
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydroxyl improving the activity, selectivity and stability of supported Ni single atoms for selective semi-hydrogenation†
Minzhen
Jian
a,
Jin-Xun
Liu
 *a and
Wei-Xue
Li
*ab
*a and
Wei-Xue
Li
*ab
aDepartment of Chemical Physics, School of Chemistry and Materials Science, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: wxli70@ustc.edu.cn; jxliu86@ustc.edu.cn
bHefei National Laboratory for Physical Sciences at the Microscale, Hefei, Anhui 230026, China
First published on 7th July 2021
Abstract
Atomically dispersed metal catalysts with high atomic utilization and selectivity have been widely studied for acetylene semi-hydrogenation in excess ethylene among others. Further improvements of activity and selectivity, in addition to stability and loading, remain elusive due to competitive adsorption and desorption between reactants and products, hydrogen activation, partial hydrogenation etc. on limited site available. Herein, comprehensive density functional theory calculations have been used to explore the new strategy by introducing an appropriate ligand to stabilize the active single atom, improving the activity and selectivity on oxide supports. We find that the hydroxyl group can stabilize Ni single atoms significantly by forming Ni1(OH)2 complexes on anatase TiO2(101), whose unique electronic and geometric properties enable high performance in acetylene semi-hydrogenation. Specifically, Ni1(OH)2/TiO2(101) shows favorable acetylene adsorption and promotes the heterolytic dissociation of H2 achieving high catalytic activity, and it simultaneously weakens the ethylene bonding to facilitate subsequent desorption showing high ethylene selectivity. Hydroxyl stabilization of single metal atoms on oxide supports and promotion of the catalytic activity are sensitive to transition metal and the oxide supports. Compared to Co, Rh, Ir, Pd, Pt, Cu, Ag and Au, and anatase ZrO2, IrO2 and NbO2 surfaces, the optimum interactions between Ni, O and Ti and resulted high activity, selectivity and stability make Ni1(OH)2/TiO2(101) a promising catalyst in acetylene hydrogenation. Our work provides valuable guidelines for utilization of ligands in the rational design of stable and efficient atomically dispersed catalysts.
Introduction
Raw ethylene generated by catalytic cracking generally contains 1% acetylene, which has to be removed or reduced to a few ppm to avoid downstream catalyst poisoning and the degradation of polyethylene quality during ethylene polymerization.1–5 The selective hydrogenation of trace acetylene toward ethylene is the most widely used route in the removal of acetylene from the ethylene feed. The major side reaction in acetylene hydrogenation is the generation of undesirable ethane by ethylene hydrogenation and the coupling of C2 species to a green oil blocking the active site. Correspondingly, an efficient catalyst for selective acetylene hydrogenation requires suppressing complete hydrogenation and inhibiting C–C coupling and carbon deposition.Pd is considered as a state-of-the-art catalyst for selective hydrogenation of acetylene, but its high cost and poor selectivity toward ethylene inevitably limits its widespread application.6,7 Belonging to the same group as Pd in the periodic table, Ni metal also shows excellent hydrogenation activity.8,9 The earth abundant and low cost Ni can be considered as an alternative to Pd-based catalysts. However, Ni metal can oligomerize hydrocarbon reactants, which subsequently reduces catalytic selectivity in acetylene hydrogenation towards ethylene.10–12 Improved selectivity for acetylene hydrogenation towards ethylene can be achieved by alloying Ni with inactive metals, such as Zn,13–17 Cu,18–20 Au,21–23 Ga24–26 and Sn,27,28 to form bimetallic particles. Nevertheless, the resulting bimetallic catalysts with the presence of an extended ensemble are not free from the formation of ethane and oligomers. Previous studies revealed that three neighboring Ni atoms can trigger oligomerization,8,29 and isolation of the Ni atoms would suppress oligomerization and improve coke resistance.30 Therefore, oligomer and coke formation is expected to decrease dramatically on atomically dispersed Ni-based catalysts, which might be promising for acetylene semi-hydrogenation.
Atomically dispersed catalysts received wide attention due to their complete exposure of the active metal and demonstrated high activity and selectivity in various reactions.40,41 Compared to traditional nanocatalysts on supports, the confined environment of atomically dispersed catalysts with distinct electronic and geometric structures influences reactant adsorption, activation, reaction on the surface as well as the desorption of desired products dramatically, for achieving the corresponding catalytic activity and selectivity. Compared to stronger adsorption of acetylene than ethylene on palladium particles, acetylene adsorption on atomically dispersed catalysts is weak,31 which would lower the overall activity of acetylene semi-hydrogenation, considering rather low acetylene partial pressure under practical reaction conditions. Improving acetylene bonding but not ethylene bonding, which would prevent ethylene desorption and decrease corresponding selectivity otherwise, is a prerequisite. There have been considerable efforts devoted to modifying the chemical environment of single metal atoms with altered electronic and geometric structures to achieve better activity and selectivity.32–37 To the best of our knowledge, there are still only a few experimental studies reported on acetylene semi-hydrogenation over the atomically dispersed Ni catalyst,38,39 let alone on the chemical modification of atomically dispersed Ni catalysts but addressed in present work theoretically.
Atomically dispersed metal catalysts might often suffer from poor H2 activation due to their relatively weak bonding compared to alternative reactants and/or intermediates involved in hydrogenation reactions. Efficient activation of H2 without triggering the undesirable side reaction is another prerequisite.42 Zheng and co-workers43 found that on an ethylene glycolate (EG)-stabilized Pd single atom on ultrathin TiO2 nanosheets, heterolytic H2 dissociation occurs at the Pd–O interface with extremely high activity. The Karim group44 found that a CO–Ir complex anchored on a MgAl2O4 support is the active site for CO oxidation at low temperature. New interfacial sites formed for the metal–ligand complexes on supports, found for hydroxyl (OH) as well,45–47 could be explored. Moreover, the presence of ligands can improve the stability of atomically dispersed metal catalysts by forming energetically more favorable complexes;47–50 It is one of the critical aspects for practical application. Though OH is widely present in catalytic systems, the question of whether and how OH could improve H2 activation and acetylene semi-hydrogenation, in addition to stability, over a Ni single atom catalyst remains open.
Supports play a vital role in enhancing the stability and catalytic performance of a single atom catalyst. The anatase titanium oxide (TiO2) phase, which is often used in acetylene semi-hydrogenation,51,52 is well known for its high-reactivity in photocatalysis and stability in nanocrystals due to the exposure of more (101) facet with a low surface free energy.53–56 In the present work, we studied OH modulated Ni single atom catalysts on anatase TiO2(101) for acetylene semi-hydrogenation by density functional theory (DFT) calculations. We first studied how Ni1 atoms were stabilized in different Ni1-hydroxyl complexes on TiO2 under steaming conditions. Energetically favorable Ni1(OH)2/TiO2 with right energetics for acetylene adsorption and ethylene desorption was identified. Facile H2 activation and selective hydrogenation promoted by the OH ligand were revealed. We clarify the origin of the high performance of Ni1(OH)2/TiO2 in acetylene semihydrogenation by comparing with other transition metal atoms (Co, Rh, Ir, Pd, Pt, Cu, Ag and Au) and oxide supports (ZrO2, NrO2 and IrO2). The present work provides valuable insights into the critical role of ligands in stabilizing atomically dispersed metal atoms on supports and enhancing the catalytic activity and selectivity.
Computational methods
Spin-polarized periodic DFT calculations were performed by using the Vienna Ab initio Simulation Package (VASP).57,58 The exchange-correlation interaction is described by the optB86b-vdW functional.59 The GGA+U approach was used to treat the strong on-site Coulomb interaction of localized electrons in TiO2, NbO2, ZrO2 and IrO2 with corresponding U values of 3, 2, 4, 1 eV for Ti, Nb, Zr and Ir, respectively, according to previous literature.60–62 The core electrons were represented by the projector augmented wave (PAW) method63 and the Kohn–Sham valence states [Ti(3d2, 4s2), Zr(4d2, 5s2), Nb(4p6, 4d4, 5s1), Ir(4f14, 5d7, 6s2), Ni(4s2, 3d8), O(2s2, 2p4), C(2s2, 2p2), and H(1s1)] were expanded in a plane-wave basis set with a kinetic energy cutoff of 400 eV. The convergence threshold for electronic self-consistent interactions is 10−5 eV. Spin-polarized calculations were incorporated in the geometry optimization and transition state search. Structure optimization and transition state search were converged to the extent that the maximum residual force was 0.02 eV Å−1 and 0.05 eV Å−1 in all relaxed degrees of freedom, respectively. Transition states were determined by the climbing image nudged elastic band (CI-NEB) method64,65 and improved dimer method,66 then verified to possess only one vibrational mode with a negative curvature in the direction of the bond breaking or forming process. The vibrational frequencies and corresponding normal modes were calculated based on the numerical calculations of the second derivatives of the potential energy surface based on the harmonic oscillator approximation. For the vibrational frequencies and modes calculations, all the Ni1(OH)2 active center and adsorbates were allowed to relax. The adsorbates and the Ni coordinated with adsorbates were allowed to relax during the frequency calculations on the Ni (111) surface.A p(2 × 3) slab including six O–Ti–O atomic layers was used to model the anatase TiO2(101) surface, where the top three O–Ti–O atomic layers in the supercell including the Ni atom were fully relaxed. The crystal phases with the same space group as anatase TiO2 were chosen for the other metal oxides (ZrO2, NbO2, and IrO2) employing the same supercell and layer as TiO2(101). The vacuum space perpendicular to the surface was 20 Å, which was enough to avoid interactions between the neighboring slabs. The artificial mirror interaction along the z-direction was avoided by dipole correction. The surface Brillouin zone was sampled on a (3 × 3 × 1) Monkhorst–Pack k-point grid. The adsorption energy without Gibbs free energy correction was calculated by ΔEads = Etot – Eslab − Egas, where Etot and Eslab refer to the energy of the slab with adsorbates and the energy of the clean slab, respectively, and Egas refers to the energy of the involved gas phase adsorbates in a neutral state. The reaction energy and activation energy barrier without Gibbs free energy correction were calculated as ΔErxn = EFS – EIS and Ea = ETS – EIS, where EIS, EFS and ETS refer to the energy of the initial state (IS), final state (FS) and corresponding transition state (TS), respectively.
The Gibbs free energies of all the species were corrected by:
| G(T) = EDFT + ZPE + H(T) − TS(T) | (1) |
The entropy and enthalpy of the adsorbed species at temperature T were estimated according to the harmonic oscillator approximation following the equation:
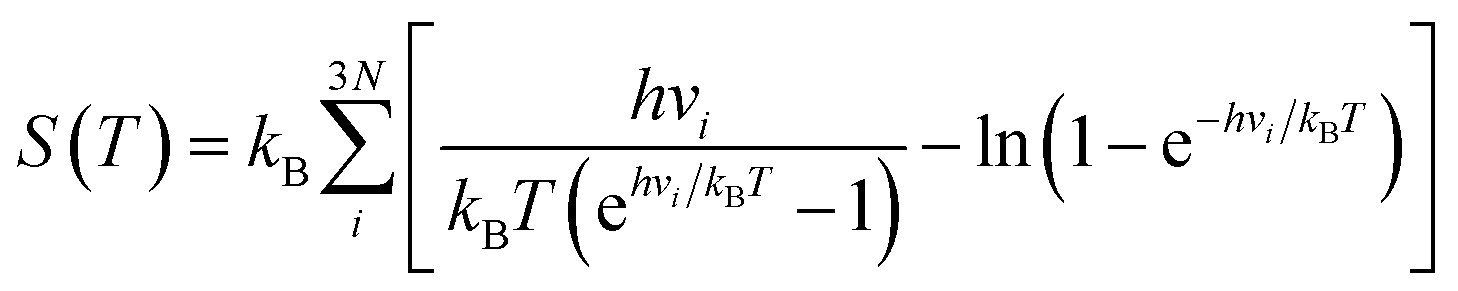 | (2) |
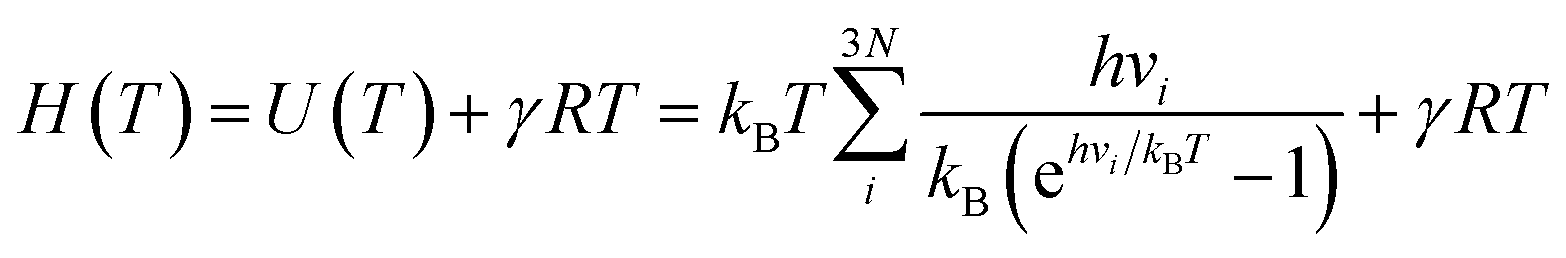 | (3) |
Thus, the Gibbs free energy change for the surface elementary reaction and adsorption was calculated as:
| ΔG = ΔE + ΔZPE + ΔH − TΔS | (4) |
Results and discussion
Structure and stability of Ni1/TiO2 and Ni1(OH)2/TiO2 catalysts
An efficient catalyst for selective semi-hydrogenation should consist of spatially isolated sites to avoid C–C coupling or C–C cleavage in the formation of green oil or coke. In the present work, the atomically dispersed non-noble Ni metal on the anatase-TiO2(101) surface, namely Ni1/TiO2, was studied. The most stable structure for Ni single atom adsorption on TiO2 is given in Fig. S1.† The Ni single atom prefers to anchor on the hollow site by binding three O atoms in TiO2(101) with the binding energy of −3.62 eV, which is in line with a previous report67 and similar to the value of single Pt or Pd atom adsorption on the anatase TiO2(101) surface.68,69 Hydroxyl (OH) is a common species on the oxide surface, especially in the presence of H2 and/or steaming conditions. Therefore, OH can be used as a suitable candidate ligand to modulate the stability and electronic structure of Ni atoms. When treating Ni1/TiO2 with steam, one H2O molecule can be decomposed at the Ni site forming two OH groups binding with the Ni atom, namely Ni1(OH)2/TiO2 (Fig. S1†). H2O dissociation at the Ni1/TiO2 catalyst to generate two OH ligands is exothermic by −0.35 eV with the activation barrier of 0.96 eV (Fig. 1A). Therefore, the formation of the stable Ni1(OH)2/TiO2 form is thermodynamically and kinetically feasible under the reaction conditions at T = 360 K and P(H2O) = 1 atm. The two OH ligands combined with the Ni atom are in a linear configuration in the Ni1(OH)2/TiO2 catalyst leaving side space for acetylene hydrogenation. The stabilities of Ni1/TiO2, Ni1(OH)_H/TiO2 and Ni1(OH)2/TiO2 as a function of temperature and pressure of H2O are shown in Fig. 1B. Ni1/TiO2 is only present at a high temperature or low pressure of H2O. However, Ni1(OH)_H/TiO2 and Ni1(OH)2/TiO2 can be formed at a low temperature and high pressure of H2O. Regardless of the temperature and pressure of water, the Gibbs free energy difference between the formation of Ni1(OH)_H/TiO2 and Ni1(OH)2/TiO2 is 0.01 eV, indicating Ni1(OH)_H/TiO2 and Ni1(OH)2/TiO2 can interconvert with each other easily and coexist. To ensure the stability of the Ni1(OH)2/TiO2 catalyst in the hydrogenation reaction, the partial pressure of H2O should be at least 10−3 atm.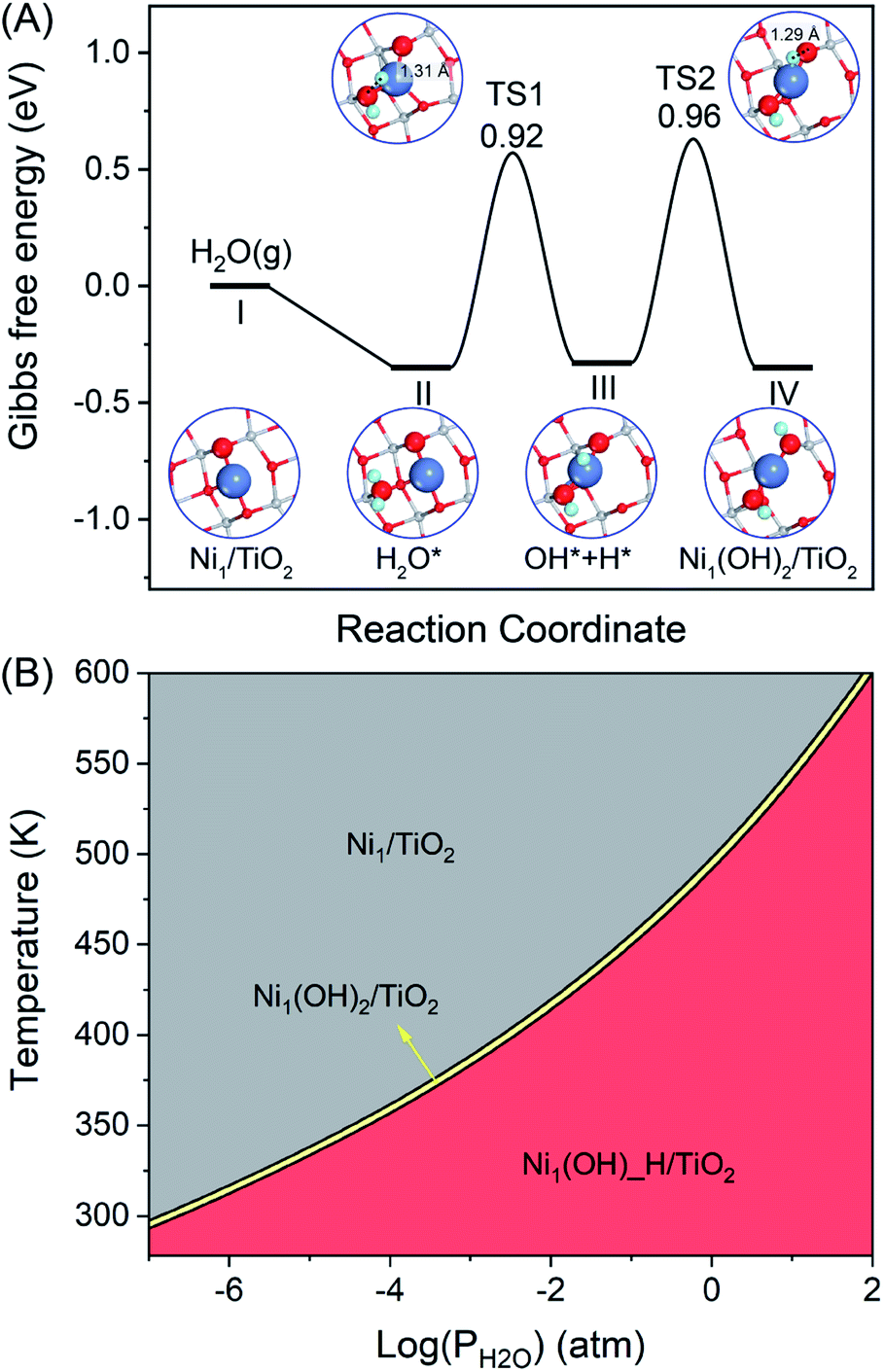 | ||
| Fig. 1 (A) Gibbs free energy diagram and corresponding configurations for H2O dissociation on Ni1/TiO2(101) in the formation of Ni1(OH)2/TiO2(101) at T = 360 K and P(H2O) = 1 atm. Blue, grey, red and green balls are nickel, titanium, oxygen and hydrogen atoms, respectively. (B) Stability diagrams (difference in Gibbs free energy ΔG) of the Ni1/TiO2, Ni1(OH)_H/TiO2 and Ni1(OH)2/TiO2 catalysts as a function of temperature and partial pressure of H2O. | ||
Although the Ni adatom is bound to the TiO2(101) surface strongly with the binding energy of −3.62 eV, it has high mobility to aggregate into large nanoparticles on the TiO2(101) surface with a low diffusion barrier.70 Furthermore, H2 molecule heterolytically dissociates on Ni1/TiO2 is thermal neutral (−0.07 eV) that H2 dissociative adsorption cannot compete with acetylene/ethylene adsorption, a fact that limits the overall reaction activity. However, the OH ligand bridges the Ni adatom and Ti on the oxide support with a higher atomic Ni diffusion barrier of 1.19 eV (Fig. S2†). The hydroxyl groups on the Ni1/TiO2 surface arises from the dissociation of steam, which stabilizes the single-atom Ni due to the strong interactions between Ni and OH. The two H in OH bridged Ni and Ti atoms are stable with the diffusion energies of 1.22 eV and 2.22 eV, respectively (Fig. S2†). Our calculation results are in line with many theoretical calculations and experimental studies which elucidate that transition metal atoms can be stabilized on the TiO2(101) surface via the interaction with excess O originating from the OH groups.32,71,72 A large number of studies have shown that the presence of OH not only stabilizes single atom catalysts but also serves as an intermediate in the chemical reaction cycle.45–47,73 The function of OH ligands, which can also serve as an intermediate in the chemical reaction cycle,45–47,73 in the stable Ni1(OH)2/TiO2 catalyst for catalyzing acetylene hydrogenation is extensively studied in the present work.
Adsorption of acetylene and ethylene
The adsorption strength and competitive adsorption of acetylene and ethylene determine the activity and selectivity of acetylene hydrogenation. Acetylene prefers to adsorb on the Ni (111) surface following the μ-bridge adsorption mode with the Gibbs free energy of −2.12 eV, whereas ethylene favors the perpendicular-bridge adsorption mode with the adsorption strength −0.57 eV weaker under reaction conditions (Fig. S3 and Table S1†). Ethylene adsorption is insensitive to the number of bound Ni atoms ranging from 1, 2 to 3. In contrast, the acetylene adsorption strength decreases with the decreasing number of bound Ni atoms ranging from 1 to 4. As a result, the adsorption difference between ethylene and acetylene at their most stable configurations is −1.55 eV, and becomes −0.08 eV in the π-bonded adsorption mode. The great change in competitive deficiency between acetylene and ethylene adsorption will have a tremendous impact on the selectivity of the acetylene hydrogenation reaction over Ni1 catalysts, where acetylene and ethylene have the same π-bonded adsorption configuration.Similar to the same π-bonded adsorption configuration on the Ni (111) surface, ethylene and acetylene have similar adsorption energies under reaction conditions (Gads = −0.68 eV vs. −0.43 eV, Fig. 2 and Table S2†) on Ni1/TiO2. Therefore, Ni1/TiO2 will not be a good catalyst in acetylene hydrogenation because of the competitive hydrogenation of ethylene and acetylene resulting in poor selectivity of acetylene hydrogenation towards ethylene. Different from Ni1/TiO2, acetylene is adsorbed much more strongly than ethylene on the Ni1(OH)2/TiO2 catalyst by 0.56 eV, conforming to the catalyst screening rule for acetylene semi-hydrogenation14,74 which states that the large difference in acetylene and ethylene adsorption strength results in high selectivity in acetylene semi-hydrogenation towards ethylene. The presence of OH ligands modulates the electronic structure of the Ni atom in Ni1(OH)2/TiO2, which has different catalytic behavior as compared with Ni1/TiO2, namely, acetylene adsorbs at the top of the Ni single atom in Ni1/TiO2 and at the side of the Ni single atom in Ni1(OH)2/TiO2 (Fig. 2B and Table S2†).
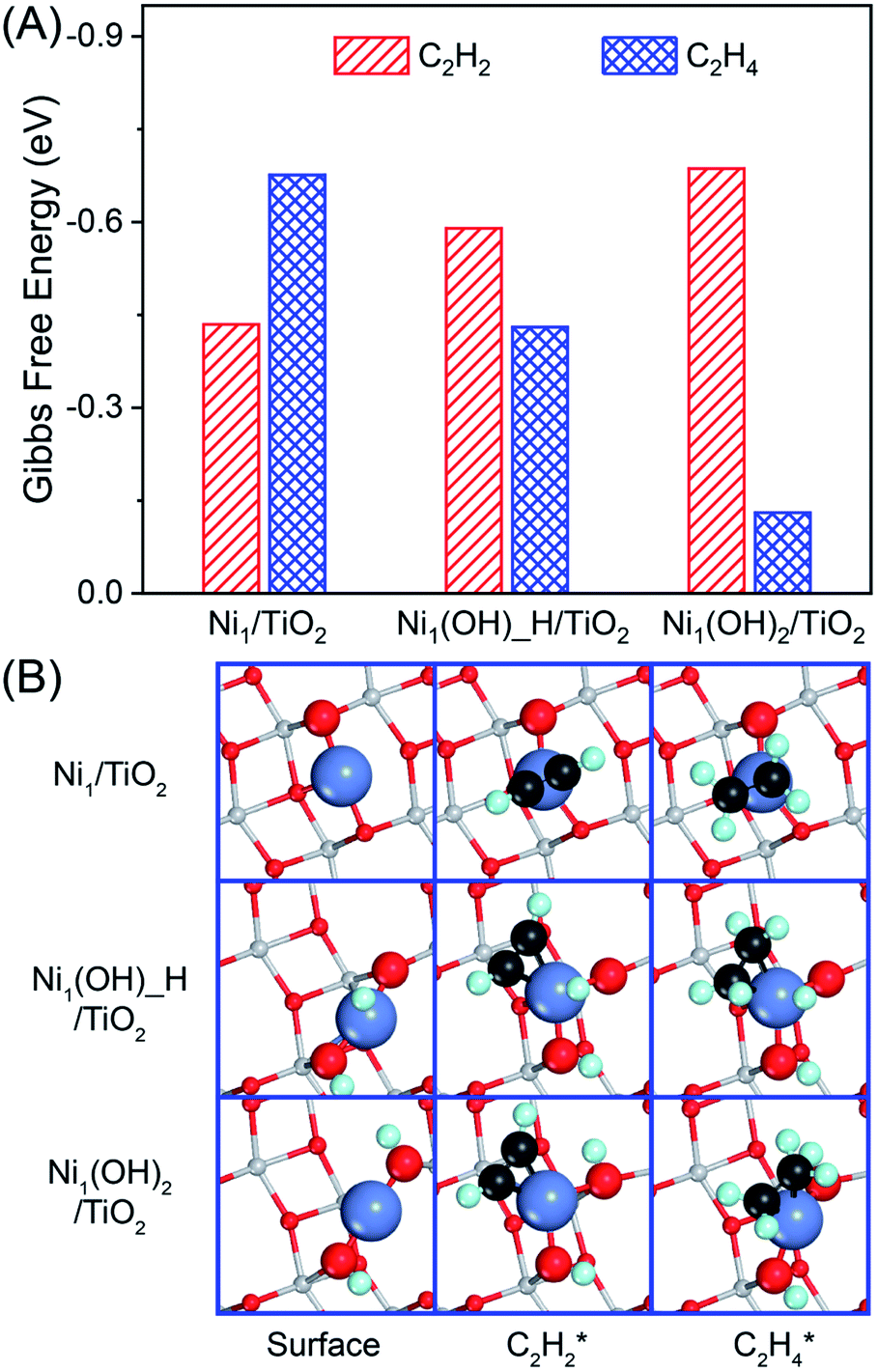 | ||
| Fig. 2 Calculated adsorption energies with Gibbs free energy correction on reaction conditions (A) and the corresponding adsorption configurations (B) of C2H2 and C2H4 on Ni1/TiO2, Ni1(OH)_H/TiO2 and Ni1(OH)2/TiO2. Reaction conditions: T = 360 K, P = 0.1 MPa with 0.5% C2H2, 25% C2H4 and 5% H2. Color code: Ni (blue), C (black), H (green), Ti (gray), O (red). | ||
To reveal the reason behind the adsorption energies of acetylene and ethylene having an opposite trend on Ni1/TiO2, and Ni1(OH)2/TiO2, we resort to integrated COHP(ICOHP), a parameter that describes the bonding strength between reactants and the surface (Table S3†), and decompose the adsorption energy into the adsorption induced structure distortion cost Edis and the chemical bonding energy gain Eb between the adsorbate and catalyst (Fig. S4†). For ethylene on Ni1/TiO2 and Ni1(OH)2/TiO2, the corresponding – ICOHP is 1.59 eV and 1.65 eV. Comparable – ICOHP values indicate similar bonding strengths Eb between ethylene and the two catalysts (−2.00 eV, and −1.96 eV), in contrast to their large difference in Eads, −0.79 eV for Ni1(OH)2/TiO2vs. −1.39 eV for Ni1/TiO2. The less exothermic Eads of ethylene on the former one is due to its larger distortion cost Edis, 1.17 eV vs. 0.62 eV. For acetylene adsorption, though the distortion cost on Ni1(OH)2/TiO2 (2.58 eV) remains larger than that on Ni1/TiO2 (0.76 eV), the corresponding – ICOHP is much larger, 2.25 eV vs. 1.69 eV. In fact, the calculated bonding energy Eb of acetylene on Ni1(OH)2/TiO2 is −4.02 eV, significantly stronger than that on Ni1/TiO2 (−1.92 eV). This results in an overall more exothermic adsorption Eads, −1.44 eV for Ni1(OH)2/TiO2vs. −1.16 eV for Ni1/TiO2, with an opposite trend to ethylene adsorption.
Hydrogenation of acetylene over the Ni1(OH)2/TiO2 catalyst
We studied the mechanism of acetylene hydrogenation towards ethane over the Ni1(OH)2/TiO2 catalyst. According to our DFT calculation results (Table S2†), the Ni single atom sites in the Ni1(OH)2/TiO2 catalyst prefer to adsorb acetylene rather than H2 and ethylene. The strongly competitive adsorption of acetylene compensates for the deficiency of the partial pressure of acetylene, improving the activity and selectivity of acetylene semi-hydrogenation thermodynamically. This is consistent with the rules for screening catalysts in previous literature.14,74 For an acetylene presorbed Ni1(OH)2/TiO2 catalyst, there are fewer active sites and less space for dissociation of physisorbed H2 into atomic H species. Therefore, H atoms in OH ligands can serve as the hydrogen source in acetylene hydrogenation. Through systematic evaluation (Fig. 3, S5 and S6†), the optimal potential energy surface and the corresponding configurations involved in acetylene hydrogenation are shown in Fig. 3 and S5;† H in the bridged OH firstly diffuses to the acetylene adsorbed Ni single atom endothermically (ΔG = 0.12 eV) with an activation barrier of 0.49 eV (I → II). After, the reaction of acetylene with the H at the Ni single atom site is highly exothermic (ΔG = −0.78 eV) and much feasible with the activation barrier of 0.09 eV (II → III). Then, the formed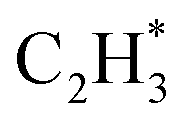 (* refers to the adsorption site) species can react with the physisorbed H2 molecule generating
(* refers to the adsorption site) species can react with the physisorbed H2 molecule generating 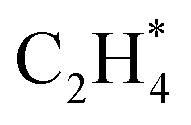 and leaving the second H atom still bound to the Ni single adatom (IV → V). This step is facile and highly exothermic, with an activation barrier of 0.33 eV and a reaction energy of −0.63 eV. Finally, the H atom bound to the Ni single atom site diffuses to the O atom forming OH (V → VI) endothermically by 0.28 eV with an activation energy barrier of 0.29 eV to recover the Ni1(OH)2/TiO2 structure adsorbing ethylene. Although
and leaving the second H atom still bound to the Ni single adatom (IV → V). This step is facile and highly exothermic, with an activation barrier of 0.33 eV and a reaction energy of −0.63 eV. Finally, the H atom bound to the Ni single atom site diffuses to the O atom forming OH (V → VI) endothermically by 0.28 eV with an activation energy barrier of 0.29 eV to recover the Ni1(OH)2/TiO2 structure adsorbing ethylene. Although 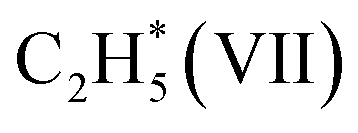 has a lower Gibbs free energy in the potential energy surface, it will not block the active site because the reverse reaction of
has a lower Gibbs free energy in the potential energy surface, it will not block the active site because the reverse reaction of 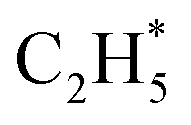 dehydrogenation to
dehydrogenation to 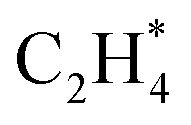 has an activation barrier of 0.60 eV (Fig. S7†) which is still low to overcome at the typical temperature (360 K) for the acetylene hydrogenation reaction. The formed
has an activation barrier of 0.60 eV (Fig. S7†) which is still low to overcome at the typical temperature (360 K) for the acetylene hydrogenation reaction. The formed 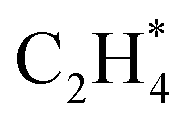 prefers desorption rather than further hydrogenation towards ethane due to the higher activation barriers in the formation of
prefers desorption rather than further hydrogenation towards ethane due to the higher activation barriers in the formation of 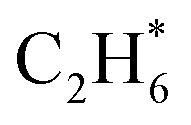 species than that of ethylene desorption by 0.13 eV. Therefore, the Ni1(OH)2/TiO2 catalyst exhibits high selectivity in acetylene hydrogenation towards ethylene. The migration of H from the OH ligand to Ni, which has the highest activation for acetylene hydrogenation towards ethylene over the Ni1(OH)2/TiO2 catalyst, could be the rate-determining step in acetylene hydrogenation.
species than that of ethylene desorption by 0.13 eV. Therefore, the Ni1(OH)2/TiO2 catalyst exhibits high selectivity in acetylene hydrogenation towards ethylene. The migration of H from the OH ligand to Ni, which has the highest activation for acetylene hydrogenation towards ethylene over the Ni1(OH)2/TiO2 catalyst, could be the rate-determining step in acetylene hydrogenation.
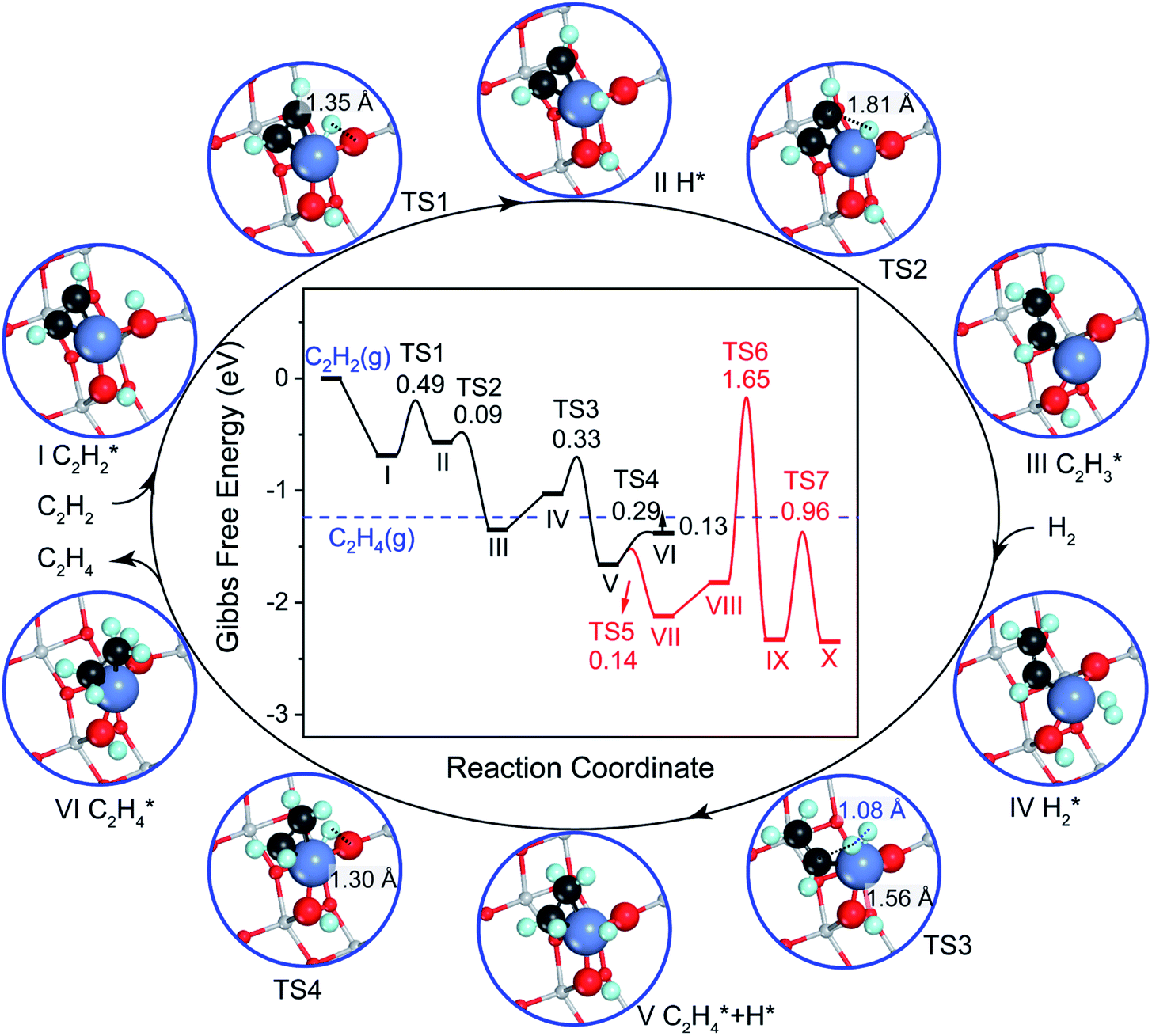 | ||
| Fig. 3 Gibbs free energy diagram and corresponding configurations for acetylene hydrogenation towards ethane on the Ni1(OH)2/TiO2(101) catalyst. The Gibbs free energy was corrected at 360 K in a total pressure of 0.1 MPa, for 0.5% C2H2, 25% C2H4 and 5% H2, respectively. The configurations involved in ethylene hydrogenation to ethane following the red curve in the potential energy diagram are given in Fig. S4.† All the energies are calculated with respect to acetylene and the hydrogen molecule in the gas phase. The blue dashed line represents the ethylene in the gas phase. The distances between C and H atoms in the transition states are indicated in Å. | ||
The charge states of Ni, OH and TiO2 surfaces during acetylene hydrogenation were analysed. Only 0.38e transfers from Ni to the support/OH ligand in the Ni1(OH)2/TiO2 system (Table S4†), where some of the electron 0.25e localize at lattice oxygen in TiO2 and 0.15e localizes on six-coordinated Ti underneath the Ni atom in the Ni1(OH)2/TiO2 catalyst. The charge state of Ni, Ti underneath the Ni atom and O coordinated with Ni varies less than 0.20e during the acetylene hydrogenation cycle such that no polarons could migrate during the entire acetylene hydrogenation reaction. These results clearly show that the magnetic moments of Ti and O atoms have a slight change before and after loading the Ni single atom as well as during the hydrogenation cycle in Ni1(OH)2/TiO2 catalytic systems. The surface coverage of H on metal oxides and the effect of hydrogenation of the metal oxide surface, which is likely to occur under the hydrogenation procedure, on the catalytic performance were also systemically evaluated. We found that the presence of surface H does not have a great influence on the activity and selectivity of acetylene hydrogenation (Fig. S8 and S9†).
The single atom catalyst Ni1(OH)2/TiO2 has different activity and selectivity of acetylene hydrogenation towards ethylene as compared with a nanoparticle catalyst represented with a Ni (111) surface. The Gibbs free energy diagram and corresponding configurations for acetylene hydrogenation over the Ni (111) surface are shown in Fig. S10.† On the Ni (111) surface, the ethylene hydrogenation barrier is comparable to its desorption energy such that ethylene is not the dominant product in acetylene hydrogenation. Therefore, the Ni (111) surface has a lower selectivity in acetylene hydrogenation towards ethylene as compared with the Ni1(OH)2/TiO2 structure. Among others, the effective hydrogenation barrier on Ni (111) is much higher than that on Ni1(OH)2/TiO2 by 0.45 eV (Fig. 4), which could have originated from the stronger acetylene and ethylene adsorption strength on Ni (111) as compared with the Ni1(OH)2/TiO2 catalyst. As a result, Ni1(OH)2/TiO2 has superior performance to Ni nanoparticles not only in selectivity but also in reactivity.
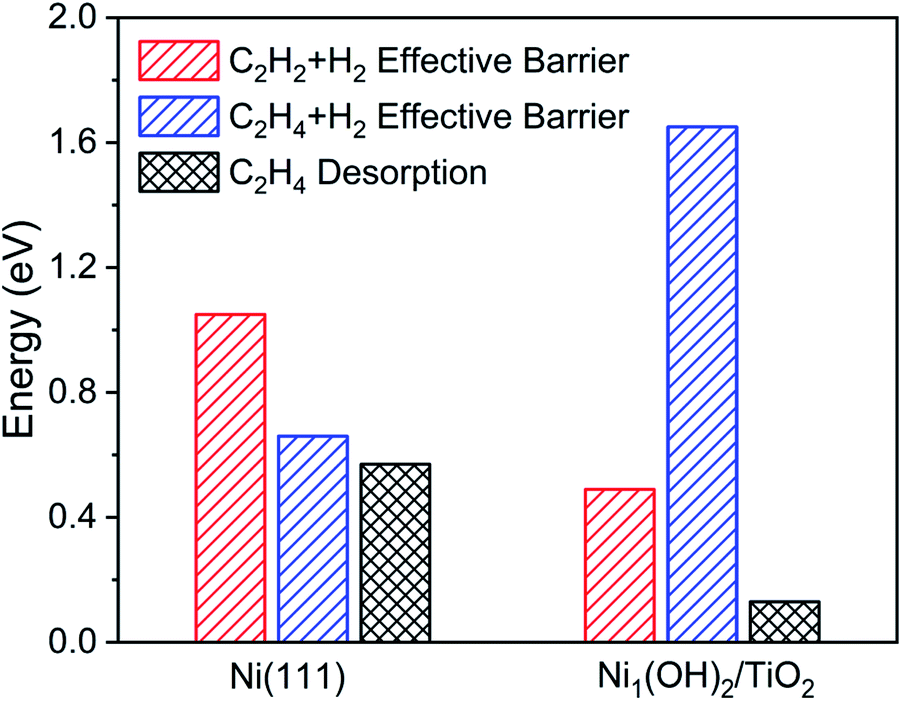 | ||
| Fig. 4 The activity and selectivity comparisons between Ni (111) and Ni1(OH)2/TiO2. The activity is represented by the effective hydrogenation barriers of acetylene. The selectivity for ethylene is estimated by comparing the difference between the desorption energies of ethylene and the effective hydrogenation barriers of ethylene. Reaction conditions: T = 360 K, P = 0.1 MPa with 0.5% C2H2, 25% C2H4 and 5% H2. | ||
As stated above, the hydrogenation of acetylene in the formation of 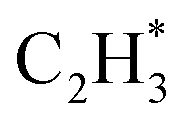 has an even lower activation barrier than
has an even lower activation barrier than 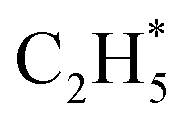 over the Ni1(OH)2/TiO2 catalyst, because the Ni adatom and adsorbed
over the Ni1(OH)2/TiO2 catalyst, because the Ni adatom and adsorbed 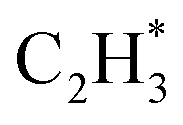 and
and 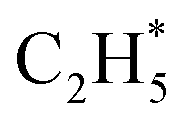 construct an interface that fulfills the formation of Frustrated Lewis Pairs (FLP)75,76 and
construct an interface that fulfills the formation of Frustrated Lewis Pairs (FLP)75,76 and 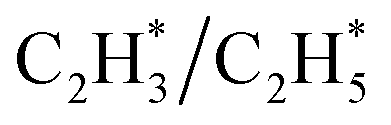 can serve as a basic ligand to accelerate the heterolytic dissociation of H2. The hydrogenation of
can serve as a basic ligand to accelerate the heterolytic dissociation of H2. The hydrogenation of 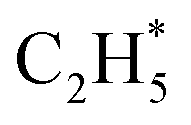 has a higher activation barrier than that of
has a higher activation barrier than that of  by 1.32 eV over the Ni1(OH)2/TiO2 catalyst, which is mainly attributed to their different basicity values. The basicity of C in
by 1.32 eV over the Ni1(OH)2/TiO2 catalyst, which is mainly attributed to their different basicity values. The basicity of C in 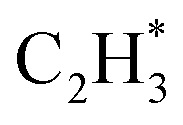 is more than that in
is more than that in 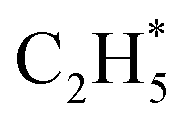 reflected from the Bader charge analysis, +0.34e for C in
reflected from the Bader charge analysis, +0.34e for C in 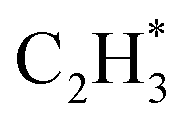 and +0.18e for the C in
and +0.18e for the C in 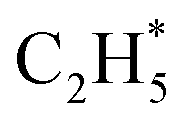 (Fig. S11A and B†), which results in a much lower activation barrier for
(Fig. S11A and B†), which results in a much lower activation barrier for 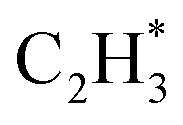 hydrogenation due to the strong electrostatic interactions between H and Ni at the corresponding transition state. Homolytic H2 activation on the Ni site in pristine Ni1(OH)2/TiO2 is inhibited due to a strongly endothermic reaction energy of 2.09 eV (Fig. S11C†), and the heterolytic dissociation of H2 is aided by ligand dominant acetylene hydrogenation activity.
hydrogenation due to the strong electrostatic interactions between H and Ni at the corresponding transition state. Homolytic H2 activation on the Ni site in pristine Ni1(OH)2/TiO2 is inhibited due to a strongly endothermic reaction energy of 2.09 eV (Fig. S11C†), and the heterolytic dissociation of H2 is aided by ligand dominant acetylene hydrogenation activity.
The OH formed by H2O dissociation anchors on the single Ni atom, inhibiting the migration and aggregation of the single Ni adatom. Modulating the electronic structure of Ni atoms by OH ligands enhances the adsorption strength of acetylene accompanied by weakening ethylene adsorption strength, which enlarges the adsorption energy difference between acetylene and ethylene, and improves the activity and selectivity of the catalyst thermodynamically. Hydrogen dissociative adsorption is often the rate-limiting step for single atom catalysts due to the limited space available for H2 adsorption and activation. The formation of the  interface aided by adsorbed acetylene combined with H in the OH ligand led to the formation of effective Frustrated Lewis Pairs (FLP) and enabled more facile heterolytic dissociation of H2, thus promoting the reactivity kinetically. Since stabilization of the Ni1(OH)2 complexes occurs on the whole TiO2 surfaces, rather than on limited defect or step edge sites, a high loading of active Ni1(OH)2/TiO2 can be achieved, one of the central challenges in single atom catalysts. All these together make Ni1(OH)2/TiO2 a promising, stable, highly loaded, active and selective single atom catalyst for partial hydrogenation.
interface aided by adsorbed acetylene combined with H in the OH ligand led to the formation of effective Frustrated Lewis Pairs (FLP) and enabled more facile heterolytic dissociation of H2, thus promoting the reactivity kinetically. Since stabilization of the Ni1(OH)2 complexes occurs on the whole TiO2 surfaces, rather than on limited defect or step edge sites, a high loading of active Ni1(OH)2/TiO2 can be achieved, one of the central challenges in single atom catalysts. All these together make Ni1(OH)2/TiO2 a promising, stable, highly loaded, active and selective single atom catalyst for partial hydrogenation.
Comparison with other transition metals and more oxide supports
To reveal the uniqueness of Ni1(OH)2/TiO2, we considered other transition metal catalysts including Co, Rh, Ir, Ni, Pd, Pt, Cu, Ag, and Au, and more oxide surfaces including Ni1(OH)2/MO2 (M = Zr, Ti, Nb and Ir). According to the optimal structure of the Ni single atom bound with two OH ligands in Ni1(OH)2/TiO2, we further studied various M1(OH)2/TiO2 catalysts (M = Co, Cu, Rh, Pd, Ag, Ir, Pt and Au) which have different electronic structures as compared with Ni1(OH)2/TiO2. The stabilities of M1(OH)2/TiO2 catalysts were first evaluated based on their formation energies which reflects the ability in the formation of the M1(OH)2/TiO2 catalyst by steam treatment of the nanoparticles. The formation of M1(OH)2/TiO2 can be calculated using:| Ef = ETM1(OH)2/TiO2 − ENP − ETiO2 − EH2O | (5) |
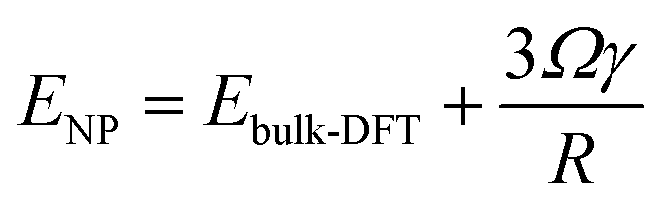 | (6) |
Positive values of M1(OH)2/TiO2 formation energies indicate that the formation of atomically dispersed catalysts is thermodynamically unfavorable, vice versa. Among all nine considered metals, Cu1(OH)2/TiO2 is energetically most favorable, followed by Ni1(OH)2/TiO2. However Ir1(OH)2/TiO2 is least stable and highly endothermic. The stability of M1(OH)2/TiO2 is determined by the interactions between the metal single atom and O (Table S5 and Fig. S12†). We note that although formation of Cu1(OH)2/TiO2 is most favorable, the ethylene adsorption is stronger than that of acetylene, a fact that prevents effective acetylene hydrogenation (Table S6†).
The support effect on the stability of Ni1(OH)2/MO2 (M = Zr, Ti, Nb and Ir) catalysts was further studied. Similar to Ni1(OH)2/TiO2, the stability of Ni1(OH)2/MO2 catalysts was defined as below:
| Ef = ENi1(OH)2/MO2 − ENP − EMO2 − EH2O | (7) |
It can be found that the Ni1(OH)2/IrO2 catalyst has the highest stability, followed by Ni1(OH)2/NbO2 and Ni1(OH)2/TiO2 catalysts. However Ni1(OH)2/ZrO2 is least stable among all four different metal oxide supported Ni single atom catalysts (Fig. 5B). The stability order of Ni1(OH)2/MO2 catalysts is opposite to the order of the metal oxide formation energy (IrO2 < NbO2 < TiO2 < ZrO2). This could have originated from the fact that the stronger the interactions between metal and O in the host oxide surface, the less the energy gain to form a chemical bond toward Ni. For Ni1(OH)2/IrO2, we found that acetylene and ethylene tend to adsorb at the exposed Ir atom, rather on the Ni1 sites (Table S6†). A similar observation was made for Ni1(OH)2/NbO2. The corresponding reactivity for acetylene hydrogenation is out of the scope of the present work.
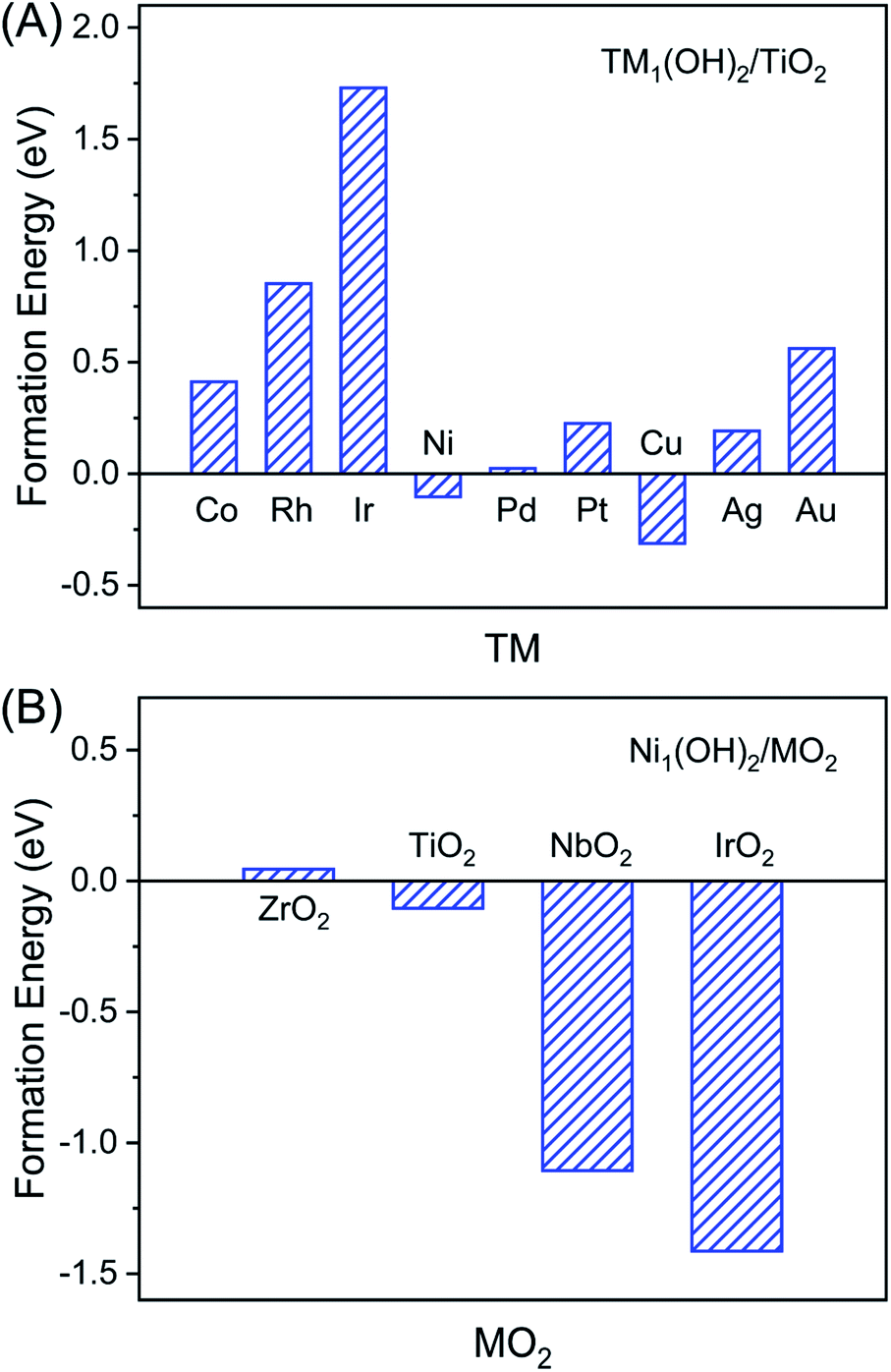 | ||
| Fig. 5 The formation energies of TM1(OH)2/TiO2 (TM = Co, Cu, Ni, Rh, Pd, Ag, Ir, Pt and Au) (A) and Ni1(OH)2/MO2 (M = Zr, Ti, Nb and Ir) (B) by steam-treated corresponding metal nanoparticles. | ||
Conclusions
DFT calculations reveal that hydroxyls present widely in catalytic systems can stabilize Ni single atoms on the anatase TiO2(110) surface efficiently. The resulting Ni1(OH)2/TiO2 catalyst shows high activity for acetylene hydrogenation because its unique electronic and geometric structures form favourable bonds with acetylene and dissociates H2 actively at the same time. Importantly, its weak bonding to ethylene facilitates the subsequent desorption achieving high selectivity toward ethylene. The uniqueness of Ni1(OH)2/TiO2 is attributed to the synergy of stabilizing single transition metal atoms and modulating acetylene and ethylene adsorption. Our work demonstrates a promising strategy to utilize appropriate ligands forming new interfacial sites to mitigate competitive adsorption and molecular activation over atomically dispersed metal catalysts and revealed insights that could be used as guidelines for design of highly stable atomically dispersed catalysts for acetylene hydrogenation on metal oxide materials without sacrificing activity.Author contributions
Wei-Xue Li and Jin-Xun Liu led the conceptualization, design of DFT calculations, analysis and validation of the results. Minzhen Jian contributed to the DFT calculations and data analysis. All the authors participated in writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Key Technologies R&D Program of China (2018YFA0208603), the National Natural Science Foundation of China (91945302), the Chinese Academy of Sciences Key Project (QYZDJ-SSW-SLH054), the start-up funds of University of Science and Technology of China (KY2060000171), USTC Research Funds of the Double First-Class Initiative (YD2060002012) and high-performance computational resources provided by the University of Science and Technology of China (http://scc.ustc.edu.cn).Notes and references
- A. Borodziński and G. C. Bond, Catal. Rev., 2006, 48, 91 CrossRef.
- M. Takht Ravanchi, S. Sahebdelfar and S. Komeili, Rev. Chem. Eng., 2018, 34, 215 CrossRef CAS.
- F. Zaera, ACS Catal., 2017, 7, 4947 CrossRef CAS.
- A. Borodziński and G. C. Bond, Catal. Rev., 2008, 50, 379 CrossRef.
- S. A. Nikolaev, L. N. Zanaveskin, V. V. Smirnov, V. A. Averyanov and K. L. Zanaveskin, Russ. Chem. Rev., 2009, 78, 231 CrossRef CAS.
- A. J. McCue and J. A. Anderson, Front. Chem. Sci. Eng., 2015, 9, 142 CrossRef CAS.
- A. Bos and K. Westerterp, Chem. Eng. Process., 1993, 32, 1 CrossRef CAS.
- B. Bridier, N. Lopez and J. Perez-Ramirez, Dalton Trans., 2010, 39, 8412 RSC.
- D. L. Trimm, I. O. Y. Liu and N. W. Cant, Appl. Catal., A, 2010, 374, 58 CrossRef CAS.
- R. T. Vang, K. Honkala, S. Dahl, E. K. Vestergaard, J. Schnadt, E. Laegsgaard, B. S. Clausen, J. K. Norskov and F. Besenbacher, Nat. Mater., 2005, 4, 160 CrossRef CAS.
- D. L. Trimm, I. O. Y. Liu and N. W. Cant, J. Mol. Catal. A: Chem., 2008, 288, 63 CrossRef CAS.
- D. L. Trimm, I. O. Y. Liu and N. W. Cant, J. Mol. Catal. A: Chem., 2009, 307, 13 CrossRef CAS.
- C. S. Spanjers, J. T. Held, M. J. Jones, D. D. Stanley, R. S. Sim, M. J. Janik and R. M. Rioux, J. Catal., 2014, 316, 164 CrossRef CAS.
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sørensen, C. H. Christensen and J. K. Nørskov, Science, 2008, 320, 1320 CrossRef CAS.
- D. L. Trimm, N. W. Cant and I. O. Y. Liu, Catal. Today, 2011, 178, 181 CrossRef CAS.
- C. S. Spanjers, R. S. Sim, N. P. Sturgis, B. Kabius and R. M. Rioux, ACS Catal., 2015, 5, 3304 CrossRef CAS.
- Y. Niu, X. Huang, Y. Wang, M. Xu, J. Chen, S. Xu, M. G. Willinger, W. Zhang, M. Wei and B. Zhang, Nat. Commun., 2020, 11, 3324 CrossRef CAS PubMed.
- S. Zhou, L. Kang, X. Zhou, Z. Xu and M. Zhu, Nanomaterials, 2020, 10, 509 CrossRef CAS.
- Y. Liu, J. Zhao, J. Feng, Y. He, Y. Du and D. Li, J. Catal., 2018, 359, 251 CrossRef CAS.
- H. Liu, M. Chai, G. Pei, X. Liu, L. Li, L. Kang, A. Wang and T. Zhang, Chin. J. Catal., 2020, 41, 1099 CrossRef CAS.
- M. Chai, X. Liu, L. Li, G. Pei, Y. Ren, Y. Su, H. Cheng, A. Wang and T. Zhang, Chin. J. Catal., 2017, 38, 1338 CrossRef CAS.
- S. A. Nikolaev and V. V. Smirnov, Catal. Today, 2009, 147, S336 CrossRef CAS.
- S. A. Nikolaev, V. V. Smirnov, A. Y. Vasil'kov and V. L. Podshibikhin, Kinet. Catal., 2010, 51, 375 CrossRef CAS.
- Y. Cao, H. Zhang, S. Ji, Z. Sui, Z. Jiang, D. Wang, F. Zaera, X. Zhou, X. Duan and Y. Li, Angew. Chem., Int. Ed. Engl., 2020, 59, 11647 CrossRef CAS PubMed.
- D. M. Rao, S. T. Zhang, C. M. Li, Y. D. Chen, M. Pu, H. Yan and M. Wei, Dalton Trans., 2018, 47, 4198 RSC.
- Q. Li, Y. Wang, G. Skoptsov and J. Hu, Ind. Eng. Chem. Res., 2019, 58, 20620 CrossRef CAS.
- A. Onda, T. Komatsu and T. Yashima, Phys. Chem. Chem. Phys., 2000, 2, 2999 RSC.
- Y. Liu, X. Liu, Q. Feng, D. He, L. Zhang, C. Lian, R. Shen, G. Zhao, Y. Ji, D. Wang, G. Zhou and Y. Li, Adv. Mater., 2016, 28, 4747 CrossRef CAS.
- D. Teschner, J. Borsodi, A. Wootsch, Z. Révay, M. Hävecker, A. Knop-Gericke, S. D. Jackson and R. Schlögl, Science, 2008, 320, 86 CrossRef CAS PubMed.
- M. Akri, S. Zhao, X. Li, K. Zang, A. F. Lee, M. A. Isaacs, W. Xi, Y. Gangarajula, J. Luo, Y. Ren, Y. T. Cui, L. Li, Y. Su, X. Pan, W. Wen, Y. Pan, K. Wilson, L. Li, B. Qiao, H. Ishii, Y. F. Liao, A. Wang, X. Wang and T. Zhang, Nat. Commun., 2019, 10, 5181 CrossRef PubMed.
- F. Huang, Y. Deng, Y. Chen, X. Cai, M. Peng, Z. Jia, P. Ren, D. Xiao, X. Wen, N. Wang, H. Liu and D. Ma, J. Am. Chem. Soc., 2018, 140, 13142 CrossRef CAS PubMed.
- L. DeRita, J. Resasco, S. Dai, A. Boubnov, H. V. Thang, A. S. Hoffman, I. Ro, G. W. Graham, S. R. Bare and G. Pacchioni, Nat. Mater., 2019, 18, 746 CrossRef CAS.
- Y. Pan, Y. Chen, K. Wu, Z. Chen, S. Liu, X. Cao, W.-C. Cheong, T. Meng, J. Luo, L. Zheng, C. Liu, D. Wang, Q. Peng, J. Li and C. Chen, Nat. Commun., 2019, 10, 1 CrossRef.
- W. Liu, L. Zhang, X. Liu, X. Liu, X. Yang, S. Miao, W. Wang, A. Wang and T. Zhang, J. Am. Chem. Soc., 2017, 139, 10790 CrossRef CAS.
- C. Martinez-Macias, P. Serna and B. C. Gates, ACS Catal., 2015, 5, 5647 CrossRef CAS.
- H. Yan, X. Zhao, N. Guo, Z. Lyu, Y. Du, S. Xi, R. Guo, C. Chen, Z. Chen, W. Liu, C. Yao, J. Li, S. J. Pennycook, W. Chen, C. Su, C. Zhang and J. Lu, Nat. Commun., 2018, 9, 3197 CrossRef PubMed.
- P. Liu and N. Zheng, Natl. Sci. Rev., 2018, 5, 636 CrossRef CAS.
- X. Dai, Z. Chen, T. Yao, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong and S. Wei, Chem. Commun., 2017, 53, 11568 RSC.
- Y. Chai, G. Wu, X. Liu, Y. Ren, W. Dai, C. Wang, Z. Xie, N. Guan and L. Li, J. Am. Chem. Soc., 2019, 141, 9920 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981 CrossRef CAS PubMed.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2019, 120, 683 CrossRef PubMed.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Science, 2012, 335, 1209 CrossRef CAS PubMed.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Science, 2016, 352, 797 CrossRef CAS PubMed.
- Y. Lu, J. Wang, L. Yu, L. Kovarik, X. Zhang, A. S. Hoffman, A. Gallo, S. R. Bare, D. Sokaras, T. Kroll, V. Dagle, H. Xin and A. M. Karim, Nat. Catal., 2018, 2, 149 CrossRef.
- L. Cao, W. Liu, Q. Luo, R. Yin, B. Wang, J. Weissenrieder, M. Soldemo, H. Yan, Y. Lin, Z. Sun, C. Ma, W. Zhang, S. Chen, H. Wang, Q. Guan, T. Yao, S. Wei, J. Yang and J. Lu, Nature, 2019, 565, 631 CrossRef CAS PubMed.
- M. Jian, C. Zhao and W. X. Li, ChemPhysChem, 2020, 21, 2417 CrossRef CAS.
- H. Jeong, G. Lee, B. S. Kim, J. Bae, J. W. Han and H. Lee, J. Am. Chem. Soc., 2018, 140, 9558 CrossRef CAS PubMed.
- R. Addou, T. P. Senftle, N. O'Connor, M. J. Janik, A. C. T. van Duin and M. Batzill, ACS Nano, 2014, 8, 6321 CrossRef CAS PubMed.
- G. S. Parkinson, Z. Novotny, G. Argentero, M. Schmid, J. Pavelec, R. Kosak, P. Blaha and U. Diebold, Nat. Mater., 2013, 12, 724 CrossRef CAS PubMed.
- S. Tosoni and G. Pacchioni, Surf. Sci., 2017, 664, 87 CrossRef CAS.
- S. Riyapan, Y. Boonyongmaneerat, O. Mekasuwandumrong, H. Yoshida, S.-I. Fujita, M. Arai and J. Panpranot, J. Mol. Catal. A: Chem., 2014, 383, 182 CrossRef.
- S. Riyapan, Y. Boonyongmaneerat, O. Mekasuwandumrong, P. Praserthdam and J. Panpranot, Catal. Today, 2015, 245, 134 CrossRef CAS.
- M. Lazzeri, A. Vittadini and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 155409 CrossRef.
- C. Arrouvel, M. Digne, M. Breysse, H. Toulhoat and P. Raybaud, J. Catal., 2004, 222, 152 CrossRef CAS.
- A. S. Barnard, P. Zapol and L. A. Curtiss, Surf. Sci., 2005, 582, 173 CrossRef CAS.
- A. S. Barnard and P. Zapol, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 235403 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 54, 11169 CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83 Search PubMed.
- A. O'Hara, T. N. Nunley, A. B. Posadas, S. Zollner and A. A. Demkov, J. Appl. Phys., 2014, 116 Search PubMed.
- F. G. Sen, A. Kinaci, B. Narayanan, S. K. Gray, M. J. Davis, S. K. R. S. Sankaranarayanan and M. K. Y. Chan, J. Mater. Chem. A, 2015, 3, 18970 RSC.
- H.-Y. T. Chen, S. Tosoni and G. Pacchioni, Surf. Sci., 2016, 652, 163 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901 CrossRef CAS.
- J. Kastner and P. Sherwood, J. Chem. Phys., 2008, 128, 014106 CrossRef PubMed.
- Y. Wang, Y. Su, M. Zhu and L. Kang, RSC Adv., 2015, 5, 16582 RSC.
- Y. Han, C.-j. Liu and Q. Ge, J. Phys. Chem. B, 2006, 110, 7463 CrossRef CAS PubMed.
- J. Zhang, M. Zhang, Y. Han, W. Li, X. Meng and B. Zong, J. Phys. Chem. C, 2008, 112, 19506 CrossRef CAS.
- A. Alghannam, C. L. Muhich and C. B. Musgrave, Phys. Chem. Chem. Phys., 2017, 19, 4541 RSC.
- H. V. Thang and G. Pacchioni, J. Phys. Chem. C, 2019, 123, 7271 CrossRef CAS.
- H. V. Thang, G. Pacchioni, L. DeRita and P. Christopher, J. Catal., 2018, 367, 104 CrossRef CAS.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Science, 2017, 358, 1419 CrossRef CAS PubMed.
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sørensen, C. H. Christensen and J. K. Nørskov, Angew. Chem., 2008, 120, 9439 CrossRef.
- G. Lu, P. Zhang, D. Sun, L. Wang, K. Zhou, Z.-X. Wang and G.-C. Guo, Chem. Sci., 2014, 5, 1082 RSC.
- J. L. Fiorio, N. López and L. M. Rossi, ACS Catal., 2017, 7, 2973 CrossRef CAS.
- R. Ouyang, J. X. Liu and W. X. Li, J. Am. Chem. Soc., 2013, 135, 1760 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03087f |
| This journal is © The Royal Society of Chemistry 2021 |