
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrifluoromethyl substitution enhances photoinduced activity against breast cancer cells but reduces ligand exchange in Ru(II) complex†
Austin P.
Lanquist
a,
Sayak
Gupta
b,
Kathlyn F.
Al-Afyouni
a,
Malik
Al-Afyouni
a,
Jeremy J.
Kodanko
 *b and
Claudia
Turro
*a
*b and
Claudia
Turro
*a
aDepartment of Chemistry and Biochemistry, The Ohio State University, Columbus, OH 43210, USA. E-mail: turro.1@osu.edu
bDepartment of Chemistry, Wayne State University, Detroit, MI 48208, USA. E-mail: jkodanko@chem.wayne.edu
First published on 16th August 2021
Abstract
A series of five ruthenium complexes containing triphenyl phosphine groups known to enhance both cellular penetration and photoinduced ligand exchange, cis-[Ru(bpy)2(P(p-R-Ph)3)(CH3CN)]2+, where bpy = 2,2′-bipyridine and P(p-R-Ph)3 represent para-substituted triphenylphosphine ligands with R = –OCH3 (1), –CH3 (2) –H (3), –F (4), and –CF3 (5), were synthesized and characterized. The photolysis of 1–5 in water with visible light (λirr ≥ 395 nm) results in the substitution of the coordinated acetonitrile with a solvent molecule, generating the corresponding aqua complex as the single photoproduct. A 3-fold variation in quantum yield was measured with 400 nm irradiation, Φ400, where 1 is the most efficient with a Φ400 = 0.076(2), and 5 the least photoactive complex, with Φ400 = 0.026(2). This trend is unexpected based on the red-shifted metal-to-ligand charge transfer (MLCT) absorption of 1 as compared to that of 5, but can be correlated to the substituent Hammett para parameters and pKa values of the ancillary phosphine ligands. Complexes 1–5 are not toxic towards the triple negative breast cancer cell line MDA-MB-231 in the dark, but 3 and 5 are >4.2 and >19-fold more cytotoxic upon irradiation with blue light, respectively. A number of experiments point to apoptosis, and not to necrosis or necroptosis, as the mechanism of cell death by 5 upon irradiation. These findings provide a foundation for understanding the role of phosphine ligands on photoinduced ligand substitution and show the enhancement afforded by –CF3 groups on photochemotherapy, which will aid the future design of photocages for photochemotherapeutic drug delivery.
Introduction
Ruthenium(II) polypyridyl complexes are characterized by strong absorption throughout the visible spectral region, long lived triplet excited states, and intense emission.1–4 As such, these complexes exhibit useful photophysical properties for many applications, such as luminescent sensors,5,6 photoswitches,7,8 molecular machines,9 photoredox catalysis,10,11 and solar energy conversion.12–17 Ruthenium(II) complexes have also been shown to act as agents for photodynamic therapy (PDT) through the sensitization of 1O2,18–21 and for the photoinduced release of therapeutics with spatiotemporal control, photochemotherapy (PCT).22–25 Whereas PDT requires a stable complex, the latter features the dissociation of a ligand upon irradiation with low energy, red or near-IR light.26,27 In addition, ruthenium(II) complexes that are able to both produce 1O2 and release a drug molecule upon irradiation have been shown to be significantly more active against cancer cells than the analogous complexes that only accomplish one of these functions.28–32 The introduction of a phosphine ligand in the coordination sphere of ruthenium(II) complexes has been shown to increase the photodissociation yield of monodentate ligands that are typically not photolabile.33 In addition, cationic compounds with a triphenyl phosphine substituent have been shown to enhance cellular uptake,34,35 leading to an interest in the investigation of divalent ruthenium triphenyl phosphine complexes for PCT.In general, the photoinduced ligand dissociation in ruthenium(II) complexes is attributed to the thermal population of dissociative ligand-field (3LF) state(s) from the lowest energy triplet metal-to-ligand charge-transfer (3MLCT) excited state.36–38 The relative energies of the 3LF and 3MLCT excited states can be tuned through synthetic modifications, which result in changes in the efficiency of photoinduced ligand exchange.36–38 For example, changes in steric hindrance around the ruthenium center achieved through the placement of methyl groups on the 6- and 6′-positions of 2,2′-bipyridine (bpy), play a key role in the yield of ligand photosubstitution. This effect is believed to arise from the distortion from the pseudo-octahedral geometry around the metal, lowering the energy of the 3LF state(s) and making them more readily accessible.36,39–41
In addition to lowering the energy of the 3LF state(s) to increase the quantum yield of ligand exchange in ruthenium(II) complexes, our group recently discovered another factor that plays a role in CH3CN photodissociation. In a series of 12 complexes of the type [Ru(tpy)(L)(CH3CN)]n+ (tpy = 2,2′:6′,2′′-terpyridine), where L represents a neutral (n = 2) or an anionic (n = 1) bidentate ligand, the quantum yields for the photosubstitution of the CH3CN ligand for a water solvent molecule upon 400 nm irradiation, Φ400, resulted in the unexpected increase of ligand exchange with the increasing electron-donating character of the bidentate ligand, despite the lower 3MLCT energy across the series.42 Calculations revealed that as the π-donating ability of the bidentate ligand increased, the relative amount of metal character in the highest occupied molecular orbital (HOMO) decreased. As such, a strong inverse correlation between the % Ru(d) character of the HOMO and the Φ400 values was observed. In contrast, the opposite trend was reported for the photoisomerization of sulfoxides in the series [Ru(bpy)2(P(p-R-Ph)2)(PhSOCH3)]2+ (R = –H, –OMe, –CF3) and [Ru(tpy)(L)(dmso)]n+, where L = bpy (n = 2), N,N,N′,N′-tetramethylethylene-1,2-diamine (tmen, n = 2), acetylacetonate (acac, n = 1), or oxalate (ox, n = 0).43–45 In these complexes, the yield of S → O sulfoxide isomerization decreased with increasing electron donation of the ancillary ligand. Therefore, in contrast to the [Ru(tpy)(L)(CH3CN)]n+ series, a greater ruthenium contribution to the HOMO resulted in greater photoactivity in the sulfoxide complexes. This difference highlights the need for a systematic approach toward further understanding the structure–function relationships for tuning the electronic character of the ancillary ligands to control photochemical processes.
The present work focuses on the photoinduced CH3CN ligand exchange in the series cis-[Ru(bpy)2(P(p-R-Ph)3)(CH3CN)]2+ shown in Fig. 1, in which five para-substituted triphenylphosphine ligands, P(p-R-Ph)3, are used to tune the electronic structure of the complexes, where R = –OCH3 (1), –CH3 (2), –H (3), –F (4), and –CF3 (5). Triphenylphosphine was chosen as the ancillary ligand within this series because of its ability to serve as both a σ-donor and π-acceptor, and these properties are highly tunable through the para-substitution of the phenyl rings with electron-donating or -withdrawing groups.2,46 Furthermore, this series of complexes serves as a model system for caged ruthenium platforms for the delivery of drugs with terminal nitrile functional groups, including cysteine protease inhibitors associated with tumor metastasis.47–52 Additionally, Ru(II) complexes with ancillary phosphine ligands have been shown to play a critical role in the photo-triggered release of amine-based neurotransmitters, such as gamma-aminobutyric acid (GABA) and glutamate,33,48 and, importantly fluorination has been shown to aid in cellular uptake and overall activity of PDT cancer agents.53 Herein, we show that 1–5 are not toxic toward the triple negative breast cancer cell line MDA-MB-231 in the dark, but complexes 3 and especially –CF3-substituted 5 exhibit significant enhancement of cytotoxicity upon irradiation with blue light, shown to trigger apoptosis.
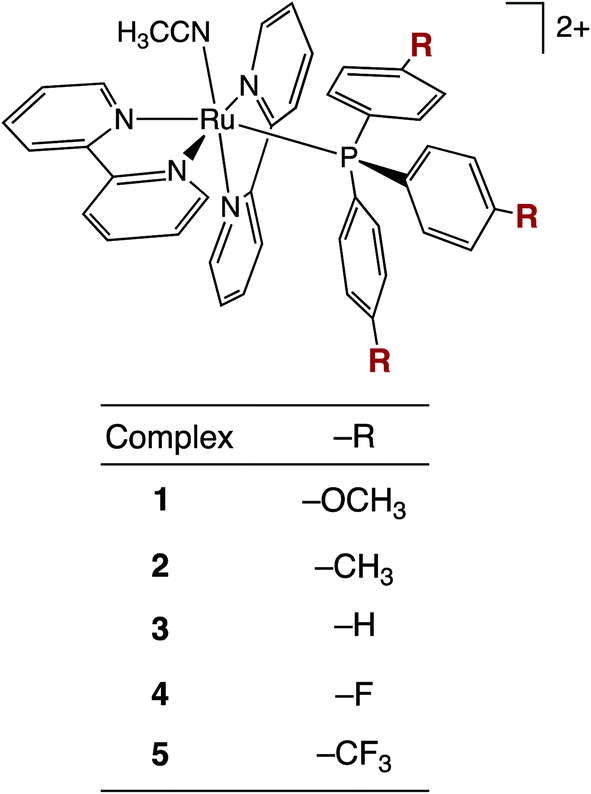 | ||
| Fig. 1 Structural representation of complexes 1–5. | ||
Experimental section
Materials
The phosphine ligands P(p-R-Ph)3 (R = –OCH3, –CH3, –H, –F, –CF3) were purchased from Aldrich, were used without further purification, and were stored in an inert atmosphere. All solvents were used as received from Aldrich and Fisher. The precursor cis-Ru(bpy)2Cl2 was prepared following a of series steps outlined in a previous literature report.54 Complexes 1–5, with general formula [Ru(bpy)2(P(p-R-Ph)3)(CH3CN)](PF6)2, where R = –OCH3 (1), –CH3 (2), –H (3), –F (4), and –CF3 (5), were prepared from the corresponding [Ru(bpy)2(P(p-R-Ph)3)Cl](PF6) precursor, where R = –OCH3 (1a), –CH3 (2a), –H (3a), –F (4a), –CF3 (5a), as described below.[Ru(bpy)2(P(p-OCH3-Ph)3)Cl](PF6) (1a)
Cis-Ru(bpy)2Cl2 (0.14 g, 0.029 mmol) was dissolved in 30 mL water/ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v:v) under N2 atmosphere; this solution was transferred via cannula into a 50 mL round bottom flask containing tris(2-methoxyphenyl)phosphine (0.210 g, 0.595 mmol) and was refluxed for 5 h under a N2 atmosphere. The color changed from purple to red during this time and, upon cooling to room temperature, the mixture was filtered to remove excess ligand. The filtrate was removed under vacuum and the resulting residue was redissolved in a minimal amount of acetone and added dropwise into 20 mL of a saturated aqueous solution of NH4PF6, forming a red participate. The solid was collected by vacuum filtration and washed with diethyl ether (5 mL × 3). The crude product was purified by column chromatography using neutral alumina stationary phase, and acetonitrile/toluene (1:1, v:v) as the eluent. The red fraction was collected, dried under vacuum, and dissolved in a minimal amount of acetone. The concentrated solution was then added dropwise to diethyl ether (30 mL), resulting the precipitation of a red solid that was then filtered and further washed with ether. Yield = 0.21 g (0.020 mmol, 68%). 1H NMR (600 MHz) in (CD3)2CO, δ/ppm (mult., coupling, integration): 9.39 (d, J = 6.49 Hz, 1H), 9.33 (d, J = 5.82 Hz, 1H), 8.73 (d, J = 8.38 Hz, 1H), 8.65 (d, J = 7.68 Hz, 1H), 8.56 (d, J = 7.68 Hz, 1H), 8.46 (d, J = 8.06 Hz, 1H), 8.19 (td, J = 8.33, 1.30 Hz, 1H), 8.12–8.03 (m, 2H), 7.84 (td, J = 8.51, 1.30 Hz, 1H), 7.54 (td, J = 6.85, 1.33 Hz, 1H), 7.50 (d, J = 5.35 Hz, 1H), 7.46 (td, J = 6.63, 1.46 Hz, 1H), 7.39–7.34 (m, 8H), 7.00 (td, J = 6.71, 1.31 Hz, 1H), 6.77–6.74 (m, 6H), 3.79 (s, 9H). 31P NMR (600 MHz) in (CD3)2CO, δ = 40.28 ppm.
:1, v:v) under N2 atmosphere; this solution was transferred via cannula into a 50 mL round bottom flask containing tris(2-methoxyphenyl)phosphine (0.210 g, 0.595 mmol) and was refluxed for 5 h under a N2 atmosphere. The color changed from purple to red during this time and, upon cooling to room temperature, the mixture was filtered to remove excess ligand. The filtrate was removed under vacuum and the resulting residue was redissolved in a minimal amount of acetone and added dropwise into 20 mL of a saturated aqueous solution of NH4PF6, forming a red participate. The solid was collected by vacuum filtration and washed with diethyl ether (5 mL × 3). The crude product was purified by column chromatography using neutral alumina stationary phase, and acetonitrile/toluene (1:1, v:v) as the eluent. The red fraction was collected, dried under vacuum, and dissolved in a minimal amount of acetone. The concentrated solution was then added dropwise to diethyl ether (30 mL), resulting the precipitation of a red solid that was then filtered and further washed with ether. Yield = 0.21 g (0.020 mmol, 68%). 1H NMR (600 MHz) in (CD3)2CO, δ/ppm (mult., coupling, integration): 9.39 (d, J = 6.49 Hz, 1H), 9.33 (d, J = 5.82 Hz, 1H), 8.73 (d, J = 8.38 Hz, 1H), 8.65 (d, J = 7.68 Hz, 1H), 8.56 (d, J = 7.68 Hz, 1H), 8.46 (d, J = 8.06 Hz, 1H), 8.19 (td, J = 8.33, 1.30 Hz, 1H), 8.12–8.03 (m, 2H), 7.84 (td, J = 8.51, 1.30 Hz, 1H), 7.54 (td, J = 6.85, 1.33 Hz, 1H), 7.50 (d, J = 5.35 Hz, 1H), 7.46 (td, J = 6.63, 1.46 Hz, 1H), 7.39–7.34 (m, 8H), 7.00 (td, J = 6.71, 1.31 Hz, 1H), 6.77–6.74 (m, 6H), 3.79 (s, 9H). 31P NMR (600 MHz) in (CD3)2CO, δ = 40.28 ppm.
[Ru(bpy)2(P(p-OCH3-Ph)3)Cl](PF6) (2a)
The procedure used to prepare 1a was followed using cis-Ru(bpy)2Cl2 (0.13 g, 0.028 mmol) and tris-methylphenylphosphine (0.20 g, 0.064 mmol). Yield = 0.22 g (0.021 mmol, 76%). 1H NMR (600 MHz) in (CD3)2CO, δ/ppm (mult., coupling, integration): 9.39 (d, J = 5.77 Hz, 1H), 9.33 (d, J = 5.57 Hz, 1H), 8.70 (d, J = 7.30 Hz, 1H), 8.64 (d, J = 7.82 Hz, 1H), 8.54 (d, J = 7.81 Hz, 1H), 8.45 (d, J = 7.03 Hz, 1H), 8.19 (td, J = 7.87, 1.40 Hz, 1H), 8.09–8.05 (m, 2H), 7.83 (td, J = 7.75, 1.39 Hz, 1H), 7.53 (td, J = 6.51, 1.58 Hz, 1H), 7.44–7.33 (m, 10H), 7.04–7.00 (m, 6H), 7.69 (td, J = 7.81, 1.35 Hz, 1H), 2.28 (s, 9H). 31P NMR (600 MHz) in (CD3)2CO, δ = 42.09 ppm.[Ru(bpy)2(P(Ph)3)Cl](PF6) (3a)
The procedure used to prepare 1a was followed using cis-Ru(bpy)2Cl2 (0.23 g, 0.048 mmol) and triphenylphosphine (0.24 g, 0.091 mmol). Yield = 0.432 g (0.0432 mmol, 89%). 1H NMR (600 MHz) in (CD3)2CO, δ/ppm (mult., coupling, integration): 9.38 (d, J = 5.55 Hz, 1H), 9.33 (d, J = 5.00 Hz, 1H), 8.72 (d, J = 8.76 Hz, 1H), 8.66 (d, J = 8.56 Hz, 1H), 8.57 (d, J = 8.23 Hz, 1H), 8.47 (d, J = 9.26 Hz, 1H), 8.20 (td, J = 7.66, 1.35 Hz, 1H), 8.10–8.05 (m, 2H), 7.83 (td, J = 7.47, 1.90 Hz, 1H), 7.54 (td, J = 7.18, 1.90 Hz, 1H), 7.52–7.50 (m, 6H), 7.45 (d, J = 5.84 Hz, 1H), 7.43–7.37 (m, 3H), 7.36–7.33 (m, 3H), 7.24–7.20 (m, 6H), 6.95 (td, J = 7.09, 1.35 Hz, 1H). 31P NMR (600 MHz) in (CD3)2CO, δ = 44.25 ppm.[Ru(bpy)2(P(p-F-Ph)3)Cl](PF6) (4a)
The procedure used to prepare 1a was followed using cis-Ru(bpy)2Cl2 (0.19 g, 0.039 mmol) and tris-p-flourophenylphosphine (0.23 g, 0.073 mmol). Yield = 0.34 g (0.032 mmol, 83%). 1H NMR (600 MHz) in (CD3)2CO, δ/ppm (mult., coupling, integration): 9.38 (d, J = 5.20 Hz, 1H), 9.28 (d, J = 5.19 Hz, 1H), 8.74 (d, J = 8.21 Hz, 1H), 8.68 (d, J = 9.26 Hz, 1H), 8.60 (d, J = 9.21 Hz, 1H), 8.50 (d, J = 9.14 Hz, 1H), 8.22 (td, J = 7.97, 1.36 Hz, 1H), 8.14 (td, J = 7.29, 1.01 Hz, 1H), 8.09 (td, J = 7.80, 1.70 Hz, 1H), 7.89 (td, J = 8.31, 1.52 Hz, 1H), 7.60–7.51 (m, 9H), 7.44–7.39 (m, 2H), 7.05–7.00 (m, 7H). 31P NMR (600 MHz) in (CD3)2CO, δ = 43.81 ppm.[Ru(bpy)2(P(p-CF3-Ph)3)Cl](PF6) (5a)
The procedure used to prepare 1a was followed using cis-Ru(bpy)2Cl2 (0.15 g, 0.031 mmol) and tris-p-triflouromethylphenylphosphine (0.25 g, 0.053 mmol). Yield = 0.32 g (0.0269 mmol, 86%) 1H NMR (600 MHz) in (CD3)2CO, δ/ppm (mult., coupling, integration): 9.38 (d, J = 5.55 Hz, 1H), 9.33 (d, J = 5.00 Hz, 1H), 8.72 (d, J = 8.76 Hz, 1H), 8.66 (d, J = 8.56 Hz, 1H), 8.57 (d, J = 8.23 Hz, 1H), 8.47 (d, J = 9.26 Hz, 1H), 8.20 (td, J = 7.66, 1.35 Hz, 1H), 8.10–8.05 (m, 2H), 7.83 (td, J = 7.47, 1.90 Hz, 1H), 7.54 (td, J = 7.18, 1.90 Hz, 1H), 7.52–7.50 (m, 6H), 7.45 (d, J = 5.84 Hz, 1H), 7.43–7.37 (m, 3H), 7.36–7.33 (m, 3H), 7.24–7.20 (m, 6H), 6.95 (td, J = 7.09, 1.35 Hz, 1H). 31P NMR (600 MHz) in (CD3)2CO, δ = 42.95 ppm.[Ru(bpy)2(P(p-OCH3-Ph)3)(CH3CN)](PF6)2 (1)
Cis-[Ru(bpy)2(P(p-OCH3-Ph)3)Cl](PF6) (1a; 0.21 g, 0.22 mmol) was dissolved in an acetonitrile:water mixture (1:1, 20 mL) and was refluxed overnight in the dark. The solution turned from red to yellow. After cooling to room temperature, the solvent was reduced under vacuum and the mixture was added dropwise to a saturated aqueous solution of NH4PF6. The resulting solid was filtered and washed with water and diethyl ether. Yield = 0.21 g (0.19 mmol, 85%). 1H NMR (600 MHz) in (CD3)2CO, δ/ppm (mult., coupling, integration): 9.21 (d, J = 6.28 Hz, 1H), 8.98 (d, J = 6.28 Hz, 1H), 8.85 (d, J = 8.42 Hz, 1H), 8.65 (d, J = 7.78 Hz, 1H), 8.55 (d, J = 8.44 Hz, 1H), 8.46 (d, J = 6.64 Hz, 1H), 8.19 (td, J = 7.91, 1.21 Hz, 1H), 8.12–8.05 (m, 2H), 7.85 (td, J = 8.51, 1.44 Hz, 1H), 7.73 (td, J = 5.56, 1.48 Hz, 1H), 7.60–7.53 (m, 4H), 7.30 (td, J = 6.30, 1.22 Hz, 1H), 7.19–7.14 (m, 6H), 6.90–6.87 (m, 6H), 3.83 (s, 9H), 2.48 (s, 3H). 31P NMR (600 MHz) in (CD3)2CO, δ = 42.15 ppm. ESI-TOF (+): [M2+ − 1PF6]+m/z = 952.14, [M2+ − 2PF6] m/z = 383.07.
[Ru(bpy)2(P(p-CH3-Ph)3)(CH3CN)](PF6)2 (2)
A procedure analogous to that for the preparation of 1 was followed using cis-[Ru(bpy)2(P(p-CH3-Ph)3)Cl](PF6) (2a; 0.25 g, 0.24 mmol), which resulted in 0.22 g (0.21 mmol, 89%) or 2. 1H NMR (600 MHz) in (CD3)2CO, δ/ppm (mult., coupling, integration): 9.20 (d, J = 5.79 Hz, 1H), 9.94 (d, J = 5.28 Hz, 1H), 8.85 (d, J = 8.23 Hz, 1H), 8.78 (d, J = 8.01 Hz, 1H), 8.59 (d, J = 7.62 Hz, 1H), 8.56 (d, J = 8.23 Hz, 1H), 8.38 (td, J = 7.05, 1.45 Hz, 1H), 8.23 (td, J = 8.04, 1.76 Hz, 1H), 8.18 (td, J = 7.88, 1.45 Hz, 1H), 8.08 (td, J = 7.89, 1.14 Hz, 1H), 7.73 (td, J = 6.96, 1.95 Hz, 1H), 7.57–7.56 (m, 1H), 7.55–7.50 (m, 4H), 7.26 (td, J = 6.70, 1.66 Hz, 1H), 7.18–7.12 (m, 12H), 2.48 (s, 3H), 2.33 (s, 9H). 31P NMR (600 MHz) in (CD3)2CO, δ = 43.94 ppm. ESI-TOF (+): [M2+ − 1PF6]+m/z = 904.13, [M2+ − 2PF6] m/z = 379.58, [M2+ − ACN − 2PF6] m/z = 359.07.[Ru(bpy)2(P(Ph)3)(CH3CN)](PF6)2 (3)
A procedure analogous to that for the preparation of 1 was followed using cis-[Ru(bpy)2(P(Ph)3)Cl](PF6) (3a; 0.200 g, 0.234 mmol), which resulted in 0.23 g (0.23 mmol, 96%) of 3. 1H NMR (600 MHz) in (CD3)2CO, δ/ppm (mult., coupling, integration): 9.22 (d, J = 5.52 Hz, 1H), 8.99 (d, J = 5.78 Hz, 1H), 8.86 (d, J = 7.77 Hz, 1H), 8.79 (d, J = 8.32 Hz, 1H), 8.61 (d, J = 7.90 Hz, 1H), 8.58 (d, J = 8.22 Hz, 1H), 8.40 (td, J = 8.01, 1.30 Hz, 1H), 8.24 (td, J = 7.86, 1.51 Hz, 1H), 8.17 (td, J = 7.16, 1.21 Hz, 1H), 8.09 (td, J = 8.01, 1.21 Hz, 1H), 7.75 (td, J = 6.67, 1.32 Hz, 1H), 7.60–7.46 (m, 7H), 7.38–7.34 (m, 6H), 7.34–7.28 (m, 6H), 7.27–7.24 (m, 1H), 2.49 (s, 3H). 31P NMR (600 MHz) in (CD3)2CO, δ = 44.30 ppm. ESI-TOF (+): [M2+ − 1PF6]+m/z = 862.1 (calc. m/z = 862.1), [M2+ − 2PF6] m/z =, 358.5 (calc. m/z = 358.7), [M2+ − ACN − 2PF6] m/z = 338.1 (calc. m/z = 338.1).[Ru(bpy)2(P(p-F-Ph)3)(CH3CN)](PF6)2 (4)
A procedure analogous to that for the preparation of 1 was followed using cis-[Ru(bpy)2(P(p-F-Ph)3)Cl](PF6) (4a; 0.234 g, 0.257 mmol), which resulted in 0.23 g (0.22 mmol, 84%) of 4. 1H NMR (600 MHz) in (CD3)2CO, δ/ppm (mult., coupling, integration): 9.20 (d, J = 5.16 Hz, 1H), 9.02 (d, J = 5.50 Hz, 1H), 8.88 (d, J = 7.91 Hz, 1H), 8.80 (d, J = 7.92 Hz, 1H), 8.66–8.63 (m, 2H), 8.42 (td, J = 7.92, 1.33 Hz, 1H), 8.27–8.22 (m, 2H), 8.12 (td, J = 8.25, 1.32 Hz, 1H), 7.78 (td, J = 7.25, 1.59 Hz, 1H), 7.62–7.59 (m, 2H), 7.58–7.55 (m, 2H), 7.41–7.35 (m, 6H), 7.32 (td, J = 6.79, 1.37 Hz, 1H), 7.17–7.12 (m, 6H), 2.51 (s, 3H). 31P NMR (600 MHz) in (CD3)2CO, δ = 45.86. ESI-TOF (+): [M2+ − 1PF6]+m/z = 916.06, [M2+ − 2PF6] m/z = 385.55, [M2+ − ACN − 2PF6] m/z = 365.04.[Ru(bpy)2(P(p-CF3-Ph)3)(CH3CN)](PF6)2 (5)
A procedure analogous to that for the preparation of 1 was followed using cis-[Ru(bpy)2(P(p-CF3-Ph)3)Cl](PF6) (5a; 0.252 g, 0.238 mmol), which resulted in 0.26 g (0.22 mmol, 93%) of 5. 1H NMR (600 MHz) in (CD3)2CO, δ/ppm (mult., coupling, integration): 9.28 (d, J = 5.58 Hz, 1H), 9.03 (d, J = 5.41 Hz, 1H), 8.90 (d, J = 7.91 Hz, 1H), 8.81 (d, J = 7.95 Hz, 1H), 8.68–8.64 (m, 2H), 8.44 (td, J = 7.93, 1.53 Hz, 1H), 8.28 (td, J = 7.93, 1.65 Hz, 1H), 8.24 (td, J = 7.92, 1.38 Hz, 1H), 8.13 (td, J = 7.88, 1.42 Hz, 1H), 7.79 (td, J = 6.76, 1.15 Hz, 1H), 7.74–7.69 (m, 12H), 7.62–7.52 (m, 4H), 7.26 (td, J = 6.76, 1.30 Hz, 1H), 2.56 (s, 3H). 31P NMR (600 MHz) in (CD3)2CO, δ = 49.52 ppm. ESI-TOF (+): [M2+ − 1PF6]+m/z = 1066.07, [M − 2PF6] m/z = 460.55, [M2+ − ACN − 2PF6] m/z = 440.04.Instrumentation and methods
1H and 31P NMR spectra were measured on a Bruker Advanced III HD 600 MHz. The 1H-NMR peaks were referenced to the residual protonated acetone solvent at 2.05 ppm, while the 31P-NMR resonances were referenced to that of phosphoric acid at δ = −48.32 ppm. Electrospray ionization (ESI) mass spectrometry was conducted on a Bruker MicroTOF mass spectrometer. Samples were dissolved in acetonitrile and went through a series of dilutions prior to injection. Crystals suitable for single crystal X-ray diffraction were grown using slow diffusion of diethyl ether into acetonitrile at room temperature.Cyclic voltammetry (CV) experiments were conducted using a BASi CV-50W potentiostat with a glassy carbon disc (3 mm) as the working electrode, platinum wire as the auxiliary electrode, and Ag/AgCl (NaCl) as the reference electrode. CVs were recorded in CH3CN containing 0.1 M NBu4PF6 under an N2 atmosphere. The scan rate was 100 mV s−1 and the potentials were referenced to the ferrocene-ferrocenium couple; ferrocene was added at the end of each experiment as an internal reference. Electronic absorption spectra were recorded on a Hewlett Packard 8453 diode array spectrometer and emission spectra were collected on a Horiba Fluormax-4 fluorimeter. Photolysis and quantum yield measurements were conducted using a 150 W Xe arc lamp (USHIO) as a light source that was contained in a MilliArc lamp house unit powered by an LPS-220 power supply equipped with an LPS-221 igniter (PTI). The energy of the light reaching each sample was controlled using a 400 nm bandpass filter for quantum yield measurements, but steady-state photochemistry was typically monitored using a 395 nm long-pass filter and samples for 1H NMR photolysis were irradiated with a 450 nm LED. The lamp photon flux for quantum yield measurements was determined using potassium ferrioxalate as an actinometer.55 Samples were dissolved in water with <5% acetone. Quantum yield measurements were conducted in triplicate and standard deviation was used as the error.
Crystals suitable for single crystal X-ray diffraction were grown using slow diffusion of diethyl ether into acetonitrile at room temperature. Diffraction patterns were collected using Mo Kα radiation at 100 K using a Bruker X8 diffractometer equipped with a kappa geometry goniometer, graphite monochromator, and an APEX-II CCD. The frames were integrated with the Bruker SAINT software package62, and the data were corrected for absorption effects using the multiscan method (SADABS) within Olex2. Structural data was solved using ShelXT. Density functional theory (DFT) calculations started with the geometry optimizations using the PBE functional, the SDD basis set for Ru, and the TZVP basis set on all other atoms gave structures in excellent agreement with experimentally determined crystal structures.
LogP values were determined as described previously.56 Solutions of each complex were prepared in octanol (2 mL, 100 μM) and combined with deionized water (2 mL) in glass vials. The vials were capped, wrapped in aluminum foil, shaken (5 min), and allowed to settle (24 h). After 24 h, relative concentrations of each complex in the water and octanol layers were determined spectrophotometrically using absorbance values at the corresponding MLCT absorption maximum. LogP was calculated using the quotient of the absorbance.
The cell viability of 1–5 was determined by plating MDA-MB-231 cells in a 96 well plate at a density of 7000 cells per well in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FBS and 1000 units per mL penicillin/streptomycin. The 96 well plates were incubated overnight in a 37 °C humidified incubator ventilated with 5% CO2. The media was then aspirated off and then quadruplicate wells were treated with DMEM supplemented with 10% FBS and 1000 units per mL penicillin/streptomycin containing different concentrations (30 μM to 500 nM) of the synthesized complexes in 1% DMSO. Plates containing wells with no cells were designated as blank wells whereas wells with cells that were not treated with the compound but only with DMEM supplemented with 10% FBS and 1000 units per mL penicillin/streptomycin containing 1% DMSO (vehicle) were designated as control wells. The plates were then again incubated in a 37 °C humidified incubator ventilated with 5% CO2. After 1 h of incubation, the cells were either irradiated with blue light (tirr = 20 min, λirr = 460–470 nm, 56 J cm−2) or kept in the dark. The plates were then incubated in a 37 °C humidified incubator with 5% CO2 for 72 h. After incubation, 10 μL of MTT reagent (5 mg mL−1 in PBS) was added to each well of the 96 well plate and incubated in a 37 °C humidified incubator ventilated with 5% CO2 for 2 h. After 2 h the media was aspirated off and 100 μL of DMSO was added. The plates were then shaken for 20 min to ensure complete dissolution of the purple formazan crystals. Absorbance of each well was then measured at 570 nm. The mean absorbance values of the blank wells were calculated and subtracted from absorbance values for each well treated with a certain concentration of a compound. The absorbance of the control wells was also taken and subtracted with the average of the blank wells. The mean of these corrected control absorbances were then calculated. Viability of the cells was finally determined by dividing the corrected absorbance of the compound wells by the mean corrected absorbance of the blank wells and expressing the mean of the ratio as a percentage value. The percent viability was plotted against the log of concentration (in molarity) of the compounds and the antilog of the concentration value at 50% viability gave us the EC50 of the complex against MDA-MB-231 cells.
The mechanism of cell death for 5 was studied in MDA-MB-231 cells. MDA-MB-231 cells were seeded at a density of 7000 cells per well in DMEM supplemented with 10% FBS and 1000 units per mL penicillin/streptomycin and allowed to incubate for 24 h in a 37 °C humidified incubator ventilated with 5% CO2. After 24 h the media was aspirated off and the plates and were then pretreated with 50 μL of DMEM supplemented with 10% FBS and 1000 units per mL penicillin/streptomycin containing 20 μM necrostatin (NEC), 20 μM of Z-VAD-FMK (Z-VAD) and 5 mM of N-acetyl cysteine (NAC) in sextuplicate. The plates also contained blank wells with no cells and control wells. One column of wells in the plate along with the blank and control wells were treated with 50 μL of DMEM supplemented with 10% FBS and 1000 units per mL penicillin/streptomycin with 1% DMSO (vehicle). The plates were incubated in a 37 °C humidified incubator ventilated with 5% CO2 for 1 h. The plates were then taken out of the incubator and 50 μL of DMEM supplemented with 10% FBS and 1000 units per mL penicillin/streptomycin containing complex 5 (4 μM) was added to the column of wells that were pretreated with vehicle along with half the wells pretreated with 20 μM necrostatin, 20 μM of Z-VAD-FMK or 5 mM of N-acetyl cysteine. Vehicle (50 μL) was added to rest of the wells, pretreated with 20 μM necrostatin, 20 μM of Z-VAD-FMK or 5 mM of NAC along with the blank and control wells. The plates were then incubated in a 37 °C humidified incubator ventilated with 5% CO2 for 1 h. At the end of incubation, the plates were either irradiated with blue light (tirr = 20 min, λirr = 460–470 nm, 56 J cm−2) or left in the dark for 20 minutes after which they were incubated in a in a 37 °C humidified incubator ventilated with 5% CO2 for 72 h. The viability of the cells was studied using MTT assay after 72 h.
For the Fluorescence Assisted Cell Sorting Studies (FACS), MDA-MB-231 cells were plated at 100000 cells per plate in six 60 mm2 cell culture dishes containing DMEM supplemented with 10% FBS and 1000 units per mL penicillin/streptomycin. After plating, the cells were incubated at 37 °C and 5% CO2 overnight (18 h). After incubation, plates were treated with 3 mL of DMEM supplemented with 10% FBS and 1000 units per mL penicillin/streptomycin containing complex 5 (10 μM) in duplicate, 1% DMSO (vehicle) in triplicate or [Ru(thd)(tpy)(py)]PF6 (6 μM, tpy = 2,2':6′,2′′-terpyridine, thd = 2,2,6,6-tetramethylheptane-3,5-dione, py = pyridine) and set to incubate at 37 °C and 5% CO2 for 1 h, after which time, one plate of treated with vehicle and complex 5 was irradiated with blue light (tirr = 20 min, λirr = 460–470 nm, 56 J cm−2) while the rest of the plates were kept in the dark. After 20 min all the plates were incubated 37 °C and 5% CO2 for 72 h. After 70 h one of the plates containing vehicle was taken and vehicle was replaced with 500 mM H2O2 in PBS and allowed to incubate for further 2 h. At the end of 72 h, the media from each plate was saved in a 15 mL falcon tube. The cells were detached from each plate via trypsinization and added to the previously removed media. The cells were centrifugated (600 g, 8 min) to pellet the cells. The supernatant was decanted, and the pellet was washed once with PBS (2 mL) and once with Annexin V binding buffer (2 mL). After the final wash, the supernatant was decanted, and the pellet was suspended in 100 μL of Annexin V binding buffer. A solution of Annexin V (5 μL, 1 mg mL−1) was added to the cell suspension. The suspension was incubated at room temperature for 15 min. After incubation, 2 mL of Annexin V binding buffer was added to the tube and the suspension was centrifuged (600 g, 5 min). The supernatant was decanted, and cells were suspended in 200 μL of Annexin V binding buffer. A solution of propidium iodide (5 μL, 12 μM) was added to the cell suspension and incubated at room temperature for 15 min. The cell suspension was diluted with 1 mL of Annexin V binding buffer. The suspension was passed through a metal mesh filter (30 μm) from Celltrics (Kobe, Hyogo Prefecture, Japan) into a small sample tube. Flow cytometric analysis was performed on a Sysmex Cyflow Space fluorescence-assisted cell sorter. Data were processed using FCS Express.fcs processing software by De Novo software (Boulder, Co).
Results and discussion
Synthesis and characterization
Complexes 1–5 were synthesized from the starting material cis-Ru(bpy)2Cl2, which allowed for the stepwise introduction of the desired phosphine ligand followed by a CH3CN molecule. The 1H and 31P NMR spectra of 1–5 are shown in the ESI (Fig. S1 and S2†). The 1H NMR spectra display the expected peak integration values and splitting patterns in the aromatic region corresponding to the two bipyridine ligands.57,58 Additionally, no signal was observed at δ ≥ 9.5 ppm, indicative of the presence of the cis-Ru(bpy)2Cl2 precursor. The 31P NMR spectra of 1–5 display two peaks, one singlet assigned to the phosphine bound to the ruthenium center and the a septet that corresponds to the phosphorus atom in the PF6− counterions coupled to six fluorine atoms, consistent with those reported for related ruthenium complexes.59 There is a clear trend in the chemical shift of the bound phosphine signal across the series as a function of the substituents, where electron-withdrawing substituents result in deshielding of the phosphine atoms, leading to signals that shift from 42.2 ppm in 1 to 49.5 ppm in 5.The ORTEP diagrams from X-ray diffraction of single crystals of 1 and 2 are shown in Fig. 2, and those of 3–5 are displayed in the ESI (Fig. S3†), along with crystallographic details and selected bond lengths and angles for 1–5 (Tables S1 and S2†). The structures of 1–5 confirmed the presence of two 2,2′-bipyridine ligands coordinated to the metal in a cis-disposition, with the corresponding phosphine and acetonitrile ligands positioned cis to each other. Minimal variation of the cone angle of the substituted phosphine ligands is observed, consistent with previous literature reports.58,60,61 Moreover, there are no significant changes in the CH3CN–Ru–P bond angles or the Ru–NCCH3 bond lengths in 1–5, suggesting that the variation in phosphine substitution does not result in a significant structural effect on the ground state of these complexes. Therefore, it is expected that any differences observed in the photophysical properties and photochemistry across the series would not be related to steric effects that results from the variation of the substituents on the phosphine ligand.
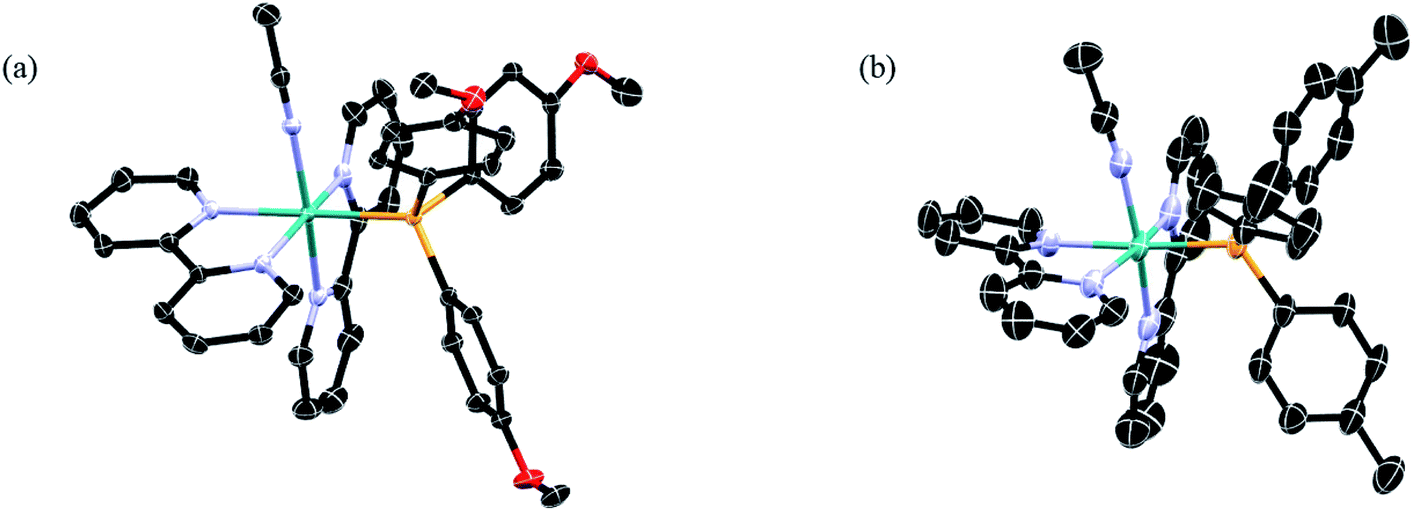 | ||
| Fig. 2 Thermal ellipsoid plots of (a) 1 and (b) 2; H atoms, PF6− ions, and co-crystallized solvent molecules omitted for clarity (ellipsoids drawn at 50% probability). | ||
Photophysical properties and electrochemistry
The electronic absorption spectra of 1–5 were recorded in CH3CN at room temperature (Fig. 3), and the absorption maxima (λabs) and molar extinction coefficients (ε) are presented in Table 1. Complexes 1–5 featured a strong absorption at ∼285 nm that is independent of the substituent on the phosphine ligand assigned as arising from a ligand-centered bpy 1ππ* transition, consistent with other Ru(II) complexes containing bpy ligands.62 Additionally, a broad absorption band is present in 1–5 with maxima at ∼400 nm was attributed to Ru(dπ) → bpy(π*) metal-to-ligand charge transfer (MLCT), consistent with that observed for cis-[Ru(bpy)2(CH3CN)2]2+ at 425 nm in CH3CN.58 The blue shift in the 1MLCT absorption energies observed in 1–5 upon substitution of one CH3CN ligand in cis-[Ru(bpy)2(CH3CN)2]2+ with a phosphine is expected based on related phosphine-containing Ru(II) complexes. For example, the 1MLCT peak shifts from 427 nm in cis-[Ru(bpy)2(CH3CN)2]2+ to 388 nm in 5 in H2O. The small shifts observed in the 1MLCT maxima of 1–5 are attributed to differences in the electron donating or withdrawing character of the phosphine, which are expected to affect the energy of the Ru(dπ) orbitals. Complex 1 exhibits the lowest energy 1MLCT transition with a maximum at 410 nm, which can be compared to those of 3 at 400 nm and 5 at 390 nm. These results are consistent with the stabilization of the Ru(dπ) orbitals with increasing electron withdrawing character of the substituent on the phosphine ligand.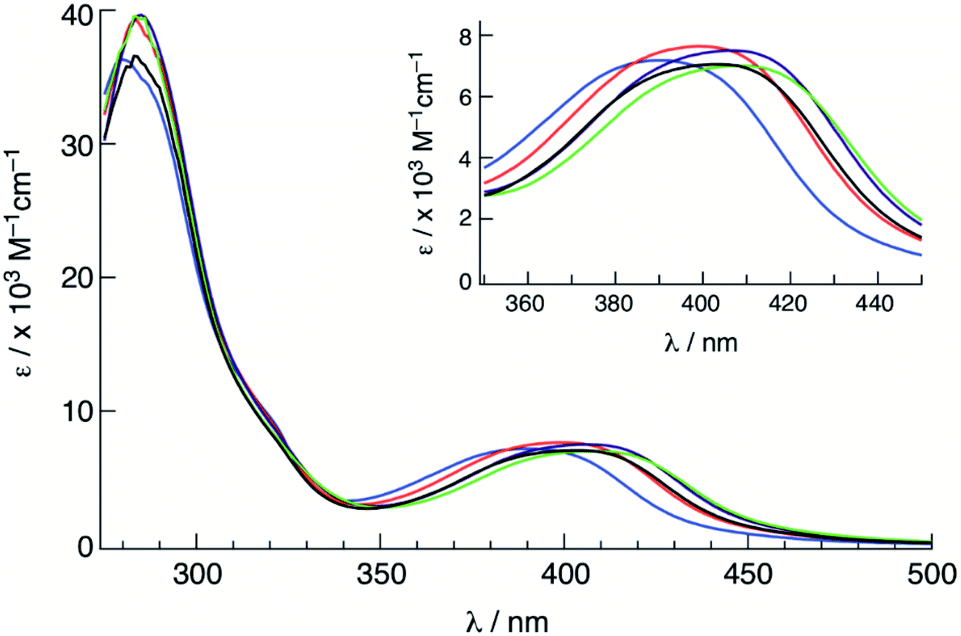 | ||
| Fig. 3 Electronic absorption spectra of 1 (green), 2 (purple), 3 (black), 4 (red) and 5 (blue) in CH3CN. | ||
| Complex | λ abs /nm (ε/×103 M−1 cm−1) | E 1/2 /V | Φ 400 |
|---|---|---|---|
| a In CH3CN. b In CH3CN (0.1 M Bu4NPF6), vs. Ag/AgCl, scan rate = 100 mV s−1. c In water (<5% acetone) at 298 K, λirr = 400 nm. | |||
| 1 | 286 (39), 410 (7.2) | +1.52, −1.31, −1.49 | 0.077(2) |
| 2 | 285 (39), 406 (7.0) | +1.54, −1.31, −1.51 | 0.072(3) |
| 3 | 283 (35), 402 (7.0) | +1.53, −1.34, −1.53 | 0.065(1) |
| 4 | 285 (39), 399 (7.2) | +1.60, −1.32, −1.50 | 0.057(1) |
| 5 | 280 (35), 390 (7.2) | +1.70, −1.26, −1.44 | 0.025(2) |
Room temperature luminescence was not detected, but 1MLCT excitation of 1–5 resulted in broad emission at 77 K in CH3CN (Fig. S4–S8†), assigned as arising from the Ru(dπ) → bpy(π*) 3MLCT excited state. The maximum of the lowest energy transition, E00, ranged from 525 to 540 nm (2.30–2.36 eV) with no significant shift across the series, indicative similar 3MLCT energies. Additionally, the emission spectra of each complex exhibited two lower intensity peaks at ∼580 and ∼630 nm, which correspond to a vibronic progression with an energy separation of ∼1200 cm−1 typical for Ru(II) polypyridyl complexes associated with aromatic ring vibrations of the bpy ligands.1,3,4
The cyclic voltammograms of 1–5 are shown in Fig. 4 and the observed half-wave reduction potentials, E1/2, are listed in Table 1. Two reversible reduction waves were observed in 1–5 at about −1.3 V and −1.5 V in CH3CN vs. Ag/AgCl (0.1 M Bu4PF6). These reduction potentials are relatively invariant to the substituent on the phosphine ligand and are assigned as arising from the consecutive reduction of the two bipyridine ligands within each complex.62 The reduction of bpy ligands bound to Ru(II) have been previously reported at similar potentials, for example at −1.26 V and −1.46 V vs. Ag/AgCl in [Ru(bpy)2(PPh3)(CH3CN)]2+.63 Complexes 1–5 also exhibit one reversible oxidation within the measurement window at approximately +1.55 V vs. Ag/AgCl that is dependent on the phosphine substituent (Table 1 and Fig. 4) and is assigned to the RuIII/II couple, based on published data for related Ru(II) polypyridyl complexes.5,64 In agreement with the energy of the 1MLCT absorption across the series, the electron donating substituents –OCH3 in 1 and –CH3 in 2 shifts the RuIII/II couple to more negative potentials, +1.52 V and +1.54 V, respectively, making them easier to oxidize than 3. In contrast, the –F and –CF3 electron withdrawing substituents make these complexes more difficult to oxidize, with E1/2(RuIII/II) values of +1.60 V for 4 and +1.70 V for 5. It should also be noted that the RuIII/II oxidation potentials of 1–5 are all more positive than that measured for cis-[Ru(bpy)2(CH3CN)2]2+ under similar conditions, consistent with the blue-shifted 1MLCT absorption maxima of the former complexes relative to the latter arising from the presence of the phosphine.
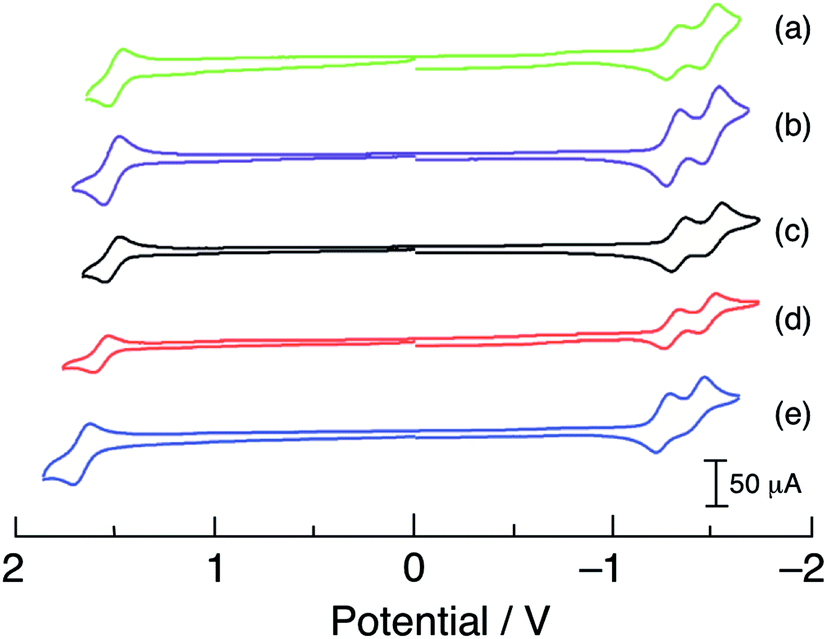 | ||
| Fig. 4 Cyclic voltammograms of (a) 1, (b) 2, (c) 3, (d) 4, and (e) 5 in CH3CN vs. Ag/AgCl (0.1 M TBAPF6, scan rate = 100 mV s−1). | ||
Electronic structure calculations
The energies of the highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) of 1–5 calculated using density functional theory (DFT) are shown in Fig. 5. Comparisons of selected calculated and experimental bond lengths and angles are listed in Tables S3–S7† and the atomic coordinates as detailed in Tables S8–S12.† In each complex, the LUMO is centered on the bpy(π*) orbitals and only small changes in energy are predicted across the entire series, consistent with the relatively constant value measured for the first reduction potential across the series (Table 1). In general, the electron density of the HOMO of each complex was centered on the ruthenium ion. However, in 4 and 5 a greater degree of mixing is calculated between the Ru(dπ) orbital and P–C(σ*) orbitals on the phosphine ligand, resulting in greater stabilization of the Ru-based HOMO in these complexes. In this manner, the electron donating ability of the phosphine substituents plays a role on the energy of the HOMO across the series, in good agreement with the observed trend in the metal-centered oxidation in 1–5 (Table 1). The HOMO–LUMO energy gaps are significantly smaller in 1 and 2 as compared to those calculated for 4 and 5, consistent with the observed blue shift in the increasing energy of the 1MLCT absorption from 1 to 5.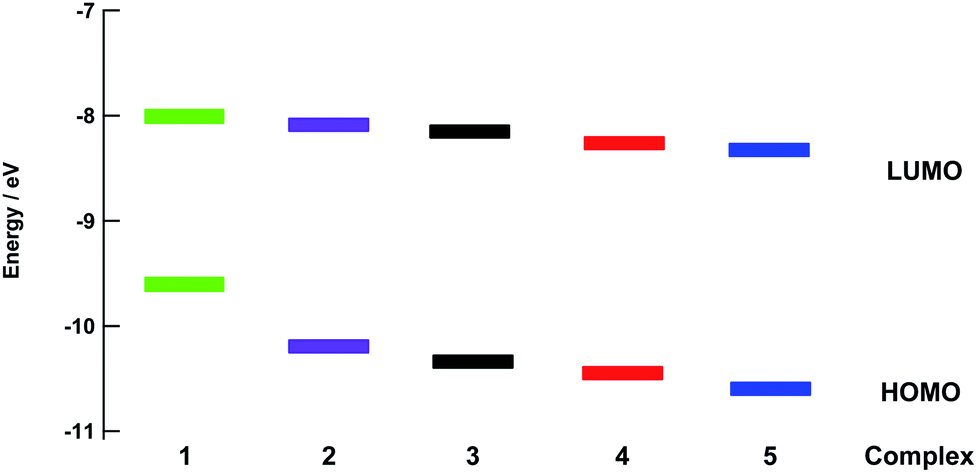 | ||
| Fig. 5 Calculated energies of the HOMO and LUMO for 1–5. | ||
Photochemistry
The ligand substitution photochemistry of complexes 1–5 was investigated using electronic absorption and NMR spectroscopies. The irradiation of 1 in water (<5% acetone) with visible light, λirr ≥ 395, results in a decrease of the 1MLCT band centered at 410 nm with the concomitant growth of a new peak at 440 nm (Fig. 6). A similar red-shift in the 1MLCT absorption peak was reported for the substitution of CH3CN with a solvent H2O molecule to generate the corresponding aqua Ru(II) complex.58 The complete conversion occurred within 80 seconds with two isosbestic points at 355 nm and 420 nm, consistent with the formation of a single photoproduct. Similar changes to the electronic absorption spectra are observed upon the irradiation of 2–4 in H2O (λirr > 395 nm), as shown in Fig. S9.† No spectral changes were observed for 1–5 in water when the samples were kept in the dark at room temperature under otherwise identical conditions (Fig. S10†), consistent with their thermal stability.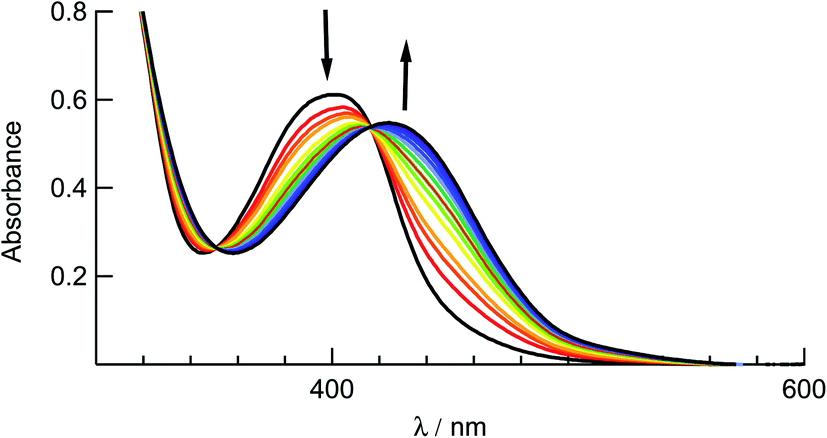 | ||
| Fig. 6 Changes to the electronic absorption spectrum of 1 upon irradiation (tirr = 0–80 s) in water (<5% acetone, λirr > 395 nm). | ||
Additionally, changes to the 1H NMR spectrum of 3 upon irradiation (λirr ≥ 395 nm) in CD3CN resulted in a decrease of the resonance of coordinated CH3CN at 2.55 ppm with the concomitant increase of that attributed to free acetonitrile at 1.96 ppm, with no other spectral changes (Fig. S11†). A similar 31P NMR experiment in CD3CN did not lead to any changes or shifts in the triphenylphospine resonance upon irradiation (λirr ≥ 395 nm). These results are consistent with the exchange of CH3CN for the deuterated solvent and show that the photochemistry arises solely from ligand exchange.
It should be noted that longer irradiation times were necessary for ligand exchange to take place for 2–5 as compared to 1 under similar experimental conditions. This dependence is evident in the ∼3-fold decrease in the quantum yield for ligand exchange, Φ400, across the series listed in Table 1, from 0.077(2) in 1 to 0.025(2) in 5 (λirr = 400 nm). These data show that the quantum yield of ligand exchange is dependent on the substituent on the ancillary phosphine ligand, however, it is an unexpected trend based on the lower energy 1MLCT absorption of 1 as compared to 5. Instead, the quantum yields of 1–5 can be correlated with the Hammett para parameter (σp) corresponding to the substituent on each phosphine ligand and the results are shown in Fig. 7.65 The linear relationship between Φ400 and σp (R = 0.98) suggests that the difference in the quantum yield values across the series is strongly correlated to the electronic nature of the phosphine despite being ancillary to the Ru(dπ) → bpy(π*) 1MLCT transition and the CH3CN photodissociation. Similarly, the pKa values of each substituted phosphine ligand can also be correlated with the Φ400 values (Fig. S12†), as the pKa values are an excellent representation of the changing electronic character across the series and are now known to serve a substitute for Hammett parameters.2,46,66
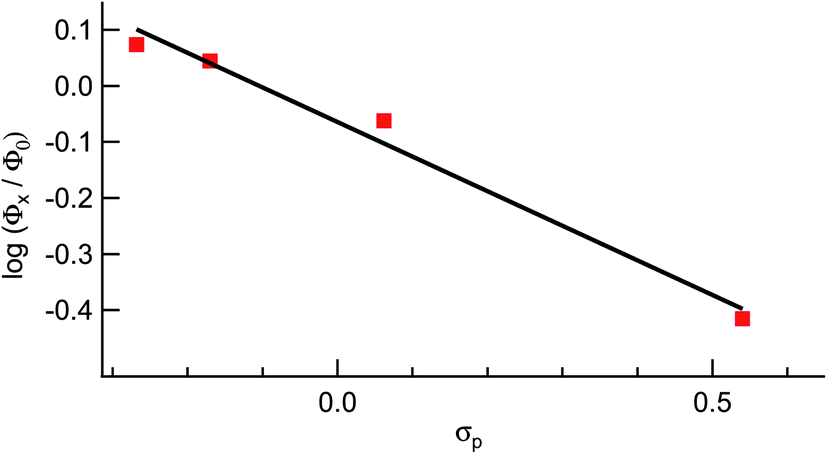 | ||
| Fig. 7 Hammett parameter plot of the relative values of Φ400 for 1, 2, 4, and 5, Φx, relative to that of 3, Φ0, as a function of σp values. | ||
Indeed, the fact that the value of Φ400 increases as the 1MLCT absorption shifts to lower energy is counter to the commonly accepted viewpoint, since a lower in energy 1MLCT absorption is indicative of a lower probability of the population of the dissociative 3LF excited state(s) from the 3MLCT.25,38,42 However, recent work has shown that the photodissociation of acetonitrile behaves differently than other ligands, such as pyridine and sulfoxides. In particular, it has been theorized that the strength of the Ru–NCCH3 bond relies on the ability of ruthenium to backbond into the CH3CN(π*) orbitals, which in turn is highly dependent on the electron density on the ruthenium metal center.42,67 The removal of electron density in the HOMOs of 1 and 2 through interaction with the P–C(σ*) orbital of the phospine ligands weakens the Ru–P bond,68 especially in the 3MLCT excited state where an electron is removed from the metal, resulting in more efficient ligand dissociation in these complexes as compared to 4 and 5. It should be noted that dynamic effects should also be considered, such as changes to the orbital/state energies as the Ru–P bond elongates in the 3MLCT potential energy surface. Thus, from a combination of spectroscopic and electrochemical data, the observed increase of Φ400 with additional electron donating groups likely arises from the electronic nature of each phosphine ligand and is not due to steric differences among the complexes.
Cell viability and cell death mechanism
In order to understand how photochemical reactivity influences the biological behavior of complexes 1–5, the compounds were evaluated against triple-negative breast cancer MDA-MB-231 cells upon irradiation and under dark conditions. The MDA-MB-231 cell line was chosen to benchmark phosphine-derived complexes 1–5 against other photoactive Ru(II) complexes we have evaluated in the past.30,56,69 Half effective concentration or EC50 were determined, which is the measure of the concentration of compound needed to produce 50% of viable cells as compared to the with vehicle control (1% DMSO in DMEM), which was defined as 100% viability. A lower EC50 value of a compound corresponds to a greater toxicity and the curves for each complex in the dark and upon irradiation are shown in Fig. S13–S17.†MDA-MB-231 cells were incubated with 1–5, then the cells were left in the dark or irradiated with 460–470 nm light (tirr = 20 min, 56 J cm−2). After 72 h, cell viability was assessed using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay. Interestingly, 1–5 were not toxic against MDA-MB-231 cells in the dark under the limits of our experimental conditions, limited by the maximum concentration permitted by solubility in the growth media, 30 μM (Table 2). However, upon irradiation with blue light, toxicity was significantly increased for 3 and 5. Complex 3 showed an ECI50 value of 7.0 ± 1.4 μM under irradiation as compared to ECD50 >30 μM in the dark, resulting in a phototherapeutic index, PI, greater than 4.2 (Table 2). Complex 5, bearing –CF3 substituents, was >3-fold more potent than 3, with ECI50 = 1.6 ± 0.3 μM upon irradiation and a PI > 19 (Table 2). Importantly, this level of activity is ∼10-fold greater than that of oxaliplatin and cisplatin against MDA-MB-231 cells, and more potent than a number of promising new platinum and ruthenium compounds.70,71 Photoactivated 5 exhibits a 3-fold lower ECI50 value against MDA-MB-231 cells than [Ru(tpy)(Me2dppn)(py)](PF6)2 (Me2dppn = dimethylbenzo[i]dipyrido[3,2-a:2′,3′-c]phenazine), with ECI50 = 4.6 ± 0.5 μM, a lead compound reported by us previously that generates 1O2 and releases pyridine.56
P values of 1–5 in MDA-MB-231 cells in the dark, ECD50, and upon irradiation, ECI50 along with the phototherapeutic index (PI)
| Complex | ECD50a | ECI50a | PIb | LogPc |
|---|---|---|---|---|
|
a Average of three independent experiments.
b PI = ECD50/ECI50.
c LogP = octanol:water partition coefficient, determined by shake flask method, 298 ± 3 K, results are average of three independent experiments, errors are standard deviations.
d Not determined.
|
||||
| 1 | >30 | >30 | — | |
| 2 | >30 | >30 | — | −0.042 ± 0.010 |
| 3 | >30 | 7.0 ± 1.4 | >4.2 | −0.098 ± 0.002 |
| 4 | >30 | >30 | — | 0.50 ± 0.01 |
| 5 | >30 | 1.6 ± 0.3 | >19 | −0.29 ± 0.01 |
As described earlier, when solutions of 1–5 are irradiated in water, the CH3CN ligand in each complex is substituted with a solvent H2O molecule, resulting in the corresponding Ru(II) mono-aqua complex. It is evident from the data that irradiation triggers growth inhibition effects for 3 and 5, which suggests the Ru(II) aqua complexes [Ru(bpy)2(L)(H2O)]2+ where L = PPh3 and P(p-CF3Ph)3 are major contributors towards toxicity in the MDA-MB-231 cells. Additional experiments will be required in the future to gain further understanding of the differences in activities among the complexes, including cellular penetration and subcellular localization, especially because complex 5 exhibits the lowest ligand exchange quantum yield in the series (Table 1), but the greatest photocytotoxicity (Table 2). Furthermore, cellular toxicity did not show a positive correlation with LogP values (octanol:water partition coefficients, Table 2), which are a measure of compound lipophilicity. Higher LogP values are often associated with greater cellular uptake for Ru(II) compounds.72 In this case, complex 5 was the most hydrophilic and also the most cytotoxic. It should be pointed out, however, that the related complexes cis-[Ru(bpy)2(CH3CN)2]2+ and [Ru(tpy)(CH3CN)3]2+ (tpy = 2,2':6′,2′′-terpyridine), both of which generate mono-aqua complexes upon irradiation and bis-aqua products under further photolysis, are not cytotoxic against in the dark or upon irradiation.29,52 This finding is consistent with the phosphine ligands in 3 and 5, PPh3 and P(p-CF3-Ph)3, respectively, providing enhanced cellular uptake and/or mitochondrial localization for increased activity.34,35,53
The EC50 experiments indicate that 5 is the most potent inducer of triple-negative breast cancer cell death, and also displayed the highest PI value. In order to gain more insight into the cellular behavior of complex 5 in MDA-MB-231 cells, the mode of cell death was investigated. Various pathways are known to induce cell death. Some of the major causes of cell death are apoptosis, necrosis, necroptosis and cell death due to production of reactive oxygen species (ROS). Apoptosis is a form of programmed cell death and is known to be controlled by caspases.73 Cellular changes that include plasma membrane blebbing, cell shrinkage and condensation of chromatin are known to be some characteristic features of apoptosis.74 ROS species such as superoxide radical, hydroxyl radical, and hydrogen peroxide are known to be produced due to dysfunction of the mitochondria.75 ROS generation results in degradation of proteins, lipids, and nucleic acids, eventually leading to cell death by inducing apoptosis.76 Necrosis is a form of passive or uncontrolled form of cell death, characterized by the rupturing of the plasma membrane. Necroptosis is a form of programmed necrosis known to begin as apoptosis but to end like necrosis, mediated by receptor-interacting protein kinase 1 (RIPK1) enzyme.77 Necrostatin (NEC) inhibits cell death that proceeds via necroptosis,78 Z-VAD-FMK (ZVAD) inhibits apoptosis through caspase inactivation, and N-acetyl cysteine (NAC) inhibits cell death due to the production of ROS.79,80 Cell rescue, or an increase in the cellular viability when cells are cotreated with agents and respective cell death inhibitors, NEC, ZVAD or NAC, as compared to treatment with the agents alone, can support the action of individual cell death mechanism pathways.
Control experiments confirmed when MDA-MB-231 cells were treated with NEC (20 μM), ZVAD (20 μM) or NAC (5 mM) alone, no toxicity was observed under dark or blue light irradiation (Fig. S18 and S19†). Cotreatment of the cells with NEC or NAC and complex 5 (2 μM) under irradiation (tirr = 20 min, λirr = 460–470 nm, 56 J cm−2) did not cause any increase in cellular viability as compared to 5 alone (Fig. 8). However, cotreatment of the cells with complex 5 and ZVAD (20 μM) showed statistically significant (P < 0.05) increase in cellular viability (Fig. 8). In contrast, no changes in cell viability are observed with the co-treatment of 5 with NEC, ZVAD, or NAC when kept in the dark (Fig. S20†). The increase in cellular viability on cotreatment of the cells with complex 5 and ZVAD is consistent with complex 5 inducing apoptosis in MDA-MB-231 cells upon irradiation.
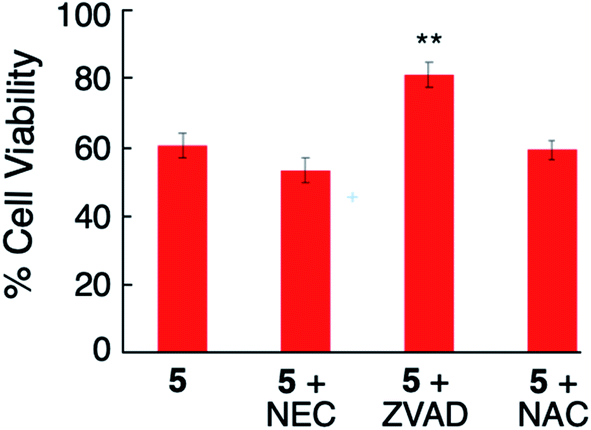 | ||
| Fig. 8 Viability of MDA-MB-231 cells treated with 5 (2 μM) alone and co-treated with necrostatin (NEC), Z-VAD-FMK, N-acetyl cysteine (NAC) followed by irradiation (tirr = 20 min, λirr = 460–470 nm, 56 J cm−2). P values are vs. 2 μM 5; ***P < 0.01 **P < 0.05 *P < 0.10. | ||
In order to gain more evidence that complex 5 causes cell death through apoptosis, fluorescence-assisted cell sorting (FACS) was utilized. In these experiments, MDA-MB-231 cells were treated with vehicle (1% DMSO) alone as a negative control, complex 5, H2O2 as a necrosis positive control, or [Ru(thd)(tpy)(py)]PF6 (thd = 2,2,6,6-tetramethylheptane-3,5-dione, py = pyridine), which serves as a positive control for apoptosis.81 Cells were treated with vehicle or 5 (10 μM) for 1 h, then irradiated with blue light (tirr = 20 min, λirr = 460–470 nm, 56 J cm−2) or left in the dark for the same amount of time. The cells were stained with Annexin V and propidium iodide (PI) 72 h after light irradiation and were analyzed by FACS. During apoptosis, phosphatidylserine is translocated from the inner to the outer leaflet of the cell membrane which facilitates binding with Annexin V.82 Propidium iodide is non-permeable in healthy cells but can enter cells when the integrity of the outer cell membrane is compromised, which occurs during necrosis.14 Cells treated with vehicle alone under irradiation (Fig. 9a) or dark (Fig. 9b) conditions, under dark conditions were viable, showing minimal staining with Annexin V or propidium iodide (>97% of population of cells in lower left quadrant). Cells treated with [Ru(thd)(tpy)(py)]PF6 (6 μM, Fig. 9c) showed Annexin V staining but not PI uptake (>14% lower right quadrant), consistent with apoptosis, whereas cells treated with H2O2 (500 mM, Fig. 9d) showed heavy Annexin V and PI staining consistent with necrosis (>85% of population in upper right quadrant). Cells treated with 5 (10 μM, Fig. 9e) under dark conditions, showed Annexin V staining but not PI uptake (>26% of population in lower right coordinate) similar to that of Fig. 9c, with a shift to significant Annexin V staining (>64% of population in the lower right coordinate) upon irradiation (Fig. 9f). The shifting of the cell population from the lower left to the lower right quadrant under dark vs. irradiation conditions is consistent with complex 5 inducing apoptosis in MDA-MB-231 cells.
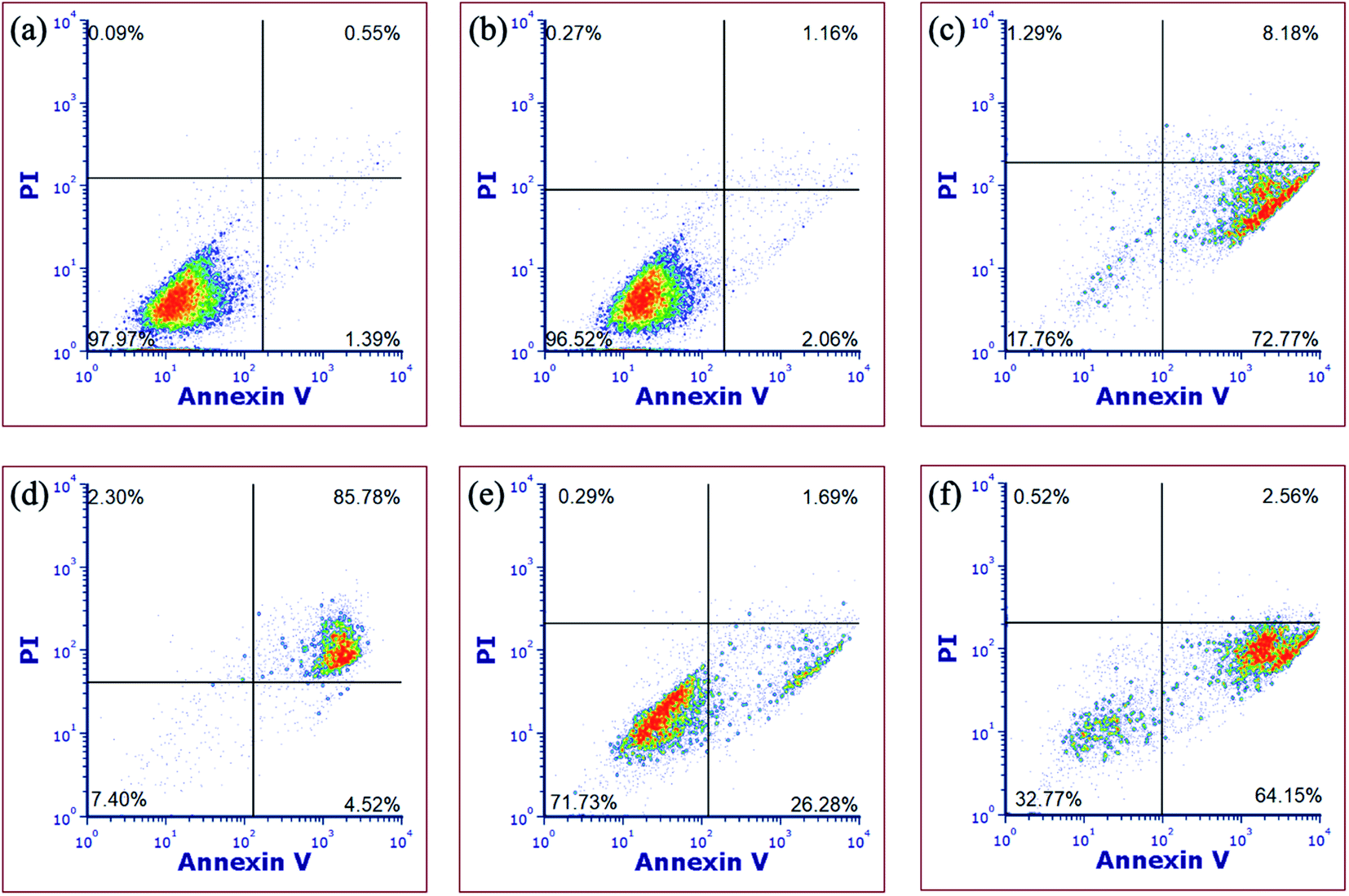 | ||
| Fig. 9 Flow cytometric analysis of MDA-MB-231 cells after 72 h treatment using Annexin V/propidium iodide staining, showing (a) vehicle followed by irradiation, (b) vehicle without irradiation, (c) [Ru(thd)(tpy)(py)]PF6 (6 μM), (d) H2O2 (500 mM); (e) 5 (10 μM) without irradiation and (f) 5 (10 μM) with irradiation. Data are indicative of three independent experiments. | ||
Conclusion
A series of phosphine-containing Ru(II) complexes of the type cis-[Ru(bpy)2(P(p-R-Ph)3)(CH3CN)](PF6)2, where R = –OCH3 (1), –CH3 (2), –H (3), –F (4), and –CF3 (5), was synthesized and their ability undergo photoinduced CH3CN ligand exchange was evaluated. Upon irradiation with visible light, the CH3CN ligand exchanges with solvent water molecules, resulting in the formation of the corresponding aqua complex with quantum yields, Φ400, ranging from 0.077(2) in 1 to 0.025(2) in 5. Complex 1 proved to be the most photoactive although its 1MLCT absorption possesses the lowest energy maximum and calculated HOMO–LUMO gap across the series. The para Hammett parameter for the substituent on the phosphine ligand and the phosphine pKa values correlate linearly with the value of Φ400, pointing to an electronic influence on the photochemistry and providing evidence of the role of electron density on the metal to weaken the Ru–NCCH3 bond through reduced π-backbonding. While 1–5 are nontoxic towards a triple negative breast cancer cell line, complexes 3 and 5 exhibit >4.2- and >19-fold increase in toxicity upon irradiation. A number of experiments show that the photoinduced cell death by 5 is a result of apoptosis, not necrosis or necroptosis. This work provides a new strategy for the design of ruthenium complexes for the photoinduced release of cytotoxic ruthenium fragments that are damaging to cancer cells, as well as nitrile-derived therapeutics.Author contributions
Austin P. Lanquist synthesized the complexes and measured the photochemistry, Kathlyn F. Al-Afyouni performed calculations, Malik Al-Afyouni helped in solving crystal structures, Sayak Gupta conducted cellular measurements and Jeremy J. Kodanko and Claudia Turro directed the project and finalized the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for a grant from the National Science Foundation (CHE-2102508) for partial support of this work.References
- T. J. Meyer, Acc. Chem. Res., 1978, 44, 61–82 Search PubMed.
- M. P. Mitoraj and A. Michalak, Inorg. Chem., 2010, 49, 578–582 CrossRef CAS PubMed.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Top. Curr. Chem., 2007, 117–214 CrossRef CAS.
- J. C. Hidalgo-Acosta, M. A. Méndez, M. D. Scanlon, H. Vrubel, V. Amstutz, W. Adamiak, M. Opallo and H. H. Girault, Chem. Sci., 2015, 6, 1761–1769 RSC.
- R. Forster, P. Bertoncello and T. Keyes, Annu. Rev. Anal. Chem., 2009, 2, 359–385 CrossRef CAS PubMed.
- A. W. King, L. Wang and J. J. Rack, Acc. Chem. Res., 2015, 48, 115–1122 CrossRef PubMed.
- B. A. McClure, E. R. Abrams and J. J. Rack, J. Am. Chem. Soc., 2010, 132, 5428–5436 CrossRef CAS PubMed.
- P. Ceroni, A. Credi and M. Venturi, Chem. Soc. Rev., 2014, 43, 4068–4083 RSC.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785–9789 CrossRef CAS PubMed.
- G. F. Manbeck and K. J. Brewer, Coord. Chem. Rev., 2013, 257, 1660–1675 CrossRef CAS.
- J. J. Concepcion, J. W. Jurss, M. K. Brennaman, P. G. Hoertz, A. O. T. Patrocinio, N. Y. Murakami Iha, J. L. Templeton and T. J. Meyer, Acc. Chem. Res., 2009, 42, 1954–1965 CrossRef CAS PubMed.
- J. Zhu, W. A. Maza and A. J. Morris, J. Photochem. Photobiol., A, 2017, 344, 64–77 CrossRef CAS.
- B. Pashaei, H. Shahroosvand, M. Graetzel and M. K. Nazeeruddin, Chem. Rev., 2016, 116, 9485–9564 CrossRef CAS PubMed.
- L. Hammarström and O. Johansson, Coord. Chem. Rev., 2010, 254, 2546–2559 CrossRef.
- M. D. Turlington, M. D. Brady and G. J. Meyer, ACS Appl. Energy Mater., 2021, 4, 745–754 CrossRef CAS.
- C. Reichardt, S. Monro, F. H. Sobotta, K. L. Colón, T. Sainuddin, M. Stephenson, E. Sampson, J. Roque III, H. Yin, J. C. Brendel, C. G. Cameron, S. McFarland and B. Dietzek, Inorg. Chem., 2019, 58, 3156–3166 CrossRef CAS PubMed.
- R. N. Pickens, B. J. Neyhouse, D. T. Reed, S. T. Ashton and J. K. White, Inorg. Chem., 2018, 18, 11616–11625 CrossRef PubMed.
- H. Yin, M. Stephenson, J. Gibson, E. Sampson, G. Shi, T. Sainuddin, S. Monro and S. A. McFarland, Inorg. Chem., 2014, 53, 4548–4559 CrossRef CAS PubMed.
- (a) Y. Liu, R. Hammitt, D. A. Lutterman, L. E. Joyce, R. P. Thummel and C. Turro, Inorg. Chem., 2009, 48, 375–385 CrossRef CAS PubMed; (b) Y. Sun, L. E. Joyce, N. M. Dickson and C. Turro, Chem. Commun., 2010, 46, 2426–2428 RSC; (c) Y. Sun, M. El Ojaimi, R. Hammitt, R. P. Thummel and C. Turro, J. Phys. Chem. B, 2010, 114, 14664–14670 CrossRef CAS PubMed.
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC.
- T. N. Singh and C. Turro, Inorg. Chem., 2004, 43, 7260–7262 CrossRef CAS PubMed.
- J. K. White, R. H. Schmehl and C. Turro, Inorg. Chim. Acta, 2017, 454, 7–20 CrossRef CAS PubMed.
- J. D. Knoll and C. Turro, Coord. Chem. Rev., 2015, 282, 110–126 CrossRef PubMed.
- M. H. Al-Afyouni, T. N. Rohrabaugh Jr, K. F. Al-Afyouni and C. Turro, Chem. Sci., 2018, 9, 6711–6720 RSC.
- L. M. Loftus, K. F. Al-Afyouni and C. Turro, Chem.–Eur. J., 2018, 24, 11550–11553 CrossRef CAS PubMed.
- J. D. Knoll, B. A. Albani and C. Turro, Acc. Chem. Res., 2015, 48, 2280–2287 CrossRef CAS PubMed.
- B. A. Albani, B. Peña, N. A. Leed, N. A. B. G. De Paula, C. Pavani, M. S. Baptista, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2014, 136, 17095–17101 CrossRef CAS PubMed.
- K. Arora, M. Herroon, M. H. Al-Afyouni, N. P. Toupin, T. N. Rohrabaugh Jr, L. M. Loftus, I. Podgorski, C. Turro and J. J. Kodanko, J. Am. Chem. Soc., 2018, 140, 14367–14380 CrossRef CAS PubMed.
- T. N. Rohrabaugh Jr, K. A. Collins, C. Xue, J. K. White, J. J. Kodanko and C. Turro, Dalton Trans., 2018, 47, 11851–11858 RSC.
- J. D. Knoll, B. A. Albani and C. Turro, Chem. Commun., 2015, 51, 8777–8780 RSC.
- (a) O. Filevich and R. Etchenique, Photochem. Photobiol. Sci., 2013, 12, 1565–1570 CrossRef CAS PubMed; (b) L. Zayat, M. G. Noval, J. Campi, C. I. Calero, D. J. Calvo and R. Etchenique, ChemBioChem, 2007, 8, 2035–2038 CrossRef CAS PubMed.
- (a) A. A. Khan, K. S. Allemailen, A. Almatroudi, S. A. Almatroodi, M. A. Alsahli and A. H. Rahmani, J. Drug Delivery Sci. Technol., 2021, 61, 102315 CrossRef CAS; (b) M. Ali, L. Dondaine, A. Adolle, C. Sampaio, F. Chotard, P. Richard, F. Denat, A. Bettaieb, P. Le Gendre, V. Laurens, C. Goze, C. Paul and E. Bodio, J. Med. Chem., 2015, 58, 4521–4528 CrossRef CAS PubMed.
- R. S. Correa, L. M. Bomfim, K. M. Oliveira, D. R. M. Moreira, M. B. P. Soares, J. Ellena, D. P. Bezerra and A. A. Batista, J. Biol. Inorg Chem., 2019, 198, 110751 CAS.
- Q. Sun, S. Mosquera-Vazquez, Y. Suffren, J. Hankache, N. Amstutz, L. Lawson Daku, E. Vauthey and A. Hauser, Coord. Chem. Rev., 2015, 282, 87–99 CrossRef.
- W. F. Wacholtz, R. A. Auerbach and R. H. Schmehl, Inorg. Chem., 1986, 25, 227–234 CrossRef CAS.
- P. C. Ford, Coord. Chem. Rev., 1982, 44, 61–82 CrossRef CAS.
- K. Nisbett, Y. J. Tu, C. Turro, J. J. Kodanko and H. B. Schlegel, Inorg. Chem., 2018, 57, 231–240 CrossRef CAS PubMed.
- Q. Sun, S. Mosquera-Vazquez, L. Lawson Daku, L. Guénée, H. A. Goodwin, E. Vauthey and A. Hauser, J. Am. Chem. Soc., 2013, 135, 13660–13663 CrossRef CAS PubMed.
- J. D. Knoll, B. A. Albani, C. Durr and C. Turro, J. Phys. Chem. A, 2014, 118, 10603–10610 CrossRef CAS PubMed.
- L. M. Loftus, K. F. Al-Afyouni, T. N. Rohrabaugh Jr, J. C. Gallucci, C. E. Moore, J. J. Rack and C. Turro, J. Phys. Chem. C, 2019, 123, 10291–10299 CrossRef CAS.
- G. K. Kosgei, D. J. Breen, R. W. Lamb, M. Y. Livshits, L. A. Crandall, C. J. Ziegler, C. E. Webster and J. J. Rack, J. Am. Chem. Soc., 2018, 140, 9819–9822 CrossRef CAS PubMed.
- D. A. Lutterman, A. A. Rachford, J. J. Rack and C. Turro, J. Phys. Chem. Lett., 2010, 23, 3371–3375 CrossRef.
- D. A. Lutterman, A. A. Rachford, J. J. Rack and C. Turro, J. Phys. Chem. A, 2009, 113, 11002–11006 CrossRef CAS PubMed.
- J. A. Tossell, J. H. Moore and J. c. Giordan, J. Inorg. Chem., 1985, 24, 1100–1103 CrossRef CAS.
- T. Respondek, R. N. Garner, M. K. Herroon, I. Podgorski, C. Turro and J. J. Kodanko, J. Am. Chem. Soc., 2011, 133, 17164–17167 CrossRef CAS PubMed.
- L. Zayat, O. Filevich, L. M. Baraldo and R. Etchenique, Philos. Trans. R. Soc., A, 2013, 371, 1–12 CrossRef PubMed.
- A. Li, R. Yadav, J. K. White, M. K. Herroon, B. P. Callahan, I. Podgorski, C. Turro, E. E. Scott and J. J. Kodanko, Chem. Commun., 2017, 53, 3673–3676 RSC.
- T. Respondek, R. Sharma, M. K. Herroon, R. N. Garner, J. D. Knoll, E. Cueny, C. Turro, I. Podgorski and J. J. Kodanko, ChemMedChem, 2014, 9, 1306–1315 CrossRef CAS PubMed.
- R. N. Garner, J. C. Gallucci, K. R. Dunbar and C. Turro, Inorg. Chem., 2011, 50, 9213–9215 CrossRef CAS PubMed.
- M. A. Sgambellone, A. David, R. N. Garner, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2013, 135, 11274–11282 CrossRef CAS PubMed.
- (a) N. A. M. Pereira, M. Laranjo, B. F. O. Nascimento, J. C. S. Simões, J. Pina, B. D. P. Costa, G. Brites, J. Braz, J. S. Seixas de Melo, M. Pineiro, M. F. Botelho and T. M. V. D. Pinho e Melo, RSC Med. Chem., 2021, 12, 615–627 RSC; (b) B. Prucelik, A. Sulek, A. Drozd, G. Stochel, M. M. Pereira, S. M. A. Pinto, L. G. Aranut and J. M. Dabrowski, Int. J. Mol. Sci., 2020, 21, 2786 CrossRef PubMed; (c) B. Pucelik, I. Gurol, V. Ahsen, F. Dumoulin and J. M. Dabrowski, Eur. J. Med. Chem., 2016, 124, 284–298 CrossRef CAS PubMed.
- B. P. Sullivan, D. J. Salmon and T. J. Meyer, Inorg. Chem., 1978, 17, 3334–3341 CrossRef CAS.
- H. J. Kuhn, S. E. Braslavsky and R. Schmidt, Pure Appl. Chem., 1989, 61, 187–210 CAS.
- N. P. Toupin, S. Nadella, S. J. Steinke, C. Turro and J. J. Kodanko, Inorg. Chem., 2020, 59, 3919–3933 CrossRef CAS PubMed.
- J. V. Caspar and T. J. Meyer, J. Am. Chem. Soc., 1983, 105, 5583–5590 CrossRef CAS.
- (a) Y. Liu, D. B. Turner, T. N. Singh, A. M. Angeles-Boza, A. Chouai, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2009, 131, 26–27 CrossRef CAS PubMed; (b) J. T. Warren, W. Chen, D. H. Johnston and C. Turro, Inorg. Chem., 1999, 38, 6187–6192 CrossRef CAS PubMed.
- S. V. Litke, A. Y. Ershov and T. J. Meyer, J. Phys. Chem. A, 2014, 118, 6216–6222 CrossRef CAS PubMed.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- N. Fey, A. G. Orpen and J. N. Harvey, Coord. Chem. Rev., 2009, 253, 704–722 CrossRef CAS.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- J. Huang, J. Chen, H. Gao and L. Chen, Inorg. Chem., 2014, 53, 9570–9580 CrossRef CAS PubMed.
- N. Nickita, M. J. Belousoff, A. I. Bhatt, A. M. Bond, G. B. Deacon, G. Gasser and L. Spiccia, Inorg. Chem., 2007, 46, 8638–8651 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- C. A. Streuli, Anal. Chem., 1960, 32, 985–998 CrossRef CAS.
- L. M. Loftus, J. J. Rack and C. Turro, Chem. Commun., 2020, 56, 4070–4073 RSC.
- A. G. Orpen and N. G. Connelly, Organometallics, 1990, 9, 1206–1210 CrossRef CAS.
- S. D. Ramalho, R. Sharma, J. K. White, N. Aggarwal, A. Chalasani, M. Sameni, K. Moin, P. C. Vieira, C. Turro, J. J. Kodanko and B. F. Sloane, PLoS One, 2015, 10, e0142527 CrossRef PubMed.
- N. Muhammad, C.-P. Tan, U. Nawaz, J. Wang, F.-X. Wang, S. Nasreen, L.-N. Ji and Z. W. Mao, Inorg. Chem., 2020, 59, 12632–12642 CrossRef CAS PubMed.
- S. Acharya, M. Maji, M. P. Chakraborty, I. Bhattacharya, R. Das, A. Gupta and A. Mukherjee, Inorg. Chem., 2021, 60, 3418–3430 CrossRef CAS PubMed.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams and J. M. Kelly, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC.
- A. Gorelick-Ashkenazi, R. Weiss, L. Sapozhnikov, A. Florentin, L. Tarayrah-Ibraheim, D. Dweik, K. Yacobi-Sharon and E. Arama, Nat. Commun., 2018, 9, 1–15 CrossRef CAS PubMed.
- G. Häcker, Cell Tissue Res., 2000, 301, 5–17 CrossRef PubMed.
- E. Georgieva, D. Ivanova, Z. Zhelev, R. Bakalova, M. Gulubova and I. Aoki, Anticancer Res., 2017, 37, 5373–5381 CAS.
- M. Redza-Dutordoir and D. A. Averill-Bates, Biochim. Biophys. Acta, 2016, 1863, 2977–2992 CrossRef CAS PubMed.
- J. Yuan, P. Amin and D. Ofengeim, Nat. Rev. Neurosci., 2019, 20, 19–33 CrossRef CAS PubMed.
- W. Zheng, A. Degterev, E. Hsu, J. Yuan and C. Yuan, Bioorg. Med. Chem. Lett., 2008, 18, 4932–4935 CrossRef CAS PubMed.
- N. J. Waterhouse, D. M. Finucane, D. R. Green, J. S. Elce, S. Kumar, E. S. Alnemri, G. Litwack, K. Khanna, M. F. Lavin and D. J. Watters, Cell Death Differ., 1998, 5, 1051–1061 CrossRef CAS PubMed.
- F. Malik, A. Kumar, S. Bhushan, S. Khan, A. Bhatia, K. A. Suri, G. N. Qazi and J. Singh, Apoptosis, 2007, 12, 2115–2133 CrossRef CAS PubMed.
- S. Lee, X. Meng, K. Flatten, D. Loegering and S. Kaufmann, Cell Death Differ., 2013, 20, 64–76 CrossRef CAS PubMed.
- A. M. Rieger, K. L. Nelson, J. D. Konowalchuk and D. R. Barreda, J. Visualized Exp., 2011, 2597 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and crystallographic data, emission spectra, photolysis data, dark controls, DFT structures and atomic coordinates, cell assay control experiments, EC50 curves. CCDC 2009188, 2005084, 2009191, 2009189 and 2009190. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03213e |
| This journal is © The Royal Society of Chemistry 2021 |