
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSecond-sphere effects on H2O2 activation by non-heme FeII complexes: role of a phenol group in the [H2O2]-dependent accumulation of FeIVO vs. FeIIIOOH†
Jean-Noël
Rebilly
*,
Christian
Herrero
,
Katell
Sénéchal-David
,
Régis
Guillot
 ,
Tanya
Inceoglu
,
Hélène
Maisonneuve
and
Frédéric
Banse
*
,
Tanya
Inceoglu
,
Hélène
Maisonneuve
and
Frédéric
Banse
*
Université Paris-Saclay, CNRS, Institut de Chimie Moléculaire et des Matériaux d'Orsay (ICMMO), 91405 Orsay Cedex, France. E-mail: jean-noël.rebilly@universite-paris-saclay.fr; frederic.banse@universite-paris-saclay.fr
First published on 17th November 2021
Abstract
Redox metalloenzymes achieve very selective oxidation reactions under mild conditions using O2 or H2O2 as oxidants and release harmless side-products like water. Their oxidation selectivity is intrinsically linked to the control of the oxidizing species generated during the catalytic cycle. To do so, a second coordination sphere is used in order to create a pull effect during the activation of O2 or H2O2, thus ensuring a heterolytic O–O bond cleavage. Herein, we report the synthesis and study of a new non-heme FeII complex bearing a pentaazadentate first coordination sphere and a pendant phenol group. Its reaction with H2O2 generates the classical FeIIIOOH species at high H2O2 loading. But at low H2O2 concentrations, an FeIVO species is generated instead. The formation of the latter is directly related to the presence of the 2nd sphere phenol group. Kinetic, variable temperature and labelling studies support the involvement of the attached phenol as a second coordination sphere moiety (weak acid) during H2O2 activation. Our results suggest a direct FeII → FeIVO conversion directed by the 2nd sphere phenol via the protonation of the distal O atom of the FeII/H2O2 adduct leading to a heterolytic O–O bond cleavage.
Introduction
Redox metalloenzymes carry out oxidation reactions at room temperature with high efficiency.1 In cytochrome P450 or Rieske dioxygenases, O2 is activated by sequential electron and proton transfers from a reductase to generate the active species: a (P˙+)FeIVO species in cytochrome P450 (“compound I”)2–7 and a proposed cis FeV(O)(OH) or FeIV(O)(OH) species in the case of Rieske dioxygenases.8–13 In both cases, an FeIIIOOH intermediate is the precursor that results from O2 reductive activation. Such an intermediate can be generated with non-heme iron complexes using the peroxide shunt pathway14–17 and was demonstrated to be involved in oxidation reactions.14,18,19 Usually, when non-heme FeII complexes are used as precursors, they react in a two-step process: (i) FeII is oxidized by a 1st equivalent of H2O2 through a homolytic process to yield a {FeIIIOH; HO˙} pair, with HO˙ readily reacting with either the solvent20 or another FeII complex21 and (ii) the resulting FeIIIOH is involved in a substitution reaction with a 2nd equivalent of H2O2 to yield FeIIIOOH.21–25 The H2O2 activation pathway can be modulated by taking inspiration from natural systems. In peroxidases and catalases, a heterolytic activation of H2O2 is observed in a single step (FeIII to formally FeVO). To achieve this, a histidine, assisted by an arginine or asparagine residue in the 2nd sphere26 shuttles the proton of FeIII-bound H2O2 from the proximal to the distal O atom in order to polarize the O–O bond, create a pull effect and ensure the heterolytic cleavage of the O–O bond.5,6,27 In peroxidases, a water molecule located between the histidine and the FeIII–H2O2 moiety further facilitates the proton transfer by reinforcing the H-bond network.28Inspired by this strategy, we report here a new non-heme FeII complex based on a pentaazadentate coordination sphere incorporating a phenol group in the 2nd coordination sphere. Remarkably, a switch in the H2O2 activation mechanism is observed depending on reaction conditions: at high H2O2 loading, the classical FeIII(OOH) species is formed, while FeIVO is predominantly accumulated at low H2O2 concentration. We demonstrate by thorough kinetic studies that the latter forms faster than FeIII(OOH), and that FeIVO forms from FeII through the heterolytic activation of an FeII/H2O2 adduct assisted by the 2nd sphere acidic phenol.
Results and discussion
Ligand synthesis
L5PhOH (Chart 1) was synthesized from mpL42 (Scheme S2†) and 2,4-di-tert-butyl-6-(azidomethyl)phenol by the CuAAC methodology using CuSO4/sodium ascorbate in tBuOH/water in 67% yield (Schemes S1–S3 and Fig. S1–S6†). | ||
| Chart 1 Structure of the ligands mentioned in this paper. | ||
Synthesis of the complexes
The complete experimental procedures are described in the ESI.†[(L5PhOH)FeIICl2] (1) was obtained in quantitative yield by filtration of the yellow precipitate that appears after 3 days in MeCN upon mixing equimolar quantities of L5PhOH and FeIICl2.
[(L5PhOH)FeIICl](PF6) (2) was isolated in 71% yield after addition of 1 equiv. AgPF6 to [(L5PhOH)FeIICl2] in MeCN and removal of AgCl.
[(L5PhOH)FeII(MeCN)](PF6)2 (3) was obtained in 74% yield after reaction of two equiv. AgPF6 with [(L5PhOH)FeIICl2] in MeCN. [(L5PhOH)FeII(OTf)](OTf) (4) was prepared from L5PhOH and FeII(OTf)2 in MeCN and isolated in 82% yield.
All complexes were characterized by UV-vis spectroscopy, cyclic voltammetry (CV) and ESI-MS (Fig. S7–S21†).
Solid state studies
Single crystals of [(L5PhOH)FeIICl](PF6) (2) were obtained by slow diffusion of tBuOMe into a methanolic solution of the complex. The same procedure applied to complex (3) led to single crystals of a rearranged complex of formula [(L5PhOH)FeII(H2O)(OH)](PF6) (5).The crystal structure of the molecular cation [(L5PhOH)FeCl]+ of (2) (Fig. 1a) shows a distorted octahedral environment and an overall binding mode very similar to that of the parent complex [(L5)FeCl](PF6).20L5PhOH acts as a pentadentate ligand with its two pyridines sitting in trans positions. The 6th position is occupied by the chloride ion. With bond distances between 2.17 and 2.30 Å, the complex is in line with structural data reported for high spin FeII centers, which is expected with a bound anion.29 In this structure, the distance between the phenol oxygen and the metal center is 6.49 Å.
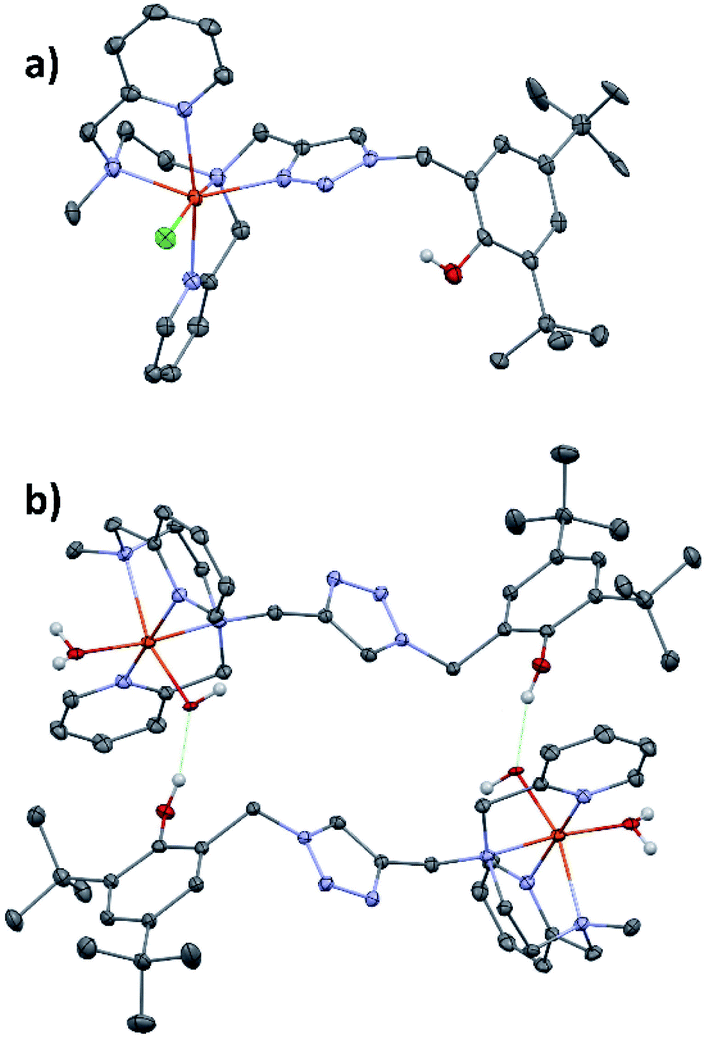 | ||
| Fig. 1 ORTEP drawing of compounds (a) [(L5PhOH)FeIICl]+ of (2) and (b) [(L5PhOH)FeII(H2O)(OH)]+ of (5). Thermal ellipsoids are shown at the 30% level. Fe is in orange-red, C in grey, N in blue, O in dark red, Cl in green. | ||
[(L5PhOH)FeII(H2O)(OH)](PF6) (5) shows a different binding mode (Fig. 1b). The metal sits in a pseudo-octahedral environment, but L5PhOH now acts as a tetradentate ligand. The two pyridines still occupy trans positions, but in this case neither the triazole nor the phenol group are bound to the metal. The coordination sphere is completed by an aquo and an hydroxo ligands. Bond distances lie in the range 2.10–2.22 Å (Fe–N) and 1.86–1.88 Å (Fe–O). In the unit cell, this structure is stabilized by intermolecular hydrogen bonds developing within supramolecular dimers: the hydroxo ligand of one complex is hydrogen-bonded to the dangling phenol group of the partner in a head-to-tail fashion.
[(L5PhOH)FeII(H2O)(OH)](PF6) (5) could not be isolated in pure form in bulk quantities. Therefore, it was not studied in solution but its structure is mentioned here to support the triazole lability discussed later.
In the following reactivity and spectroscopic studies, we will only focus on the triflate salt [(L5PhOH)FeII(OTf)](OTf) (4), as triflate, unlike chloride, is readily displaced by MeCN in the coordination sphere of iron.29
Solution studies
The CV (Fig. S20†) of [(L5PhOH)FeII(OTf)](OTf) (4) in MeCN indicates a major [(N5)FeII(MeCN)]2+ species (Eap = 1.0 V), and a very minor [(N5)FeII(OTf)]+ one (Eap = 0.69 V), identified by analogy with parents complexes, and confirming that triflate is easily substituted by MeCN.20,29 The triazole functional group is bound to the metal while the phenol group is free in the 2nd coordination sphere, as evidenced by its oxidation wave at Eap = 1.61 V.30 Furthermore, a reduction wave ascribed to the reduction of the phenolic proton is observed at Ecp = −1.55 V. Accordingly, the FeII → py MLCT observed at 379 nm (ε = 4400 L mol−1 cm−1) (Fig. S19†) shows a magnitude that is significantly higher than that of [(L5PhOH)FeIICl](PF6) (2) (385 nm, ε = 1100 L mol−1 cm−1). This is indicative of more covalent FeII-py bonds in 4, in agreement with a more pronounced low spin character. This is confirmed by magnetic moment determination by NMR in solution (Evans method, Fig. S22†). While 2 displays a high spin (S = 2) state (μeff = 5.06 μB), 4 displays a μeff of 3.3 μB, which indicates a spin state equilibrium between a high spin (S = 2) and low spin (S = 0) species. The same value of μeff is found for 3 which points toward the substitution of the anionic triflate by MeCN. This spectroscopic signature is typical of [(N5)FeII(MeCN)]2+ complexes in solution.20,29 Compound 4 in solution adopts a similar binding mode as [(L5PhOH)FeIICl](PF6), with the stronger field MeCN as exogenous ligand instead of Cl−.Spectroscopic studies of intermediates
Iodosylbenzene (1.3 equiv. dissolved in MeOH/MeCN 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15) was added to a 1 mM solution of 4 in MeCN at 293 K and the evolution of the UV-vis spectrum was followed using a stopped flow spectrophotometer (Fig. 2 and S23†). Between 0 and 0.7 s (Fig. 2a), the tail of the FeII → py MLCT (379 nm) decreases, indicating oxidation of FeII, together with the growth of a new band at 736 nm, characteristic of an FeIVO species.20,29,31,32 Based on the extinction coefficient of parent complex [(L52)FeIVO]2+ (ε = 300 L mol−1 cm−1),33 a conversion of FeII to FeIVO of 86% can be estimated. The intermediate then decays to yield a new chromophore at 627 nm, typical of a PhO− → FeIII LMCT (Fig. 2b and c).34 The FeIVO decay is better fitted to a 2nd order kinetics, which indicates a bimolecular decay process (Fig. S24†). The EPR spectrum recorded at 90 K of a solution frozen in liquid nitrogen just after mixing reactants in the tube displays a main signal in the HS region (g = 8.55, 4.3) expected for FeIII(OPh)34 and non-specific (rhombic) high spin species (Fig. S25†). In the low spin region, two species are detected with parameters g = 2.320, 2.170, 1.935 and g = 2.320, 2.145, 1.935, which can be ascribed to (N5)FeIII(OMe)2+ and (N4)FeIII(OMe)(MeCN)2+,29,35–37 together with a radical signal at g = 2.004 (Fig. 2d).
:15) was added to a 1 mM solution of 4 in MeCN at 293 K and the evolution of the UV-vis spectrum was followed using a stopped flow spectrophotometer (Fig. 2 and S23†). Between 0 and 0.7 s (Fig. 2a), the tail of the FeII → py MLCT (379 nm) decreases, indicating oxidation of FeII, together with the growth of a new band at 736 nm, characteristic of an FeIVO species.20,29,31,32 Based on the extinction coefficient of parent complex [(L52)FeIVO]2+ (ε = 300 L mol−1 cm−1),33 a conversion of FeII to FeIVO of 86% can be estimated. The intermediate then decays to yield a new chromophore at 627 nm, typical of a PhO− → FeIII LMCT (Fig. 2b and c).34 The FeIVO decay is better fitted to a 2nd order kinetics, which indicates a bimolecular decay process (Fig. S24†). The EPR spectrum recorded at 90 K of a solution frozen in liquid nitrogen just after mixing reactants in the tube displays a main signal in the HS region (g = 8.55, 4.3) expected for FeIII(OPh)34 and non-specific (rhombic) high spin species (Fig. S25†). In the low spin region, two species are detected with parameters g = 2.320, 2.170, 1.935 and g = 2.320, 2.145, 1.935, which can be ascribed to (N5)FeIII(OMe)2+ and (N4)FeIII(OMe)(MeCN)2+,29,35–37 together with a radical signal at g = 2.004 (Fig. 2d).
 | ||
| Fig. 2 Evolution of the UV-vis spectrum of a 1 mM solution of [(L5PhOH)FeII(OTf)](OTf) (4) in MeCN upon addition of 1.3 equiv. PhIO at 293 K: growth of the FeIVO species (a) and its decay to yield a FeIII(OPh) species (b). Time traces at 627 and 736 nm (c). X band EPR spectrum in MeCN (90 K) of a mixture of [(L5PhOH)FeII(OTf)](OTf) (4) in MeCN and 1.2 equiv. PhIO and simulated spectrum as the sum of 3 components (d) (component A: g = 2.320, 2.170, 1.935; component B: g = 2.320, 2.145, 1.935; radical: g = 2.004 in a A/B ratio of 1/0.4). These LS signals represent 25% of the initial FeII. | ||
The formation and decay of [(L5PhOH)FeIVO]2+ (and its analogue without the phenol group [(L5)FeIVO]2+) can be followed by ESI-MS. The time evolution of the mass spectrum with the (L5PhOH) complex shows the decay of the starting FeII complex to yield FeIII complexes, with the transient formation of FeIVO within a 10 s window in line with the half-life determined by UV-vis spectroscopy (Fig. S28–S31†). Interestingly, unlike the case of the (L5) complex (Fig. S32 and S33†) where the peaks correspond to a single ion, the peaks of FeIV and FeIII species with (L5PhOH) are split (Fig. S29–S31†). The features can be nicely simulated as the superimposition of the isotopic patterns of (LPhOH5)Fe and  species, where
species, where  results from phenol H atom abstraction by an oxidizing species.
results from phenol H atom abstraction by an oxidizing species.
All these observations can be rationalized by the mechanism proposed in Scheme 1. O-atom transfer from PhIO to FeII generates an FeIVO intermediate, which is capable of electron and proton abstraction from the 2nd sphere phenol. Indeed, [(L5PhOH)FeIVO]2+ decays with a half-life t1/2 = 3.9 s vs. t1/2 = 34.8 s for [(L5)FeIVO]2+ deprived of phenol (Fig. S23†). The 2nd order decay of the intermediate suggests a bimolecular process, which is in line with the geometric constraints of the complex (the phenol moiety can only engage in intermolecular interactions with a metal center). This reaction between the metal and the phenol yields FeIII(OH), readily converted to FeIII(OMe) in the presence of MeOH, and a phenoxyl radical on the neighboring complex, which are both detected by EPR (Fig. 2 and S25–S27†) and ESI-MS (Fig. S28, S30, and S31†). In these conditions, the radical is reduced quickly to phenolate (by remaining FeII, or by oxidation of the solvent or an oxidizable site of the bound ligand) which undergoes further stabilisation as an intermolecular FeIII(OPh) species detected by UV-vis spectroscopy. The time evolution of the radical is detailed in Fig. S26, S27, and Scheme S4.† For the sake of simplicity, we represent the decay product as a dimeric structure but it could be another oligomeric form (Scheme 1). Interestingly, reactivity studies provide further support to the proposed mechanism: the reaction of 4 with 2 equiv. PhIO and cyclooctene (800 equiv.) yields cyclooctene oxide, confirming the formation of a reactive species capable of epoxidation. Furthermore, the yield is decreased by 27% when compared to the same reaction carried out with [(L5)FeII(OTf)](OTf), not bearing the 2nd sphere phenol group, in line with the interception of FeIVO by the phenol (Table S1†).
 | ||
| Scheme 1 Proposed mechanism for the growth and decay of the FeIVO species generated with iodosylbenzene. S stands for MeCN. Note (*): the FeIII(OH) species indicated in this scheme are readily displaced in the presence of MeOH towards their FeIII(OMe) counterparts (detected by EPR and ESI-MS). For clarity, these equilibria are omitted here. | ||
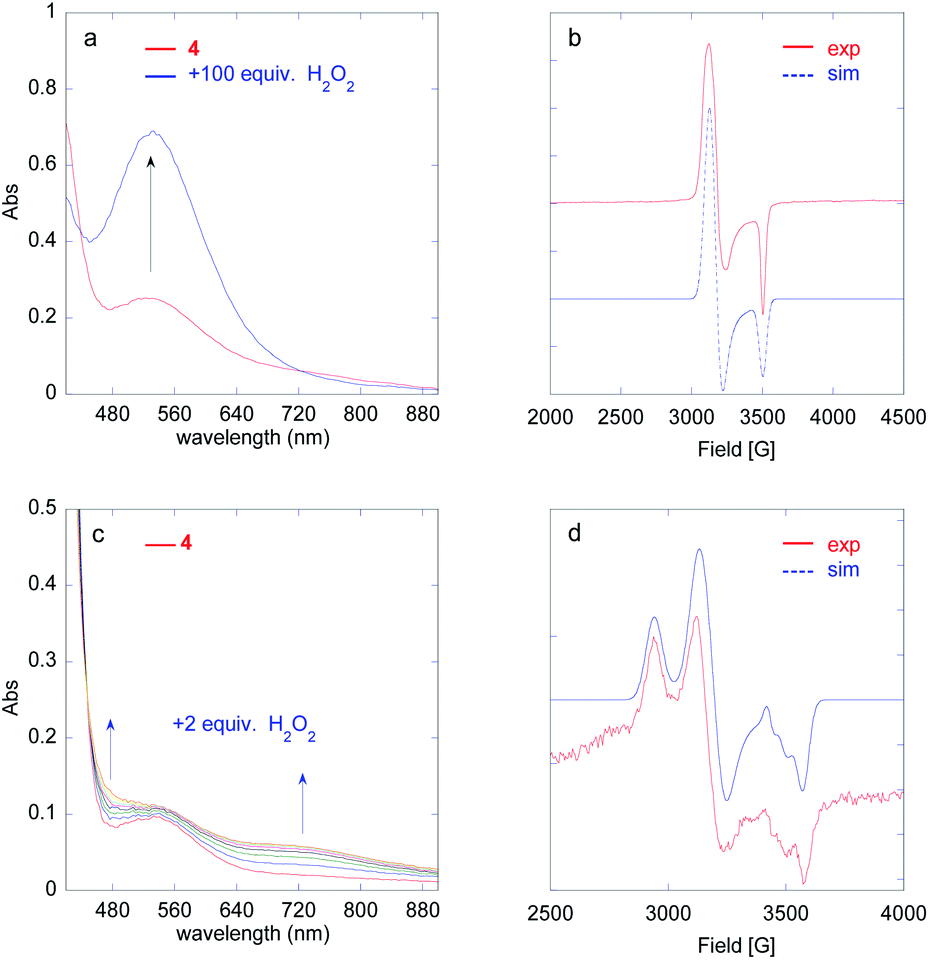 | ||
| Fig. 3 Generation of FeIII(OOH) (a) and FeIVO (c) species followed by UV-vis spectroscopy upon addition of 100 equiv. (a) or 2 equiv. (c) H2O2 to a 1 mM solution of [(L5PhOH)FeII(OTf)](OTf) (4) in MeCN at 293 K. X band EPR spectrum in MeCN (90 K) of the solution drawn at the maximum of the 530 nm band ((b), 100 equiv. H2O2) and at the maximum of the 730 nm band ((d), 2 equiv. H2O2). Experimental spectrum (red) and simulation (blue) with g = 2.205, 2.165, 1.965. (b). Experimental spectrum (red) and simulation (blue) as a sum of 3 components. The LS signals are residual and only represent 7% of the initial FeII (d): component A (FeIII(OH)): g = 2.340, 2.140, 1.928; component B ((N4)FeIII(OOH)(MeCN)2+): g = 2.205, 2.165, 1.965; radical: g = 2.004. | ||
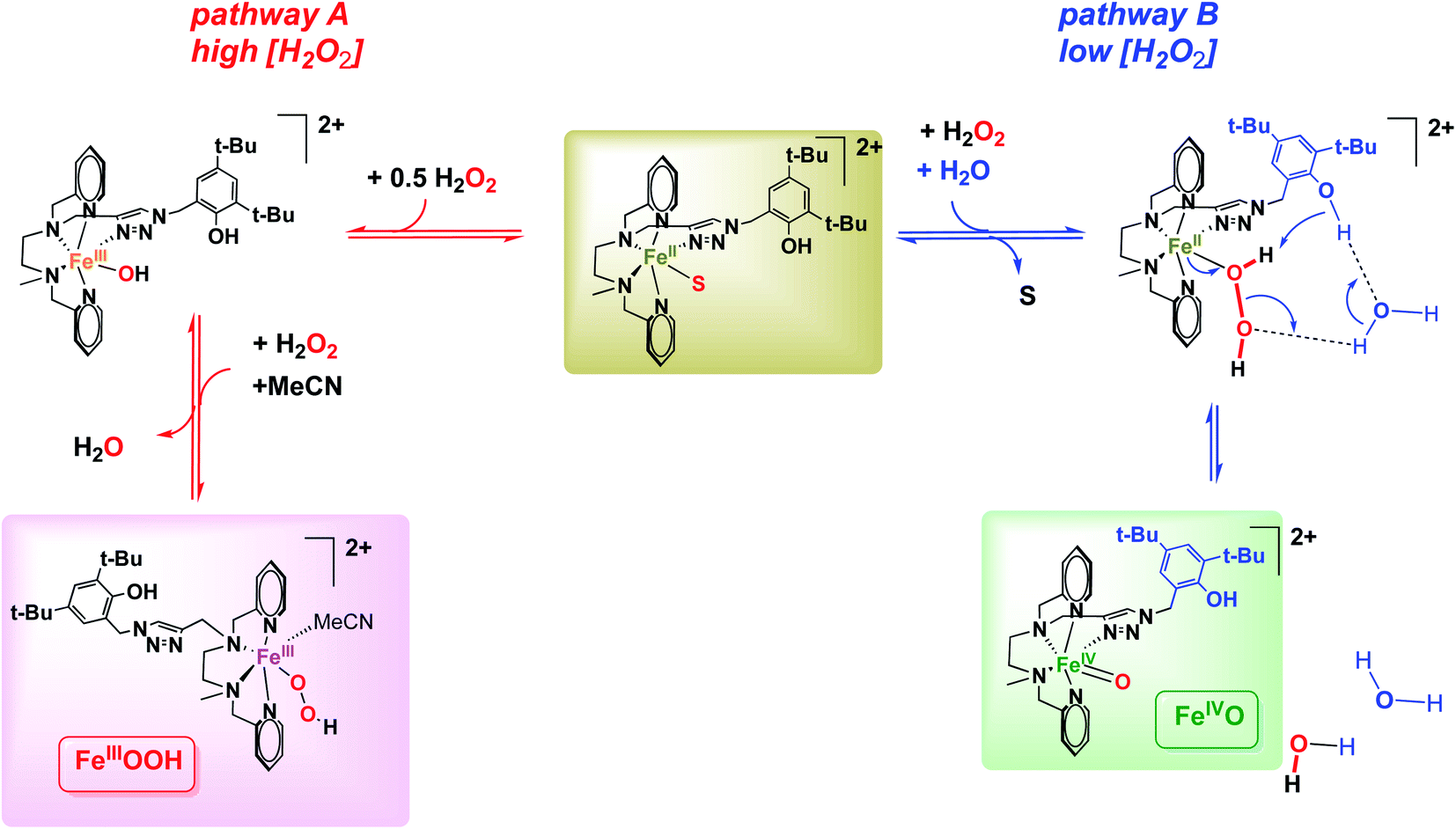 | ||
| Scheme 2 Proposed mechanisms for the activation of H2O2 to FeIII(OOH) (pathway A) and FeIVO species (pathways B), depending on H2O2 concentration. S stands for MeCN. | ||
The growth of FeIII(OOH) was fitted at 530 nm using a monoexponential kinetics law. The rate constant for the formation of FeIII(OOH) shows a linear dependence on H2O2 concentration (Fig. S38†), in line with a bimolecular reaction between the iron complex and the oxidant as previously observed for the parent [(L5)FeIII(OOH)]2+ species.20
 | ||
Fig. 4 HR-ESI-MS spectrum (red) recorded upon mixing [(L5PhOH)FeII(OTf)](OTf) in MeCN with 2 equiv. H2O2 at 293 K. Summed simulation (a, blue), isolated simulations: 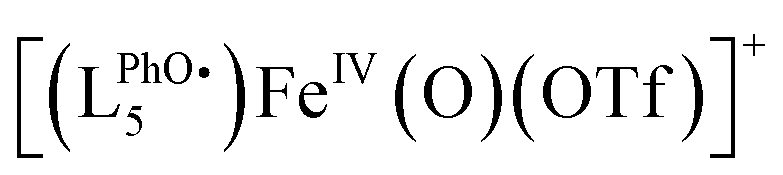 : 775.2421 (b), [(LPhOH5)FeIV(O)(OTf)]+: 776.2499 (c), [(LPhOH5)FeIII(OH)(OTf)]+: 777.2572 (d). : 775.2421 (b), [(LPhOH5)FeIV(O)(OTf)]+: 776.2499 (c), [(LPhOH5)FeIII(OH)(OTf)]+: 777.2572 (d). | ||
The spectrum obtained upon mixing 2 eq. H2O2 with the FeII complex in MeCN is dominated by a massif around m/z = 760 corresponding to the starting [(LPhOH5)FeII(OTf)]+ ion and its oxidized form  . But a second massif around m/z = 776 corresponds to the overlay of three ions: [(LPhOH5)FeIV(O)(OTf)]+, its oxidized form
. But a second massif around m/z = 776 corresponds to the overlay of three ions: [(LPhOH5)FeIV(O)(OTf)]+, its oxidized form  and its decay product [(LPhOH5)FeIII(OH)(OTf)]+ (Fig. S40 and S41†), confirming the formation of the FeIVO intermediate. When the reaction is carried out using H218O2, a similar massif is obtained, but with an increased intensity of the peak at m/z = 778.2549 corresponding to the insertion of 18O (Fig. S42 and S43†). This confirms that the oxo species results from the activation of H2O2.
and its decay product [(LPhOH5)FeIII(OH)(OTf)]+ (Fig. S40 and S41†), confirming the formation of the FeIVO intermediate. When the reaction is carried out using H218O2, a similar massif is obtained, but with an increased intensity of the peak at m/z = 778.2549 corresponding to the insertion of 18O (Fig. S42 and S43†). This confirms that the oxo species results from the activation of H2O2.
In order to get more insight into the formation of the FeIVO species, the evolution of FeIVO absorption was fitted at 730 nm using a biexponential A → B → C law describing its formation and its subsequent decay (Fig. S44†).
The formation rate of the FeIVO species shows a linear dependence on [H2O2] (Fig. S44†). This is in line with a bimolecular process between the FeII complex and H2O2. Furthermore, FeIVO forms much faster than FeIII(OOH). Indeed, the observed rate constant for the formation of FeIVO (k1obs (730 nm)) is nearly 5 times that of FeIII(OOH) (k1obs (530 nm), Fig. S44h†). This comparison indicates that FeIVO does not derive from FeIII(OOH) upon homolytic O–O bond cleavage.
Such an activation of H2O2 by FeII complexes to form FeIVO species has been reported in slightly acidic aqueous medium43 or in the presence of external or preorganized internal bases,33,44,45 which act as a proton shuttle. In these instances H2O2 binds to FeII and the base shuttles a proton from the proximal to the distal O atom, generating an H2O leaving group and favoring a heterolytic O–O bond cleavage to generate FeIVO (Table 1). This process competes with the usual sequence of (i) FeII oxidation to FeIII(OH); (ii) formation of FeIII(OOH) and H2O via OH/H2O2 substitution.
| O–O cleavage | System | ΔH≠ kJ mol−1 | ΔS≠ J mol−1 K−1 |
|---|---|---|---|
| This work | [(L5PhOH)FeII(OTf)](OTf) + 2H2O2 in MeCN | 28 ± 2 | −144 ± 5 |
| Heterolytic FeII → FeIVO | [FeII(NHBn-tpen)]2+ + H2O2 in MeOH45 | 16 | −180 |
| [FeII(L52)]2+ + H2O2 + NEt3 in MeOH33 | 16 | −156 | |
| [FeII(bispidineN5)]2+ + H2O2 in water43 | 34 | −150 | |
| [FeII(TMC)]2+ + H2O2 + 2,6-lutidine in MeCN44 | 29 | −144 | |
| Homolytic FeIII(OOH) → FeIVO + (HO˙) | [FeIII(OOH)(L52)]2+ (ref. 14) | 81 | −1 |
| [FeIII(OOH)(bppc)]2+ (ref. 48) | 53 | −68 | |
| [FeIII(OOH)(TMC)]2+ (ref. 49) | 56 | −75 | |
| [FeIII(OOH)(N4Py)]2+ (ref. 49) | 53 | −121 |
In order to confirm the heterolytic O–O cleavage scenario, we carried out in depth kinetic studies. Eyring plot analysis for the formation of FeIVO using 2 equiv. H2O2 (Fig. S45†) gives activation parameters ΔH≠1obs = 28 ± 2 kJ mol−1; ΔS≠1obs = −144 ± 5 J K−1 mol−1 in line with those reported for the heterolytic activation of H2O2 by FeII complexes to form FeIVO species (Table 1).43–45 By comparison, the parameters obtained for the generation of FeIVO (alongside HO˙) by homolytic cleavage of the O–O bond of FeIIIOOH species are very different.
In the absence of the 2nd sphere phenol moiety (with the parent [(L5)FeII(OTf)](OTf), Fig. S46†), no such FeIVO formation is observed, as testified by the absence of 730 nm chromophore upon reaction with H2O2. This suggests that the 2nd sphere phenol group must be involved in the heterolytic H2O2 activation into FeIVO. Given the spatial constraints of the ligand backbone, this process would have to be assisted by a water molecule in the case of an intramolecular process, or alternatively, the activation process could be intermolecular. However, the latter hypothesis can be discarded as the growth of the FeIVO is fitted to a monoexponential law, in line with an intramolecular process.
To further support this mechanistic proposition, we carried out stopped flow experiments in the presence of additional H2O/D2O using MeCN/H2O 96:4 (v/v) as solvent instead of neat MeCN. Interestingly, using D2O instead of H2O slightly lowers the FeIVO rate of formation (Fig. S47†). The kinetic isotope effect (KIE = k1obs(H2O)/k1obs(D2O)) determined under these conditions is 1.4 (Fig. S48 and Table S2†). By comparison, among examples of FeII to FeIVO conversion assisted by a base, KIE values of 3.7 (ref. 44) and 5.6 (ref. 45) were observed.
All of these observations point to a fast heterolytic cleavage of H2O2 by the FeII complex which requires the assistance of the surrounding phenol moiety. It can be rationalized using pathway B (Scheme 2): FeII(H2O2) is activated by protonation via a network of hydrogen bonds between the distal O atom of the adduct and the phenol moiety likely assisted by water molecules as proton relays. The H2O/D2O normal kinetic effect (KIE = 1.4) suggests the cleavage of a O–H(D) bond in the rate determining step of FeIVO formation. Pathway B (Scheme 2) supports this KIE value as it proposes a PhO–H(D) bond cleavage assisted by H2O/D2O in a Grotthuss-type mechanism. The same trend was already reported for model systems displaying an FeII(H2O2) to FeIVO conversion.44,45
The time evolution of the EPR signal shows a monotonic decrease of the low spin signal, while the high spin signal increases in a first step and decreases in the next (Fig. S51a–d†). This is in line with FeIVO decaying to give a high spin FeIII complex, which then dimerizes. The double integration of the signal remains low throughout the process when compared to the high H2O2 loading (Fig. S51e–g†).
In this experiment (2 equiv. H2O2vs. FeII), the accumulation of FeIVO is only ca. 20% (based on the 730 nm absorbance) and thus a significant amount of H2O2 (formally, 9 equiv. vs. FeIVO) and FeII remains in solution: the latter can be seen from the remaining intensity of the FeII → py MLCT at low wavelength, at the apex of FeIVO (Fig. S52†).
The decay rate k2obs (730 nm) increases linearly with [H2O2] up to 50 equiv. vs. FeII (Fig. S44g†).50 This observation strongly suggests that FeIVO essentially decays via a reaction with H2O2, in agreement with previously reported results.33,41,42 The slope gives an estimate of the 2nd order rate k2 = k2obs/[H2O2] = 77 L mol−1 s−1, which is much larger than the bimolecular decay rate (reaction with phenol) discussed above (k = 7.8 L mol−1 s−1, Fig. S24†). Eqn (1) could rationalize the [H2O2] dependence of the FeIVO decay rate.
 | (1) |
The resulting FeIII(OH) could then dimerize to give oxo-bridged EPR-silent species (2), while the second product of reaction (1), 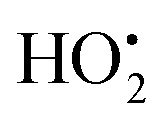 , could then react with remaining FeII to generate FeIII(OOH) (3), disproportionate (4), or react with a 2nd FeIVO species leading FeIII(OH) and O2, as already reported (5).42
, could then react with remaining FeII to generate FeIII(OOH) (3), disproportionate (4), or react with a 2nd FeIVO species leading FeIII(OH) and O2, as already reported (5).42
| 2FeIII(OH) ⇆ FeIII–O–FeIII + H2O | (2) |
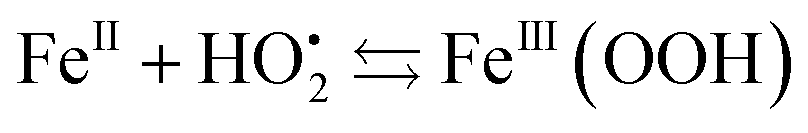 | (3) |
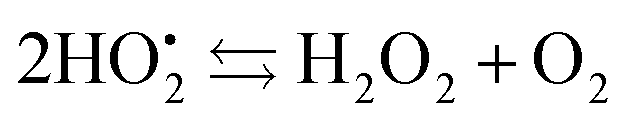 | (4) |
 | (5) |
This set of reactions is in line with the LS residual signals observed by EPR (FeIII(OH) and FeIII(OOH)) and with the final signature in UV-vis spectroscopy (FeIII–O–FeIII). It also fits with the time evolution of the double integration of the EPR signals during FeIVO decay which remains very weak throughout the process and finally decreases when no H2O2 remains and reaction (2) becomes predominant (Fig. S51†).
In the presence of excess H2O2, FeIII(OOH) can be formed via reactions (6) and (7):
| FeII + H2O2 ⇆ FeIII(OH) + HO˙ | (6) |
| FeIII(OH) + H2O2 ⇆ FeIII(OOH) + H2O | (7) |
Discussion
The heterolytic activation of H2O2 by non porphyrinic FeII model complexes is rather scarce. Reported examples involve the addition of an exogenous base,44 the role of which is to shuttle the proton from the proximal to the distal O atom in the FeII/H2O2 adduct. A similar activation was reported in slightly acidic conditions, with H2O likely playing the role of the proton shuttle.43Incorporation of a base within the complex scaffold has been shown to greatly enhance the selectivity in favor of the quantitative formation of FeIVO.45 Conversely, with a complex displaying the same 1st coordination sphere, with an analogous but exogenous base, a clear competition is observed between the formation of FeIVO and FeIII(OOH), evidencing the role of preorganization of the 2nd sphere.33
In using [(L5PhOH)FeII(OTf)](OTf), a weakly acidic group is incorporated in the 2nd coordination sphere (pKa of standard phenol is 29.1 in MeCN51). The activation parameters derived from kinetic studies (Eyring plots) indicate a FeIVO formation mechanism similar to that reported for the above mentioned examples: the distal O atom of the FeII/H2O2 adduct is protonated by the phenol in the rate determining step. Kinetic studies performed in the presence of H2O or D2O are also in agreement with this process. However, the phenol moiety is located too far in the ligand backbone to directly protonate the distal O atom of the adduct. Water molecules are likely to act as a relay between them.
The behavior of this complex can be compared to that of its counterpart lacking the phenol group, [(L5)FeII(OTf)](OTf). In this latter case, no FeIVO species is detected upon H2O2 addition, only FeIII(OOH) forms and the mechanism was proposed to be described by eqn (6) and (7).20
Together, these data support a mechanism analogous to that described for the heterolytic activation of H2O2 by FeII complexes to form FeIVO species (Table 1).43–45
Conclusion
We report a new ligand L5PhOH and its FeII complexes. In MeCN, complex [(L5PhOH)FeII(OTf)](OTf) displays a structure with the ligand bound in a pentadentate fashion and the phenol group unbound as a 2nd coordination sphere, as in the structure of complex [(L5PhOH)FeIICl](PF6).A FeIVO species is generated with PhIO, and quickly decays by reacting with the dangling phenol group. In reactions involving large amounts of H2O2 (100 equiv.), a FeIII(OOH) species is generated, similarly to parent complex missing the phenol [(L5)FeII(OTf)](OTf). However, a different behavior between the two complexes is observed at low H2O2 concentration. For [(L5PhOH)FeII(OTf)](OTf), an FeIVO species is formed predominantly, which is not observed for [(L5)FeII(OTf)](OTf). This is ascribed to a “pull” mechanism induced by the acidic 2nd sphere phenol, which favors a heterolytic O–O bond cleavage of H2O2 and a direct FeII to FeIVO conversion. This activation is supported by the activation parameters of the process and kinetic isotope effects in the presence of H2O/D2O.
Author contributions
JNR and FB designed the research, JNR, CH, KSD, RG, TI and HM performed the experimental work, JNR analyzed the data, JNR and FB wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by a public grant overseen by the French National Research Agency (ANR) as part of the “Investissements d’Avenir” program (Labex charmmmat, reference: ANR-11-LABX-0039-grant).Notes and references
- I. Bertini, A. Sigel and H. Sigel, Handbook of metalloproteins, Marcel Dekker, Inc, New York, 2001 Search PubMed.
- I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Chem. Rev., 2005, 105, 2253–2277 CrossRef CAS PubMed.
- B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed.
- P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef CAS PubMed.
- M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2888 CrossRef CAS PubMed.
- T. L. Poulos, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS PubMed.
- I. Bertini, H. B. Gray, E. I. Stiefel and J. S. Valentine, Biological Inorganic Chemistry, Structure and Reactivity, University Science Books, Sausalito, CA, 2007 Search PubMed.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed.
- S. M. Resnick, K. Lee and D. T. Gibson, J. Ind. Microbiol. Biotechnol., 1996, 17, 438–457 CrossRef CAS.
- E. Carredano, A. Karlsson, B. Kauppi, D. Choudhury, R. E. Parales, J. V. Parales, K. Lee, D. T. Gibson, H. Eklund and S. Ramaswamy, J. Mol. Biol., 2000, 296, 701–712 CrossRef CAS PubMed.
- A. Karlsson, J. V. Parales, R. E. Parales, D. T. Gibson, H. Eklund and S. Ramaswamy, Science, 2003, 299, 1039–1042 CrossRef CAS PubMed.
- M. D. Wolfe and J. D. Lipscomb, J. Biol. Chem., 2003, 278, 829–835 CrossRef CAS PubMed.
- M. Tarasev and D. P. Ballou, Biochemistry, 2005, 44, 6197–6207 CrossRef CAS PubMed.
- A. Thibon, V. Jollet, C. Ribal, K. Sénéchal-David, L. Billon, A. B. Sorokin and F. Banse, Chem.–Eur. J., 2012, 18, 2715–2724 CrossRef CAS PubMed.
- A. Company, L. Gomez, X. Fontrodona, X. Ribas and M. Costas, Chem.–Eur. J., 2008, 14, 5727–5731 CrossRef CAS PubMed.
- K. Chen, M. Costas, J. H. Kim, A. K. Tipton and L. Que, J. Am. Chem. Soc., 2002, 124, 3026–3035 CrossRef CAS PubMed.
- O. Cussó, X. Ribas and M. Costas, Chem. Commun., 2015, 51, 14285–14298 RSC.
- A. S. Faponle, F. Banse and S. P. de Visser, J. Biol. Inorg Chem., 2016, 21, 453–462 CrossRef CAS PubMed.
- A. S. Faponle, M. G. Quesne, C. V. Sastri, F. Banse and S. P. de Visser, Chem.–Eur. J., 2015, 21, 1221–1236 CrossRef CAS PubMed.
- J.-N. Rebilly, W. Zhang, C. Herrero, H. Dridi, K. Sénéchal-David, R. Guillot and F. Banse, Chem.–Eur. J., 2020, 26, 659–668 CrossRef CAS PubMed.
- K. Chen, M. Costas and L. Que Jr, J. Chem. Soc., Dalton Trans., 2002, 672–679, 10.1039/B108629D.
- V. Balland, F. Banse, E. Anxolabehere-Mallart, M. Ghiladi, T. A. Mattioli, C. Philouze, G. Blondin and J. J. Girerd, Inorg. Chem., 2003, 42, 2470–2477 CrossRef CAS PubMed.
- S. V. Kryatov, E. V. Rybak-Akimova and S. Schindler, Chem. Rev., 2005, 105, 2175–2226 CrossRef CAS PubMed.
- A. Hazell, C. J. McKenzie, L. P. Nielsen, S. Schindler and M. Weitzer, J. Chem. Soc., Dalton Trans., 2002, 310–317, 10.1039/B103844N.
- M. R. Bukowski, P. Comba, C. Limberg, M. Merz, L. Que Jr and T. Wistuba, Angew. Chem., Int. Ed., 2004, 43, 1283–1287 CrossRef CAS PubMed.
- H.-P. Hersleth, U. Ryde, P. Rydberg, C. H. Görbitz and K. K. Andersson, J. Inorg. Biochem., 2006, 100, 460–476 CrossRef CAS PubMed.
- C. D. Putnam, A. S. Arvai, Y. Bourne and J. A. Tainer, J. Mol. Biol., 2000, 296, 295–309 CrossRef CAS PubMed.
- P. Vidossich, G. Fiorin, M. Alfonso-Prieto, E. Derat, S. Shaik and C. Rovira, J. Phys. Chem. B, 2010, 114, 5161–5169 CrossRef CAS PubMed.
- N. Ségaud, J.-N. Rebilly, K. Sénéchal-David, R. Guillot, L. Billon, J.-P. Baltaze, J. Farjon, O. Reinaud and F. Banse, Inorg. Chem., 2013, 52, 691–700 CrossRef PubMed.
- T. Osako, K. Ohkubo, M. Taki, Y. Tachi, S. Fukuzumi and S. Itoh, J. Am. Chem. Soc., 2003, 125, 11027–11033 CrossRef CAS PubMed.
- O. V. Makhlynets and E. V. Rybak-Akimova, Chem.–Eur. J., 2010, 16, 13995–14006 CrossRef CAS PubMed.
- M. Martinho, F. Banse, J. F. Bartoli, T. A. Mattioli, P. Battioni, O. Horner, S. Bourcier and J. J. Girerd, Inorg. Chem., 2005, 44, 9592–9596 CrossRef CAS PubMed.
- A. Bohn, C. Chinaux-Chaix, K. Cheaib, R. Guillot, C. Herrero, K. Sénéchal-David, J.-N. Rebilly and F. Banse, Dalton Trans., 2019, 48, 17045–17051 RSC.
- R. Mayilmurugan, E. Suresh and M. Palaniandavar, Inorg. Chem., 2007, 46, 6038–6049 CrossRef CAS PubMed.
- J. J. Girerd, F. Banse and A. J. Simaan, Struct. Bonding, 2000, 97, 145 CrossRef CAS.
- M. Martinho, P. Dorlet, E. Riviere, A. Thibon, C. Ribal, F. Banse and J.-J. Girerd, Chem.–Eur. J., 2008, 14, 3182–3188 CrossRef CAS PubMed.
- O. Y. Lyakin, I. Prat, K. P. Bryliakov, M. Costas and E. P. Talsi, Catal. Commun., 2012, 29, 105–108 CrossRef CAS.
- V. Balland, D. Mathieu, N. Pons-Y-Moll, J. F. Bartoli, F. Banse, P. Battioni, J. J. Girerd and D. Mansuy, J. Mol. Catal. A: Chem., 2004, 215, 81–87 CrossRef CAS.
- A. Thibon, J.-F. Bartoli, R. Guillot, J. Sainton, M. Martinho, D. Mansuy and F. Banse, J. Mol. Catal. A: Chem., 2008, 287, 115–120 CrossRef CAS.
- M. Martinho, G. Blain and F. Banse, Dalton Trans., 2010, 39, 1630–1634 RSC.
- J. Chen, A. Draksharapu, D. Angelone, D. Unjaroen, S. K. Padamati, R. Hage, M. Swart, C. Duboc and W. R. Browne, ACS Catal., 2018, 8, 9665 CrossRef CAS PubMed.
- J. J. Braymer, K. P. O'Neill, J.-U. Rohde and M. H. Lim, Angew. Chem., Int. Ed., 2012, 51, 5376–5380 CrossRef CAS PubMed.
- J. Bautz, M. R. Bukowski, M. Kerscher, A. Stubna, P. Comba, A. Lienke, E. Munck and L. Que, Angew. Chem., Int. Ed., 2006, 45, 5681–5684 CrossRef CAS PubMed.
- F. Li, J. England and L. Que Jr, J. Am. Chem. Soc., 2010, 132, 2134–2135 CrossRef CAS PubMed.
- K. Cheaib, M. Q. E. Mubarak, K. Sénéchal-David, C. Herrero, R. Guillot, M. Clémancey, J.-M. Latour, S. P. deVisser, J.-P. Mahy, F. Banse and F. Avenier, Angew. Chem., 2019, 131, 864–868 CrossRef.
- S. Taktak, S. V. Kryatov, T. E. Haas and E. V. Rybak-Akimova, J. Mol. Catal. A: Chem., 2006, 259, 24–34 CrossRef CAS.
- S. Taktak, S. V. Kryatov and E. V. Rybak-Akimova, Inorg. Chem., 2004, 43, 7196–7209 CrossRef CAS PubMed.
- Q. Zhang and C. R. Goldsmith, Inorg. Chem., 2014, 53, 5206–5211 CrossRef CAS PubMed.
- L. V. Liu, S. Hong, J. Cho, W. Nam and E. I. Solomon, J. Am. Chem. Soc., 2013, 135, 3286–3299 CrossRef CAS PubMed.
- The behaviour at higher H2O2 loading is due to the concomitent formation of the FeIIIOOH chromophore in large quantities at these concentrations: k2obs (730 nm) becomes a composite of FeIVO decay and FeIIIOOH formation and is thus not reliable to discuss the fate of FeIVO. Below 50 mM, the absorption of FeIIIOOH at 730 nm can be neglected and k2obs (730 nm) reflects FeIVO decay alone.
- E. Raamat, K. Kaupmees, G. Ovsjannikov, A. Trummal, A. Kütt, J. Saame, I. Koppel, I. Kaljurand, L. Lipping, T. Rodima, V. Pihl, I. A. Koppel and I. Leito, J. Phys. Org. Chem., 2013, 26, 162–170 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1992915 and 1992916. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03303d |
| This journal is © The Royal Society of Chemistry 2021 |