
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcid-catalysed liquid-to-solid transitioning of arylazoisoxazole photoswitches†
Luuk
Kortekaas
 *a,
Julian
Simke
a,
Niklas B.
Arndt
a,
Marcus
Böckmann
b,
Nikos L.
Doltsinis
b and
Bart Jan
Ravoo‡
*a
*a,
Julian
Simke
a,
Niklas B.
Arndt
a,
Marcus
Böckmann
b,
Nikos L.
Doltsinis
b and
Bart Jan
Ravoo‡
*a
aCenter for Soft Nanoscience and Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Busso-Peus-Straße 10, 48149, Münster, Germany. E-mail: kortekaas@uni-muenster.de; b.j.ravoo@uni-muenster.de
bInstitute for Solid State Theory and Center for Multiscale Theory & Computation, Westfälische Wilhelms-Universität Münster, Wilhelm-Klemm-Str. 10, 48149, Münster, Germany
First published on 19th July 2021
Abstract
Molecular photoswitches play a vital role in the development of responsive materials. These molecular building blocks are particularly attractive when multiple stimuli can be combined to bring about physical changes, sometimes leading to unexpected properties and functions. The arylazoisoxazole molecular switch was recently shown to be capable of efficient photoreversible solid-to-liquid phase transitions with application in photoswitchable surface adhesion. Here, we show that the arylazoisoxazole forms thermally stable and photoisomerisable protonated Z- and E-isomers in an apolar aprotic solvent when the pKa of the applied acid is sufficiently low. The tuning of isomerisation kinetics from days to seconds by the pKa of the acid not only opens up new reactivity in solution, but also the solid-state photoswitching of azoisoxazoles can be efficiently reversed with selected acid vapours, enabling acid-gated photoswitchable surface adhesion.
Introduction
Over the past decades, molecular switches have become ubiquitous in the development of smart materials, rendering reversible changes in material function upon input stimuli.1,2 In addition to designing new molecular switches and the synthetic optimisation of existing structures, the deeper understanding of their fundamental responsivity has led to a vast body of research on small molecular photoswitches, most notably on azobenzenes,3,4 spiropyrans5,6 and diarylethenes.7 These exemplary molecular photoswitches have proven to be excellent building blocks for integration in materials owing in particular to their multi-responsiveness, reacting not only to light,8,9 but also, e.g., heat,10 mechanical input,11 and pH-changes.12 It is particularly attractive when there is synergy between distinct stimuli that leads to new functionality, as is the case for the photo- and pH-response in spiropyrans, where weak acids slow down the thermal reversion and, as more recently shown, strong acids can open up to new photophysics in the protonated isomers.6,13 Conversely, for the azobenzene photoswitch, the interaction with acids has been long shown to catalyse rather than retard the reversion of Z-to E-isomer (Scheme 1).14–20 The extent of catalytic reversion depends on the exact acid strength and the amount added, as well as the solvent type in conjunction with its limiting pKa.21I.e., in 1938 Hartley already reported that addition of several drops of acetic acid to water accelerates the optical Z-to E-conversion to several seconds,14 while Ciccone et al. found no effect upon addition of acetic acid to Z-azobenzene in aqueous ethanol.15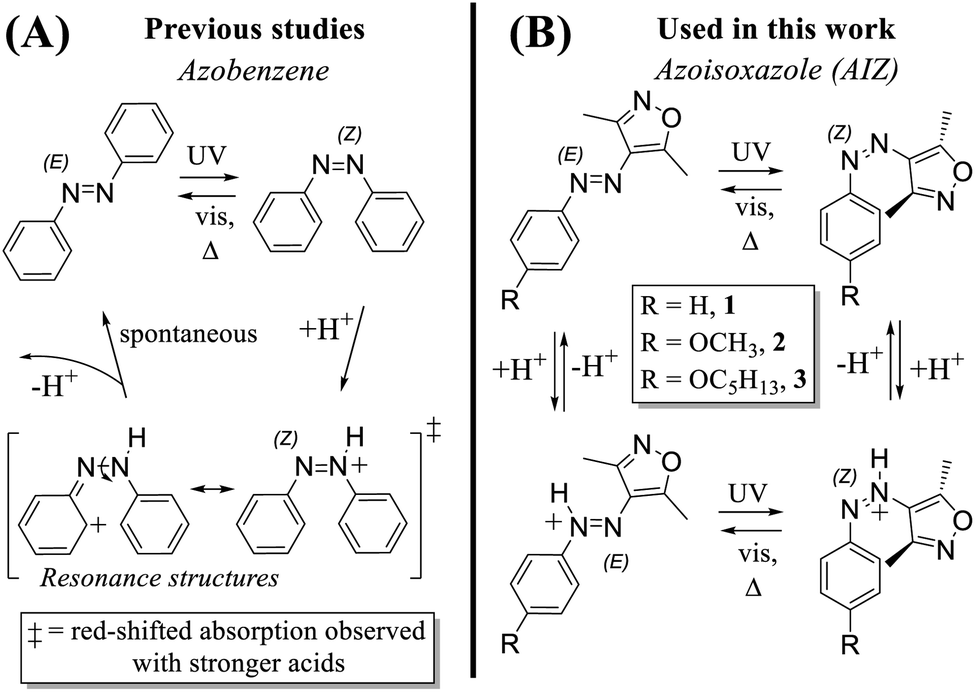 | ||
| Scheme 1 (A) Acid-catalysed Z-to E-reversion reported for azobenzenes. The isomerisation occurs spontaneously, a lingering red-shifted azonium absorption is observed when strong acids are used. (B) The (pH-gated) photochromism and acidochromism of azoisoxazole reported here. We note that the depicted protonation sites are based solely on an extensive theoretical analysis (see below). | ||
Acid-catalysed reversion rates in azobenzenes were reported over the years to be independent of ionic strength of the acids,15,16 with the isomerisation step of the Z-to E-cation determined as the rate determining step.17 The accelerated isomerisation is commonly attributed to the mesomerism of the azonium ion weakening the azo double bond,14–17,19 an effect that was observed to be stronger when electron-rich substituents are conjugated to the azo double bond.14,16 Interestingly, despite the strong pH dependence of the Z-isomer, the absorption of the plain E-azobenzene was found to be independent of aqueous HCl between pH 1 and 13.16 Indeed, the formation of azonium species in some (para-) substituted azobenzenes was observed to require much harsher conditions, such as a 6 M HClO4 solution,17,20 or gas phase ionisation.22
Alternatively, aminoazobenzenes, such as the methyl orange pH-indicator, have been shown to yield red-shifted azonium ions on account of resonance contributions from the amino-substituent,19 with 4,4′-diaminoazobenzenes even protonating up to pH 4.23 Such red-shifting of absorption through azonium formation runs not only parallel to the strategy of appending auxiliary functional groups,24–27 it also gives rise to new isomerisation kinetics altogether. The group of Woolley, for example, discovered that the pKa of the azo-moiety is significantly increased owing to hydrogen-bonding in tetra-ortho-methoxy azobenzenes, such that the protonated species is preferred at physiological conditions.28–31 Their red-shifted absorption opens up to E-to Z-isomerisation with NIR light with lifetimes in the order of milliseconds to seconds,28–30 leading to application in various smart materials.32–35 Importantly, the tetra-ortho substitution was found to be vital in raising their pKa to such extent, while a symmetric shift of two methoxy units to the meta-position caused a pKa drop of 5 units.29
Despite the rapidly grown interest in photochromically superior azoheterocycles as an alternative to azobenzenes,36 only few contributions have been concerned with the generation and stabilisation of their azonium species.37–39 For example, Fuchter et al. showed that for azopyrazole40–42 (AAP) the acid-catalysis of azobenzenes is retained when trichloroacetic (CCl3COOH), trifluoroacetic (CF3COOH) and hydrochloric acid (HCl) are applied, and that the transient Z-azonium AAP could even be observed during catalysis.37 In further work, ortho-substitution of the azopyrazole benzene ring analogous to the work of Woolley et al. was found to have an effect on the lifetime of Z-azopyrazoles also, albeit without mention of its effects on acid-catalysis.38 More recently, Walther et al. succeeded in demonstrating the general acid-catalysis of water-soluble azopyrazoles, opening up to a tunability of Z-isomer half-lives over 5 orders of magnitude depending on the pH value.39 This retention of the acid catalysis in the AAP azoheterocycle brings about important implications for azoheterocycles in general, as their superior photophysics, and their vastly increased thermal half-lives in particular, give rise to a significantly larger control over half-life times without the need for synthetic adjustments.
Azoisoxazoles (AIZs), as a relatively new type of azoheterocycle, show great promise as components in smart materials owing to the photoinduced reversible solid-to-liquid phase transition demonstrated recently.43 Due to the sterically induced out-of-plane bending of the Z-isomer the intermolecular interactions are incapacitated, leading to a drastic decrease in melting point and consequent liquefication of the azoisoxazole upon UV-induced isomerisation. Previously, we have utilised this remarkable retention of photophysics in the solid state to develop photoreversible low molecular weight (LMW) adhesives, cohering glass surfaces at 30.8 N cm−2 with rapid debonding upon UV-irradiation.44 Although the control over the bonding/debonding process is remarkably swift and reversible even upon physical breaking of the cohesion, the photochemical approach requires the use of at least one transparent surface. In this contribution, we seek to expand on the promising reusable photophysical features of these low molecular weight azoisoxazoles by exploring the possibility to kinetically control their solidification with protons. Though we demonstrate that the acid-induced isomerisation takes place with select acids for azoisoxazoles also, the terms “weak acid” and “strong acid” turn out to be imperative while use of acids stronger than the conventional HCl and CF3COOH, i.e., trifluoromethanesulfonic acid (CF3SO3H, pKa = 0.7 in acetonitrile45), yield steady state Z-azonium (Z-AIZH+) and E-azonium (E-AIZH+) species as observed by UV/vis and 1H NMR spectroscopy. Furthermore, the isomerisation kinetics are not only pKa-tuneable in solution, but also in the pristine liquefied Z-form by exposure to acid vapours. Ultimately, it empowers the versatile adhesive with a rapid, non-destructive trigger for reversibly conjoining surfaces with remarkable adhesive strengths irrespective of transparency, and showcases the further potential that the added pH-gated control opens up to for this already widely versatile class of photochromes.
Results and discussion
Azonium formation in azoheterocycles
Contrary to the catalysed azo-reversion with conventional acids such as CF3COOH and HCl, we observe that the addition of the more acidic trifluoromethanesulfonic acid (CF3SO3H) to azoisoxazole AIZ 1 causes a shift to a new steady-state absorption (Fig. S1†). Comparably, the analogous unsubstituted AAP 4 (ref. 41) also shows a steady-state absorption upon addition of CF3SO3H (Fig. S2†) and, furthermore, distinct azonium chemical shifts in 1H NMR spectroscopy (Fig. S3,† for AIZ spectra see below, “Thermally stable Z-and E-protonation with strong acids”). Although the addition of CF3SO3H to the unsubstituted AIZ 1 yields a new absorption at 315 nm, the alkoxy AIZ 2 and 3 azonium ions (AIZH+) exhibit strong red-shifted absorptions instead (Fig. 1), while undergoing catalytic Z-to E-reversion in response to HCl and CF3COOH. Hence, it is possible to capitalise on varying responses and response times by judicious choice and quantity of acid, opening up to a wider range of control over previously established as well as new physical properties.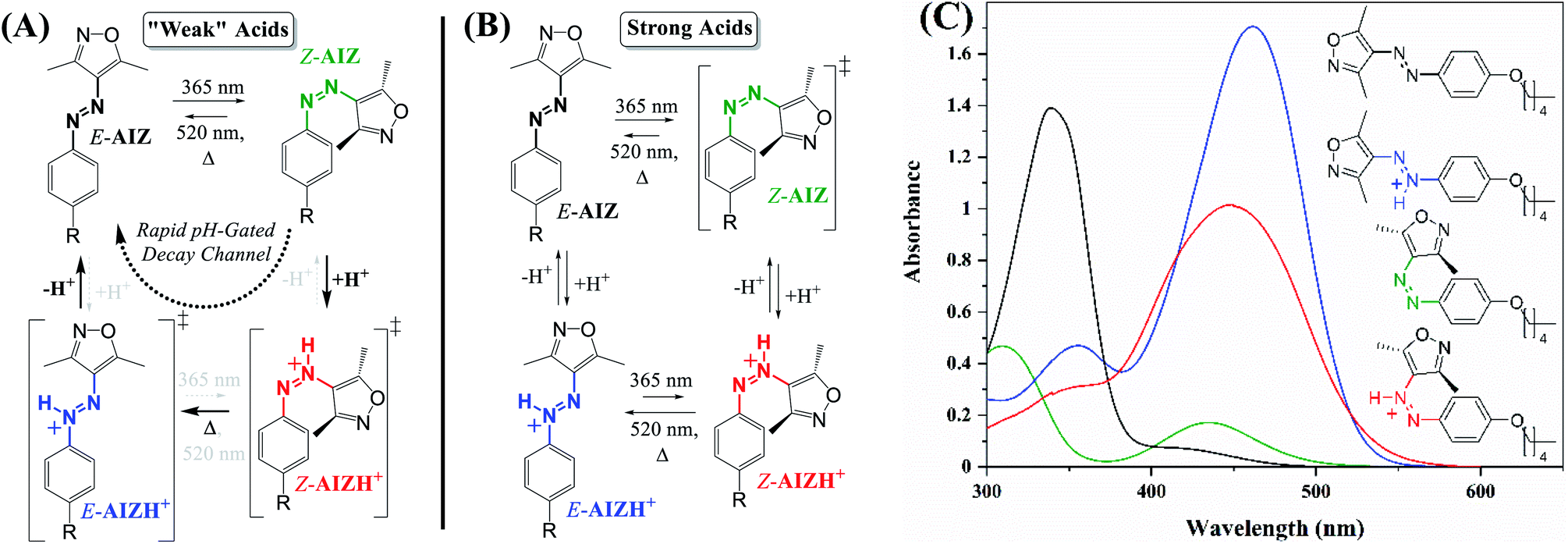 | ||
| Fig. 1 Mechanistic pathways of AIZ for (A) acid catalysed Z-to E-reversion with use of relatively weak acids (HCl, CF3COOH) and (B) pH-gated photochromism with strong acids (i.e., CF3SO3H). ‡ = not observed. (C) UV/vis absorption of E-AIZ 3 (70 μM in acetonitrile) at thermal equilibrium (black) and after irradiation at 365 nm in the Z-isomer (green). Addition of 1500 and 1000 eq. of CF3SO3H under these conditions yields E-AIZH+ (blue) and Z-AIZH+ (red), respectively. DFT predicts protonation adjacent to the heterocycle (Fig. S4†), causing a break in symmetry in each of the AIZ isomers and greatly increasing the probability of the lower energy n–π* transition similar to the changes observed upon E-to Z-isomerisation. For AIZ 2, see Fig. S5.† For a comparison to theoretical TD-DFT spectra, see Fig. S6 and S7.† | ||
Accelerated Z-to E-reversion with weak acids
Owing to mesomerism,46 the uncatalysed half-life of the 4-alkoxy-substituted Z-AIZs 2 and 3 in solution was determined to be 7 d (ref. 44) rather than the 45 d for Z-AIZ 1.43 Although we observe no reactivity of Z-AIZ 2 and 3 to a large excess of acetic acid (CH3COOH, 5000 equivalents in acetonitrile, Fig. S8†), addition of CF3COOH and HCl cause the half-life to diminish to the (sub)second timescale at near-stoichiometric amounts already (Fig. S9 and S10†). DFT calculations of activation free energies confirm that the Z-to E-barrier is substantially reduced in the azonium species from 1.29 eV (versus 1.11 eV experimentally44) to 0.86 eV (Fig. 2A). Importantly, out of three possible N-protonation sites, the azo-N at the phenyl ring is lowest in energy for the E-form, and the N-azo at the heterocycle for the Z-form (Fig. S11,† the protonated heterocycle being highest in both cases; we note that our calculations on AAP 4 are in line with those by Fuchter et al.,37 Fig. S12†). The greatly reduced thermal Z/E barrier accessible at excess HCl or CF3COOH causes the generated Z-AIZH+ isomer to rapidly equilibrate with E-AIZH+. However, although the pKa of HCl is sufficient to interact with the Z-form, any E-AIZH+ generated is spontaneously deprotonated due to its significantly increased acidity. The thermodynamically trapped unprotonated E-AIZ (barrier of 1.86 eV to Z-AIZ) that results from this energetically favourable protonation–isomerisation–deprotonation sequence is thus continuously and, moreover, rapidly formed owing to Le Chatelier's principle (Fig. 1A; further supported by calculations taking into account conjugate bases, Fig. S13†). | ||
| Fig. 2 (A) Relative Gibbs free energies of the unprotonated and protonated isomers of AIZ 2 and their transition states. For details on the DFT calculations see the experimental section. (B) UV/vis absorption spectra of AIZ 3 (70 μM in acetonitrile) with 0.05 eq. of HCl (black solid line). The thermal reversion is sped up through the acid-catalysed reversion on the right, hence the PSS365nm (radiant flux 1.2 W) is compromised through spontaneous reversal via the azonium ions (black dashed lines, spectra every 2 min). Note also that the isosbestic point at 403 nm, where the molar absorptivity of both azonium species is significantly higher (Fig. 1C), rules out a steady state generation of the Z-AIZH+ isomer. | ||
Notably, catalytic amounts of HCl or CF3COOH cause the reversion to occur gradually, allowing for the thermal reversion to be moderated as described for azobenzenes and azopyrazoles before (Fig. 2B, S14†).14–20,37,39 Though the tuneable half-life ranges from several seconds at stoichiometric equivalents of HCl to minutes at catalytic amounts, the higher pKa and solubility of trifluoroacetic acid allow for more control at superstoichiometric quantities (Fig. S15†), yielding varying well-tuneable half-lives (Table 1). The reduction in half-life eventually stagnates upon increasing the acid content, analogous to the earlier reports on the deviation of first-order kinetics in azobenzenes.14,15,17,37,39 We note, however, that this is likely due to the weak but increasing involvement of the E-azonium isomer, not least because an increase to 5000 eq. CF3COOH causes the half-life to significantly rise again. In other words, despite the mismatch in pKa the E-AIZH+ becomes energetically more feasible in presence of thousands of possible (transient) proton sources, increasing its life-time and thus delaying the catalytic process.
| CF3COOH | Thermal half-life | Rate constant | |
|---|---|---|---|
| 0 eq. (ref. 44) | 7 d | — | 0.00115 × 10−3 s−1 |
| 2 eq. | 338.4 s | ±46.3 | 2.05 × 10−3 s−1 |
| 10 eq. | 242.1 s | ±11.1 | 2.86 × 10−3 s−1 |
| 50 eq. | 118.8 s | ±18.9 | 5.84 × 10−3 s−1 |
| 200 eq. | 43.5 s | ±8.2 | 15.94 × 10−3 s−1 |
| 1000 eq. | 24 s | ±1.2 | 28.93 × 10−3 s−1 |
| 2000 eq. | 25.2 s | ±0.5 | 27.51 × 10−3 s−1 |
| 5000 eq. | 59.9 s | ±3.5 | 11.57 × 10−3 s−1 |
Theoretical analysis of the Z → E thermal conversion
The large change in thermal reversion rates upon protonation was investigated in further detail through locating the accessible regime of the corresponding potential energy hypersurfaces employing DFT. For azobenzenes, thermal reversion pathways have been subject to many theoretical and experimental studies.4,47–50 The molecular motion during azo-isomerisation in general is typically described as comprising rotation and inversion, and certain combinations thereof (Scheme S1†).4 Classically, quantum chemical calculations rely on locating individual stationary points (minima, transition states) and lack information on how these points are dynamically connected. Here, we adopt a point of view more suited to a dynamical interpretation and scan the potential energy surface. Fig. 3 presents relaxed energy profiles starting out of the minimum energy valley of AIZ 2 at varying CNNC dihedral angles and C(5-ring)NN bond angles.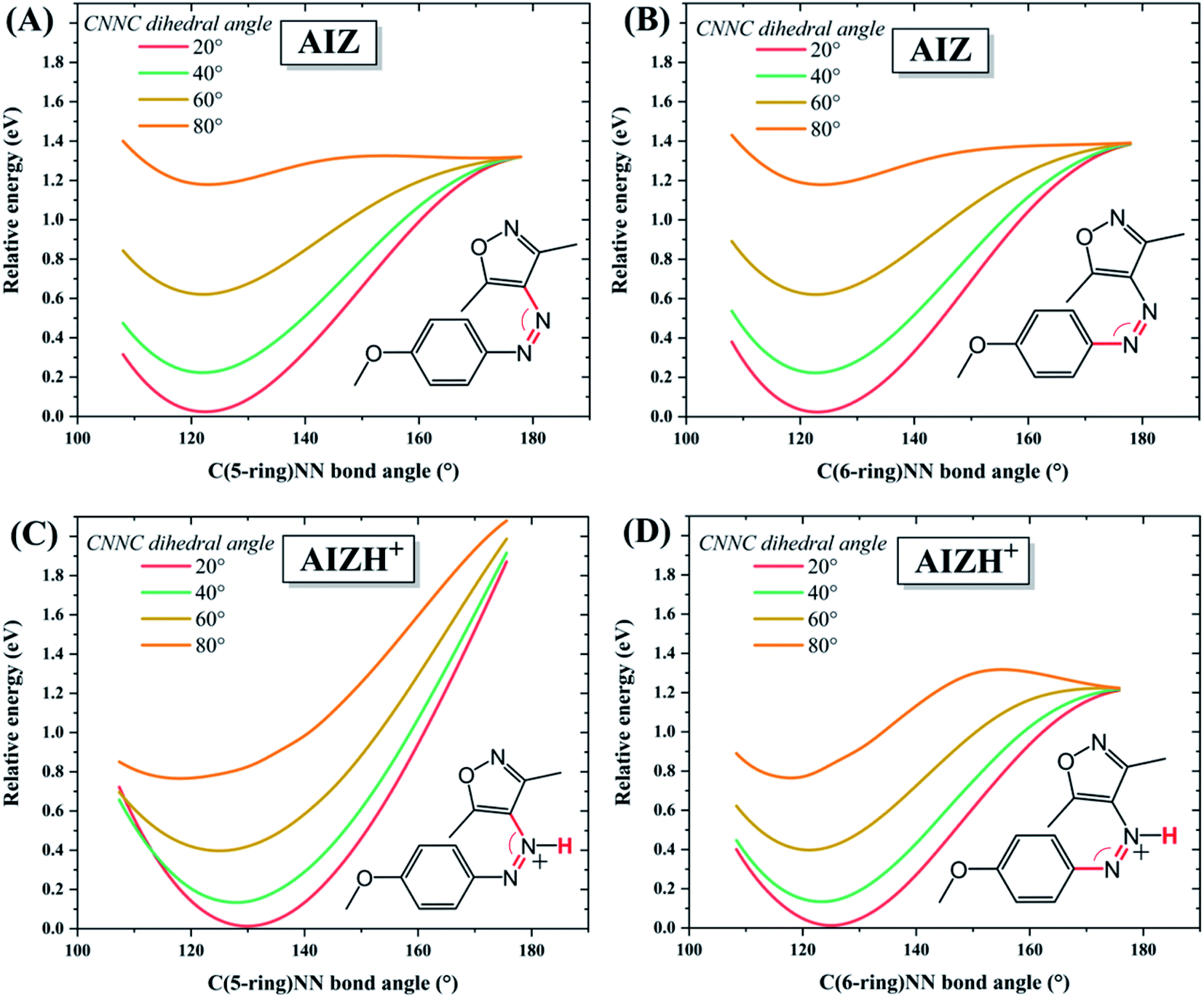 | ||
| Fig. 3 Relative potential energies of (A and B) AIZ 2 and (C and D) AIZH+2 along the coordinates of the dihedral angle CNNC (fixed at the values specified), and either (A and C) C(5-ring)NN bond angle or (B and D) C(6-ring)NN bond angle. The amount of data was reduced here for better visualisation, additional data at 10° intervals of CNNC dihedral angle is reported in Fig. S16.† | ||
Accordingly, for the neutral as well as the protonated AIZ species, the minimum energy pathway is dominated by an increase of the dihedral angle CNNC (commonly viewed as ‘rotation’) up to 80° whereupon an opening of the bond angle CNN becomes feasible. While the neutral AIZ allows bond angle opening to both sides, for the protonated AIZ opening at the non-protonated side is feasible only. Hence, the mechanism involved is labelled ‘rotation-assisted inversion’ for the neutral and ‘(pure) rotation’ for the protonated AIZ species (Scheme S1†). The corresponding results for the (neutral isomers of) AAP 4 and the parent compound azobenzene are effectively the same as for the unprotonated AIZ 2 (Fig. S17†), and thus constitute a general feature of the azo moiety, in line with recent experimental49 and theoretical50 reports. Protonation of Z-AIZ can occur at either of the two azo-nitrogens, adjacency to the 5-ring being energetically favoured by less than 0.1 eV (Fig. S11†). Additionally, the likelihood of protonation via the solvent, investigated by minimum energy path calculations for proton transfer from either azo-nitrogen to a single explicit molecule of acetonitrile, CH3COO−, CF3COO−, or CF3SO3− (Fig. S13†), further supports our experimental observations. The acidity of CH3COOH is far too weak to protonate AIZ, and while for CF3COOH protonation of AIZ is considerably more likely, the proton still slightly favours the conjugate base of the acid. In the case of CF3SO3H, however, the equilibrium strongly shifts to AIZ. Concerning a possible shift of the proton from one azo-nitrogen to the other, Fig. S13† reveals that a frequent exchange should take place via CF3COO− due to a very shallow potential landscape for both isomers, whereas for CF3SO3− it is only feasible for the E-isomer, though less frequent than for CF3COO−.
Thermally stable Z-and E-protonation with strong acids
Although substitution patterns play a vital role in increasing the pKa of E-azonium towards physiological conditions,19,23,28–30 the access to a larger range of molecular properties in less complex existing azo-structures is highly advantageous. To achieve this, the pKa of the acid needs to be near the low pKa of the azo-bond. Whereas neither of the azonium isomers are observed by UV/vis spectroscopy even at 500 equivalents of HCl, addition of increasing amounts of CF3SO3H to E-AIZ 3 causes a stepwise formation of the E-AIZH+ azonium with saturation at about 1500 equivalents (Fig. S18†). Accordingly, by considering the equilibrium being practically exclusively between E-AIZ and E-AIZH+ and assuming full dissociation of CF3SO3H, the pKa of the E-isomer is approximated to be −2.48 ± 0.16 by UV/vis spectroscopy.Interestingly, a sharp cut-off is observed for protonation in 1H NMR spectroscopy instead, transitioning from unprotonated to protonated between 5 and 6 equivalents of CF3SO3H (Fig. S19†). We note, however, that adding multiple equivalents of CF3SO3H to 3.3 mM AIZ in 1H NMR spectroscopy (versus 70 μM in UV/vis) results in a much lower pH and thus increased possibilities of interaction. This observation was made by the group of Fuchter also while protonating an AAP with HCl, requiring several orders of magnitude less equivalents at 1H NMR concentration.37 Presumably, the high interaction is also the reason for the sharper cut-off in chemical speciation than in UV/vis absorbance spectroscopy.
Alternatively, addition of CF3SO3H at the PSS365nm (97% Z-isomer) yields an additional red-shifted absorption in UV/vis spectroscopy (Fig. S20†) as well as distinct Z-AIZH+ chemical shifts in 1H NMR spectroscopy (Fig. 4, S21, Table S1†). Importantly, neither the Z-AIZH+ nor E-AIZH+ chemical shifts are observed upon addition of equal amounts of CF3SO3Li to AIZ 3 (Fig. S22†). Furthermore, whereas near-equivalent amounts of CF3SO3H destabilise the Z-AIZ already, its presence is unaltered with 15 eq. of CF3SO3Li, excluding a significant role of ionic strength on chemical speciation. The Z-AIZH+ species generated in presence of CF3SO3H, unlike the unprotonated Z-form, is metastable at room temperature and reverts to a thermodynamic equilibrium with 94% E-AIZH+ over time. The larger thermodynamic ratio of the Z-to E-azonium in presence of CF3SO3H in 1H NMR spectroscopy (6% Z-form as opposed to ca. 1% in absence of a proton source44) is in line with the change in energetic barriers observed by DFT (Fig. 2A). Moreover, only 2 new sets of signals are observed here, their kinetics being indicative of the E-and Z-azonium observed in UV/vis spectroscopy. Additionally, double protonation would result in blue-shifting of absorptions (Fig. S23†), and no such features are observed upon addition of a large range of acid equivalents (Fig. 5). Instead, an interplay between protonation/deprotonation and reversible Z/E-photo- and thermal isomerisation opens up to four distinct regimes of kinetic control in this acidity range. At near-stoichiometric equivalents primarily E-AIZ and Z-AIZH+ are observed (Fig. 5A), differing from catalysis with weak acids as it transiently yields a strongly absorbing Z-AIZH+ isomer upon UV-irradiation (Fig. 1B). Upon increasing the equivalent CF3SO3H to ca. 30 eq. the absorption of the red-shifted Z-AIZH+ reaches a fully photo- (Fig. 5B) and thermoreversible (Fig. S24†) steady state, while a further increase to ca. 500 eq. leads to a more significant share of E-AIZH+ and a limitation of absorption changes primarily to the UV-range (Fig. 5C). At ca. 800 eq. of CF3SO3H, the absorption is dominated by the azonium species, their large overlap in absorption providing subtle photoreversible changes in the UV- and visible range (Fig. 5D). Thus, we find that although the molecular components of the abovementioned four photochemical systems are chemically identical, the relative composition provides tuneable control over various absorptions without the need for synthetic alterations.
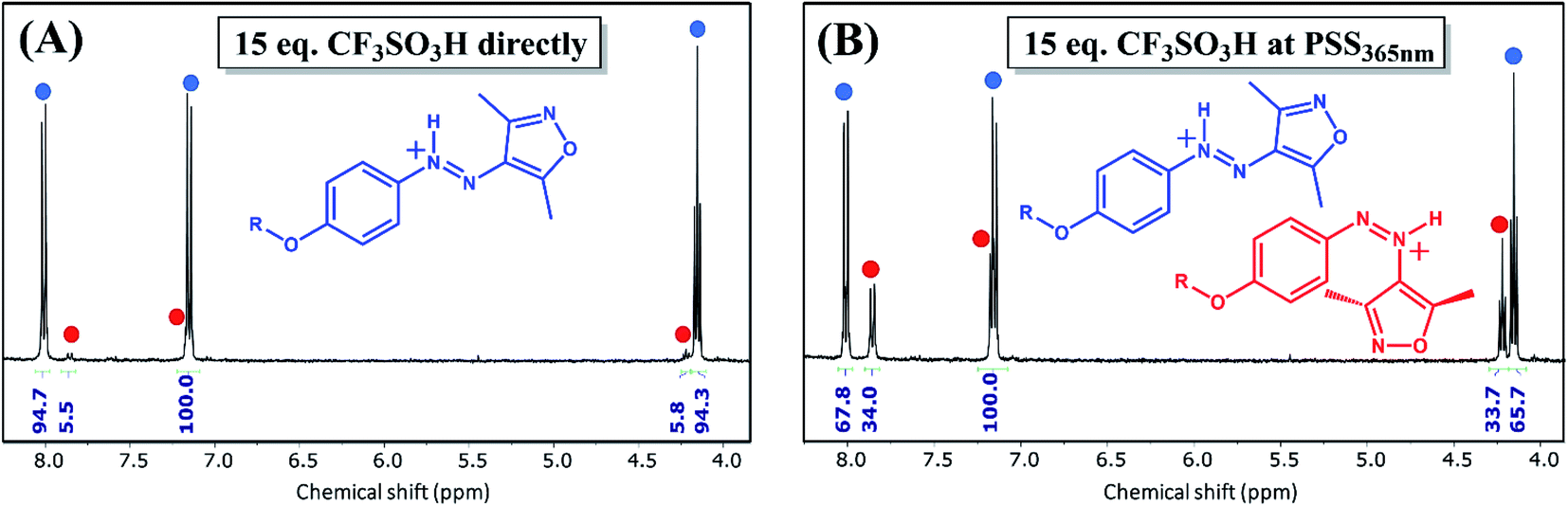 | ||
| Fig. 4 1H NMR spectroscopy of AIZ 3 (3.3 mM in CD3CN) (A) after immediate addition of 15 eq. of CF3SO3H and (B) after addition of 15 eq. of CF3SO3H to the PSS365nm (radiant flux 1.2 W). Although the required number of CF3SO3H equivalents to form the azonium species is much smaller at 1H NMR than UV/vis concentration, such stronger interaction at higher concentration ranges has been noted before.37 Note that the delay between addition of CF3SO3H and measuring causes the predominantly present Z-isomer at the PSS365nm to revert largely to the E-azonium. For a full comparison with chemical shifts of the unprotonated forms see Table S1 and Fig. S21.† | ||
 | ||
| Fig. 5 UV/vis absorption spectra of AIZ 3 (70 μM in acetonitrile) giving rise to varying photocontrol over azo-speciation at 365 nm (radiant flux 1.2 W, teal arrows) and at 520 nm (radiant flux 87 lm, orange arrows) depending on the amount of CF3SO3H present. 20 steps shown for (B)–(D), ‡ = not clearly observed. Since only 2 species are predominant at 30 and 800 equivalents of CF3SO3H, this allows for approximation of the PSSs under these circumstances by comparing the combined absorption at λ = 340 and 460 nm with those observed in Fig. 1C: (B) PSS365nm ≈ 62% Z-AIZH+ and 38% E-AIZ, PSS520nm ≈ 20% Z-AIZH+ and 80% E-AIZ; (D) PSS365nm ≈ 37% Z-AIZH+ and 63% E-AIZH+, PSS520nm ≈ 12% Z-AIZH+, 88% E-AIZH+. | ||
Acid-catalysed liquid-to-solid phase transitioning
Although observed in few AAPs also,51,52 AIZs exhibit a much higher tendency to undergo solid state switching with reversible solid-to-liquid phase transitions, owing to a drastic change in melting point upon the out-of-plane bending in the Z-form.43 Fascinatingly, the pKa dependence of the liquefied AIZ is completely orthogonal to that in solution, showing for all three AIZs a swift catalytic reversion in response to CF3COOH vapours, accompanied by a delayed liquid-to-solid phase transition (Video S1†) that was also observed photochemically.44 Analogous to the solution behaviour, CH3COOH fumes yield no response in phase as well as absorption, while CF3SO3H vapours are found to be sufficiently acidic to protonate either isomer, merely resulting in slow Z/E-equilibration (Video S2†). Slow dispersion of the CF3SO3H in air occurs over time, however, ultimately leading to resolidification over ca. 45 min as stoichiometric quantities of acid still induce catalysed reversion (Fig. 5A). Only trifluoroacetic acid and hydrochloric acid (Video S2†) cause an immediate red-shift in absorption followed up by a blue-shift (Fig. S25†), indicative of transient formation of the metastable red-shifted Z-AIZH+ isomer.Acid-catalysed adhesion
The retention of acid-catalysis in the liquefied Z-isomer followed by the delayed recrystallisation of the E-isomer opens up further opportunities in the application of AIZs as adhesives (Fig. 6A). The delay in nucleation of several seconds after the near-instant acid-catalysis with CF3COOH vapours provides time to press the surfaces together, while the resulting flux of still liquid E-isomer favourably delays the recrystallisation further and allows for quick readjustments still (Video S3†). Accordingly, the acid-catalysed curing yields glass-to-glass adhesive strengths of 25.8 ± 4.8 N cm−2 and PMMA-to-PMMA adhesion at 21.9 ± 2.5 N cm−2 compared to 29.7 ± 0.1 N cm−2 and 22.9 ± 1.2 N cm−2, respectively, that were achieved photochemically.44 The larger error margin is due to the adventitious coverage of the acid-catalysis sometimes leaving residual Z-isomer, most notably upon the more rapid acid catalysis with HCl which yields an overall lower glass-to-glass adhesive strength of 15.9 ± 3.0 N cm−2. The photochemical debonding in the acid-cured adhesives is fully retained, nevertheless, with identically swift response times of photochemical debonding, i.e., ca. 0.5 seconds, under near-maximum loads (Fig. 6B, Video S4†). To further probe the influence of acid on adhesive strength, introduction of CF3COOH during complete thermal liquefication and resolidification, i.e., adding vapours after heating past the AIZ melting point and immediately conjoining the slides, was found to yield a thermal adhesive strength of 13.9 ± 1.1 N cm−2. Although the thermal adhesion of 19.6 N cm−2 without acid vapours was previously also shown to be lower than in the photochemical pathway,44 the adhesive strength is affected by acid vapours more strongly in the thermal pathway. Thus it seems that the efficiency of the catalytic reversion plays a key role in attaining adhesive strengths nearing those obtained photochemically.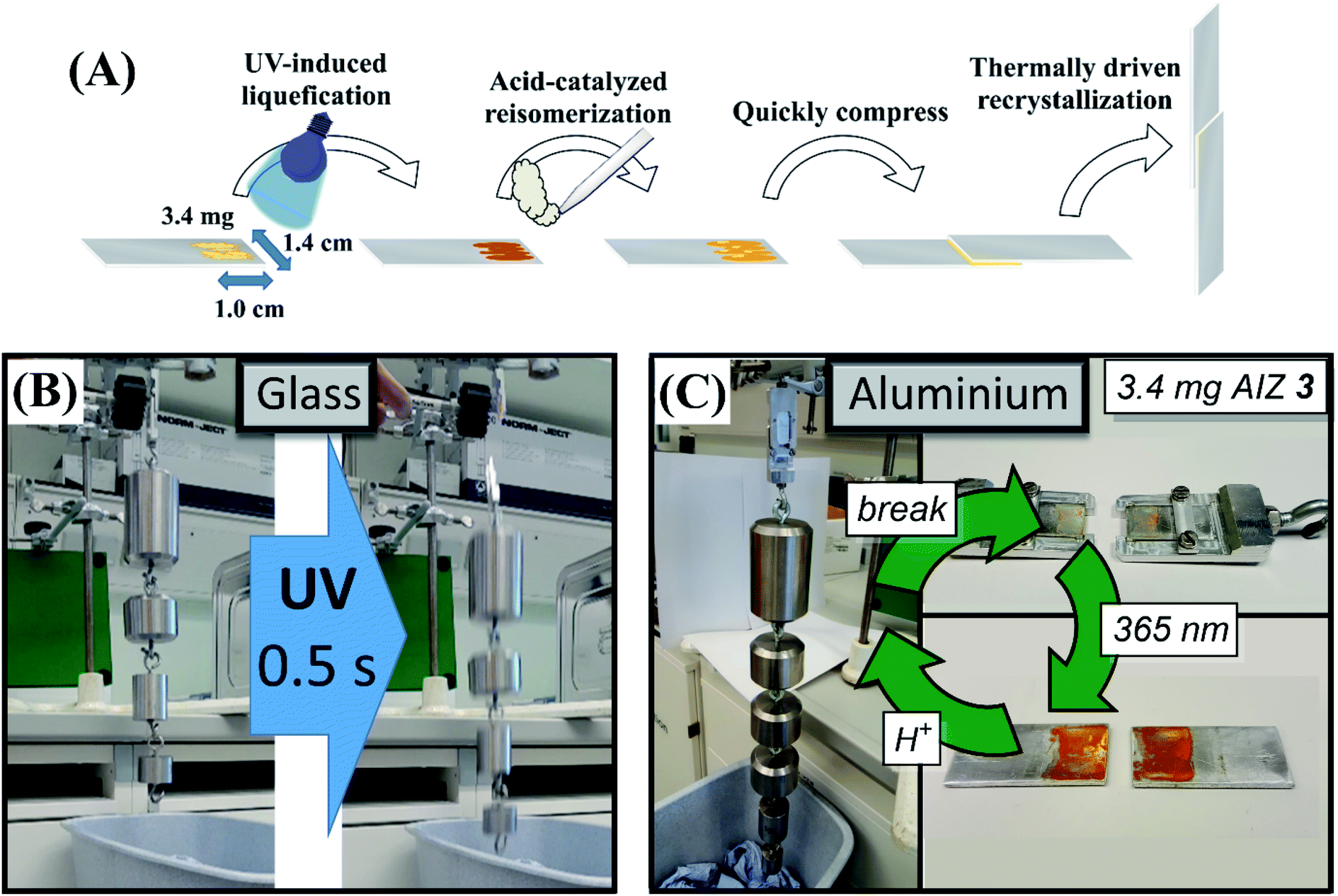 | ||
| Fig. 6 (A) Strategy for acid-catalysed adhesion independent of the transparency of substrates. Acid-catalysis by CF3COOH acid vapours causes near-instant Z-to E-reisomerisation of photoliquefied AIZ 3. The several seconds that separate the catalytic recovery of E-AIZ and the advent of nucleation sites for recrystallisation provide time to press slides together before the solidification takes place. (B) Suspension of glass slides conjoined with 2.2 mg of compound 3 at a 1.0 by 1.4 cm overlapping surface area, resulting in an adhesive strength of 25.8 ± 4.8 N cm−2 (shown here to hold 3.8 kg). The UV-induced debonding44 is unaffected by the acid-catalysis over 5 cycles of acid-catalysed bonding and photochemical debonding (Video S4†), fully retaining response times of ca. 0.5 s at near-maximum loads. (C) Suspension of aluminium slides conjoined with 3.4 mg AIZ 3 at a 1.0 by 1.4 cm overlapping surface area, capable of holding a load of 35.8 ± 3.5 N cm−2 (over 15 samples, shown here to hold 5.7 kg). Although the non-transparency rules out photochemical annealing, acid-catalysed bonding can be repeatedly performed at 35.0 ± 2.8 N cm−2 over 5 cycles of physical debonding. | ||
Interestingly, the lingering presence of trace acid vapours between adhered slides also invokes a self-healing mechanism in the AIZ adhesive (Video S5†), persisting after 10 days still (Video S6†). The debonding is found to be unaffected by the self-healing, as the latter occurs over a longer timescale than the near-instant photoliquefication of the interfacial thin layer that dictates the adhesive strength. The mechanism of the acid-catalysed reversion thus additionally holds promise for increased resilience against exposure to ambient UV. Furthermore, while the photoreversible nature of the solid-to-liquid phase transition requires the use of at least transparent surface to harden the versatile adhesive at a range of metallic and polymeric substrates,44 the acid-catalysed recrystallisation is wavelength independent and opens up to non-transparent surfaces being conjoined also.
In this way, the acid-catalysed pathway further broadens the scope to completely non-transparent substrates with, e.g., a pair of aluminium slides being able to bear 35.8 ± 3.5 N cm−2 (Fig. 6C, S26†). Though the adhesion of non-transparent surfaces cannot be reversed with light, physical separation of such surfaces (i.e., breaking of the cohesion by force, or heating over the melting point of the adhesive44) still yields the chemically unaltered AIZ 3. Accordingly, a pair of aluminium slides was repeatedly glued together to successfully yield an adhesive strength of 35.0 ± 2.8 N cm−2 over 5 bonding/debonding cycles without increasing the amount of adhesive agent.
Conclusions
Previously, the arylazoisoxazole class of azo switches was shown to possess promising photophysical characteristics in solution as well as the solid-state. The solid-state E-to Z-AIZ photoswitching accompanied by a reversible solid-to-liquid phase transition made the photoswitch extremely suitable for application in fully photocontrolled reversible adhesion. Here, we demonstrate that the acid-catalysed Z-to E-AIZ reversion in solution is tuneable by the pKa of the acid employed. We show that although such acid-catalysis was observed for azobenzenes and azopyrazoles before, the key to this reactivity is that the acid strength is intermediate to the Z-and E-isomer. Commonly employed acids possess a pKa lower than only that of Z-AIZH+, thermodynamically driving the isomerisation over the significantly lower Z-AIZH+ to E-AIZH+ energy barrier as confirmed by DFT. Conversely, when an acid is employed that possesses a pKa that is lower than both the Z-and E-isomer, such as CF3SO3H, it leads to a gradual establishment of the equilibrium between the now thermally stable E-AIZH+ and Z-AIZH+, giving rise to several regimes of photoreversible control over a 4-state switching cycle. Most interestingly, the acid catalysis observed in the solution studies applies to the pristine liquefied Z-isomer also, providing a chemical trigger for rapid and wavelength-independent hardening of the adhesive with HCl or CF3COOH vapours. Moreover, although the E-isomer has no strong enough interaction with CF3COOH vapours, traces of the acid remain after the acid-catalysed adhesion and provide a continual self-healing mechanism without distorting the adhesive properties. The non-destructive chemical hardening of the adhesive and the fundamental understanding thereof opens up further opportunities in the use of the azoisoxazole class of photoswitches, not least the successful adhesion of non-transparent substrates. Furthermore, the adhesive shows excellent reusability after physical breaking of cohesion between non-transparent substrates, retaining its “green” application. Ultimately, the expanded understanding of the acid-reactivity in azoisoxazoles and the demonstrated advance in application have broad implications for the pH-tuneable functionality of the myriad of existing azobenzenes and azoheterocycles.Data availability
The datasets supporting this article have been uploaded as part of the ESI.Author contributions
LK conceptualised the work and refined it with the help of JS, NBA and BJR. JS performed the synthesis and characterisation of compounds 1–4. LK performed the primary investigation, NBA supported on the solution studies. MB performed the theoretical investigation under supervision of NLD. BJR provided supervision and reviewed and edited the draft, LK wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for funding by the Alexander von Humboldt Foundation (postdoctoral fellowship to LK) and the Deutsche Forschungsgemeinschaft (project Z01 in CRC 858 to MB and NLD and project A01 in CRC 1459 to BJR).Notes and references
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Adv. Mater., 2020, 32, 1905966 CrossRef CAS PubMed.
- C. Buten, L. Kortekaas and B. J. Ravoo, Adv. Mater., 2020, 32, 1904957 CrossRef CAS PubMed.
- M. Baroncini, G. Ragazzon, S. Silvi, M. Venturi and A. Credi, Pure Appl. Chem., 2015, 87, 537–545 CAS.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- Z. L. Pianowski, Chem. - Eur. J., 2019, 25, 5128–5144 CrossRef CAS.
- A. Fihey, A. Perrier, W. R. Browne and D. Jacquemin, Chem. Soc. Rev., 2015, 44, 3719–3759 RSC.
- A. Abdollahi, H. Roghani-Mamaqani, B. Razavi and M. Salami-Kalajahi, Polym. Chem., 2019, 10, 5686–5720 RSC.
- M. Stratigaki and R. Göstl, ChemPlusChem, 2020, 85, 1095–1103 CrossRef CAS PubMed.
- H. Nie, J. L. Self, A. S. Kuenstler, R. C. Hayward and J. Read de Alaniz, Adv. Opt. Mater., 2019, 7, 1900224 CrossRef.
- L. Kortekaas, J. Chen, D. Jacquemin and W. R. Browne, J. Phys. Chem. B, 2018, 122, 6423–6430 CrossRef CAS PubMed.
- G. S. Hartley, J. Chem. Soc., 1938, 633–642 RSC.
- S. Ciccone and J. Halpern, Can. J. Chem., 1959, 37, 1903–1910 CrossRef CAS.
- R. Lovrien and J. C. B. Waddington, J. Am. Chem. Soc., 1964, 86, 2315–2322 CrossRef CAS.
- G. Wettermark, M. E. Langmuir and D. G. Anderson, J. Am. Chem. Soc., 1965, 87, 476–481 CrossRef CAS.
- N. J. Dunn, W. H. Humphries, A. R. Offenbacher, T. L. King and J. A. Gray, J. Phys. Chem. A, 2009, 113, 13144–13151 CrossRef CAS PubMed.
- A. M. Sanchez, M. Barra and R. H. de Rossi, J. Org. Chem., 1999, 64, 1604–1609 CrossRef CAS.
- E. Sawicki, J. Org. Chem., 1957, 22, 1084–1088 CrossRef CAS.
- L. P. Hammett, J. Am. Chem. Soc., 1928, 50, 2666–2673 CrossRef.
- M. S. Scholz, J. N. Bull, N. J. A. Coughlan, E. Carrascosa, B. D. Adamson and E. J. Bieske, J. Phys. Chem. A, 2017, 121, 6413–6419 CrossRef CAS.
- S. Stoyanov, L. Antonov, T. Stoyanova and V. Petrova, Dyes Pigm., 1996, 32, 171–185 CrossRef CAS.
- P. Weis and S. Wu, Macromol. Rapid Commun., 2018, 39, 1700220 CrossRef PubMed.
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687 CrossRef CAS PubMed.
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef.
- M. J. Hansen, M. M. Lerch, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 55, 13514–13518 CrossRef CAS.
- S. Samanta, A. Babalhavaeji, M. Dong and G. A. Woolley, Angew. Chem., Int. Ed., 2013, 52, 14127–14130 CrossRef CAS.
- M. Dong, A. Babalhavaeji, M. J. Hansen, L. Kálmán and G. A. Woolley, Chem. Commun., 2015, 51, 12981–12984 RSC.
- M. Dong, A. Babalhavaeji, C. V. Collins, K. Jarrah, O. Sadovski, Q. Dai and G. A. Woolley, J. Am. Chem. Soc., 2017, 139, 13483–13486 CrossRef CAS PubMed.
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Acc. Chem. Res., 2015, 48, 2662–2670 CrossRef CAS.
- G. Wang, D. Yuan, T. Yuan, J. Dong, N. Feng and G. Han, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2768–2775 CrossRef CAS.
- Q. Bian, M. Jin, S. Chen, L. Xu, S. Wang and G. Wang, Polym. Chem., 2017, 8, 5525–5532 RSC.
- A. K. Saydjari, P. Weis and S. Wu, Adv. Energy Mater., 2017, 7, 1601622 CrossRef.
- D. Wang, M. Wagner, H.-J. Butt and S. Wu, Soft Matter, 2015, 11, 7656–7662 RSC.
- S. Crespi, N. A. Simeth and B. König, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef CAS.
- R. S. L. Gibson, J. Calbo and M. J. Fuchter, ChemPhotoChem, 2019, 3, 372–377 CrossRef CAS.
- J. Calbo, A. R. Thawani, R. S. L. Gibson, A. J. P. White and M. J. Fuchter, Beilstein J. Org. Chem., 2019, 15, 2753–2764 CrossRef CAS PubMed.
- S. Ludwanowski, M. Ari, K. Parison, S. Kalthoum, P. Straub, N. Pompe, S. Weber, M. Walter and A. Walther, Chem. - Eur. J., 2020, 26, 13203–13212 CrossRef CAS.
- C. E. Weston, R. D. Richardson, P. R. Haycock, A. J. P. White and M. J. Fuchter, J. Am. Chem. Soc., 2014, 136, 11878–11881 CrossRef CAS PubMed.
- L. Stricker, E.-C. Fritz, M. Peterlechner, N. L. Doltsinis and B. J. Ravoo, J. Am. Chem. Soc., 2016, 138, 4547–4554 CrossRef CAS.
- L. Stricker, M. Böckmann, T. M. Kirse, N. L. Doltsinis and B. J. Ravoo, Chem. - Eur. J., 2018, 24, 8639–8647 CrossRef CAS PubMed.
- P. Kumar, A. Srivastava, C. Sah, S. Devi and S. Venkataramani, Chem. - Eur. J., 2019, 25, 11924–11932 CrossRef CAS.
- L. Kortekaas, J. Simke, D. W. Kurka and B. J. Ravoo, ACS Appl. Mater. Interfaces, 2020, 12, 32054–32060 CrossRef CAS PubMed.
- A. Kütt, T. Rodima, J. Saame, E. Raamat, V. Mäemets, I. Kaljurand, I. A. Koppel, R. Y. Garlyauskayte, Y. L. Yagupolskii, L. M. Yagupolskii, E. Bernhardt, H. Willner and I. Leito, J. Org. Chem., 2011, 76, 391–395 CrossRef PubMed.
- R. J. W. Le Fèvre and J. Northcott, J. Chem. Soc., 1953, 867–870 RSC.
- J. Fregoni, G. Granucci, M. Persico and S. Corni, Chem, 2020, 6, 250–265 CAS.
- J. Dokić, M. Gothe, J. Wirth, M. V. Peters, J. Schwarz, S. Hecht and P. Saalfrank, J. Phys. Chem. A, 2009, 113, 6763–6773 CrossRef PubMed.
- L. Zhou, L. Chen, G. Ren, Z. Zhu, H. Zhao, H. Wang, W. Zhang and J. Han, Phys. Chem. Chem. Phys., 2018, 20, 27205–27213 RSC.
- D. L. Isac, A. Airinei, M. Homocianu, N. Fifere, C. Cojocaru and C. Hulubei, J. Photochem. Photobiol., A, 2020, 390, 112300 CrossRef CAS.
- M. A. Gerkman, R. S. L. Gibson, J. Calbo, Y. Shi, M. J. Fuchter and G. G. D. Han, J. Am. Chem. Soc., 2020, 142, 8688–8695 CrossRef PubMed.
- Y. Nagai, K. Ishiba, R. Yamamoto, T. Yamada, M. Morikawa and N. Kimizuka, Angew. Chem., Int. Ed., 2021, 60, 6333–6338 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: additional UV/vis and NMR data, additional results of DFT calculations, methods. See DOI: 10.1039/d1sc03308e |
| ‡ Lead contact. |
| This journal is © The Royal Society of Chemistry 2021 |