blood cells).9,10 Consequently, it is of interest to develop new platinum(IV) drugs with superior tumor-targeting properties. One promising strategy is the use of albumin as a drug carrier.11 Albumin is the most abundant plasma protein (35–50 g L−1 in human plasma) with an average (plasma) half-life of 19 days.12,13 The multiple physiological functions of albumin also include transport of several nutrients (e.g. fatty acids).11,14 Cancer cells are often characterized by enhanced endocytic uptake of this protein as an additional source of nutrition via different pathways (macropinocytosis, clathrin-mediated or caveolin-mediated).11,15 Moreover, due to the combination of leaky blood capillaries with the absence/defect of lymphatic drainage, albumin accumulates in malignant tissue, a phenomenon also called enhanced permeability and retention (EPR) effect.11,16 Albumin targeting has already been successfully employed for several tumor-targeting strategies. The most prominent clinical examples are abraxane, an albumin paclitaxel-containing nanoparticle, with clinical approval for several cancer types (including lung, pancreas and breast cancer) and the maleimide-containing doxorubicin derivative aldoxorubicin, which has finished a phase III trial in patients with soft tissue sarcoma.11,16,17With the aim to overcome the drawbacks of oxaliplatin therapy, we have recently developed and preliminarily characterized the first maleimide-functionalized oxaliplatin(IV) prodrug KP2156, which is able to bind to Cys34 of albumin.18 Noteworthy, there is surprisingly little literature available on the in vivo behavior of platinum(IV) drugs in general (e.g. concerning the exact location and time point of prodrug activation) and even less is known about tumor-targeted derivatives. Consequently, the present study focused on the in-depth characterization of our new prodrug system in cell culture and especially in vivo using mouse models. To this end, the fate and effects of the platinum(IV) drug after intravenous application into tumor-bearing mice was followed by several methods including cutting-edge analytical tools such as laser ablation-inductively coupled plasma-time of flight mass spectrometry (LA-ICP-TOF-MS) and detection of isotope-labeled derivatives by nano-scale secondary ion mass spectrometry (NanoSIMS). We show that the superior anticancer activity of KP2156 in vivo is based on a distinctly altered pharmacokinetic profile. This not only results in prolonged plasma half-life but also effective tumor accumulation and retention of the albumin-bound prodrug in the tumor tissue (10-fold higher than oxaliplatin), where it is engulfed (and intracellularly activated) by the cancer cells via endocytosis. These data support the hypothesis that albumin binding is a potent tool to increase the tumor specificity of platinum(IV) drugs and prevent their premature activation in other compartments of the body.
Results and discussion
Characterization of albumin homeostasis of CT26 cells in cell culture and in vivo
As oxaliplatin is primarily used in the treatment of colorectal cancer, this study focused on this type of malignancy. Noteworthy, the anticancer activity of oxaliplatin is strongly dependent on an intact immune system and the drug is therefore barely active in immunodeficient mice.19–21 Consequently, the murine colon cancer model CT26 was chosen, as it allows allograft experiments in an immune-competent host. As a first step, the albumin homeostasis was characterized in this cell model in cell culture using fluorescein isothiocyanate (FITC) labeling. Flow cytometry and confocal fluorescence microscopy experiments revealed that the cellular uptake in CT26 cells is mediated via clathrin- and caveolin-dependent endocytosis (but not macropinocytosis) (Fig. 1A–C). In detail, uptake of FITC-labeled albumin was significantly reduced by methyl-β-cyclodextrin (MßCD), a compound that inhibits caveolin-dependent endocytosis by removing cholesterol from the plasma membrane,22 as well as by chlorpromazine (CHP), an inhibitor of the clathrin-dependent endocytosis by translocating clathrin from the cell surface to intracellular endosomes.23 In contrast, the macropinocytosis inhibitor 5-(N-ethyl-N-isopropyl)amiloride (EIPA) had no impact on albumin uptake. All inhibitors were applied at concentrations, which did not have an impact on cell viability (Fig. S1†). The endocytic (vesicular) albumin uptake was also indicated by the scattered distribution of the FITC-labeled albumin in the cells (Fig. 1C and S2†).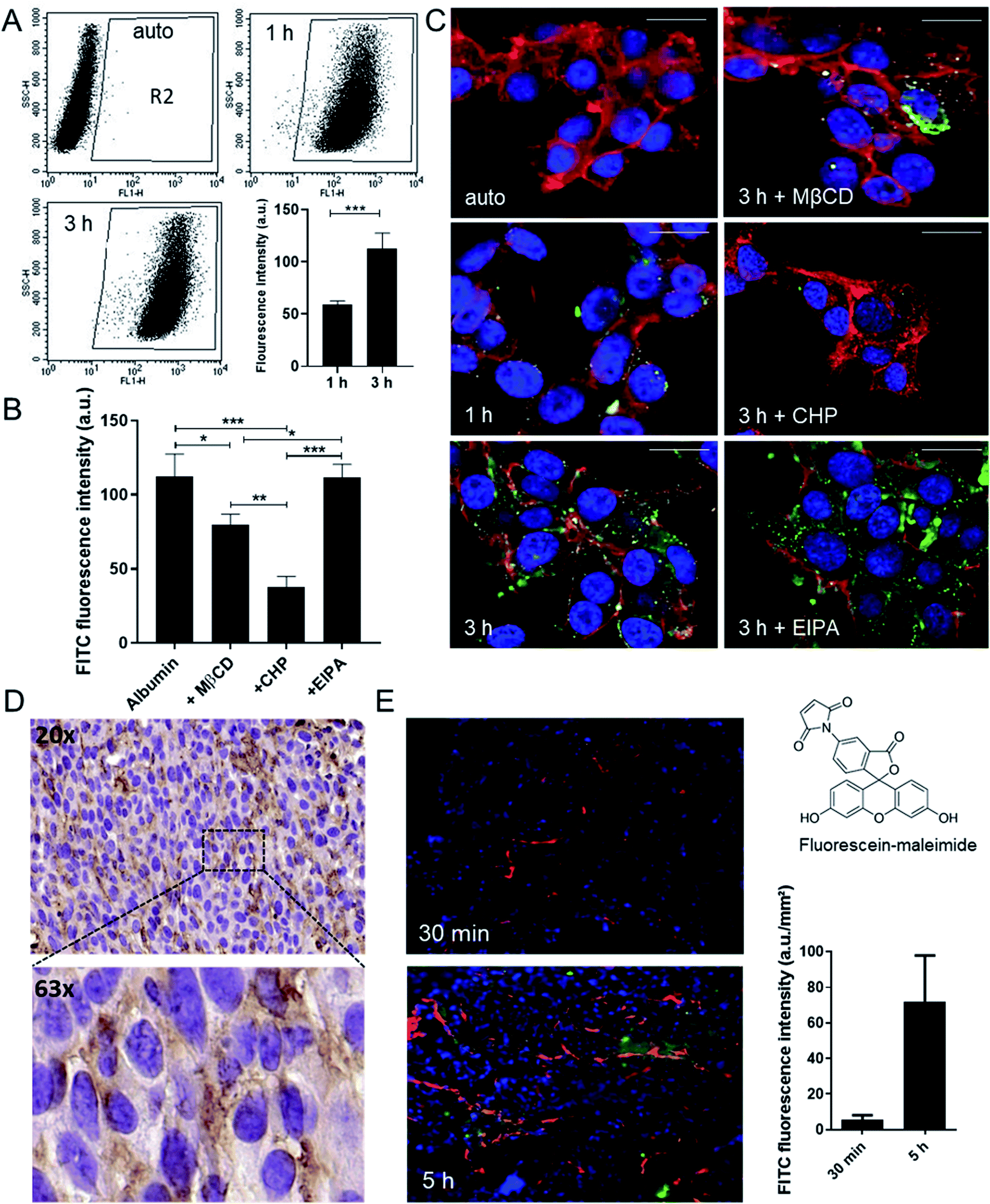 | ||
| Fig. 1 Characterization of albumin uptake in CT26 cells. (A) Cells were incubated with FITC-labeled albumin. FITC-positive cells (scatter plot, R2) and mean FITC fluorescence intensity (bar plot) were determined by flow cytometry (ex/em: 488/530 nm, fluorescence intensity was normalized to auto fluorescence control). (B) Impact of endocytosis inhibitors MβCD, CHP and EIPA (1 h pretreatment) on uptake of FITC-labeled albumin after 3 h was determined by flow cytometry. Values in (A) and (B) are means ± SD of three independent experiments. Statistical significance was tested by one-way ANOVA and Dunnett's multiple comparison test (*p < 0.05, **p < 0.01 and ***p < 0.001). (C) Uptake of FITC-labeled albumin (green) and the impact of the three endocytosis inhibitors was verified by confocal microscopy. Nuclei (blue) and membranes (red) were co-stained by DAPI and WGA, respectively. The images show an overlay of all three channels. (D) Immunohistochemical analysis of albumin content in s.c. CT26 tumors from untreated mice (microscopies with 20× and 63× objective). Nuclei and albumin were visualized by hematoxylin (violet) and 3,3′-diaminobenzidine (brown), respectively. (E) CT26-bearing mice were treated with 16.5 mg kg−1 fluorescein-labeled maleimide (green). After 30 min and 5 h the tumors were harvested followed by immunofluorescence staining of the nuclei (DAPI, blue) and the blood vessels (endomucine, red). Evaluation was done by fluorescence microscopy using a 40× objective. The images show an overlay of all three channels. Fluorescein fluorescence intensity per mm2 was calculated using Definiens software (bar plot in left panel). Values of fluorescence intensity are given as mean ± SD of two different tumor samples. | ||
To visualize accumulation of albumin in the tumor tissue in vivo, two approaches were followed: (1) albumin distribution was investigated immunohistochemically in formalin-fixed and paraffin-embedded tumor samples (Fig. 1D) and (2) fluorescein-labeled maleimide was injected to CT26 tumor-bearing mice to follow the kinetics of albumin tissue accumulation (Fig. 1E and S3†). These experiments revealed the tissue entry sites for the albumin–maleimide–fluorescein conjugate in a close proximity to the blood vessels already 30 min after the application, followed by deeper diffusion into the tumor tissue after 5 h. In line with the hypothesis of albumin-assisted accumulation of drugs into the tumor, the fluorescence signal in the malignant tissue increased over time (Fig. 1E). Similar results were obtained when applying the ex vivo pre-conjugated FITC-labelled albumin used in the cell culture experiments above (data not shown).
Albumin binding of KP2156 leads to cellular uptake via clathrin- and caveolin-dependent endocytosis
Previously, we have shown by UV-vis spectrometry that incubation of KP2156 with albumin leads to an efficient and rapid covalent binding.18 In contrast, the corresponding succinimide drug KP2157 remains unbound (for chemical structures see Fig. 2A). This could be also confirmed by size exclusion ICP-MS (SEC-ICP-MS) measurements in calf serum, where KP2156 was detected in the albumin-fraction, while KP2157 was exclusively in the low molecular weight range.18 In the current study, the impact of albumin binding on the drug uptake of cancer cells was investigated in cell culture using ICP-MS analysis. To ensure maximal albumin binding, KP2156 and, as references, KP2157 and oxaliplatin were pre-incubated in fetal calf serum (FCS) for 2 h and subsequently added to cell culture media. Noteworthy, although the used stoichiometric amounts of albumin in FCS should have ensured complete binding of KP2156 (10 μM), SEC-ICP-MS analysis indicated that only ∼60% was bound to albumin and albumin dimers (Fig. S4†). This can be explained by the naturally occurring fraction of oxidized albumin at position Cys34, which decreases the amount of accessible Cys34-SH groups for reaction with the maleimide moieties.24 The results showed that oxaliplatin was rapidly entering the cancer cells (Fig. S5†). In accordance to the literature on other platinum(IV) prodrugs,25,26 the cellular KP2157 uptake was very low (about 100-fold less than observed for oxaliplatin, Fig. 2B). This difference can be explained by a generally reduced recognition of platinum(IV) prodrugs by active import proteins (such as the organic anion transporters and the copper transporters), which are responsible for the active uptake of oxaliplatin into cancer cells.25,26 In contrast, (albumin-bound) KP2156 was rapidly transported into the cells (Fig. 2B), and resulted in 10-fold higher platinum levels than for KP2157. Subsequent inhibitor experiments showed that this effect is based on differences in the cellular drug uptake, as similar to the results obtained with FITC-labelled albumin, KP2156 uptake was mediated via clathrin- and caveolin-dependent endocytosis (but not via macropinocytosis, Fig. 2C). In contrast, neither accumulation of oxaliplatin nor KP2157 were affected by endocytosis inhibition, indicating that these processes do not play a key role in their cellular accumulation (Fig. 2C). Next, the anticancer activity of the compounds was determined after different drug exposure periods. Despite their rather rapid uptake within a few hours, the two platinum(IV) complexes obtained IC50 values of only ∼50 μM after 72 h, which is about 16-fold higher than that of oxaliplatin (∼3.5 μM). This reduced activity is in good agreement with the proposed prodrug nature of the new drugs and in line with other publications on stable oxaliplatin(IV) complexes.27,28 To analyze whether a prolonged incubation time (up to 14 days) leads to activation of the prodrugs, long-term colony formation assays with drug concentrations up to 10 μM were performed. Indeed, prolonged incubation times resulted in an at least partial alignment of the efficacy between (albumin-bound) KP2156 and oxaliplatin. At all time points, KP2156 was superior to KP2157 (Fig. 2E).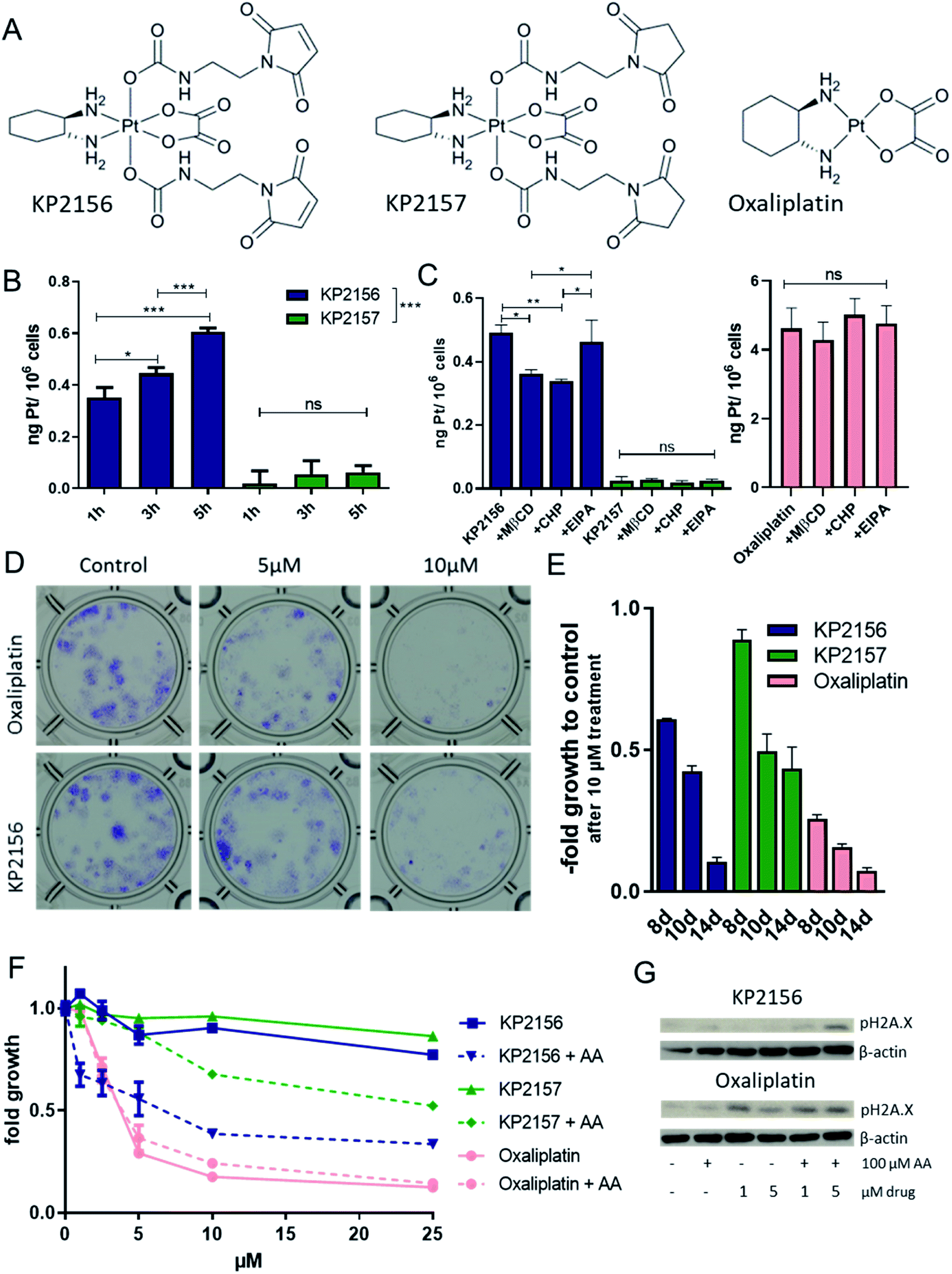 | ||
| Fig. 2 Comparison of cellular uptake and cytotoxicity of KP2156, KP2157 and oxaliplatin in cell culture. (A) Structural formulae of the used drugs. Cellular uptake of 10 μM KP2156 and KP2157 after (B) 1 h, 3 h and 5 h as well as (C) impact of the endocytosis inhibitors MβCD, CHP and EIPA (1 h pretreatment) on uptake of the drugs after 3 h in CT26 cells was determined by ICP-MS. Values in (B) and (C) are presented as means ± SD of three independent experiments. Statistical significance was tested by one-way ANOVA and Dunnett's multiple comparison Test (*p < 0.05, **p < 0.01 and ***p < 0.001). (D) Clonogenic survival after treatment with the indicated drugs after 14 days of incubation. Cell colonies were visualized by crystal violet staining. The shown figure is a representative of three independently performed experiments. (E) Cytotoxicity of the test drugs (10 μM) after 8 d, 10 d and 14 d. Crystal violet staining intensity was evaluated by fluorescence measurement. Values are given as means ± SD of one representative experiment performed in duplicates. (F) The impact of co-treatment of 100 μM ascorbic acid (AA) with the indicated drugs on the cell viability (after 72 h) as well as on (G) the induction of pH2A.X (DNA double strand break marker) after 24 h were determined via a MTT-based assay and Western blot analysis, respectively. In case of (F), values refer to means ± SD of one representative experiment performed in triplicates. | ||
As a next step, we investigated whether addition of the reducing agent ascorbic acid (AA) was able to activate the platinum(IV) prodrugs. Under cell-free conditions, 100 μM KP2157 was slowly reduced in combination with AA (10 eq.), as observed by NMR measurements over 72 h (Fig. S6; † <50% reduction after 72 h). In comparison, Satraplatin was completely reduced already after <24 h under the same conditions. For this experiments KP2157 had been used, as it is well-known that maleimides tend to slowly hydrolyze at pH 7.4 in unbound form.29 Subsequent cell culture experiments indicated that this reduction is also occurring with KP2156. In more detail, while AA had no relevant impact on the effectiveness of oxaliplatin, significant enhancement of activity was observed in case of both prodrugs (Fig. 2F). This resulted also in enhanced DNA damage of the treated cancer cells (indicated by the DNA damage marker pH2X.A, Fig. 2G). Taken together, these experiments indicate that the albumin binding of KP2156 leads to an enhanced cellular drug uptake via endocytosis followed by slow intracellular activation/reduction of the prodrug.
KP2156 exhibits strong anticancer activity in vivo based on enhanced drug accumulation in the tumor, reduced cell proliferation and induction of apoptotic cell death
We have previously shown that KP2156 has promising anticancer activity against CT26 tumors in vivo in terms of the endpoint “tumor growth”.18 To gain more insights into its efficiency and mode of action, this study compared KP2156 with oxaliplatin and KP2157 in more detail. Therefore, two experiments were performed: one with the endpoint “overall survival” in order to investigate the long-term efficiency of the drugs. In the second experiment, tissues were collected on day 12 (24 h after the last therapy) for analysis of drug uptake as well as histological and cell biological changes. All compounds were well tolerated, with a minor loss in body weight (<10%) of the KP2156-treated animals (Fig. S7A†). In agreement with previous data,18,30 KP2156 and oxaliplatin showed significant activity against the CT26 tumors (Fig. 3A, and S7†). Noteworthy, towards the end of the experiment, at around day 15, the oxaliplatin-treated tumors distinctly progressed again indicating the escape of the tumor cells from therapy. In case of KP2157, in contrast to previous reports,18 in this study no significant impact on tumor growth was observed (Fig. S7B†). Most strikingly, KP2156 not only efficiently stopped tumor growth, but induced (complete) remissions in several animals (Fig. 3A and S8†). This not only resulted in a distinctly prolonged overall survival of all KP2156-treated animals from the “survival experiment”, but also in the cure of one animal (Fig. 3B). Importantly, this mouse was in turn also able to reject tumor cells upon re-challenge with living CT26 cells (injected into the other flank of the animal) indicating the development of a drug-induced antitumor memory/vaccination effect (data not shown).20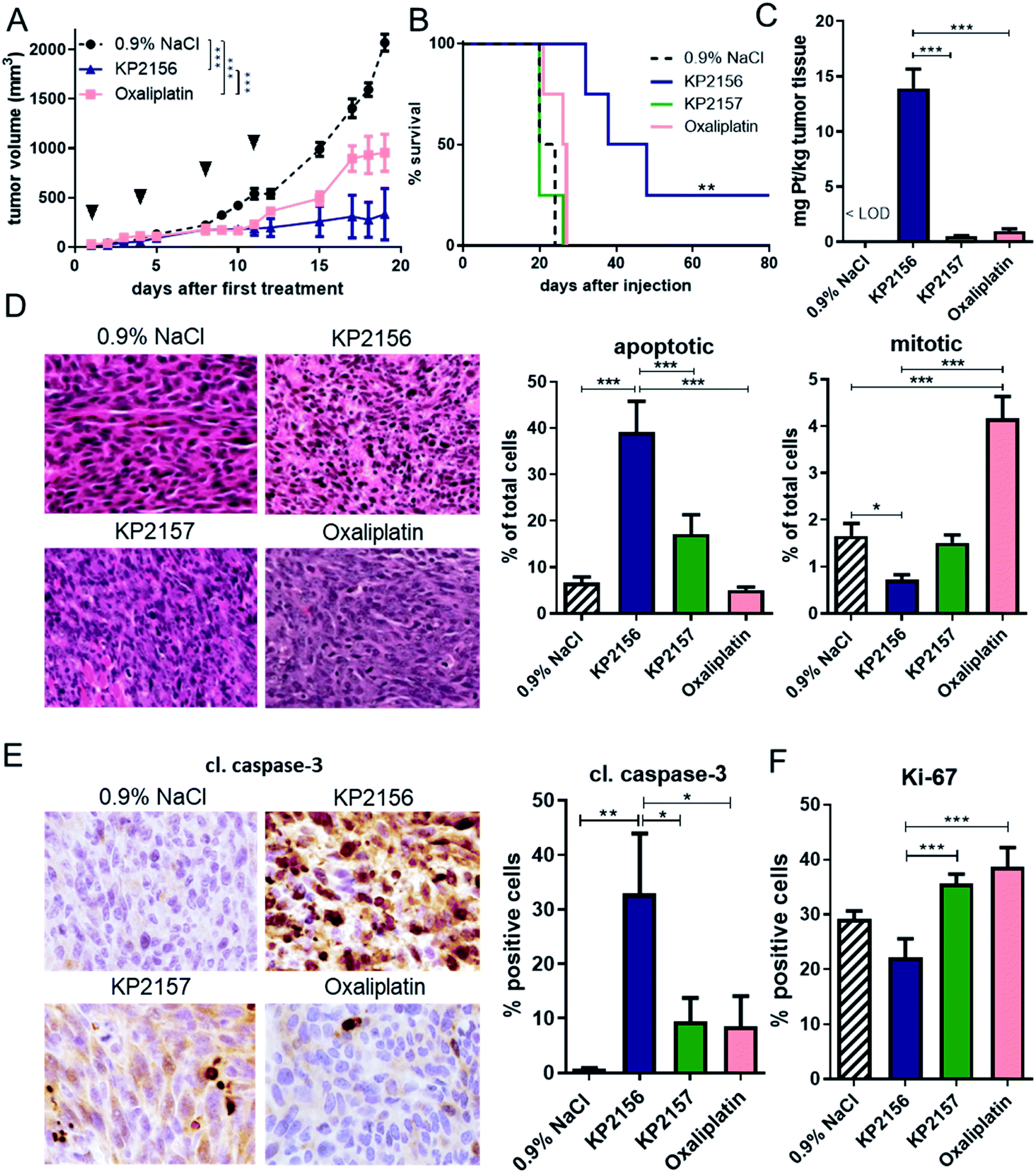 | ||
| Fig. 3 Anticancer activity of KP2156, KP2157 and oxaliplatin in vivo. CT26-bearing BALB/c mice were treated twice a week for two weeks i.v. with concentrations equimolar to 9 mg kg−1 oxaliplatin. (A) Impact on tumor growth; data are presented as means ± SEM. Statistical significance was tested by two-way ANOVA (***p < 0.001). (B) The overall survival is depicted via a Kaplan–Meier curve. Statistical significance was tested by log-rank test and Mantel–Cox posttest (**p < 0.01). In a second experiment tissues were collected on day 12 (24 h after the last therapy). The collected tissue was analyzed (C) for its platinum content by ICP-MS and (D–F) for diverse histological parameters: (D) amount of apoptotic and mitotic cells in H&E-stained samples (analyzed by counting), as well as (E) % of cl. caspase-3-positive (apoptosis marker) and (F) % of Ki-67-positive (proliferation marker) cells (analyzed by Definiens software). Values displayed in (C) and (D–F) refer to means ± SD and ± SEM, respectively. Statistical significance was tested by one-way ANOVA and Dunnett's multiple comparison test (*p < 0.05, **p < 0.01 and ***p < 0.001). | ||
In order to better understand the observed differences in activity, the tumor samples collected from the second experiment at day 12 were analyzed by ICP-MS for their platinum content (Fig. 3C). KP2156 treatment resulted in more than 14-fold higher platinum levels in the malignant tissue than oxaliplatin therapy (both compounds were applied at equimolar concentrations in the same therapy scheme). Relative to KP2157, which is unable to covalently bind to albumin in the bloodstream, the platinum levels of KP2156 were 28-fold higher. Subsequent histological analysis of the tumor tissues (Fig. 3D–F) showed that the pronounced anticancer effects of KP2156 were not only based on reduction of cell proliferation (about 50% reduction of the mitotic cell fraction as well as a tendency for a decreased expression of the proliferation marker Ki-67), but also on strong induction of apoptotic cell death (∼40% of cells with apoptotic morphology, 30% cells positive for caspase-3 cleavage). In contrast, tumors of oxaliplatin-treated animals were characterized by significant increase of mitotic cells, which could either indicate cell cycle arrest in M phase or enhanced cell proliferation due to the observed regrowth of the tumors (compare Fig. 3A). Overall, these experiments show that KP2156 has not only superior activity in comparison to oxaliplatin, but that KP2156 seems to have distinct different pharmacological properties than the respective non-targeted compounds.
Albumin binding of KP2156 leads to a distinctly prolonged terminal elimination half-life, prevention of premature drug release, and enhanced drug accumulation in the tumor
To gain more insights into the pharmacokinetics of the new albumin-targeted compound, Balb/c mice were treated once with an equimolar dose of the three test drugs and blood was collected after different time points via the facial vein (Fig. 4A). Previous spiking experiments already demonstrated rapid albumin-specific binding of KP2156 in FCS ex vivo.18 In good agreement with these data, and the SEC-ICP-MS measurements of the serum samples isolated after 30 min, all platinum in the KP2156-treated mice was detected in the albumin-containing fractions (Fig. S9†). Noteworthy, no free drug (unbound or bound to low-molecular weight plasma components) was found at any time point. Pharmacokinetic analyses revealed that KP2156 is retained in the plasma much longer in comparison to KP2157 and oxaliplatin (Table S1†).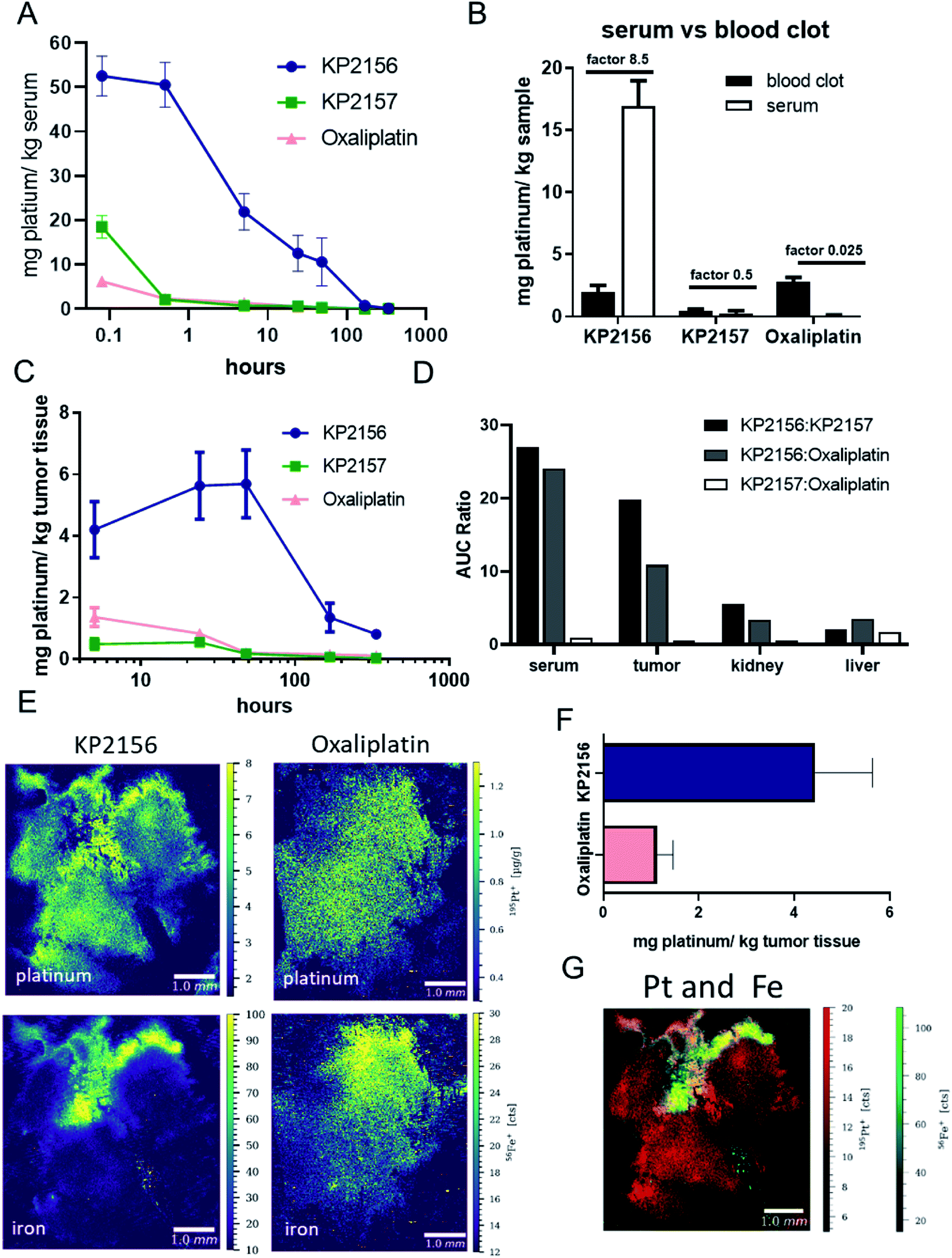 | ||
| Fig. 4 Pharmacological evaluation and intratumoral platinum distribution of the test drugs in single-dosed Balb/c mice. Animals (naive for serum pharmacokinetics, CT26-bearing for organ distribution) were treated once i.v. with concentrations equimolar to 9 mg kg−1 oxaliplatin (n = 4 in each treatment group). Serum and tissue samples were collected at different time points between 0.08 h and 336 h as well as 5 h and 336 h, respectively. The average platinum concentration in the different tissues was determined by ICP-MS. (A) Concentration–time curves of the indicated drugs in the serum of the animals. (B) Comparison of the platinum content in serum and the cellular blood fraction (blood clot) after 24 h. (C) Concentration–time curves of the drugs in the tumor. (D) AUC ratio between the drugs in different tissues. Cryosections of KP2156 and oxaliplatin (5 h) treated CT26 tumors were analyzed for diverse elements by LA-ICP-MS. (E) LA-ICP-MS images of the platinum and iron distribution. (F) The average platinum content in tumor tissue determined by LA-ICP-MS. Values refer to means ± SEM. (G) Platinum (red) to iron (green) correlation pattern (image overlay). | ||
This can already be seen at the first sampling time point (5 min), where the measured concentrations of KP2157 and oxaliplatin were drastically lower than for KP2156 (despite equimolar application, Fig. 4A). Furthermore, as studies with satraplatin indicated premature platinum(IV) reduction in the red blood cells,9,10 the platinum ratios between serum and the cellular blood compartment was assessed for the three compounds 24 h after treatment (Fig. 4B). The albumin binding of KP2156 potently prevented the drug from entering the red blood cells. In contrast, KP2157 and, in particular, oxaliplatin levels were higher in the cellular blood compartment at this time point. Together with the lack of unbound KP2156 in the serum of the treated animals, this indicates a high stability of the KP2156-albumin adduct in the circulation.
For evaluation of the body-mass balance, in a second experiment CT26-bearing mice were treated once with KP2156, KP2157 or oxaliplatin and dissected at different time points for tumor, organs (liver and kidney). Urine was simultaneously collected. All samples were analyzed for their platinum content via ICP-MS (Fig. 4C and S10†). In general, when calculating the area under the concentration–time curve (AUC) as the most robust parameter, KP2156 differed significantly from the reference compounds KP2157 and oxaliplatin (Table S1†). This parameter can be directly compared as all the compounds were administered equimolarly. More specifically, exposure of the mice to KP2156 was approximately 25-times higher as assessed by serum AUC, in comparison to the reference compounds (Fig. 4D). This effect was observed over a period of at least 168 h after drug administration with significantly higher drug concentrations for KP2156 in the systemic circulation (Fig. 4A). Moreover, this higher exposure resulted in an increased uptake by the tumor, which was 10-times higher than that of oxaliplatin (20-fold higher than KP2157; Fig. 4C and D). This effect was caused by the different uptake kinetics of the platinum drugs into the tumor: KP2156 concentrations were not only higher 5 h after drug administration, but still showed increasing drug levels up to 48 h, when reaching the maximum tumor level (Fig. 4C). At that time, the two reference compounds were widely excreted from the serum. Due to the sustained high levels of KP2156 in the blood, highly perfused organs such as kidney and liver were also enriched with exposure comparable to the tumor (Fig. 4D, S10 and Table S1†). This compares favorably with KP2157 and oxaliplatin, where tumor exposure was consistently below that of other organs indicating poor distribution into the target compartment (Fig. 4D). The tissue kinetics for all three compounds, however, were similar in liver and kidney (Fig. S10†). This overall distribution pattern revealed highly significant increases in the total clearance for KP2157 and oxaliplatin, which were at least one order of magnitude higher than that of KP2156 (Table S1†). In contrast, the terminal elimination half-life of KP2156 was approximately twice that of the other compounds (KP2156: 55.7 h; KP2157: 25.7 h; oxaliplatin: 32.5 h). In summary, the circulation time of KP2156 was strongly increased compared to the other compounds and KP2156 highly accumulated in the tumor, where significant levels could be observed even more than 300 h after treatment in the malignant tissue. Thus, when KP2156 was still distributing into the target compartment (tumor tissue) after 48 h, both reference compounds had already been widely eliminated from the circulation of the animals.
In general, this pharmacological behavior is in good agreement with published data on aldoxorubicin, a doxorubicin prodrug, which also binds to albumin via a maleimide.31 Also aldoxorubicin is characterized by a prolonged elimination half-life. Free doxorubicin has an in vivo elimination half-life in the order of minutes with a very rapid systemic clearance. Thus, already 15 min after i.v. injection the concentration of doxorubicin in blood is less than 2% of the initial amount, albumin binding prolonged this to 2.3 h.32 Noteworthy, in contrast to our investigated drugs, the enhanced tumor accumulation of aldoxorubicin already reached a plateau after 2 h with no further increase during the next 24 h.32 This indicates that the prolonged plasma half-life of KP2156 (in comparison to aldoxorubicin) leads to an even more favorable tumor accumulation profile and that the platinum(IV) prodrug concept is potentially more robust against premature activation than the acid-sensitive linker of aldoxorubicin.
Albumin binding leads to efficient transport of platinum into the tumor tissue
Since the pharmacological data showed potent KP2156 accumulation in the malignant tissue, we then analyzed the impact of albumin binding on the intratumoral drug distribution. To this end, tumors were collected after single treatment with KP2156 or oxaliplatin, snap-frozen and analyzed by LA-ICP-TOF-MS for the distribution of platinum, iron, sulfur and phosphorus (Fig. 4E–G, S11†). KP2156 showed a strong platinum signal in iron-rich regions of the tumor together with a leakage of the compound into the surrounding malignant tissue. As hemoglobin of the red blood cells represent the main source for the iron signal in the tumor, these iron-rich areas are presumably in the surrounding of blood vessels. Similar to the average platinum concentration in the tumor tissue determined by ICP-MS, the platinum levels in the KP2156 tumors were about 4-fold higher than upon oxaliplatin treatment as determined by LA-ICP-MS analysis. This indicates that the albumin-bound platinum drug is transported into the tumor via the bloodstream, which is in good agreement with the distribution data of FITC-labeled albumin shown in Fig. 1E. Subsequently, the albumin binding leads to superior retention and accumulation of the drug in the malignant tissue. To our knowledge, KP2156 is the first albumin-binding drug, which has been investigated for its intratumoral distribution on a cellular level33,34 and correlated with other elements indicative for the specific tissue compartments (iron for blood vessels, phosphorus for DNA, sulfur for protein-rich areas). Due to its intrinsic fluorescence properties in case of aldoxorubicin34 increased tumor accumulation (compared to doxorubicin) could be analyzed by fluorescence measurements (which is in line with our data) in a bone metastasis tumor model, but no information on the distribution pattern within tissue compartments were presented. There are several LA-ICP-MS studies reporting on the platinum distribution of cisplatin, oxaliplatin and satraplatin in tumor tissue.35–37 For oxaliplatin, elevated platinum levels were mainly observed at the edge of tumor tissue and in areas with connective tissue,35–37 whereas in the present study, platinum was evenly distributed in the tumor tissue after oxaliplatin treatment. The platinum pattern correlated with the phosphorus and sulfur distribution. With regard to KP2156, the authors have previously investigated the platinum distribution in CT-26 tumors of mice after treatment for 24 h.33 Already in this LA-ICP-MS study, pronounced platinum accumulation was observed in certain tumor areas, which were hypothesized to be blood vessels or well-perfused compartments. The correlation with iron-rich areas in the tumor (Fig. 4G) shown by the multi-elemental analysis experiments together with the histological data on FITC-labelled albumin further supports this hypothesis.KP2156 enters cancer cells in vivo via the endo-/lysosomal pathway
As the final step, we aimed at characterization of the uptake of the platinum compound in the tumor on a (sub-)cellular level. LA-ICP-TOF-MS imaging provides information on the localization and concentration of a wide range of elements (including Pt, Fe, S, P etc.) in tumor tissue with a spatial resolution on the cellular scale. In contrast, NanoSIMS imaging allows addressing the sub-cellular distribution of elements and specific isotopes. NanoSIMS has been successfully employed to investigate the cellular fate of the metal center simultaneously to isotopically labeled ligands of different metal-based complexes in cell culture experiments.38–41 There are few NanoSIMS studies using this approach to investigate anticancer compounds also in in vivo settings.33,42 Recently, we succeeded in determining of the subcellular platinum distribution in tumors of KP2156-treated mice.33 In the current study, a dual stable isotope-labeled analog of KP2156 (KP2603) was synthesized (Fig. S12†) in order to better understand the in vivo distribution of the metal center with respect to the ligands. To this end, the maleimide moiety and the diaminocyclohexane (DACH) ligand were 13C- and 2H-labeled, respectively (Fig. 5A). In more detail, 13C7,15N-labelled maleimidopropionic acid was synthesized from β-alanine–13C3,15N and maleic anhydride-13C4, while the deuterium-labelled oxaliplatin was synthesized in six steps starting from D10-cyclohexene based on a protocol for similar compounds.43 After oxidation of oxaliplatin to the dihydroxido species and conversion of the maleimidopropionic acid into the isocyanate, the two compounds were coupled yielding KP2603. The complex was characterized by one- and two-dimensional NMR spectroscopy, high-resolution mass spectrometry, elemental analysis and HPLC (Fig. S13†). Subsequently, the labeled compound was injected to CT26-bearing Balb/c mice. The compound was applied four times in the same scheme as used in the “overall survival” endpoint study (compare Fig. 3A). The mice were sacrificed 24 h after the last application, because at this time point the tumor contains the highest level of platinum (compare Fig. 3C). The chemically fixed tumor tissue samples were embedded in epoxy resin and prepared for consecutive investigation by transmission electron microscopy (TEM) and NanoSIMS. Images from both methods were correlated to show the accumulation of the compound on the sub-cellular scale (Fig. 5).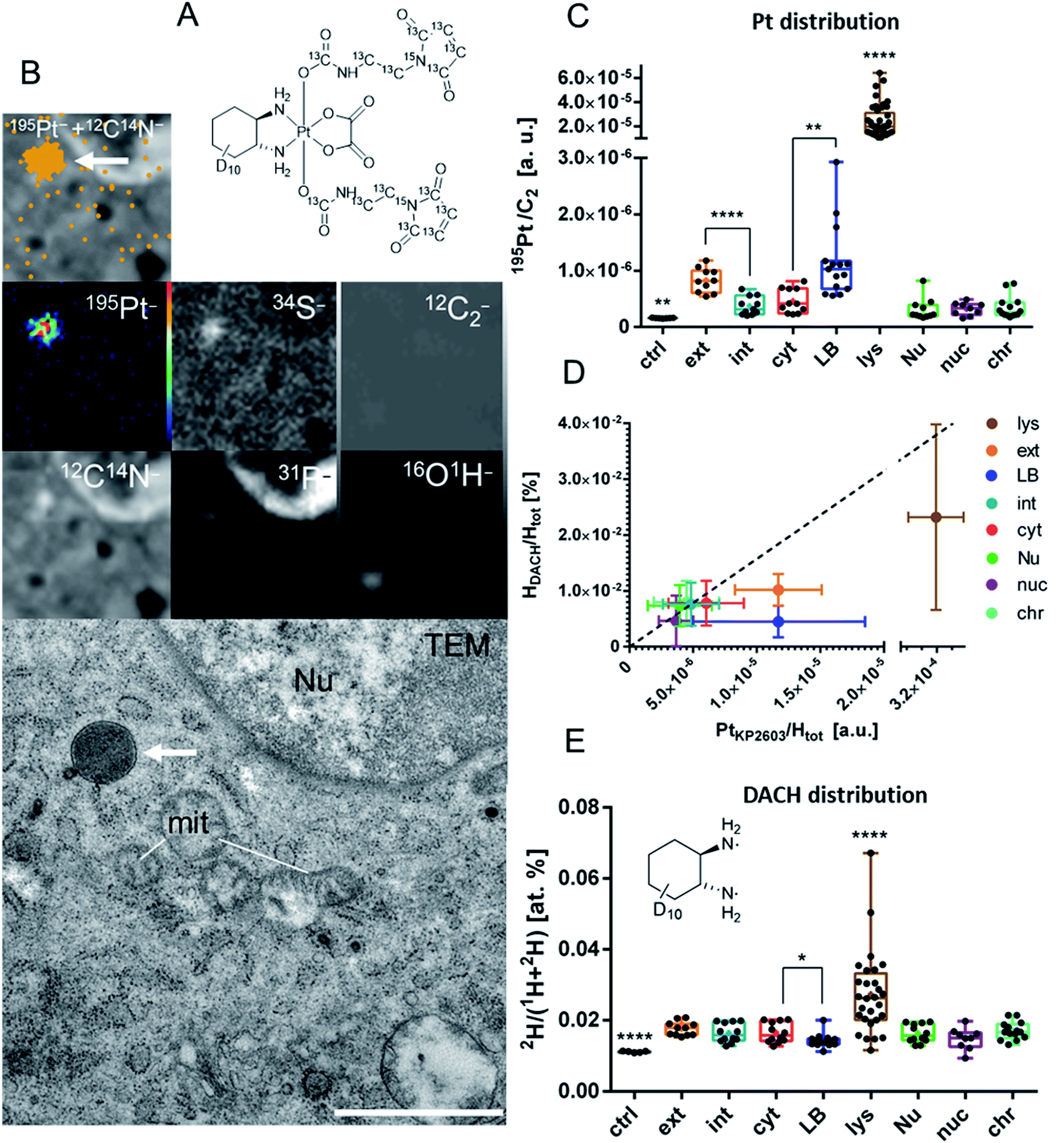 | ||
| Fig. 5 (Sub-)cellular platinum and DACH ligand distribution of the dual stable isotope-labeled KP2156 derivative KP2603 in vivo. Tumor-bearing mice were treated 4-times according to the overall survival experiment and tumors were collected after 24 h of the last dosage. (A) Structural formula of KP2603. (B) NanoSIMS 12C14N−, 195Pt−, 31P−, 34S−, 16O1H−, 12C2− secondary ion maps correlated to the electron microscopy (TEM) image of an ultra-thin section of malignant tissue. Intensities are displayed on a false color scale ranging from low intensities (black) to high intensities (red/white); in the overlay image, the 12C14N− signal intensity is displayed on a gray scale, the orange dots refer to areas with 195Pt− signal intensities of ≥1 count per pixel. The TEM micrograph displays the ultrastructure of the corresponding area within the same resin section. The white arrow indicates a platinum accumulation hotspot (lysosome). Scale bar: 1 μm. Subcellular distribution of the relative platinum content (C) and the relative DACH to platinum ratio (D) and subcellular distribution of the 2H-labeled DACH ligand (E) in CT26 cells as inferred from NanoSIMS elemental and isotopic mapping. Data points refer to individual ROI values. The box-and-whisker plots display the extreme values (min/max), median and lower/upper quartiles. Statistical analysis: Kolmogorov–Smirnov normality test followed by two-sided Student's t-test with Welch's correction or Mann–Whitney U-test (*p > 0.05, **p < 0.01, ****p < 0.0001). Abbreviations and number of ROIs per category: ctrl – negative control (average values of tumor cells from an untreated mouse, n = 7), ext – extracellular matrix (n = 10), int – intracellular compartment (whole cells, n = 11); cyt – cytoplasm (n = 11); LB – lipid bodies (n = 15); lys – lysosomes/platinum hotspots (n = 45); Nu – nucleus (n = 11); nuc – nucleolus (n = 9); chr – chromatin (n = 14). | ||
In general, a homogeneous distribution of platinum was observed within the major subcellular compartments (e.g. cytoplasm, nucleus) with a few, tiny structures responsible for compound accumulation (Fig. 5A and B). In agreement with previous data on 24 h single-treated tumors,33 platinum from KP2603 showed a tendency to accumulate in cytoplasmic, protein-rich membrane-bound organelles (platinum hotspots, Fig. 5A, B and S14†), which were identified as lysosomes based on the TEM images. In contrast, no enhanced platinum accumulation was observed in nuclear compartments (total nucleus, nucleoli, and heterochromatin). The second highest relative platinum content in the subcellular compartments of the analyzed cells was found in lipid-loaded bodies (Fig. 5B and S15†). Previously, we referred such osmiophilic lipid-accumulating protein-poor cytoplasmic structures to lipid droplets.33 The parallel detection of the 16O1H− secondary ion signal intensity helped us now to refine our previous observations: the lipid-loaded bodies showed an enhanced oxygen and hydrogen accumulation in comparison to the surrounding tissue (Fig. S15 and S16†) indicating that those organelles might in fact sequestrate oxidized lipids or represent sites of lipid peroxidation (e.g. due to H2O2 accumulation). Noteworthy, a higher platinum signal was also detected in extracellular structures (the regions between the cells proximate to the cell-limiting membranes, Fig. 5B and S15†) in comparison to the intracellular analysis areas. These regions within the tumor tissue also showed enhanced signal intensities for 12C14N− and 34S− indicating the presence of sulfur containing proteins (Fig. S15†).
The corresponding analyses of the ligand distributions in the consecutive section revealed that partially both, the 2H-labeled DACH ligands (Fig. 5E) and the 13C-labeled axial maleimide-containing ligands (Fig. S17†) end up in the lysosomes, which are also relatively enriched in platinum (Fig. 5B). This indicates that at least part of the compound arrives in the target cells without decomposition. Interestingly, the relative hydrogen to platinum content of the 2H-labeled DACH ligand was reduced in lysosomes, lipid bodies and in the extracellular matrix (Fig. 5D). In line with previous observations in adherently grown cell cultures,40 this could points towards dissociation of the tightly bound DACH ligand from the metal center, which is unexpected, as the DACH ligand is considered as being inert. One explanation for this observation could be that the compound is degraded (e.g. in lysosomal environment) and platinum is released and accumulated in lipid bodies or gets excreted from the cells. The dissociation of the non-labile NH3 ligand from the metal center was previously reported in nucleoli of cisplatin-treated cells.39 Noteworthy, this is the first report on the loss of the DACH ligand from an oxaliplatin derivative in vivo and could indicate a metabolic step, which will be a topic of follow-up studies. In contrast to platinum and deuterium, the 13C-label (Fig. S17†) was not detected outside the lysosomes. The latter suggests pronounced maleimide cleavage either prior to cell entry or as a result of the active excretion following the uptake (e.g. maleimide might be recycled with albumin, while platinum is covalently bound to biomolecules and accumulates inside the cells).
A single time-point sampling does not allow to decipher the spatio-temporal behavior of the complex in more detail. Moreover, it has to be considered that the animals have been treated several times over two weeks before sacrifice. Consequently, it would not be surprising, if the observed drug species also contain some late stage metabolites. To answer these questions, further more in-depth analysis including time-series experiments are needed. Nevertheless, the here presented data indicate that NanoSIMS analyses of dual stable isotope labeled analogs are also possible and feasible for tissue samples of treated animals. Taken together, the NanoSIMS analyses indicate that KP2156 enters the tumor cells in vivo and accumulates in the organelles of endo-/lysosomal origin. With respect to the results obtained from the cell culture experiments, this would be in line with the assumption of an albumin-mediated endocytic uptake. Thus, at least part of the albumin-bound prodrug seems to end up in the lysosomes, which suggests lysosomes as being locations for albumin degradation, reduction and platinum(II) complex release. Interestingly, we also found indications that the compound might be metabolized (intra- and/or extracellularly) to a DACH-free platinum species. Noteworthy, there are only a few studies so far, which address the intracellular or even intratumoral drug distribution of metal-based drugs or their nanoformulations in vitro44,45 or in vivo.42
Conclusion
Oxaliplatin is one of the most efficient anticancer drugs against colorectal cancer and, consequently, is part of basically all treatment schemes for the advanced stages of this disease. However, as most chemotherapeutic drugs, treatment with oxaliplatin is associated with severe adverse effects and resistance development. Consequently, the development of new derivatives with better tumor-targeting properties are urgently needed. For this, the specific characteristics of the malignant tissue need to be exploited. Albumin is the most important (nutrient) carrier protein in blood plasma and, thus, it is not surprising that cancer cells are characterized by enhanced albumin consumption. Moreover, the chaotic blood vessel architecture of the malignant tissue together with the lack of lymph drainage leads to accumulation of larger particles (EPR effect). KP2156 is a platinum(IV) prodrug, which binds via a maleimide moiety to endogenous albumin in the body. Platinum(IV) prodrugs are proposed to be activated by reduction to the active platinum(II) species (oxaliplatin in case of KP2156). However, in case of satraplatin, it was shown that premature activation already occurs in the bloodstream by reduction in the red blood cells. The here presented data support the hypothesis that KP2156 forms very stable albumin adducts in the blood stream, which are entering the cancer cells in tumor tissue by endocytosis followed by intracellular activation/reduction. Moreover, the albumin binding of KP2156 leads to a distinctly prolonged plasma half-life and consequently enhanced overall platinum treatment dose in the animals. This results in a profound and long-lasting anticancer activity of the drug. Consequently, albumin-binding platinum(IV) prodrugs are a very promising type of novel anticancer agents which should be further developed towards phase I clinical testing.Data availability
All experimental procedures, characterization and data supporting this article have been described in the manuscript and uploaded as an ESI.†Author contributions
PH, CRK and AL conceived and designed the project. PH supervised all biological evaluations. CRK designed and supervised the synthesis of KP2156 and KP2603. AL designed and interpreted the NanoSIMS analysis. HS performed main part of biological assays and part of animals experiments. HS and PH wrote the original draft. ST performed main part of analysis by div. ICP-MS methods. DG and S.v S. were involved in the cell biological assays and animal experiments. GH and LG performed analytical evaluation of the tissue samples. JM and PF synthesized the investigated compounds. NS was involved in the synthesis of the isotopically-labelled KP2603. AS, SR and MW contributed to the NanoSIMS as well as TEM measurements and data evaluation. RMM did the calculation and interpretation of pharmacological parameters. GK supervised all ICP-MS measurements. BKK and WB contributed in project design and interpretation of the gained data. All the authors discussed the results, proof-read and commented on the manuscript.Conflicts of interest
BKK, WB, CRK and PH are co-inventors of a patent on albumin-targeted platinum(IV) drugs and co-founders of the spin-off company P4 Therapeutics. All other authors declare no conflict of interest.Acknowledgements
HS and ST contributed equally to the main findings of this manuscript. HS was financed via the Austrian Science Fund (FWF) project AP32886 to PH. This work was performed in course of the FWF-funded research group FG3. Additionally, the synthesis of the labeled platinum complex, correlative imaging (NanoSIMS/TEM) experiments and corresponding sample handling were supported through FWF grant P27749 to AL. Electron microscopy work was performed at the Core Facility Cell Imaging and Ultrastructure Research, University of Vienna, member of the Vienna Life-Science Instruments (VLSI). We thank Gerhard Zeitler (Institute for Cancer Research, Vienna) for devoted animal care and Gerald Timelthaler (Institute for Cancer Research, Vienna) for help with the digital tissue analysis.References
- I. Mármol, C. Sánchez-de-Diego, A. Pradilla Dieste, E. Cerrada and M. J. Rodriguez Yoldi, Int. J. Mol. Sci., 2017, 18(1), 197 CrossRef.
- U. Olszewski and G. Hamilton, Adv. Anticancer Agents Med. Chem., 2010, 10, 293–301 CrossRef CAS.
- M. Galanski, Recent Pat. Anti-Cancer Drug Discov., 2006, 1, 285–295 CrossRef CAS.
- M. D. Hall, H. R. Mellor, R. Callaghan and T. W. Hambley, J. Med. Chem., 2007, 50, 3403–3411 CrossRef CAS.
- M. Galanski, M. A. Jakupec and B. K. Keppler, Curr. Med. Chem., 2005, 12, 2075–2094 CrossRef CAS.
- U. Jungwirth, C. R. Kowol, C. Hartinger, B. K. Keppler, W. Berger and P. Heffeter, Antioxid. Redox Signaling, 2011, 15(4), 1085–1127 CrossRef CAS.
- V. Venkatesh and P. J. Sadler, Metal ions in life sciences, 2018, vol. 18 Search PubMed.
- C. N. Sternberg, D. P. Petrylak, O. Sartor, J. A. Witjes, T. Demkow, J. M. Ferrero, J. C. Eymard, S. Falcon, F. Calabro, N. James, I. Bodrogi, P. Harper, M. Wirth, W. Berry, M. E. Petrone, T. J. McKearn, M. Noursalehi, M. George and M. Rozencweig, J. Clin. Oncol., 2009, 27, 5431–5438 CrossRef CAS.
- J. L. Carr, M. D. Tingle and M. J. McKeage, Cancer Chemother. Pharmacol., 2006, 57, 483–490 CrossRef CAS.
- J. L. Carr, M. D. Tingle and M. J. McKeage, Cancer Chemother. Pharmacol., 2002, 50, 9–15 CrossRef CAS.
- F. Kratz, J. Controlled Release, 2008, 132, 171–183 CrossRef CAS PubMed.
- A. M. Merlot, D. S. Kalinowski and D. R. Richardson, Front. Physiol., 2014, 5, 299 Search PubMed.
- J. T. Andersen, B. Dalhus, D. Viuff, B. T. Ravn, K. S. Gunnarsen, A. Plumridge, K. Bunting, F. Antunes, R. Williamson, S. Athwal, E. Allan, L. Evans, M. Bjørås, S. Kjærulff, D. Sleep, I. Sandlie and J. Cameron, J. Biol. Chem., 2014, 289, 13492–13502 CrossRef CAS.
- W. Palm and C. B. Thompson, Nature, 2017, 546, 234–242 CrossRef CAS.
- M. Kalim, J. Chen, S. Wang, C. Lin, S. Ullah, K. Liang, Q. Ding, S. Chen and J. Zhan, Drug Des., Dev. Ther., 2017, 11, 2265–2276 CrossRef CAS.
- E. Neumann, E. Frei, D. Funk, M. D. Becker, H. H. Schrenk, U. Müller-Ladner and C. Fiehn, Expert Opin. Drug Delivery, 2010, 7, 915–925 CrossRef CAS.
- M. T. Larsen, M. Kuhlmann, M. L. Hvam and K. A. Howard, Mol. Cell. Ther., 2016, 4, 3 CrossRef PubMed.
- V. Pichler, J. Mayr, P. Heffeter, O. Domotor, E. A. Enyedy, G. Hermann, D. Groza, G. Kollensperger, M. Galanksi, W. Berger, B. K. Keppler and C. R. Kowol, Chem. Commun., 2013, 49, 2249–2251 RSC.
- U. Jungwirth, D. N. Xanthos, J. Gojo, A. K. Bytzek, W. Korner, P. Heffeter, S. A. Abramkin, M. A. Jakupec, C. G. Hartinger, U. Windberger, M. Galanski, B. K. Keppler and W. Berger, Mol. Pharmacol., 2012, 81, 719–728 CrossRef CAS.
- B. Englinger, C. Pirker, P. Heffeter, A. Terenzi, C. R. Kowol, B. K. Keppler and W. Berger, Chem. Rev., 2019, 119, 1519–1624 CrossRef CAS PubMed.
- A. Tesniere, F. Schlemmer, V. Boige, O. Kepp, I. Martins, F. Ghiringhelli, L. Aymeric, M. Michaud, L. Apetoh, L. Barault, J. Mendiboure, J. P. Pignon, V. Jooste, P. van Endert, M. Ducreux, L. Zitvogel, F. Piard and G. Kroemer, Oncogene, 2010, 29, 482–491 CrossRef CAS.
- P. U. Le, G. Guay, Y. Altschuler and I. R. Nabi, J. Biol. Chem., 2002, 277, 3371–3379 CrossRef CAS PubMed.
- D. Dutta, C. D. Williamson, N. B. Cole and J. G. Donaldson, PLoS One, 2012, 7, e45799 CrossRef CAS PubMed.
- L. Turell, R. Radi and B. Alvarez, Free Radicals Biol. Med., 2013, 65, 244–253 CrossRef CAS.
- F. Arnesano, S. Scintilla and G. Natile, Angew. Chem., Int. Ed. Engl., 2007, 46, 9062–9064 CrossRef CAS PubMed.
- M. Ravera, E. Gabano, I. Zanellato, I. Bonarrigo, M. Alessio, F. Arnesano, A. Galliani, G. Natile and D. Osella, J. Inorg. Biochem., 2015, 150, 1–8 CrossRef CAS.
- S. Göschl, E. Schreiber-Brynzak, V. Pichler, K. Cseh, P. Heffeter, U. Jungwirth, M. A. Jakupec, W. Berger and B. K. Keppler, Metallomics, 2017, 9, 309–322 CrossRef PubMed.
- L. Tang, D. Cai, M. Qin, S. Lu, M. H. Hu, S. Ruan, G. Jin and Z. Wang, ACS Omega, 2020, 5, 726–734 CrossRef CAS PubMed.
- M. N. Khan, J. Pharm. Sci., 1984, 73, 1767–1771 CrossRef CAS PubMed.
- J. Mayr, P. Heffeter, D. Groza, L. Galvez, G. Koellensperger, A. Roller, B. Alte, M. Haider, W. Berger, C. R. Kowol and B. K. Keppler, Chem. Sci., 2017, 8, 2241–2250 RSC.
- M. M. Mita, R. B. Natale, E. M. Wolin, B. Laabs, H. Dinh, S. Wieland, D. J. Levitt and A. C. Mita, Invest. New Drugs, 2015, 33, 341–348 CrossRef CAS PubMed.
- P. Yousefpour, L. Ahn, J. Tewksbury, S. Saha, S. A. Costa, J. J. Bellucci, X. Li and A. Chilkoti, Small, 2019, 15, e1804452 CrossRef.
- A. A. Legin, S. Theiner, A. Schintlmeister, S. Reipert, P. Heffeter, M. A. Jakupec, J. Mayr, H. P. Varbanov, C. R. Kowol, M. Galanski, W. Berger, M. Wagner and B. K. Keppler, Chem. Sci., 2016, 7, 3052–3061 RSC.
- C. Li, Y. Zhang, G. Chen, F. Hu, K. Zhao and Q. Wang, Advanced materials, Deerfield Beach, Fla., 2017, vol. 29 Search PubMed.
- C. Carlier, B. Laforce, S. J. M. Van Malderen, F. Gremonprez, R. Tucoulou, J. Villanova, O. De Wever, L. Vincze, F. Vanhaecke and W. Ceelen, J. Pharm. Biomed. Anal., 2016, 131, 256–262 CrossRef CAS PubMed.
- S. Theiner, A. Schweikert, C. Haberler, A. Peyrl and G. Koellensperger, Metallomics, 2020, 12, 1246–1252 CrossRef CAS.
- S. Theiner, C. Kornauth, H. P. Varbanov, M. Galanski, S. Van Schoonhoven, P. Heffeter, W. Berger, A. E. Egger and B. K. Keppler, Metallomics, 2015, 7, 1256–1264 CrossRef CAS PubMed.
- R. F. S. Lee, S. Escrig, M. Croisier, S. Clerc-Rosset, G. W. Knott, A. Meibom, C. A. Davey, K. Johnsson and P. J. Dyson, Chem. Commun., 2015, 51, 16486–16489 RSC.
- A. A. Legin, A. Schintlmeister, M. A. Jakupec, M. Galanski, I. Lichtscheidl, M. Wagner and B. K. Keppler, Chem. Sci., 2014, 5, 3135–3143 RSC.
- A. A. Legin, A. Schintlmeister, N. S. Sommerfeld, M. Eckhard, S. Theiner, S. Reipert, D. Strohhofer, M. A. Jakupec, M. Galanski, M. Wagner and B. K. Keppler, Nanoscale Adv., 2021, 3, 249–262 RSC.
- L. E. Wedlock, M. R. Kilburn, R. Liu, J. A. Shaw, S. J. Berners-Price and N. P. Farrell, Chem. Commun., 2013, 49, 6944–6946 RSC.
- M. T. Proetto, C. E. Callmann, J. Cliff, C. J. Szymanski, D. Hu, S. B. Howell, J. E. Evans, G. Orr and N. C. Gianneschi, ACS Cent. Sci., 2018, 4, 1477–1484 CrossRef CAS PubMed.
- L. Habala, M. Galanski, A. Yasemi, A. A. Nazarov, N. G. von Keyserlingk and B. K. Keppler, Eur. J. Med. Chem., 2005, 40, 1149–1155 CrossRef CAS.
- D. Montagner, S. Q. Yap and W. H. Ang, Angew. Chem., Int. Ed. Engl., 2013, 52, 11785–11789 CrossRef CAS.
- J. X. Ong, H. V. Le, V. E. Y. Lee and W. H. Ang, Angew. Chem., Int. Ed. Engl., 2021, 60(17), 9264–9269 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental part, KP2603 synthesis, supplementary figures, tables. See DOI: 10.1039/d1sc03311e |
| ‡ These authors contributed equally to the main findings of this manuscript. |
| This journal is © The Royal Society of Chemistry 2021 |