
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReal-time imaging of cell-surface proteins with antibody-based fluorogenic probes†
Wenchao
Wang‡
a,
Ying
Zhang‡
a,
Hong
Zhao‡
a,
Xinlei
Zhuang
a,
Haoting
Wang
a,
Kaifeng
He
ab,
Wanting
Xu
a,
Yu
Kang
 a,
Shuqing
Chen
a,
Su
Zeng
ab and
Linghui
Qian
*ab
a,
Shuqing
Chen
a,
Su
Zeng
ab and
Linghui
Qian
*ab
aInstitute of Drug Metabolism and Pharmaceutical Analysis, Zhejiang Province Key Laboratory of Anti-Cancer Drug Research, College of Pharmaceutical Sciences, Cancer Center, Zhejiang University, Hangzhou 310058, China. E-mail: lhqian@zju.edu.cn
bHangzhou Institute of Innovative Medicine, Zhejiang University, Hangzhou 310018, China
First published on 16th September 2021
Abstract
Cell-surface proteins, working as key agents in various diseases, are the targets for around 66% of approved human drugs. A general strategy to selectively detect these proteins in a real-time manner is expected to facilitate the development of new drugs and medical diagnoses. Although brilliant successes were attained using small-molecule probes, they could cover a narrow range of targets due to the lack of suitable ligands and some of them suffer from selectivity issues. We report herein an antibody-based fluorogenic probe prepared via a two-step chemical modification under physiological conditions, to fulfill the selective recognition and wash-free imaging of membrane proteins, establishing a modular strategy with broad implications for biochemical research and for therapeutics.
Introduction
Due to the importance of cell-surface proteins in maintaining cell homeostasis as well as in dealing with pharmacological intervention,1–3 methods for selectively detecting the target protein in native environments are in high demand to illustrate its function and evaluate the therapeutic outcomes. Methods relying on genetic engineering to fuse fluorescent proteins directly or to introduce tags like SNAP or PYP proteins,4,5 peptides,6,7 or unnatural amino acids8 followed by the secondary labeling of complementary reporters have been developed. Though valuable, these methods are not feasible to monitor the dynamics of endogenously expressed proteins and may suffer from overexpression issues. Alternatively, probes suitable for the direct visualization of the endogenous protein under live-cell conditions are preferable. Ligands like small-molecule inhibitors,9,10 protein tags (e.g., cytokine/growth factor),11 and RNA aptamers12 have been employed for this purpose. Though powerful, they can only cover a narrow range of targets due to the limitation of available ligands. The specific and high-affinity binding towards antigens, along with mature production techniques, makes antibodies capable to bind virtually any immunogenic target, thus they have been widely used as a modular reagent in both basic research and biomedical applications. Fluorogenic antibody probes, offering antigen-dependent fluorescence responses, could get rid of the time-consuming washing steps and allow real-time imaging. However, the development of such probes is still in its infancy.13,14 Ueda et al. developed antibody fragment-derived “quenchbodies”, in which the genetically inserted fluorophore(s) can be quenched by intrinsic tryptophan (Trp) residues and restore upon antigen binding.15,16 Later, Hohsaka's group utilized selective N-terminal alkylation to simplify the fluorophore labeling.17 It is to be noted that such a fluorescence change is highly dependent on the amount and localization of Trp residues on the antibody, and thus efforts are still needed to develop a “universal” strategy for easy conversion of commercially available antibodies into fluorogenic probes.Antibodies have a Y-shaped structure, where the N-termini of heavy (H) and light (L) chains come together to form two identical antigen-binding sites at the tips. We envisioned that two fluorophores situated on the H/L chains of the same “Y” tip are prone to form dye–dye interaction, and it is likely to be disrupted upon antigen binding. Herein we focus on dyes that undergo self-quenching in aqueous media through H-type dimerization via π–π stacking,9,18–21 while getting restored upon disaggregation. In addition, improved signal-to-background contrast would be achieved if the dye is environment-sensitive, due to its proximity to the hydrophobic plasma membrane upon interacting with cell-surface targets. To fulfill the above requirements, sulforhodamine B (SRB) was chosen according to its optimal photophysical properties and the dimerization-induced self-quenching potency.19 Meanwhile, 2-pyridinecarboxaldehyde (2PCA) was used for N-terminal modification of antibodies under mild conditions.22,23 To realize efficient quenching and antigen-responsive “switch on” of SRB, the linker between SRB and 2PCA is critical. A two-step labeling strategy is thus proposed, where 2PCA coupled with different lengths of PEG linkers along with a terminal alkyne (2PCA–PEGn, Scheme S1†) was first used to tag the antibody. Then SRB containing an azide group (SRB-N3, Scheme S2†) was conjugated through click chemistry to form the antibody-based fluorogenic probe, Ab-PnSRB (Fig. 1a and Scheme S3†). The two-step strategy can reduce the burden on chemical synthesis as well as ensuring the labeling efficiency since PEG-containing 2PCA derivatives should have improved water solubility even at high concentrations. As a proof of concept, the epidermal growth factor receptor (EGFR) and anti-EGFR therapeutic antibodies, necitumumab (Nec) and cetuximab (Cet), were studied here. Live-cell imaging showed that after chemical labeling, both antibodies showed EGFR-dependent fluorescence, exhibiting the potential of this strategy for detecting cell-surface proteins with commercially available antibodies.
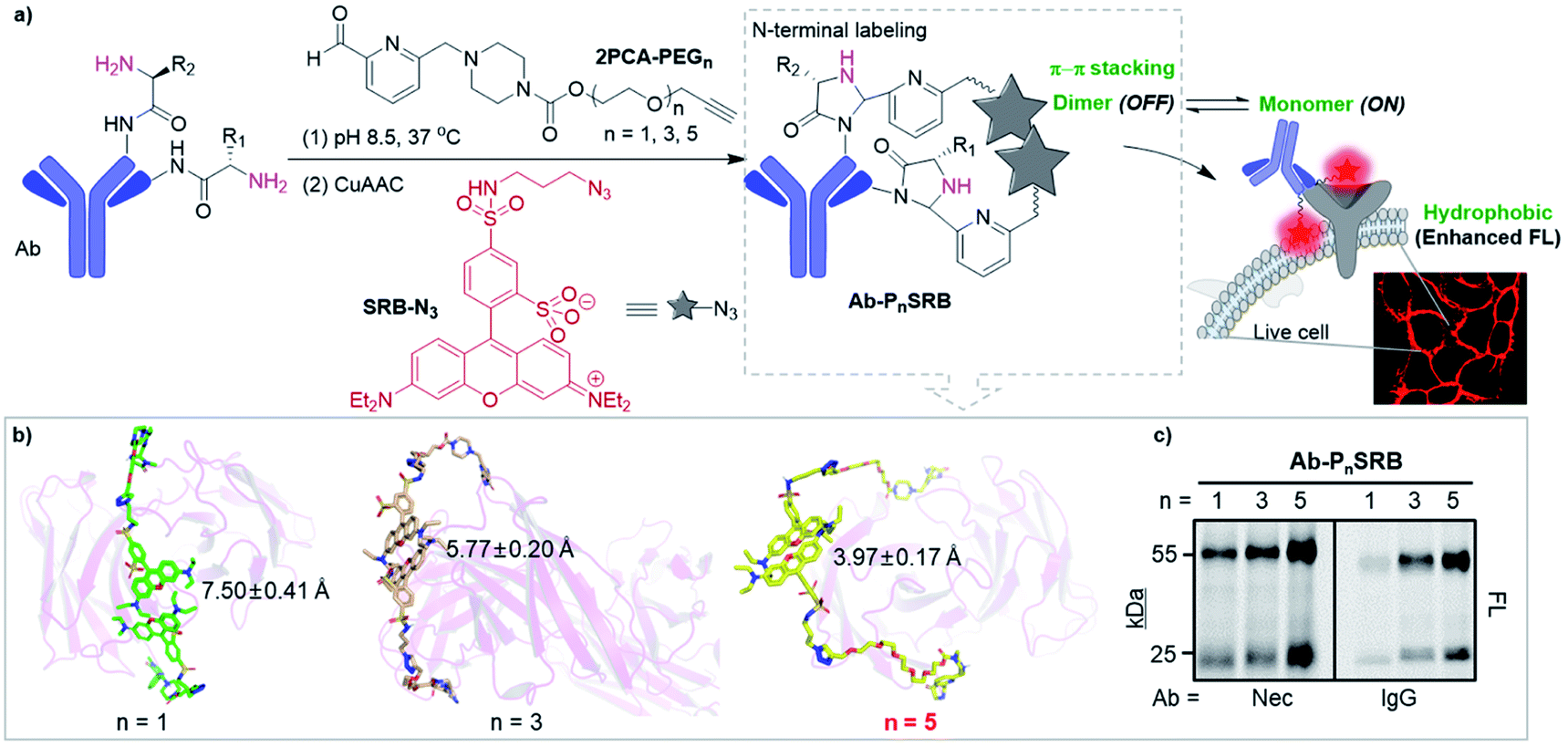 | ||
| Fig. 1 Strategy of antibody-based fluorogenic probes (Ab-PnSRB, n indicates the PEG length). (a) Schematic illustration of cell-surface protein imaging with antibody-based probes prepared by two-step labeling (details are shown in Scheme S3†). (b) The representative conformations of 2PCA–PEGn/SRB-N3 (PnSRB in short) labeled necitumumab (Nec) at the stable trajectories. Average distances and SDs between the centroid of the xanthene of two SRBs were calculated. (c) Labeling efficiency of 2PCA–PEGn/SRB-N3 towards antibodies (i.e., necitumumab and human IgG), which was visualized by in-gel fluorescent scanning (FL). | ||
Results and discussion
Initially, molecular dynamics (MD) simulation was carried out to delineate the distance between two SRBs when situated at the N-termini of H/L chains on the same tip of necitumumab and cetuximab (Fig. S1†). As shown in Fig. 1b and S2,† when n = 5, SRBs in both necitumumab and cetuximab are close to each other with a distance between 3.7 and 4.0 Å, ideal for face-to-face π–π stacking.24–26 Therefore the resulting Ab-P5SRB is expected to have the desired quenching effect in aqueous solution. In addition, the labeling experiments (Scheme S3†) exhibited that, when n = 5, the dye-to-protein ratio was the highest among the three (Fig. 1c and S3a†), estimated to be around 75% (Table S1†). It is likely due to the better water solubility of 2PCA–PEG5 than the other two, when a high concentration of the probe (10 mM) was used to promote the labeling efficiency (data not shown).22,23 Moreover, the labeling ratio of 2PCA–PEG5/SRB-N3 (PnSRB in short) towards different antibodies (i.e., IgG, Nec, and Cet) was similar (Fig. S3b; the amino acid sequences of the Fab fragments of Nec and Cet are shown in the ESI†), promising in view of universality.After confirming that the antibody retained the ability to bind its antigen after modification with P5SRB (Fig. S4†), we evaluated the photophysical properties in vitro. SRB was reported to form a quenched dimer in water when closely connected, and the π–π effect was disrupted in MeOH.19 As shown in the absorption spectra (Fig. S5a†), a blue shifted band (530 nm) appeared in water at higher concentrations, indicating the enhancement of dimeric H-aggregation.19 Along with that, an increased fluorescence discrepancy between water and MeOH as the concentration of SRB-N3 grew was observed (Fig. S5b†), likely due to the increment of SRB dimerization at higher concentrations in water. Upon the addition of MeOH to water, SRB-N3 showed fluorescence recovery similar to the classical solvatochromic fluorophore, 4-sulfamonyl-7-aminobenzoxadiazole (SBD)27 (Fig. S6a†). And stronger emission was seen in all tested organic solvents than in water for SRB-N3 and SBD (Fig. 2a and S6b†), but not for the control, rhodamine B (TER-N3; structure shown in Scheme S3†). Thus SRB-N3 is regarded as environment-sensitive as well. After being conjugated to the N-termini of antibodies, the resulting Ab-PnSRB (100 nM) showed much weaker fluorescence than the free SRB-N3 (20 nM, Fig. 2b), suggesting that obvious fluorescence quenching occurred. Since in the absorption spectra (Fig. S7a†), a higher ratio of A530 nm/A560 nm was seen from Nec-PnSRB in water than that from SRB-N3 in MeOH, the quenching is partially due to the dimeric H-aggregation. Meanwhile the background signal was still seen in comparison with the unlabeled Nec, largely due to the relatively low labeling ratio of Nec by 2PCA–PEGn. If all of the four N-terminal amines on antibodies were labeled by SRB, dimerization would occur between the H and L chain on the same tip for stronger quenching. Nevertheless, Nec-P5SRB showed no stronger fluorescence than Nec-P3SRB, even though its labeling ratio is much higher, suggesting that the quenching efficiency in Nec-P5SRB was much stronger, consistent with the MD simulation results. In contrast, the conjugation of TER-N3 to the N-termini of the antibody did not show an obvious fluorescence decrease (Fig. S7b†), confirming that the efficient quenching effect was mainly due to the photophysical properties of SRB-N3. Upon addition of recombinant EGFR, the fluorescence intensity of Nec-P5SRB increased within 30 min (Fig. 2c). In contrast, there was no “off-on” change from the negative control, IgG-P5SRB. In vitro experiments also showed that the antigen-response fluorescence enhancement was moderate and got saturated in the presence of less than 1 equivalent of EGFR, again, possibly attributed to the low labeling ratio of SRB.
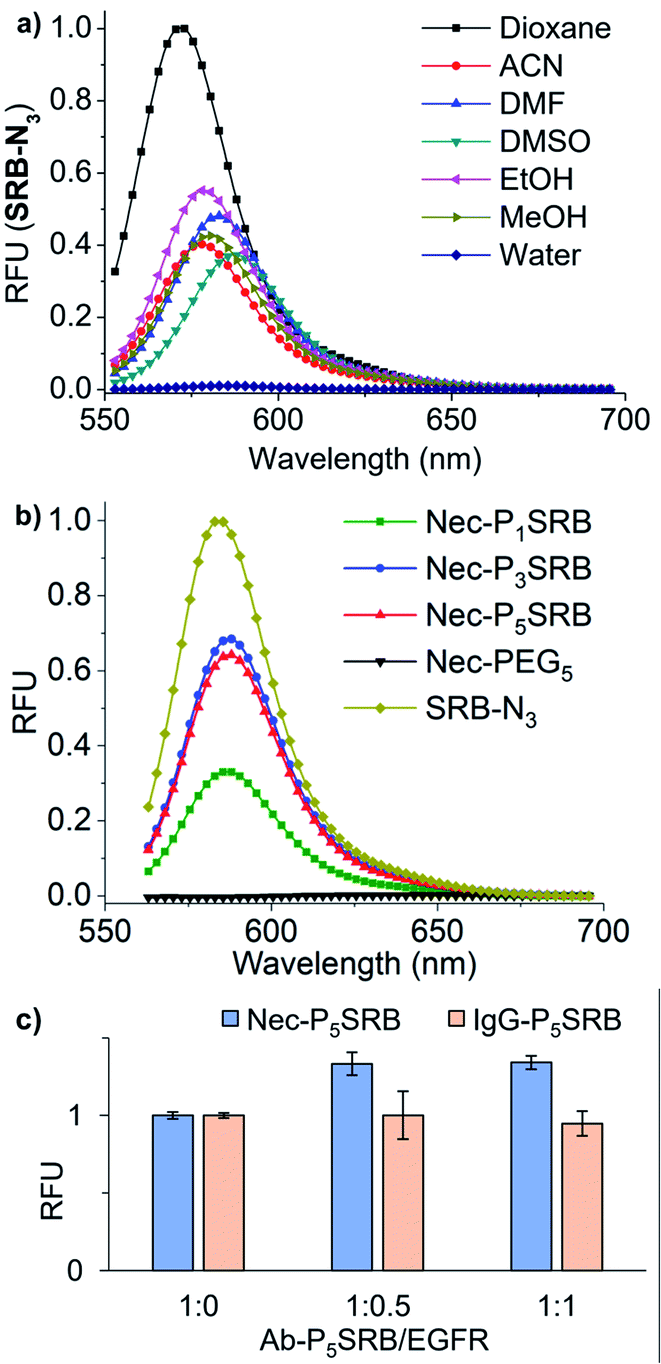 | ||
| Fig. 2 Photophysical properties of SRB-N3 and the resulting Ab-PnSRB probes. (a) Fluorescence spectra of SRB-N3 (5 μM) in different solvents (λex = 530 nm). (b) Fluorescence spectra of Nec-PnSRB (100 nM) and SRB-N3 (20 nM) in H2O (λex = 530 nm). (c) Fluorescence response of Nec-P5SRB/IgG-P5SRB (20 nM) in the presence of recombinant EGFR at different ratios (λex = 560 nm and λem = 590 nm). | ||
Afterwards, we turned our attention to live-cell imaging by using Nec-PnSRB, to check the “synergetic” switch-on potency by the hydrophobic plasma membrane. Initially, in concentration-dependent experiments, Nec-P5SRB showed detectable red fluorescence on the surface of the EGFR-positive cancer cell A431 even at a concentration as low as 20 nM (Fig. S8a†). As the probe concentration increased, all three probes showed brilliant emission, especially at a concentration of 100 nM (Fig. 3a, top), and thus we used this concentration for the subsequent live-cell imaging experiments if not specified. It is to be noted that the signal-to-background contrast of Nec-P1SRB was much lower than that of Nec-P5SRB, likely due to its weaker quenching efficiency as shown in previous in vitro results (Fig. S8b†). Meanwhile, the fluorescence signal can be fully eliminated when competed by excess Nec, and control groups including the low EGFR expression cell (i.e., HepG2) or using IgG-PnSRB (human IgG, without a specific binding target) showed no fluorescence signal (Fig. 3a, middle and bottom). These results together confirmed the EGFR-response fluorescence of Nec-P5SRB in living cells. It is worth noting that our Ab-PnSRB was designed to be quenched without binding to cell-surface EGFR. To check the background of the probe under a microscope, IgG-P5SRB and IgG-P5TER (prepared as shown in Scheme S3†) were incubated with A431, respectively, for no-wash imaging. Here TER was used as a representative “always-on” reporter for comparison. As shown in Fig. 3b, a negligible background was seen for both probes at 100 nM, while at 500 nM, strong red background was observed in IgG-P5TER treated cells. In sharp contrast, the background of the image using IgG-P5SRB remained almost dark, where the self-quenching H-aggregation and environmental sensitivity may dominate the non-fluorescent status of SRB in aqueous solution.
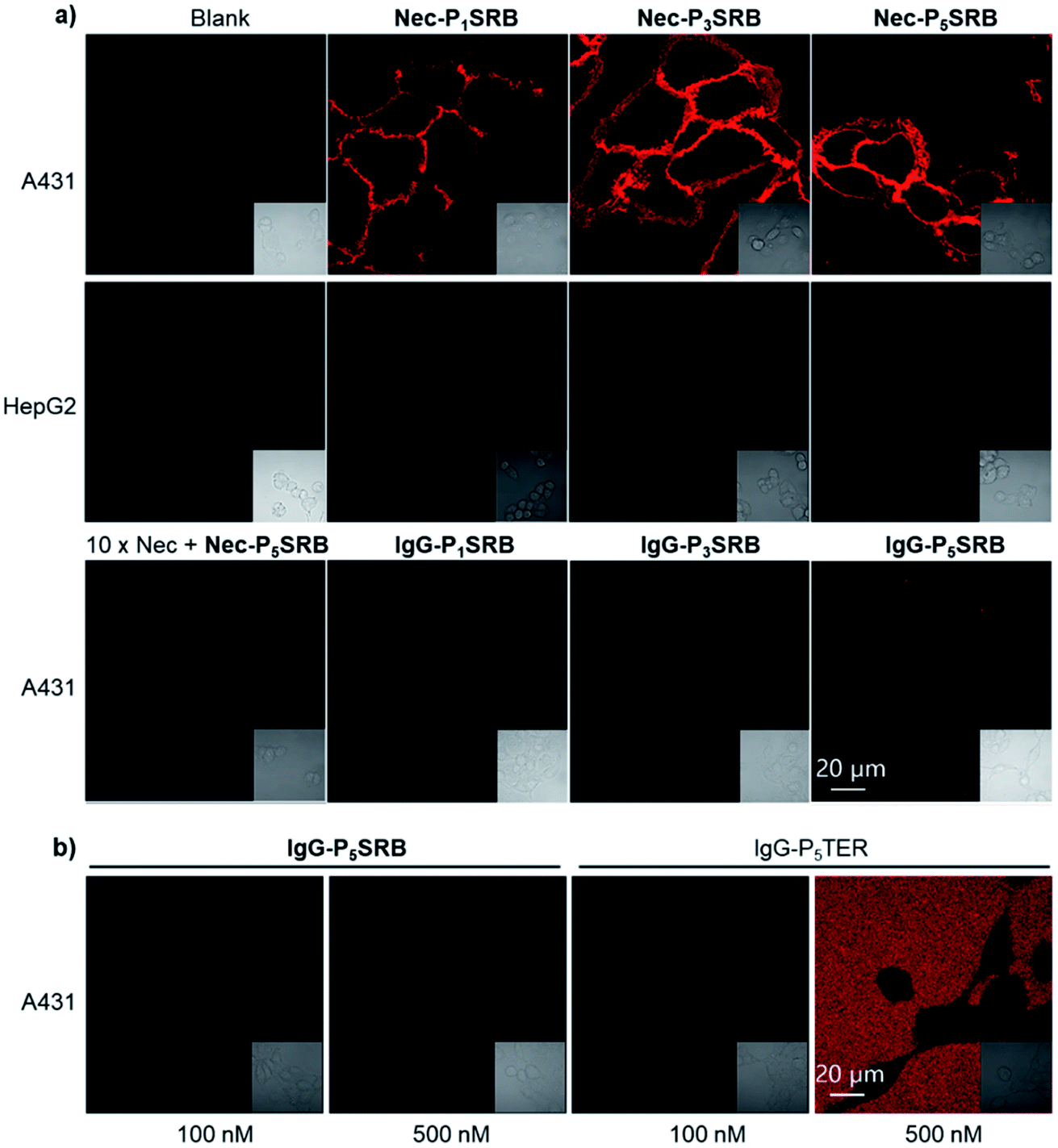 | ||
| Fig. 3 Live-cell imaging by using Ab-PnSRB (λex = 561 nm and λem = 581–639 nm). (a) Cellular performance of Nec-PnSRB (100 nM) in both EGFR-positive (A431) and EGFR-negative (HepG2) cells. 10× Nec means that cells were pretreated with 1 μM of necitumumab for 0.5 h before the addition of Nec-P5SRB. Control probes IgG-PnSRB (100 nM) were studied in parallel. (b) Comparison of the background signal from IgG-P5SRB and that from IgG-P5TER (“always-on” probe) in A431 cells without washing. | ||
With a low-background and selective fluorogenic probe in hand, real-time imaging was carried out using Nec-P5SRB (Fig. 4a and S9†). Brilliant fluorescence was detected on the cell membrane immediately after the addition of the probe. Intracellular fluorescence was detected after 5 min in some cells and became obvious in most cells after 30 min, as caused by the internalization of the antibody–EGFR complex.28,29 Taken together, we concluded that Nec-P5SRB was indeed suitable for real-time imaging of endogenous EGFR in live mammalian cells. To take one step further, endogenous EGFR expression levels in different cell lines were quantitatively analyzed by using Nec-P5SRB with FACS. Fig. 4b shows that the average fluorescence intensity in A31 cells increased linearly with the probe concentration in the range of 0 to 50 nM and got saturated at 100 nM. In A549 cells, the saturation was observed at much lower concentrations of Nec-P5SRB, while the signal remained very weak in HepG2 cells at all concentrations. Interestingly, the fluorescence readings obtained at the saturation concentration (i.e., 100 nM) from FACS showed consistent EGFR protein levels with the results obtained from WB analysis. These results demonstrated that our probe can not only indicate the dynamic localization of endogenous EGFR, but also be used for quantitative analysis.
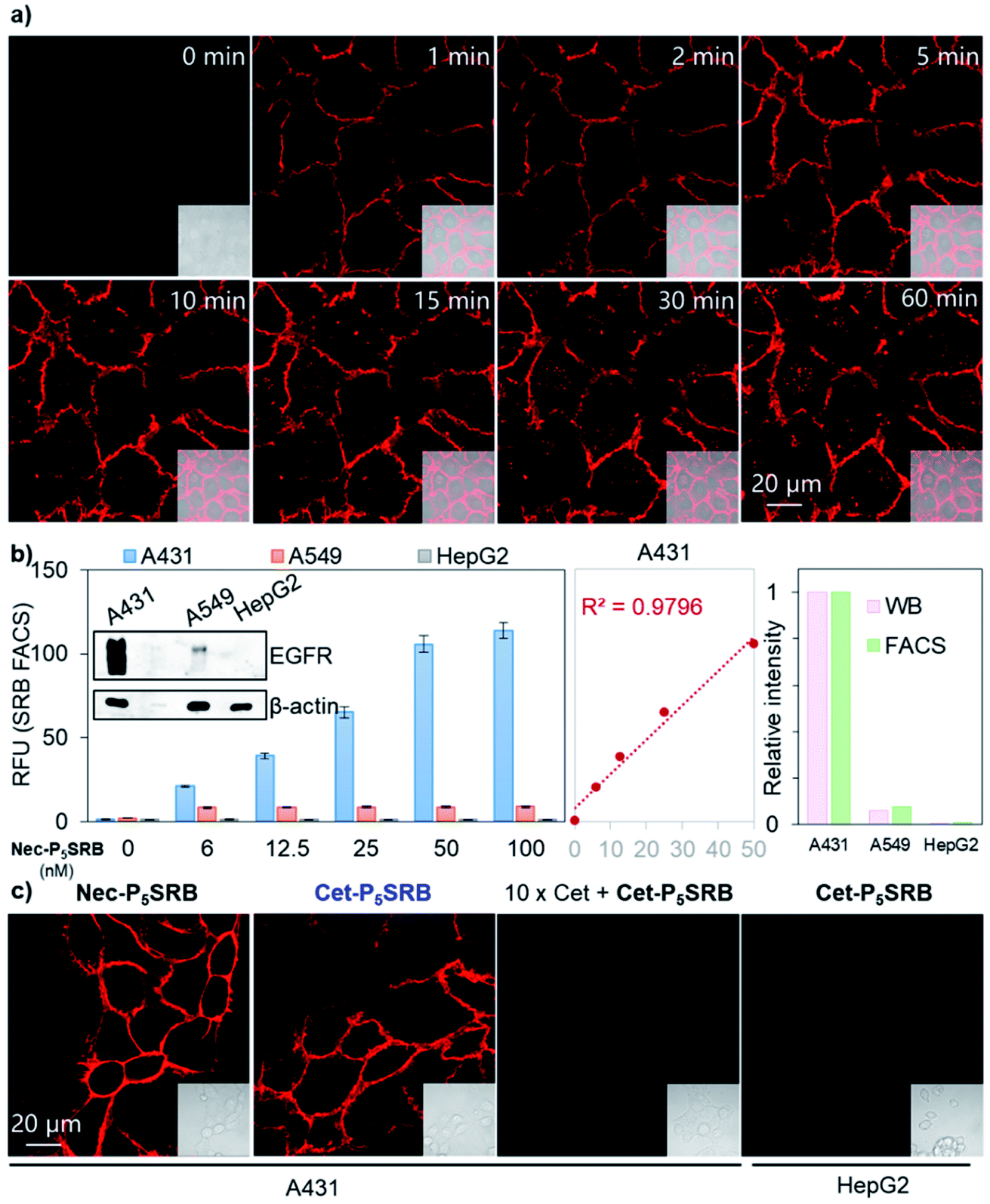 | ||
| Fig. 4 Applications of Ab-P5SRB for detecting endogenous EGFR in living cells. (a) Real-time imaging of A431 cells upon addition of Nec-P5SRB (100 nM). (b) FACS of Nec-P5SRB towards A431/A549/HepG2 (left). Western blotting (WB) analysis of EGFR in A431/A549/HepG2 (inset). Linear correlation analysis between the average fluorescence in A431 and probe's concentration (middle). Relative intensity readings extracted from WB (pink) versus those from FACS (green) (right). (c) No-wash imaging of A431/HepG2 cells obtained by using Cet-P5SRB (100 nM). 10 × Cet in the third panel means that cells were pretreated with 1 μM of cetuximab for 0.5 h before the addition of Cet-P5SRB. λex = 561 nm and λem = 581–639 nm for all cell imaging. | ||
To test the robustness and practicality of this strategy for other antibodies with different amino acid sequences, 2PCA–PEG5 labeling followed by SRB-N3 click ligation was applied to another EGFR-targeting antibody, cetuximab, under the same conditions. Identical cellular performance was seen between Cet-P5SRB and Nec-P5SRB (Fig. 4c), with minimal effect on the binding affinity of cetuximab (Fig. S10†), suggesting that this strategy has high potential as a universal method to create antibody-based fluorogenic probes towards cell-surface proteins.
Conclusions
In conclusion, we have developed an easy-operation two-step labeling strategy to create fluorogenic antibody-based probes for real-time imaging of cell surface proteins. Thanks to antibody's capability of binding to virtually any immunogenic target with exquisite specificity and high affinity, the resulting probe is highly selective and this method is promising to be applied for detecting any cell-surface protein with commercially available antibodies. Current efforts are focused on the development of probes with other colors as well as figuring out the chemistry for better labeling efficiency at N-termini.Data availability
Supporting data for this article is presented in the ESI.†Author contributions
L. Q. designed the project. W. W., Y. Z., X. Z., H. W., K. H., and W. X. carried out the experiments; H. Z. and Y. K. carried out the MD simulation and analysis. W. W., Y. Z. and H. Z. contributed equally. S. C. guided the biological experiments and the writing. L. Q. and S. Z. directed the project and wrote the manuscript. All authors commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (81903574) and the Fundamental Research Funds for the Central Universities (2019QNA7046, 2020QNA7001). We also acknowledge generous financial support from Zhejiang University. We thank Ms Haihong Hu and Ms Li Ma for managing instruments and helping with experiments. We also thank Jianyang Pan (Research and Service Center, College of Pharmaceutical Sciences, Zhejiang University) for performing NMR spectrometry.Notes and references
- L. Kuhlmann, E. Cummins, I. Samudio and T. Kislinger, Expert Rev. Proteomics, 2018, 15, 259–275 CrossRef CAS PubMed.
- D. Bausch-Fluck, U. Goldmann, S. Müller, M. van Oostrum, M. Müller, O. T. Schubert and B. Wollscheid, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E10988–E10997 CrossRef CAS PubMed.
- K. K. Leung, G. M. Wilson, L. L. Kirkemo, N. M. Riley, J. J. Coon and J. A. Wells, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 7764–7775 CrossRef CAS PubMed.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS PubMed.
- J. Gao, Y. Hori, M. Nishiura, M. Bordy, J. Hasserodt and K. Kikuchi, Chem. Lett., 2020, 49, 232–235 CrossRef CAS.
- J. Lotze, U. Reinhardt, O. Seitz and A. G. Beck-Sickinger, Mol. BioSyst., 2016, 12, 1731–1745 RSC.
- G. C. Gavins, K. Gröger, M. D. Bartoschek, P. Wolf, A. G. Beck-Sickinger, S. Bultmann and O. Seitz, Nat. Chem., 2021, 13, 15–23 CrossRef CAS PubMed.
- A. I. König, R. Sorkin, A. Alon, D. Nachmias, K. Dhara, G. Brand, O. Yifrach, E. Arbely, Y. Roichman and N. Elia, Nanoscale, 2020, 12, 3236–3248 RSC.
- Y. Jiao, J. Yin, H. He, X. Peng, Q. Gao and C. Duan, J. Am. Chem. Soc., 2018, 140, 5882–5885 CrossRef CAS PubMed.
- Q. Wu, Y. Jing, T. Zhao, J. Gao, M. Cai, H. Xu, Y. Liu, F. Liang, J. Chen and H. Wang, Nanoscale, 2020, 12, 21591–21598 RSC.
- Y. Takaoka, S. Uchinomiya, D. Kobayashi, M. Endo, T. Hayashi, Y. Fukuyama, H. Hayasaka, M. Miyasaka, T. Ueda, I. Shimada and I. Hamachi, Chem, 2018, 4, 1451–1464 CAS.
- X. Tan, T. P. Constantin, K. L. Sloane, A. S. Waggoner, M. P. Bruchez and B. A. Armitage, J. Am. Chem. Soc., 2017, 139, 9001–9009 CrossRef CAS PubMed.
- T. Hayashi, Y. Yasueda, T. Tamura, Y. Takaoka and I. Hamachi, J. Am. Chem. Soc., 2015, 137, 5372–5380 CrossRef CAS PubMed.
- K. He, S. Zeng and L. Qian, J. Pharm. Anal., 2020, 10, 397–413 CrossRef PubMed.
- R. Abe, H. Ohashi, I. Iijima, M. Ihara, H. Takagi, T. Hohsaka and H. Ueda, J. Am. Chem. Soc., 2011, 133, 17386–17394 CrossRef CAS PubMed.
- H.-J. Jeong, T. Kawamura, M. Iida, Y. Kawahigashi, M. Takigawa, Y. Ohmuro-Matsuyama, C.-I. Chung, J. Dong, M. Kondoh and H. Ueda, Anal. Chem., 2017, 89, 10783–10789 CrossRef CAS PubMed.
- K. Fukunaga, T. Watanabe, D. Novitasari, H. Ohashi, R. Abe and T. Hohsaka, Chem. Commun., 2018, 54, 12734–12737 RSC.
- I. A. Karpenko, M. Collot, L. Richert, C. Valencia, P. Villa, Y. Mély, M. Hibert, D. Bonnet and A. S. Klymchenko, J. Am. Chem. Soc., 2015, 137, 405–412 CrossRef CAS PubMed.
- F. Bouhedda, K. T. Fam, M. Collot, A. Autour, S. Marzi, A. Klymchenko and M. Ryckelynck, Nat. Chem. Biol., 2020, 16, 69–76 CrossRef CAS PubMed.
- L. Esteoulle, F. Daubeuf, M. Collot, S. Riché, T. Durroux, D. Brasse, P. Marchand, I. A. Karpenko, A. S. Klymchenko and D. Bonnet, Chem. Sci., 2020, 11, 6824–6829 RSC.
- K. T. Fam, M. Collot and A. S. Klymchenko, Chem. Sci., 2020, 11, 8240–8248 RSC.
- J. I. MacDonald, H. K. Munch, T. Moore and M. B. Francis, Nat. Chem. Biol., 2015, 11, 326–331 CrossRef CAS PubMed.
- D.-Z. Li, B.-N. Han, R. Wei, G.-Y. Yao, Z. Chen, J. Liu, T. C. W. Poon, W. Su, Z. Zhu, D. S. Dimitrov and Q. Zhao, mAbs, 2018, 10, 712–719 CrossRef CAS PubMed.
- R. Zhao and R.-Q. Zhang, Phys. Chem. Chem. Phys., 2016, 18, 25452–25457 RSC.
- L. He, P. Wang, L. He, Z. Qu, J. Luo, B. Peng, X. Tang and Y. Pei, RSC Adv., 2018, 8, 11134–11144 RSC.
- K. Carter-Fenk and J. M. Herbert, Phys. Chem. Chem. Phys., 2020, 22, 24870–24886 RSC.
- Y.-D. Zhuang, P. Y. Chiang, C.-W. Wang and K.-T. Tan, Angew. Chem., Int. Ed., 2013, 52, 8124–8128 CrossRef CAS PubMed.
- J. B. Spangler, J. R. Neil, S. Abramovitch, Y. Yarden, F. M. White, D. A. Lauffenburger and K. D. Wittrup, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13252–13257 CrossRef CAS PubMed.
- X. Shi, C. Y. Y. Yu, H. Su, R. T. K. Kwok, M. Jiang, Z. He, J. W. Y. Lam and B. Z. Tang, Chem. Sci., 2017, 8, 7014–7024 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization of all new compounds, HRMS and NMR spectra, and supplementary figures. See DOI: 10.1039/d1sc03065e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |