
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRigidochromism by imide functionalisation of an aminomaleimide fluorophore†
Jonathan T.
Husband
 a,
Yujie
Xie
a,
Thomas R.
Wilks
a,
Louise
Male
a,
Miquel
Torrent-Sucarrat
bc,
Vasilios G.
Stavros
d and
Rachel K.
O'Reilly
*a
a,
Yujie
Xie
a,
Thomas R.
Wilks
a,
Louise
Male
a,
Miquel
Torrent-Sucarrat
bc,
Vasilios G.
Stavros
d and
Rachel K.
O'Reilly
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: r.oreilly@bham.ac.uk
bDepartment of Organic Chemistry I, Universidad del País Vasco (UPV/EHU), Donostia International Physics Center (DIPC), Manuel Lardizabal Ibilbidea 3, Donostia, 20018, Spain
cIkerbasque, Basque Foundation for Science, Plaza Euskadi 5, 48009 Bilbao, Euskadi, Spain
dDepartment of Chemistry, University of Warwick, Coventry, CV4 8AL, UK
First published on 14th July 2021
Abstract
Fluorescent dyes that exhibit high solid state quantum yields and sensitivity to the mechanical properties of their local environment are useful for a wide variety of applications, but are limited in chemical diversity. We report a trityl-functionalised maleimide that displays rigidochromic behaviour, becoming highly fluorescent when immobilised in a solid matrix, while displaying negligible fluorescence in solution. Furthermore, the dye's quantum yield is shown to be sensitive to the nature of the surrounding matrix. Computational studies reveal that this behaviour arises from the precise tuning of inter- and intramolecular noncovalent interactions. This work expands the diversity of molecules exhibiting solid state environment sensitivity, and provides important fundamental insights into their design.
Introduction
Understanding how changes in the chemical and physical environment impact a material's properties is essential for the development of advanced technologies. For heterogeneous, dynamic systems, however, detecting such changes presents a formidable challenge, because bulk measurement techniques often lack the spatial resolution and sensitivity required, and are difficult to operate in parallel. In this context, fluorescence has emerged as a powerful alternative detection method: it is highly sensitive, can resolve structures down to tens of nanometers in size, can report in real time and may be readily multiplexed.1–4 Dyes whose fluorescence properties change in response to local stimuli have been employed as biological labels and probes,5 medical diagnostics,6 and photoactive materials in light-emitting devices.7 Recently, the importance of sensing changes in mechanical properties such as viscosity and rigidity has become especially clear. For example, cells have been found to sense the stiffness of their environment to determine differentiation behaviour,8 and to use mechanical stress to regulate various biological processes.9–11 Unsurprisingly, given the advantages outlined above, there has been a growing interest in the use of mechanically-sensitive (rigidochromic) fluorophores in the investigation of such phenomena.12–14 These dyes have allowed real-time visualisation of the different mechanical environments in living cells,15,16 as well as in situ probing of macromolecular self-assembly.17,18Many rigidochromic fluorescent dyes work via a restriction of intramolecular motions (RIM) mechanism, i.e., an increase in the rigidity of the local environment prevents the molecule from adopting conformations that give access to the ground state via non-radiative routes. A typical example of such a conformation is a twisted intramolecular charge transfer (TICT) complex, accessed by motion of a molecular ‘rotor’.14 In low rigidity environments, formation of the TICT complex is possible by movement of the rotor, leading to weak or negligible emission. In contrast, higher rigidity settings introduce a significant energy barrier to TICT complex formation, favouring radiative decay and a switching on of fluorescence emission. As well as changes in emission intensity, selection between these two pathways often results in changes in fluorescence lifetimes and/or emission maxima, which serve as further handles for detection. However, in order for the full potential of rigidochromic dyes to be realised, it is vital to obtain fundamental insights into their design, and to allow for expansion of their chemical diversity.
Recently, maleimide-based charge transfer fluorophores have emerged as an interesting new class of environment-sensitive dye.19,20 They are small and uncharged, but still relatively polar, and have large Stokes shifts (typically >100 nm) rendering them suitable for a range of applications including cell barcoding21 and tracking polymer self-assembly.22 However, their rigidochromic properties have so far not been investigated. Previous work has shown how straightforward alterations to the donor groups (Scheme 1) can be used to tune maleimide fluorescence properties.20 In theory, changes to the imide substituent should also affect dye behaviour. Indeed, the introduction of aromatic substituents directly adjacent to the imide has been shown to extinguish fluorescence.19,22 Of greater interest, recent publications from Tang and coworkers showed that certain aminomaleimides with alkyl imide substituents exhibit dual (solid/solution) state emission, with impressive solid state quantum yields.23 Naka and coworkers have also produced solid-state emitters by imide functionalisation of an aminomaleimide.24
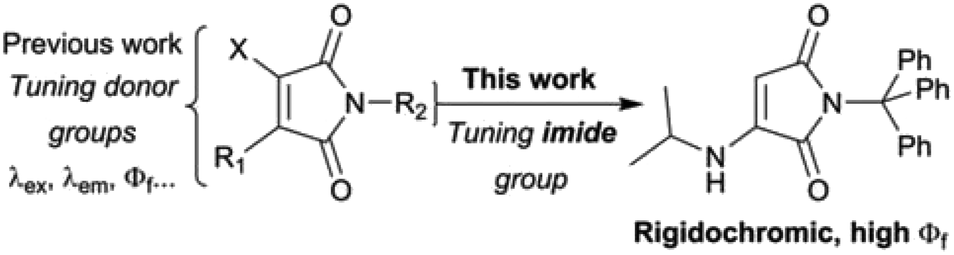 | ||
| Scheme 1 Optimisation of maleimide dye properties by tuning of the imide substituent. | ||
We reasoned that systematic tuning of the imide substituent could enable the creation of dyes with interesting new fluorescence properties. Here, we report a series of monoaminomaleimide (MAM) dyes substituted at the imine with phenyl derivatives. We find that increasing the number of phenyl rings dramatically impacts the fluorescence quantum yield. In particular, the introduction of a trityl group (three phenyl rings) results in both quenching of solution-state fluorescence and the emergence of bright solid-state fluorescence. We attribute this rigidochromic behaviour to a complex interplay between the maleimide fluorophore, the phenyl ring ‘rotors’ and the environment. In solution, the phenyl rings are free to rotate, allowing the adoption of conformations that favour non-radiative relaxation to the ground state. In the solid-state, however, the environment restricts movement of the phenyl rings, shutting down this quenching pathway and the emergence of bright fluorescence emission. The dye preserves a high solid-state quantum yield even when dispersed in a soft flexible polymer, and we further demonstrate the sensitivity of its fluorescence to the chemical nature of the solid matrix. Our work illuminates how precise tuning of inter- and intramolecular bonds can be exploited to create environment-sensitive dyes with unique and highly optimised emission properties.
Results and discussion
To obtain the dye series, mono- and diphenyl-substituted monobromomaleimide intermediates (MBMs: BzMBM and Bz2MBM) were synthesised by reaction of the appropriate mono- or dibenzylamine with bromomaleic anhydride (Fig. 1a, route Ia).25 Trityl-MBM (Bz3MBM) was obtained by reaction of bromomaleimide with trityl chloride and triethylamine (Fig. 1a, route Ib). To all MBM intermediates, isopropylamine was added to form isopropylamino-substituted MAMs (Fig. 1a, step II). N-phenyl MAM (PhMAM) and N-H MAM (HMAM) were also synthesised for comparison (Fig. 1b) using previously reported methods.19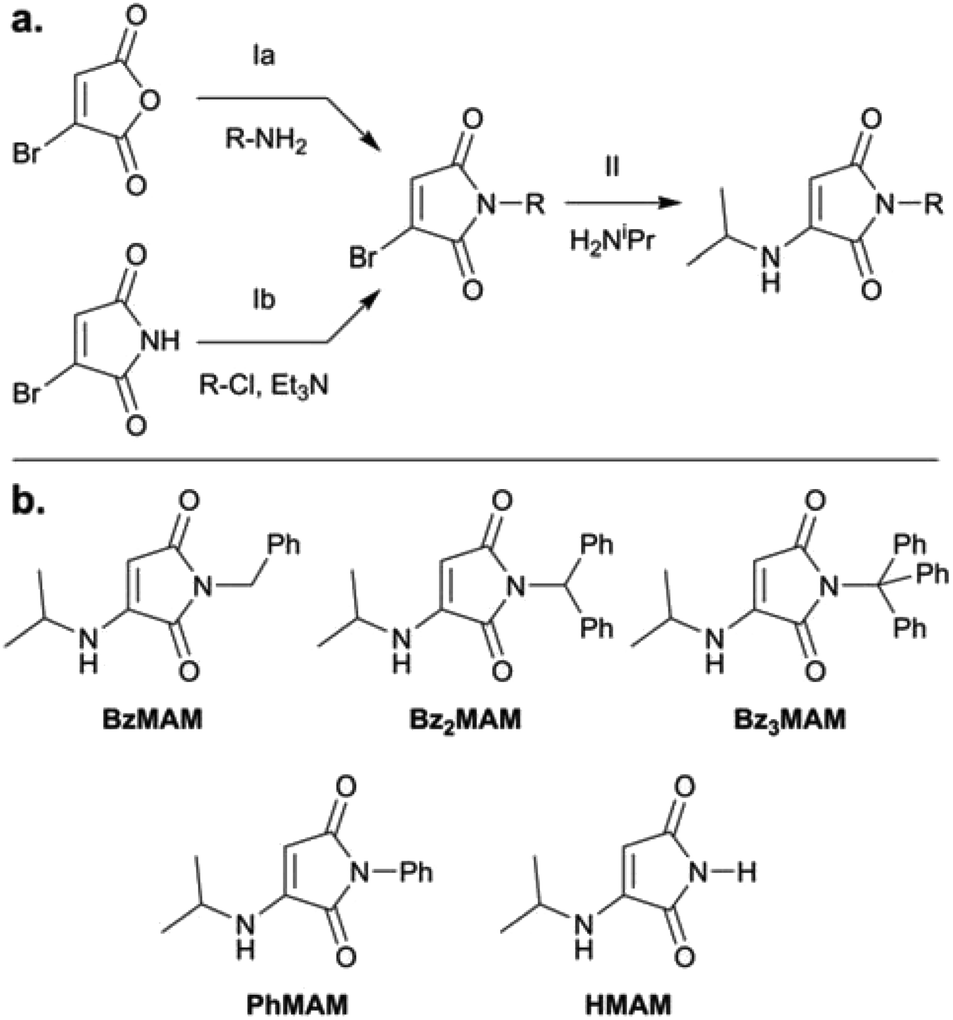 | ||
| Fig. 1 (a) Synthetic routes for the dye series. To generate the monobromomaleimide (MBM) iintermediates, route Ia was followed for all structures except Bz3MBM, when it was necessary to adopt route Ib. (b) Structures of the series of dyes used in this work. | ||
The fluorescence properties of the series were first characterised in solution, using a range of solvents with different polarities (Table 1 and Fig. 2). All dyes showed λmax emission and excitation values similar to previously reported MAMs,19,20 with excitation maxima around 350–370 nm and emission maxima around 460–480 nm (Table 1 and Fig. S14–16†).
| λ ex/λem (nm) | |||||||
|---|---|---|---|---|---|---|---|
| MeOH | DMSO | Dioxane | THF | Toluene | Et2O | Solid stateb | |
| a Not recordable. b Solid state excitation spectra were very broad, so extraction of λex was not possible. | |||||||
| HMAM | 369/489 | 357/469 | 346/452 | 346/447 | 341/443 | 341/441 | –/486 |
| PhMAM | —a | 370/505 | 361/503 | 360/501 | 358/502 | 359/500 | –/488 |
| BzMAM | 370/491 | 371/490 | 362/478 | 361/472 | 355/471 | 357/469 | –/477 |
| Bz2MAM | 367/499 | 370/484 | 359/464 | 359/462 | 355/460 | 353/456 | –/498 |
| Bz3MAM | 364/488 | 363/476 | 352/459 | 352/465 | 352/465 | 348/450 | –/472 |
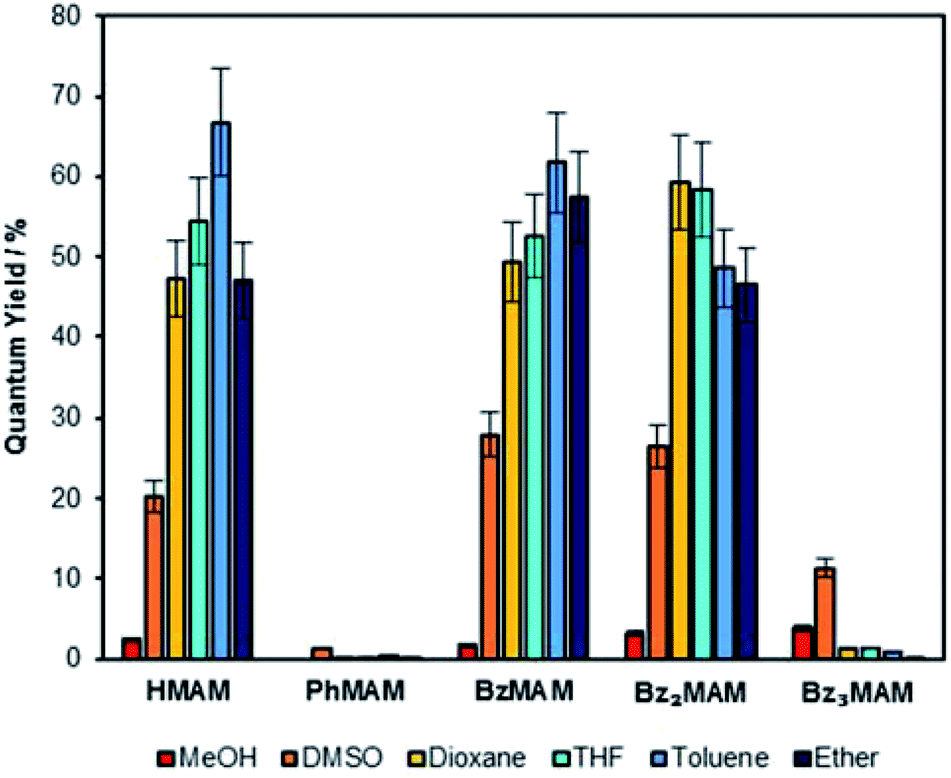 | ||
| Fig. 2 Solution-state quantum yields for the series of dyes in a range of different solvents, calculated via a reference method.26 | ||
The excitation maxima (λex) for the control dye HMAM became blue-shifted in less polar solvents. The benzyl dyes also displayed this behaviour, but the effect was less pronounced. For example, the maximum blue shift observed for the excitation maximum (Δλex) of HMAM was 28 nm (MeOH vs. Et2O), whereas for BzMAM it was 16 nm (DMSO vs. toluene). Bz2MAM and Bz3MAM showed similar maximum Δλex values of around 15–17 nm. Emission maxima (λem) were also blue-shifted in non-polar solvents, but again this effect was slightly less significant for the benzyl dyes than the control, e.g. the largest Δλem for HMAM was 48 nm while for Bz3MAM it was 38 nm (MeOH vs. Et2O in both cases). The attachment of benzyl derivatives at the imide position, therefore, appeared to reduce the impact of solvent polarity on the excitation and emission maxima of MAM dyes.
The solution-state quantum yields were also calculated via a reference method employing quinine.26 The control dyes (HMAM and PhMAM), BzMAM and Bz2MAM all followed expected trends, exhibiting Φf of 30–70% in non-polar solvents, with a significant decrease in Φf in DMSO and methanol (Fig. 2). In line with previous results,27 the observed fluorescence quenching in polar solvents was attributed to electron-driven proton transfer from the solvent to the maleimide via hydrogen bonds, and this was validated computationally (see ESI 5†). Bz3MAM, by contrast, exhibited negligible fluorescence in apolar solvents (Φf(%) <1.5%), while retaining moderate quantum yields in polar environments. This unexpected reversal of polarity-dependence suggested that Bz3MAM followed a different fluorescence mechanism to the other MAM dyes.
To give further insight into the differences in photophysical behaviour brought about by the trityl group, the dyes were next examined in the solid-state. Excitation profiles were broad, so it was not possible to extract meaningful values for λex. However, emission maxima were obtained and found to be similar to those reported for other MAMs (Table 1).19,23 Solid-state quantum yields, Φf(%), for BzMAM and Bz2MAM were measured using an integrating sphere set-up, and found to be similar to that of the control dye HMAM, at around 25% (Fig. 3). In stark contrast, Bz3MAM showed bright solid-state emission with a Φf of 74%, the highest reported to date for an aminomaleimide dye, providing further evidence for an alternative fluorescence mechanism operating in this dye.
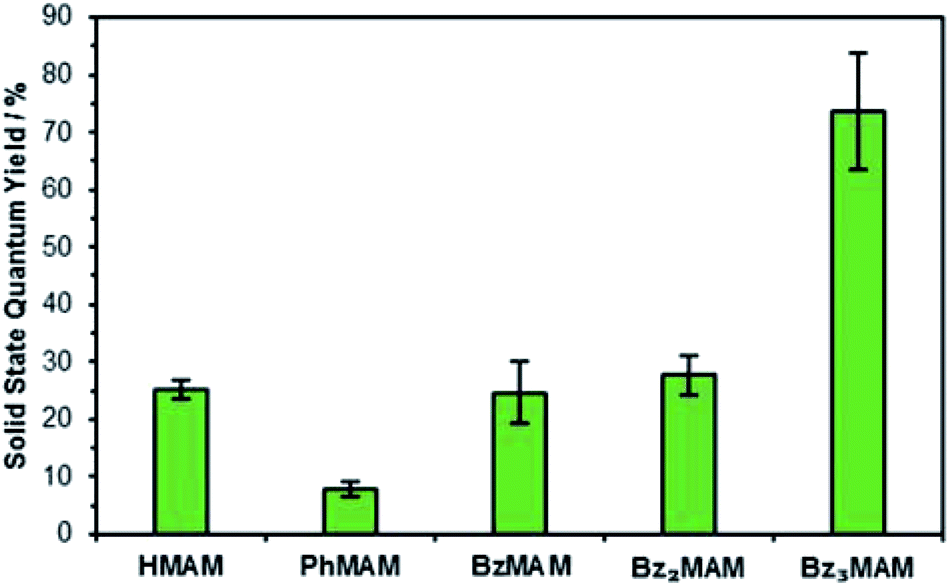 | ||
| Fig. 3 Solid-state quantum yields for the dye series, measured by an absolute method using an integrating sphere. | ||
To probe the chemical origins of the surprising fluorescence behaviour of Bz3MAM, crystal structures for the series (HMAM, BzMAM, Bz2MAM, and Bz3MAM) were obtained. HMAM and BzMAM both crystallised with anti-parallel off-centre maleimide rings, with ring–ring centroid distances of 3.65 Å in HMAM and 4.05 Å in BzMAM (Table 2). These ring orientations and spacings are consistent with π–π stacking interactions.28,29 In Bz2MAM, inter-ring centroid distances became larger at 5.04 Å, while the inter-plane distance decreased to 2.76 Å, values that are consistent with off-centre π–π stacking. Intermolecular quenching interactions, therefore, seem the most likely explanation for the moderate solid state Φf observed for these dyes. For Bz3MAM, by contrast, the shortest observed maleimide ring centroid distance was 6.46 Å, and above and below the maleimide rings sat methyl or benzyl groups, suggesting no significant maleimide π–π quenching interactions were possible. This difference was highlighted further by visualisation of noncovalent interactions (NCI) (Fig. 4, see ESI 5.1† for details).30,31 In this visualisation method, the electron densities are calculated for each crystal structure and a reduced density gradient isosurface plotted based on the second eigenvalue of the electron density Hessian, λ2. Negative values of λ2 are coloured blue, and are associated with strong attractive interactions such as hydrogen bonds, while red denotes repulsive interactions (positive λ2) such as steric clashes. Green represents intermediate values of λ2 and is associated with weak interactions such as π stacking, and this was clearly observed between the maleimide rings of HMAM, BzMAM and Bz2MAM (Fig. 4). For Bz3MAM the NCI plot became much more diffuse, suggesting the absence of any significant maleimide π–π interactions (Fig. 4, right).
| Ring plane anglea | Ring centroid distancea Å−1 | Inter plane distanceb Å−1 | |
|---|---|---|---|
| a Measured distance and angle between ring centroid of N1C1C2C3C4 and N1iC1iC2iC3iC4i (HMAMi= 1 − X, +Y, −1/2 + Z. BzMAMi = 1 − X, 1 − Y, 1 − Z. Bz2MAMi = 1 − X, 1 − Y, −Z. Bz2MAM (N101 etc.) i = −X, 2 − Y, 1 − ZBz3MAMi = +Y, +X, 1 − Z). b Measured distance between ring centroid of N1C1C2C3C4 and plane of N1iC1iC2iC3iC4i. See ESI 4.3 for more parameters and further details. | |||
| HMAM | 9.8° (±0.14) | 3.649 (±0.001) | 3.520 (±0.001) |
| BzMAM | 0.0° (±0.07) | 4.053 (±0.001) | 3.519 (±0.002) |
| Bz2MAM (N101 etc.) | 0.0° (±0.05) | 5.042 (±0.003) | 2.764 (±0.009) |
| Bz3MAM | 13.0° (±0.3) | 6.464 (±0.004) | 5.885 (±0.007) |
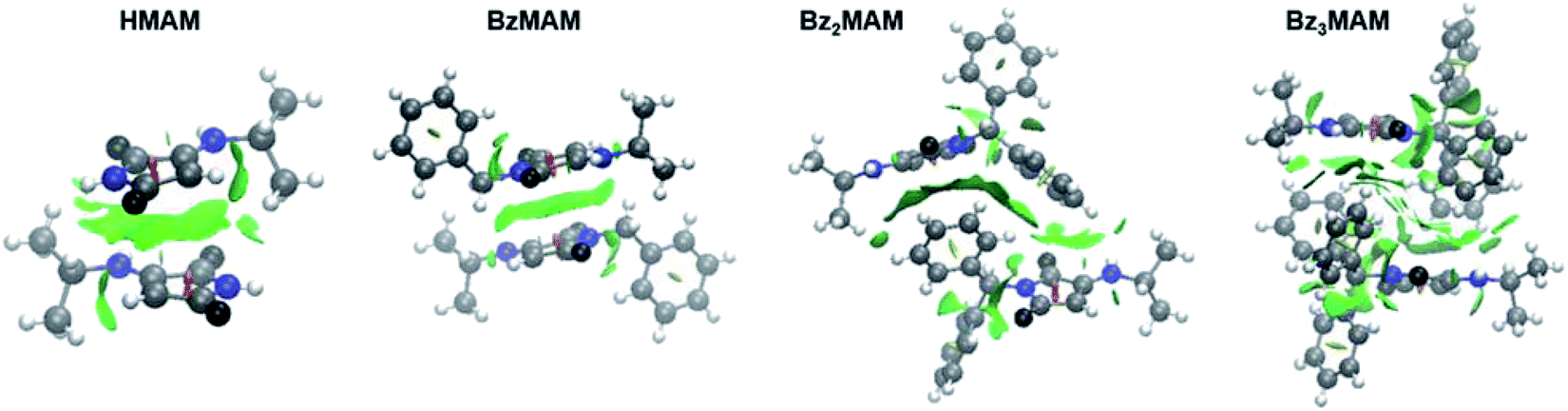 | ||
| Fig. 4 Crystal structures of HMAM, BzMAM, Bz2MAM, and Bz3MAM overlaid with CAM-B3LYP/6-311G(d,p) gradient isosurfaces with s = 0.5 and a blue–green–red colour scale from −0.05 <ρ sign(λ2) <0.05 au (see ESI 5† for further details). | ||
The conformations observed by crystal structure analysis helped to rationalise the differences in solid state Φf for the dye series, but provided no explanation for the low solution state fluorescence of Bz3MAM. To address this, the electron densities for the HOMO and natural population charges for the dyes were modelled at the CAM-B3LYP-D3BJ/6-311G(d,p) level of theory,32 with the effect of the solvent (diethyl ether) taking into account using the polarisation continuum model (PCM) (see ESI 5†). To measure the electron redistribution of the different substituents, the sum of the natural population charges of the π-conjugated structure in the aminomaleimide ring (QNBO1) was evaluated. The results (Fig. 5, ESI 5.1†) showed that for BzMAM and Bz2MAM, the majority of the electron and HOMO densities was located on the maleimide group in comparison with the rest of the molecule, with negative QNBO1 values. To our surprise, however, modelling of Bz3MAM showed that in this dye, the electron and HOMO densities were spread quite evenly across the maleimide and trityl groups, resulting in a positive value for QNBO1 (Fig. 5).
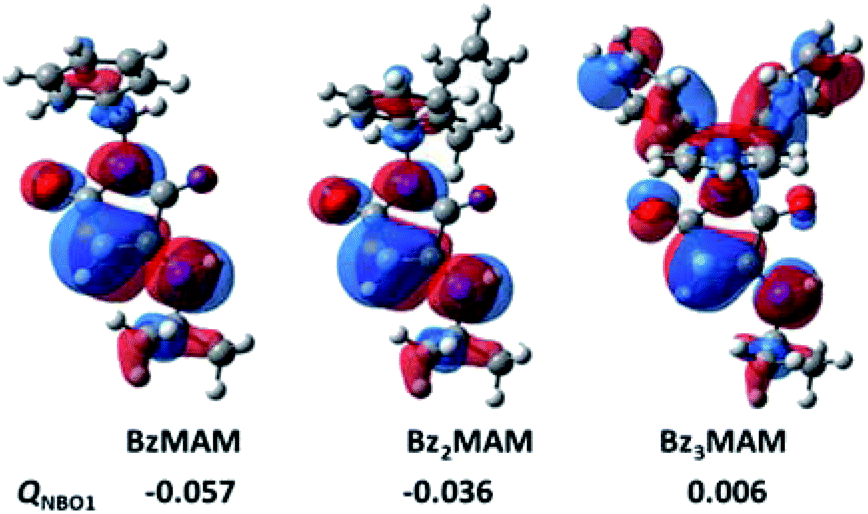 | ||
| Fig. 5 HOMO isosurfaces of the benzyl MAM dye series and the sum of the natural population charges of the π-conjugated structure in the aminomaleimide ring (QNBO1). | ||
The distribution of electron density across both the maleimide and trityl groups of Bz3MAM suggested the possibility that the phenyl rings might be involved in modulating the dye's fluorescence behaviour. Many dyes that exhibit aggregation-induced emission (AIE) and rigidochromic (viscosity-sensing) behaviour feature phenyl ‘rotors’ attached to the dye core.14,33 Upon excitation, movement of the rotor causes the molecule to adopt a conformation that favours non-radiative relaxation back to the ground state. Normally, direct conjugation is required between the dye and rotor, but in our case the electron-withdrawing nature of the trityl group appeared to be sufficient to allow delocalisation of electrons across the tertiary carbon connecting to the maleimide group. This hypothesis led us to look more directly for evidence of AIE and rigidochromism in Bz3MAM. A solution of Bz3MAM dissolved in dioxane was titrated with water (in which the dye was insoluble). As anticipated, fluorescence emission gradually increased with water content (Fig. 6), with the 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water:dioxane mixture leading to a more than sixteen-fold increase in fluorescence emission compared to pure dioxane, confirming that the dye was indeed undergoing AIE.
:1 water:dioxane mixture leading to a more than sixteen-fold increase in fluorescence emission compared to pure dioxane, confirming that the dye was indeed undergoing AIE.
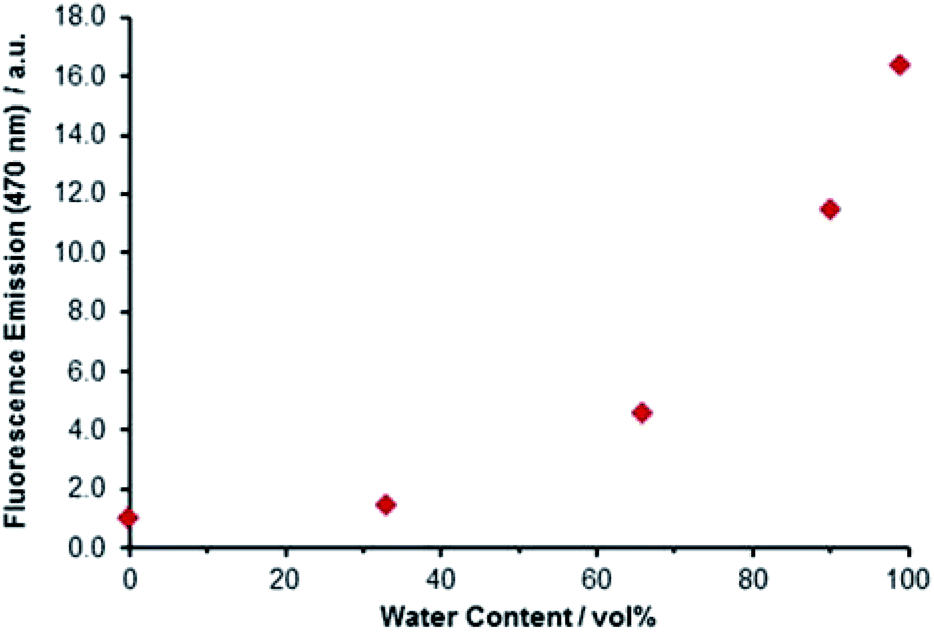 | ||
| Fig. 6 Aggregation-induced emission of Bz3MAM. A solution of Bz3MAM in dioxane was titrated with water, resulting in an increase in fluorescence upon precipitation of the dye molecule. | ||
To further test the rigidochromic behaviour of Bz3MAM, we investigated its ability to sense the nature of its solid state environment. We began by examining the effect of dispersion in the same solid matrix on Φf for all dyes in the series. Each dye was drop-cast with 12 kDa poly(ethylene glycol) (PEG) to form a dye-doped powder, and the quantum yields of these powders were then measured using an absolute method. As expected, the incorporation of HMAM, BzMAM and Bz2MAM into PEG resulted in an increase in emission, approximately two-fold. For example, the Φf of HMAM increased from 25% to 49% (Fig. 7). These data confirmed an aggregation-caused quenching (ACQ) effect in the pure solids, which we attributed to the π–π interactions revealed by the crystal structures described above. It seems reasonable to assume that, upon incorporation into a polymer, individual dye molecules are separated, reducing this intermolecular quenching effect. Bz3MAM, meanwhile, showed a negligible change in emission, presumably because quenching interactions were already prevented in the pure solid by the bulky trityl group. For comparison, we also incorporated an archetypal AIE dye, tetraphenylethylene-aldehyde (TPE),34,35 into PEG, which led to a dramatic reduction in its Φf (from 26% to 2%). In line with previous reports, this reduction most likely arose because PEG is not rigid enough to prevent TPE from accessing molecular conformations that favour non-radiative decay.36
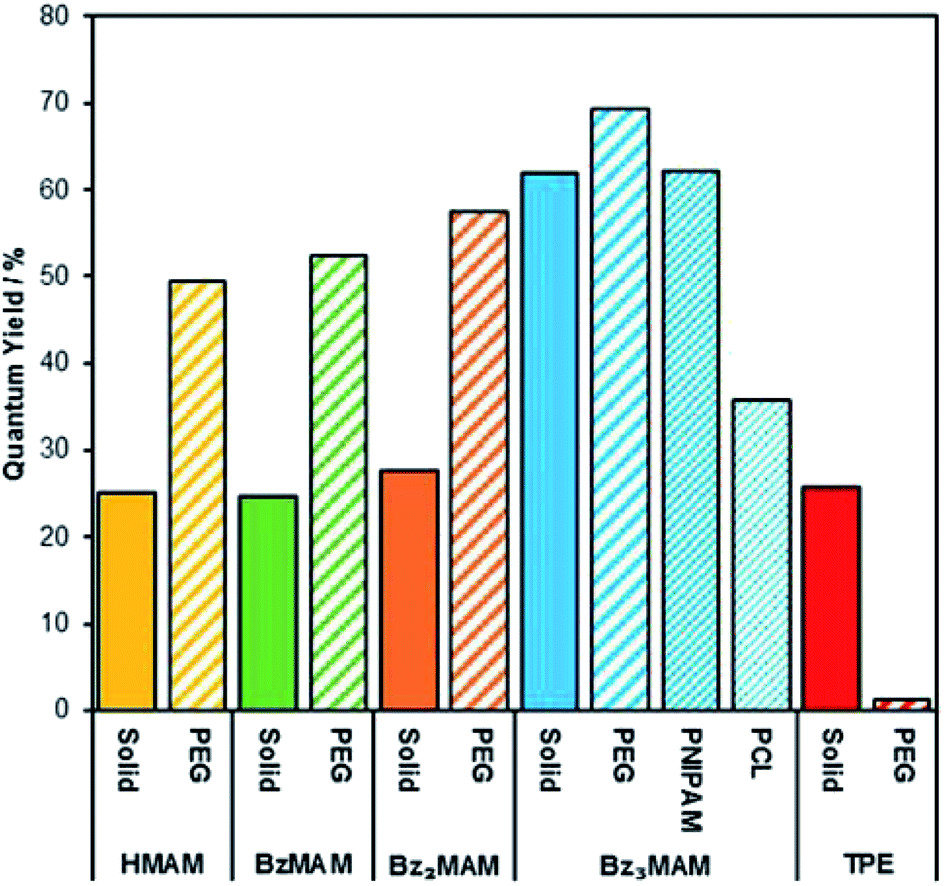 | ||
| Fig. 7 Solid-state quantum yields of the dyes before and following incorporation into different polymer matrices. | ||
To provide further evidence that delocalisation of electron density onto the trityl group was key to the dye's rigidochromic behaviour, an analogous aminobromomaleimide (ABM) dye was synthesised (Bz3ABM). Introduction of a bromine atom on the maleimide ring was expected to redistribute electron density away from the maleimide ring and trityl group, despite the mesomeric donor character of Br, which we assume is less important for these dyes. Indeed computational studies showed very little electron density residing on the trityl group in this molecule (Fig. S28† and QNBO1 value of 0.126 a.u.). As expected, Bz3ABM did not exhibit rigidochromic behaviour, and instead behaved in a similar way to BzMAM and Bz2MAM, with relatively high solution-state and moderate solid-state fluorescence quantum yields (Tables S2 and S3†).
We next investigated how variation of the polymer matrix would affect the Φf of Bz3MAM. Two further polymer environments were investigated: poly(N,N-isopropylacrylamide) (PNIPAM) and poly(caprolactone) (PCL). The solid state Φf remained high for Bz3MAM dispersed in PNIPAM, but dropped significantly – to 36% – in PCL. PEG, PNIPAM and PCL provide three distinct chemical environments, with varying polarity and numbers of H-bond donors/acceptors. The three polymers also vary in their rigidity: PEG of the molecular weight used above (12 kDa) typically has a Tg of around −20 °C,37 while the Tg of PNIPAM is much higher at ∼130 °C38 and that of PCL lower at ∼−60 °C.39 We considered the possibility that differences in the dispersibility of the dye in the different matrices was responsible for the changes in quantum yield. However, inspection of the solid state emission profiles (Fig. S17†) revealed negligible shift in the emission maximum as the matrix was changed, with all lying close to the value in DMSO (476 nm). The absence of any significant changes to the emission profiles suggested that the dye was equally effectively dispersed in all the matrices tested. We therefore propose that the sensitivity of Bz3MAM to its solid state environment arose from an interplay between the effects of rigidity and chemical functionality: variations in the environment's polarity are likely to alter the HOMO/LUMO energies (as shown by computational calculations in solution), while lower rigidity may lead to greater freedom for the trityl group to rotate, with a consequent increase in the amount of non-radiative energy dissipation. The noticeably different appearance of the PCL sample – a soft, tacky solid as opposed to the dry powders formed by PEG and PNIPAM – suggests that rigidity differences may be more important in this case, but further work is required to confirm the precise contributions of these two factors. Nevertheless, the results above clearly demonstrate the ability of the dye to sense and report on the mechanical and chemical properties of its surroundings.
The high Φf of Bz3MAM in even highly flexible polymers was surprising given the significant quenching observed for TPE, which is also thought to derive its solid state fluorescence from restriction of the motion of phenyl rotors.40 This difference in behaviour would be explained if the barrier to rotation of the phenyl groups was significantly higher for Bz3MAM than for TPE. Indeed, calculations showed that, in the gas-phase, the rotational barrier of TPE was 3 kcal mol−1 lower than Bz3MAM (see ESI 5.7†). A recent study showed how the introduction of intramolecular N–H⋯O interactions led to rigidochromic behaviour of an aniline dye,41 so we looked for evidence of this kind of supramolecular bonding. The crystal data showed that Bz3MAM exhibited noticeably shorter intramolecular carbonyl–phenyl interactions compared to the rest of the dye series (Fig. 8, top). Furthermore, significantly more intramolecular interactions were present between the phenyl rings and carbonyls, as highlighted by CAM-B3LYP/6-311G(d,p) NCI visualisation (Fig. 8, bottom). Based on these observations, we conclude that intramolecular hydrogen bonds exist between the phenyl protons and the carbonyl groups – an interaction known to exist in other aromatics and proteins42,43 – which are likely to augment the energy barrier to trityl rotation in the flexible polymer matrix. It is also possible that these intramolecular interactions mediate a certain level of through-space conjugation – as observed for other non-conjugated dyes44 – which may further explain the presence of electron density on the trityl group despite the lack of direct conjugation.
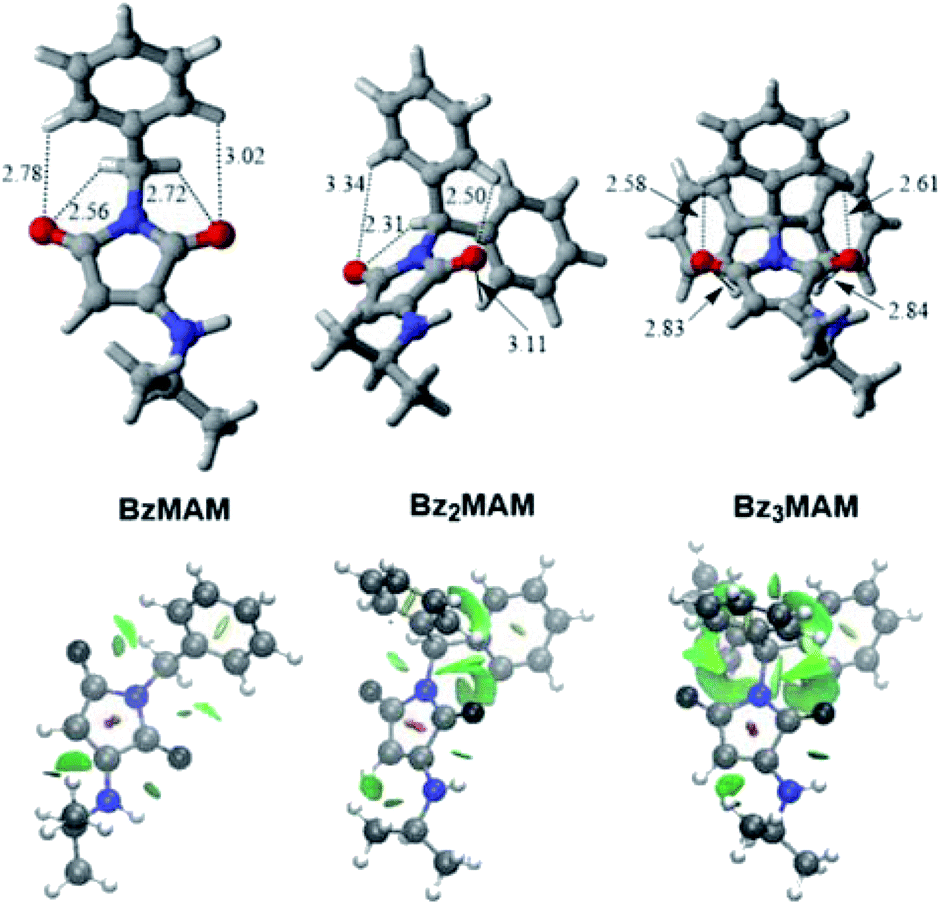 | ||
| Fig. 8 Visualisations of carbonyl–phenyl interactions for the dye series. Top: CAM-B3LYP-D3BJ/6-311G(d,p)-optimised geometries of the dyes with the most relevant distances (in Å) between the carbonyl oxygens of the maleimide ring and the hydrogens of the phenyl groups indicated. Bottom: CAM-B3LYP/6-311G(d,p) gradient isosurfaces with s = 0.5 for the dyes, with a blue-green-red colour scale from −0.05 <ρ sign(λ2) <0.05 au. | ||
We reasoned that trityl rotation might be promoted by an increase in temperature, so investigated the temperature-dependent fluorescence behaviour of Bz3MAM in DMSO. No significant variation in the fluorescence emission of the dye was observed over the range studied (20–80 °C), from which we conclude that the intramolecular interactions holding the trityl group in place were strong enough not to be disrupted even at relatively high temperatures (Fig. S19†). In addition, rigidochromic dyes often display viscosity-sensing behaviour,41 so we investigated the fluorescence of Bz3MAM in solvent mixtures of different viscosities. In particular, we were interested in the possibility that viscosity played a role in the unusually high solution state quantum yield observed for Bz3MAM in DMSO. Glycerol was therefore used as an additive to increase the viscosity of solutions of the dye in methanol. The fluorescence emission intensity of Bz3MAM remained quite stable up to 80 vol% glycerol, but then exhibited a sharp decrease of intensity in 100% glycerol, with an accompanying increase in the fluorescence lifetime (Fig. S20†). These results provide further evidence that, in solution, the chemical nature of the solvent is a much more important determinant of fluorescence behaviour than is viscosity. Furthermore, it is clear that the barrier to trityl group rotation only becomes important in much higher rigidity environments such as solids and polymer films. Future work will elucidate the dynamic range of the rigidity-sensing behaviour of these dyes, and how this may be tuned by alterations to the chemical structure, for example by substitution of the phenyl rings of the trityl group.
Taking all these data together, we propose the following model for the fluorescence behaviour of Bz3MAM, based on the well-characterised behaviour of other rigidochromic dyes incorporating molecular rotors.14 Upon photoexcitation, the molecule adopts a locally excited S1 state, which may then relax to the ground state via two principal routes (Fig. 9). In one, rotation of the trityl group causes the molecule to adopt a conformation where radiationless relaxation to the ground state is favoured, presumably by accessing a nearby conical intersection. In the second route, the locally excited state relaxes by fluorescence emission. In solution, non-radiative relaxation is favoured because the solvent provides sufficient freedom for the trityl group to rotate. Perhaps more importantly, intermolecular bonding with the solvent also influences the positions of the excited states, leading to the observed dependence of quantum yield on polarity. In the solid-state, meanwhile, the environment constricts the trityl group, preventing its rotation and thereby promoting the emissive pathway. Once again, the chemical nature (polarity, H-bonding capacity) of the surrounding matrix also influences the energies of the excited states, resulting in environment-dependent quantum yield.
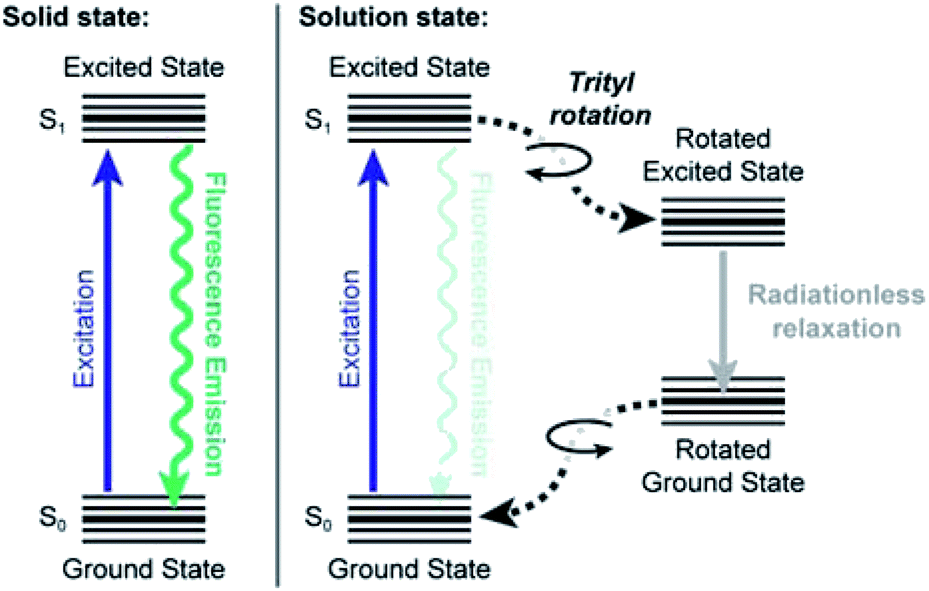 | ||
| Fig. 9 Jablonski diagram showing proposed mechanism allowing rigidochromic behaviour in Bz3MAM. The solution-state mechanism assumes an apolar solvent – in polar solvents, the situation becomes more complicated due to modification of the energies of the excited states. | ||
For rigidochromic dyes based on TICT complex formation, radiationless relaxation is thought to occur from the S1 TICT complex.14 However, TICT dyes show marked solvatochromism, with more polar solvents stabilising the TICT complex and thereby leading to reduced quantum yields. In our system, the opposite phenomenon is observed, with increased quantum yield in polar solvents compared to non-polar environments. It therefore seems unlikely that the fluorescence of Bz3MAM is mediated by TICT. An alternative mechanism operates via suppression of Kasha's rule (SOKR),45 but this relies on the dominant transition being to a state higher than S1, and our calculations (see ESI 5†) revealed only S0 to S1 transitions. Further detailed investigations are therefore required to confirm the exact mechanism of rigidochromism in Bz3MAM, but it is clear that this dye exhibits complex environment-sensitive behaviour. The results presented here provide important fundamental insights into how dye behaviour can be tuned by control over inter- and intramolecular non-covalent bonding.
Conclusions
In summary, by systematic tuning of the imide substituent we have synthesised a maleimide dye with sensitivity to its solid-state environment. It exhibits bright fluorescence when incorporated into a variety of polymer matrices, with emission intensity dependent on the nature of the matrix, while at the same time exhibiting very low fluorescence in solution. Our work expands the chemical diversity of dyes possessing this rigidochromic behaviour.This work also demonstrates how careful tuning of the strength of inter- and intramolecular bonds can result in highly desirable new properties. In the trityl-functionalised maleimide, intermolecular steric hindrance between the maleimide fluorophore and the phenyl rings is strong enough to prevent quenching when the dye is trapped in a matrix, but not when it is free in solution. The introduction of intermolecular non-covalent bonds between the dye and solvent molecules also appears to confer polarity sensitivity opposite to that seen for TICT-based rigidochromic dyes, as well as good temperature stability, which may be very useful for future sensing applications. Furthermore, this effect is achieved without direct conjugation between the parts of the molecule responsible for fluorescence due to the electron-withdrawing nature of the trityl group and the possible presence of through-space conjugation. We anticipate that the fundamental insights into dye design provided by this work will enable the creation of highly tailored fluorophores suitable for applications ranging from flexible devices to bioimaging.
Data availability
Data for this paper, are available as supplementary material online at https://doi.org/10.1039/d1sc03307g.Author contributions
Conceptualisation: JTH, YX, TRW, MTS, VGS, ROR. Experimental data collection: JTH, XY, LM, MTS. Data analysis: JTH, YX, TRW, LM, MTS, VGS, ROR. Writing JTH, YX, TRW, LM, MTS, VGS, ROR.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Ministerio de Economía y Competitividad (MINECO) of Spain (projects CTQ2016-80375-P and PID2019-104772GB-I00), Gobierno Vasco (IT1346-19) and the ERC for support (grant no. 615142). VGS thanks the Royal Society and Leverhulme Trust (LT150052). Dr Robert Keogh is thanked for helpful discussions and Chi Tsang (University of Birmingham) for high-resolution mass spectroscopy analysis. The authors acknowledge the computational resources, and technical and human support provided by the DIPC.References
- C. S. Hansel, M. N. Holme, S. Gopal and M. M. Stevens, Biomaterials, 2020, 226, 119406 CrossRef CAS.
- X. Huang, J. Song, B. C. Yung, X. Huang, Y. Xiong and X. Chen, Chem. Soc. Rev., 2018, 47, 2873–2920 RSC.
- G. Lin, M. A. B. Baker, M. Hong and D. Jin, Chem, 2018, 4, 997–1021 CAS.
- A. S. Klymchenko, Acc. Chem. Res., 2017, 50, 366–375 CrossRef CAS PubMed.
- H. Xiong, H. Zuo, Y. Yan, G. Occhialini, K. Zhou, Y. Wan and D. J. Siegwart, Adv. Mater., 2017, 29, 1700131 CrossRef PubMed.
- C. H. Heo, M. K. Cho, S. Shin, T. H. Yoo and H. M. Kim, Chem. Commun., 2016, 52, 14007–14010 RSC.
- C. Poriel and J. Rault-Berthelot, Adv. Funct. Mater., 2020, 30, 1910040 CrossRef CAS.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- Y. Chen, L. Ju, M. Rushdi, C. Ge and C. Zhu, Mol. Biol. Cell, 2017, 28, 3134–3155 CrossRef CAS PubMed.
- P. Egan, R. Sinko, P. R. Leduc and S. Keten, Nat. Commun., 2015, 6, 7418 CrossRef PubMed.
- T. J. Kirby and J. Lammerding, Nat. Cell Biol., 2018, 20, 373–381 CrossRef CAS PubMed.
- M. K. Kuimova, Phys. Chem. Chem. Phys., 2012, 14, 12671–12686 RSC.
- S. C. Lee, J. Heo, H. C. Woo, J. A. Lee, Y. H. Seo, C. L. Lee, S. Kim and O. P. Kwon, Chem. - Eur. J., 2018, 24, 13706–13718 CrossRef CAS PubMed.
- M. A. Haidekker and E. A. Theodorakis, J. Biol. Eng., 2010, 4, 1–14 CrossRef PubMed.
- L. Michels, V. Gorelova, Y. Harnvanichvech, J. W. Borst, B. Albada, D. Weijers and J. Sprakel, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 18110–18118 CrossRef CAS PubMed.
- J. T. Mika, A. J. Thompson, M. R. Dent, N. J. Brooks, J. Michiels, J. Hofkens and M. K. Kuimova, Biophys. J., 2016, 111, 1528–1540 CrossRef CAS PubMed.
- E. Lisitsyna, A. Efimov, C. Depresle, P. Cauchois, E. Vuorimaa-Laukkanen, T. Laaksonen and N. Durandin, Macromolecules, 2021, 54, 655–664 CrossRef CAS.
- R. O. Loutfy and D. M. Teegardent, Macromolecules, 1983, 16, 452–456 CrossRef CAS.
- A. B. Mabire, M. P. Robin, W. D. Quan, H. Willcock, V. G. Stavros and R. K. O'Reilly, Chem. Commun., 2015, 51, 9733–9736 RSC.
- Y. Xie, J. T. Husband, M. Torrent-Sucarrat, H. Yang, W. Liu and R. K. O'Reilly, Chem. Commun., 2018, 54, 3339–3342 RSC.
- Y. Xie, M. C. Arno, J. T. Husband, M. Torrent-Sucarrat and R. K. O'Reilly, Nat. Commun., 2020, 11, 1–9 Search PubMed.
- M. P. Robin, A. B. Mabire, J. C. Damborsky, E. S. Thom, U. H. Winzer-Serhan, J. E. Raymond and R. K. O'Reilly, J. Am. Chem. Soc., 2013, 135, 9518–9524 CrossRef CAS PubMed.
- Q. Zhu, Z. Ye, W. Yang, X. Cai and B. Z. Tang, J. Org. Chem., 2017, 82, 1096–1104 CrossRef CAS PubMed.
- K. Kizaki, H. Imoto, T. Kato and K. Naka, Tetrahedron, 2015, 71, 643–647 CrossRef CAS.
- D. Song, S. Sun, Y. Tian, S. Huang, Y. Ding, Y. Yuan and A. Hu, J. Mater. Chem. B, 2015, 3, 3195–3200 RSC.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protoc., 2013, 8, 1535–1550 CrossRef PubMed.
- M. Staniforth, W. D. Quan, T. N. V. Karsili, L. A. Baker, R. K. O'Reilly and V. G. Stavros, J. Phys. Chem. A, 2017, 121, 6357–6365 CrossRef CAS PubMed.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- J. Contreras-García, W. Yang and E. R. Johnson, J. Phys. Chem. A, 2011, 115, 12983–12990 CrossRef PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- D. D. La, S. V. Bhosale, L. A. Jones and S. V. Bhosale, ACS Appl. Mater. Interfaces, 2018, 10, 12189–12216 CrossRef CAS PubMed.
- M. Wang, G. Zhang, D. Zhang, D. Zhu and B. Z. Tang, J. Mater. Chem., 2010, 20, 1858–1867 RSC.
- G. Iasilli, A. Battisti, F. Tantussi, F. Fuso, M. Allegrini, G. Ruggeri and A. Pucci, Macromol. Chem. Phys., 2014, 215, 499–506 CrossRef CAS.
- J. A. Faucher, J. V. Koleske, E. R. Santee, J. J. Stratta and C. W. Wilson, J. Appl. Phys., 1966, 37, 3962–3964 CrossRef CAS.
- C. S. Biswas, V. K. Patel, N. K. Vishwakarma, V. K. Tiwari, B. Maiti, P. Maiti, M. Kamigaito, Y. Okamoto and B. Ray, Macromolecules, 2011, 44, 5822–5824 CrossRef CAS.
- A. Muñoz-Bonilla, M. L. Cerrada, M. Fernández-García, A. Kubacka, M. Ferrer and M. Fernández-García, Int. J. Mol. Sci., 2013, 14, 9249–9266 CrossRef PubMed.
- Z. Yang, W. Qin, N. L. C. Leung, M. Arseneault, J. W. Y. Lam, G. Liang, H. H. Y. Sung, I. D. Williams and B. Z. Tang, J. Mater. Chem. C, 2015, 4, 99–107 RSC.
- T. Beppu, S. Kawata, N. Aizawa, Y. J. Pu, Y. Abe, Y. Ohba and H. Katagiri, Chempluschem, 2014, 79, 536–545 CrossRef CAS PubMed.
- D. Ž. Veljković, G. V Janjić and S. D. Zarić, CrystEngComm, 2011, 13, 5005–5010 RSC.
- S. Scheiner, T. Kar and J. Pattanayak, J. Am. Chem. Soc., 2002, 124, 13257–13264 CrossRef CAS PubMed.
- H. Zhang, X. Zheng, N. Xie, Z. He, J. Liu, N. L. C. Leung, Y. Niu, X. Huang, K. S. Wong, R. T. K. Kwok, H. H. Y. Sung, I. D. Williams, A. Qin, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16264–16272 CrossRef CAS PubMed.
- H. Qian, M. E. Cousins, E. H. Horak, A. Wakefield, M. D. Liptak and I. Aprahamian, Nat. Chem., 2017, 9, 83–87 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03307g |
| This journal is © The Royal Society of Chemistry 2021 |