
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Expanding organofluorine chemical space: the design of chiral fluorinated isosteres enabled by I(I)/I(III) catalysis†
Stephanie
Meyer
,
Joel
Häfliger
and
Ryan
Gilmour
 *
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Correnstraße 36, 48149, Münster, Germany. E-mail: ryan.gilmour@uni-muenster.de
First published on 29th June 2021
Abstract
Short aliphatic groups are prevalent in bioactive small molecules and play an essential role in regulating physicochemistry and molecular recognition phenomena. Delineating their biological origins and significance have resulted in landmark developments in synthetic organic chemistry: Arigoni's venerable synthesis of the chiral methyl group is a personal favourite. Whilst radioisotopes allow the steric footprint of the native group to be preserved, this strategy was never intended for therapeutic chemotype development. In contrast, leveraging H → F bioisosterism provides scope to complement the chiral, radioactive bioisostere portfolio and to reach unexplored areas of chiral chemical space for small molecule drug discovery. Accelerated by advances in I(I)/I(III) catalysis, the current arsenal of achiral 2D and 3D drug discovery modules is rapidly expanding to include chiral units with unprecedented topologies and van der Waals volumes. This Perspective surveys key developments in the design and synthesis of short multivicinal fluoroalkanes under the auspices of main group catalysis paradigms.
 Stephanie Meyer | Stephanie Meyer was born in Sögel (Germany) in 1995. She completed her Bachelors degree in chemistry at the WWU Münster, which included an internship with Prof. Ian Fairlamb at the University of York (UK, 2016). She obtained her BSc degree in 2017 and, after an industrial stay at Beiersdorf AG in Hamburg, joined Prof. Andrei Yudin's group at the University of Toronto as a visiting student (Canada, 2018). Stephanie completed her Masters degree working with Prof. Ryan Gilmour (2019) and remained in the group as a doctoral student working on I(I)/I(III) catalysis. |
 Joel Häfliger | Joel Häfliger was born in Willisau (Switzerland) in 1995. He completed his undergraduate studies in chemistry at the ETH Zurich where he worked with Prof. Antonio Togni for his Master thesis on dynamic processes in ortho-substituted difluoro(phenyl)-λ3-iodanes (2019). Following an internship in chemical research at Syngenta Crop Protection AG in Stein (Switzerland, 2019), he started his PhD in the group of Prof. Ryan Gilmour in 2020. His current work is focussed on the application and development of novel iodine(I)/(III) catalysed fluorination processes. |
 Ryan Gilmour | Ryan Gilmour was born (1980) in Ayrshire, Scotland and educated at the University of St Andrews, where worked with Prof. David O'Hagan FRSE. He received his PhD from the University of Cambridge working under the direction of Prof. Andrew B. Holmes FRS. Following post-doctoral research fellowships at the Max-Planck-Institut für Kohlenforschung (with Prof. A. Fürstner) and the ETH Zürich (with Prof. P. H. Seeberger) he was appointed as Alfred-Werner-Assistant-Professor at the ETH (2008–2012). He is currently Chair of Organic Chemistry and CiMIC Professor of Chemical Biology at the Westfälische Wilhelms-Universität Münster (Germany). Gilmour is a Fellow of the Royal Society of Chemistry (2015) and a Corresponding Fellow of the Royal Society of Edinburgh (2021). |
1. Introduction
Fluorinated architectures traverse the functional small molecule landscape,1 where they manifest themselves in blockbuster drugs (1–3),2 essential agrochemicals (4–6)3 (Fig. 1) and high-performance materials such as Teflon®.4 Ubiquitous in modern society, fluorinated motifs continue to feature in the vanguard of focussed molecular design strategies5 with short perfluoroalkyl groups such as CF3 and CF(CF3)2 now enjoying “privileged” status.6,7 In a reductionist sense, the functional diversity of fluorinated materials can be attributed to the physicochemical consequences of C(sp2/sp3)-Hδ+ → C(sp2/sp3)-Fδ− structural editing8 and the new regions of chemical space that result.9 The (stereo)electronic impact of this (bio)isosterism appears subtle but, when appropriately leveraged, can induce counterintuitive conformational behaviour,10 elicit novel molecular recognition modes11 and augment stability.1,2,5 Whilst this latter consequence of fluorination has been widely lauded as a triumph in bioactive small molecule discovery, it has obvious environmental consequences.12 This is unsurprising given the conspicuous dearth of fluorinated natural products13 and, by extension, regulatory enzymes to facilitate the construction and degradation of this class of organohalogens.14 Reconciling the benefits of short, fluorinated motifs as essential modulators of health and development, with environmental considerations, continues to aggravate this complex relationship. This juxtaposition provides a powerful impetus to explore new areas of organofluorine chemical space to expand the current portfolio of drug and agrochemical discovery modules. Augmenting the current arsenal of achiral 2D and 3D motifs to include chiral 3D topologies will open up a wealth of opportunities,15 and simultaneously reduce dependence on perfluorocarbon moieties: this may allow existing degradative enzymes to be harnessed and thus mitigate environmental accumulation.16 This personal Perspective reflects on the possible motivating factors that have led to a surge of interest in the generation of short, chiral fluorinated groups and highlights the important role of I(I)/I(III) catalysis as an enabling technology in this arena.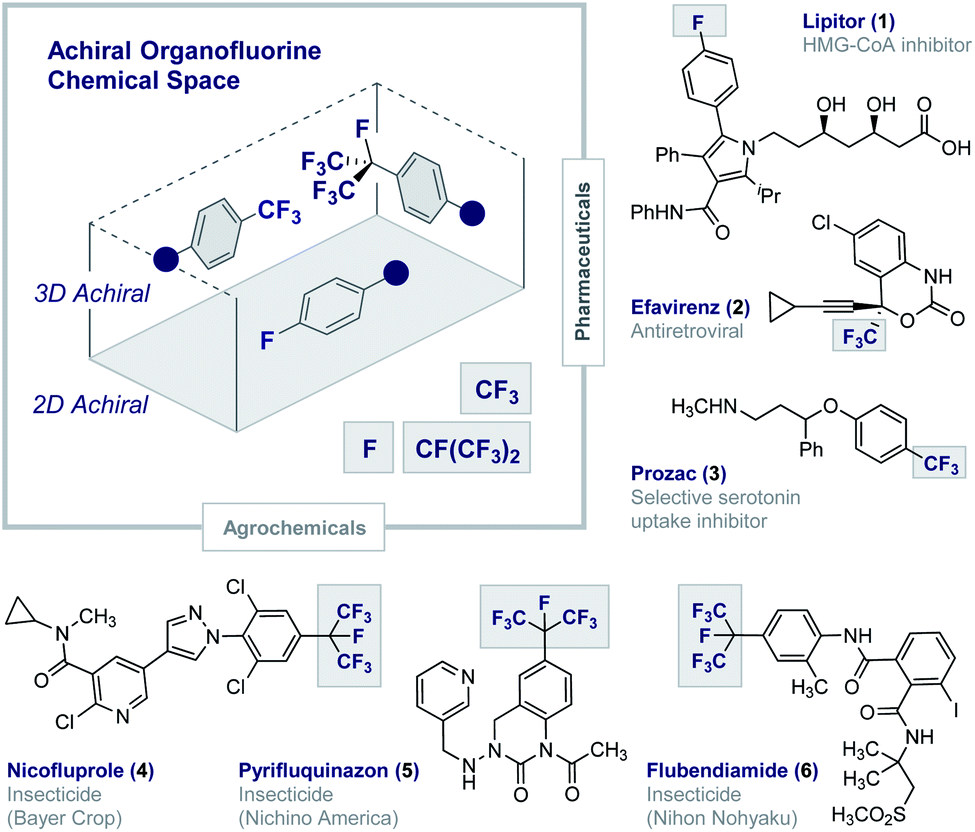 | ||
| Fig. 1 Achiral organofluorine chemical space. Selected examples of blockbuster drugs and agrochemicals containing achiral fluorinated motifs (1–6). | ||
2. Short aliphatic groups in (bio)-organic chemistry
2.1 Radioisotopes to stable isotopes
The frequency with which simple methyl groups are encountered in the natural product repertoire mirrors the success of its electronic antipode (CF3) in contemporary drug discovery. However, striking disparities in the stability of the respective isotopes of H and F render the development of a chiral CF3 group improbable. In the case of the parent methyl group, it is possible to exploit the three natural isotopes of hydrogen (1H, 2H and 3H) to generate a stereogenic center and this has been instrumental in the course of mechanistic enzymology (Fig. 2, left, the chiral methyl group).17 In addition, deuterium is regularly leveraged in drug discovery to delineate pharmacokinetic parameters18 and is now a key feature of deutetrabenazine (Austedo®) to treat Huntington's disease.19 Although fluorine has a plethora of known isotopes, it is practically and synthetically implausible to translate this into a “chiral” CF3 group. This provides an opportunity for creative endeavour in conceiving and evaluating new chemical entities based on short aliphatic groups (C1–C10). Inspiration can be gleaned in abundance from the bioactive small molecule repository (vide infra), where both linear and branched groups (e.g.tBu in ginkgolide B) are well represented. This will ultimately result in an array of new chiral entities with distinct properties that will complement the aliphatic series.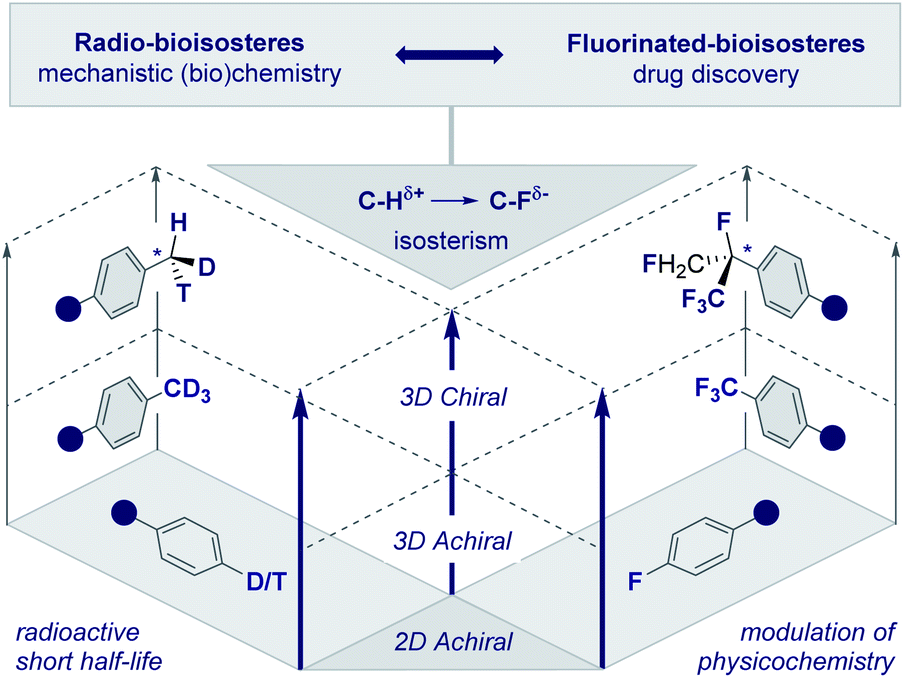 | ||
| Fig. 2 Radio- and fluorinated-bioisosteres: expanding achiral 2D and 3D motifs into chiral 3D chemical space. | ||
2.2 Expanding organofluorine chemistry beyond achiral 2D and 3D chemical space
In our quest to design short, chiral fluorine-containing groups, and having disregarded isotope discrimination blueprints from the outset, the formal oxidation of a C2 fragment was an appealing starting point. Vicinal oxidation is pervasive across the bioactive small molecule spectrum and is intimately involved in orchestrating structure–function interplay.20 Examples abound and include the immunosuppressant Rapamycin (Sirolimus) (7), the anti-tumour agents Taxol (Paclitaxel) (8) and Vinblastine (Velban) (9), and the serine palmitoyltransferase inhibitor Myriocin (Thermozymocidin) (10) (Fig. 3). It is pertinent to note that this natural product provided the inspiration for Fingolimod (Gilenya®) (11) to treat relapsing remitting multiple sclerosis.21 A conspicuous feature of these bioactive molecules is the presence of both short alkyl fragments and vicinal oxidation patterns. Indeed, this latter feature commonly occurs in the low molecular weight APIs such as the bronchodilator Salbutamol (Ventolin®) (12).22 It was envisaged that integrating these two common structural features in the development of a short, chiral fluorinated group would also provide a much-needed solution to generating a bioisostere of the vicinal diol motif. Whilst OH → F bioisosterism is well established,6vicinal difluorination strategies are comparatively underdeveloped. This is noteworthy given the interest in halogenated natural products containing contiguous halogen centres,23 including the prominent synthesis of a fluorinated analogue of the sulfolid danicalipin A by Carreira and co-workers.24 | ||
| Fig. 3 Vicinal oxidation patterns and short aliphatic groups in bioactive molecules. | ||
The conspicuous absence of selective vicinal difluorination protocols is in stark contrast with the prominence of fluorination patterns in the drug discovery process. This may reflect a limitation in synthetic organic chemistry as opposed to a lack of suitability as drug discovery modules. This echoes the sentiments expressed by former NIH Director Zerhouni that “One interesting result of the NIH Roadmap development process came when we surveyed scientists to find out what the stumbling blocks for biological sciences were. The number one stumbling block turned out to be synthetic organic chemistry.“25 As Seebach commented in his celebrated essay “Organic Chemistry: Where Next?”,26 “molecular function and activity now occupy centre stage”: realising this objective will require practitioners of organic chemistry to address deficiencies in the synthesis arsenal, such as the fundamental task of adding molecular fluorine across an alkene in a mild and selective manner. Achieving parity with vicinal chlorination and bromination, and expanding the protocol to enable the synthesis of telescoped multivicinal fluoroalkanes requires innovative solutions. This latter aspect is particularly urgent given the potential of these materials in the life sciences and materials fields (vide infra).
2.3 Multivicinal fluoroalkanes (C2–C6)
Multivicinal fluoroalkanes are an evolving class of hydrocarbon/polyfluorocarbon hybrids that are composed of repeating CHF units. The simplest member of this organohalogen class may be accessed by the programmed addition of fluorine across an alkene unit (Fig. 4).27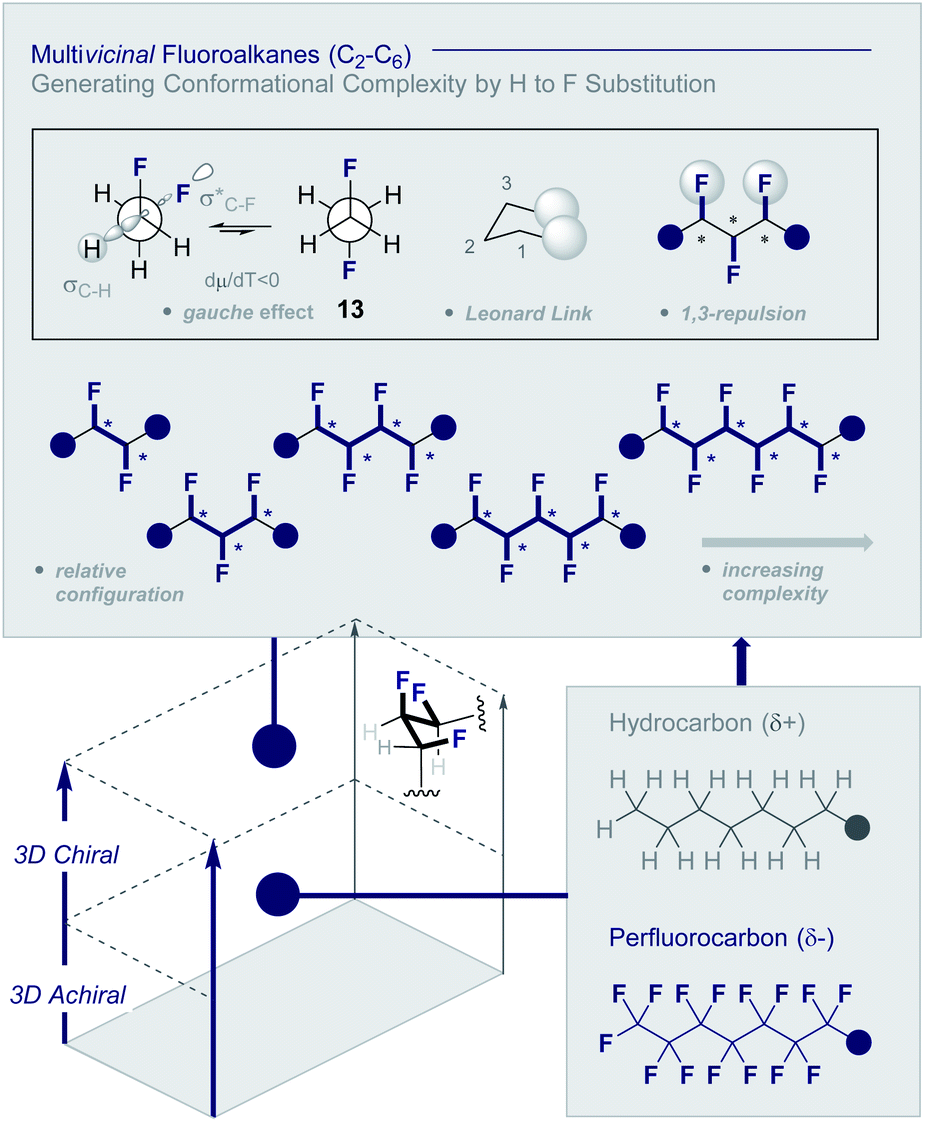 | ||
| Fig. 4 Multivicinal fluoroalkanes: merging hydrocarbons with perfluorocarbons to generate conformational diversity (* denotes a stereogenic centre). | ||
Although fluorine has a small van der Waals radius, it is highly electronegative and therefore the inclusion of multiple C (sp3)-F bonds along a carbon chain regulates conformation and physicochemistry. The relative configuration of the system gives rise to distinct topologies that manifest stabilising, second-order hyperconjugative interactions (σCH → σCF*; the venerable stereoelectronic gauche effect in 1,2-difluoroethane 13)1,10 and mitigate 1,3-repulsion.28,29 The latter acyclic conformational control aspect becomes particularly dominant in systems where n ≥3 due to formation of the venerable Leonard Link.28,30 Since each carbon homologation enables the generation of 2n stereoisomers (for n homologated carbons), these materials have the potential to significantly expand organofluorine chemical space (13): this necessarily requires the development of effective, stereocontrolled methods to facilitate synthesis. Pioneering studies, most notably by O'Hagan and co-workers,27b have culminated in the synthesis and physicochemical evaluation of several multivicinal fluoroalkane scaffolds. These elegant routes leverage (asymmetric) oxidation/stereospecific fluorodeoxygenation protocols to efficiently access the target scaffolds of interest. Applications range from the design of peptide mimics to regulate conformation (Fig. 5), through to the introduction of novel liquid crystals. Pertinent examples include the strategic use of fluorination to explore conformational effects in the neurotransmitter GABA (14, 15/16 and 17/18),31,32 to compare the erythro- and threo-diastereoisomers of 1,2-difluorodiphenylethanes and 2,3-difluorosuccinic acid derivatives,33 and to regulate the conformation of simple peptides.34–37 Augmentation to the vicinal α,β,γ-trifluoro array has been achieved and applied to the synthesis of peptides,38–40 liquid crystals41 and unnatural monosaccharides (e.g.19 and 20).42 More recently, the (terminal) tetrafluoro structural unit has been explored in analogues of the multiple sclerosis drug Gilenya® (11 – Fig. 3, 21 and 22).43 Remarkably, the O'Hagan laboratory have also reported synthesis routes to (internal) vicinal tetrafluoro-,44–46 pentafluoro- (23)47 and hexafluoro-48 motifs (24).
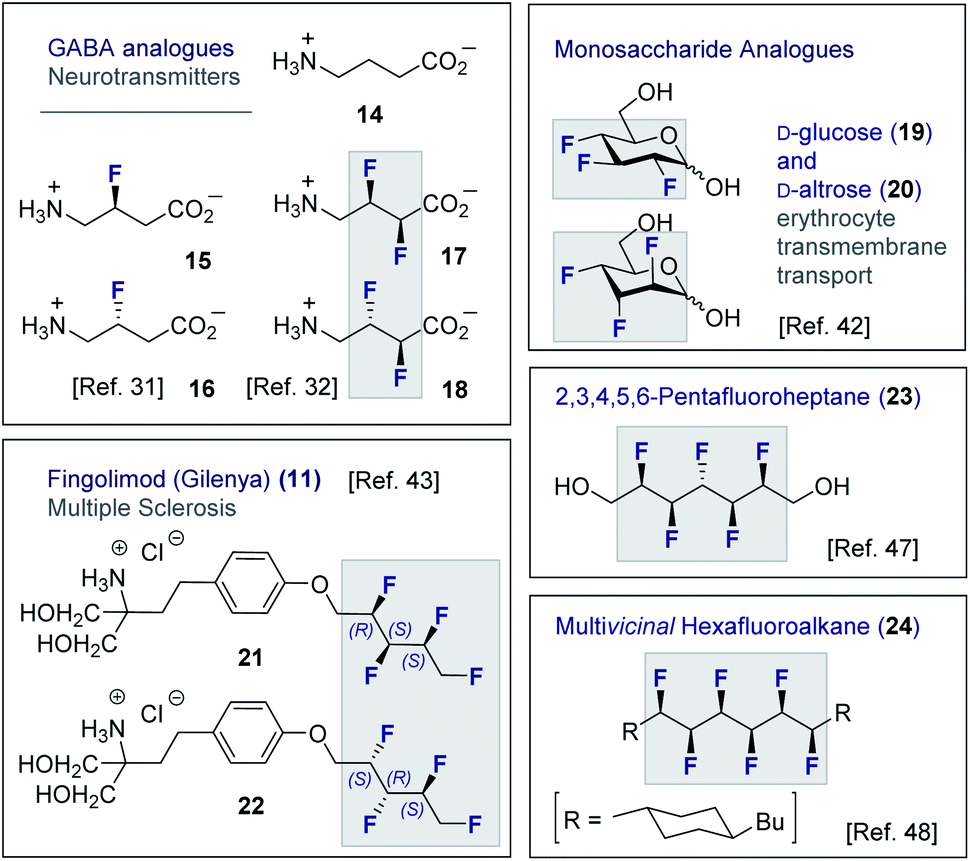 | ||
| Fig. 5 Selected examples of short, multivicinal fluoroalkane groups in functional small molecule design. | ||
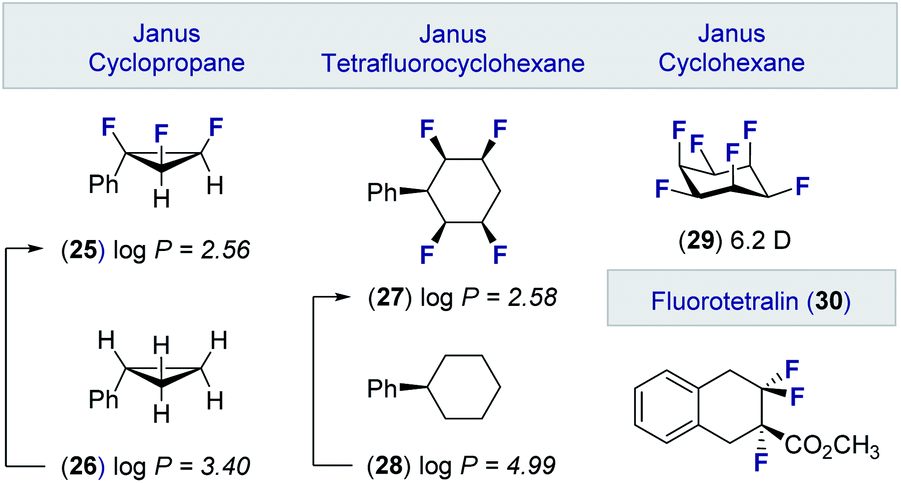
These advances in the stereocontrolled synthesis of linear multivicinal fluoroalkanes have been complemented by equally impressive synthesis campaigns to generate cyclic motifs (Fig. 6). Many of these materials, in which the fluorine atoms are in an all-syn relationship, display significantly lower log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values than the parent hydrocarbon. Examples of these facially polarised “Janus” motifs include the all-cis 1,2,3-trifluorocyclopropane 25 (cf. 26)49 and the tetrafluorocyclohexane 27 (cf. 28).50 It is interesting to note that the all-cis hexafluorocyclohexane 29 has the highest calculated dipole of any organic molecule (6.2 D).51 These materials, together with selectively fluorinated tetralins (30),52 hold great potential as drug discovery modules owing to their well-defined conformations and physicochemical profiles.53
P values than the parent hydrocarbon. Examples of these facially polarised “Janus” motifs include the all-cis 1,2,3-trifluorocyclopropane 25 (cf. 26)49 and the tetrafluorocyclohexane 27 (cf. 28).50 It is interesting to note that the all-cis hexafluorocyclohexane 29 has the highest calculated dipole of any organic molecule (6.2 D).51 These materials, together with selectively fluorinated tetralins (30),52 hold great potential as drug discovery modules owing to their well-defined conformations and physicochemical profiles.53
 | ||
| Fig. 6 Selected examples of cyclic multivicinal fluoroalkanes by O'Hagan and co-workers. | ||
3. Catalysis-based strategies to access short (≤C6), chiral fragments
The structural and physicochemical diversity intrinsic to multivicinal fluoroalkanes is expansive and renders this class of materials valuable in expanding (chiral) organofluorine chemical space. This is evident from a comparative analysis of the van der Waals radii [Å3] of common short alkyl groups with their selectively fluorinated counterparts (Fig. 7).54,55 Not only are the two partially fluorinated groups (I and II) chiral, they have volumes and 3D topologies that are complementary to structurally related aliphatic groups. Furthermore, the inclusion of short, chiral fluorinated moieties in the drug discovery portfolio redresses the current bias that favours isotropic groups over anisotropic fragments. The simplest member of the multivicinal fluoroalkane family is structure I, which is based on 1,2-difluoroethane (13). These structures are intriguing on account of the stabilising hyperconjugative interactions that give rise to the iconic gauche conformation.1,8b,10b,c This phenomenon can be rationalised by invoking stabilising σC–H → σ*C–F interactions and gives rise to a temperature-dependent dipole moment (dμ/dT <0) (Fig. 6, left). The gauche effect is a unique feature of fluorinated materials and is not observed in the corresponding chloro- or bromo-systems due to overriding repulsion.56 Collectively, these structural features are compelling arguments for the development of efficient strategies to allow small chiral groups to be assessed in the context of contemporary drug discovery.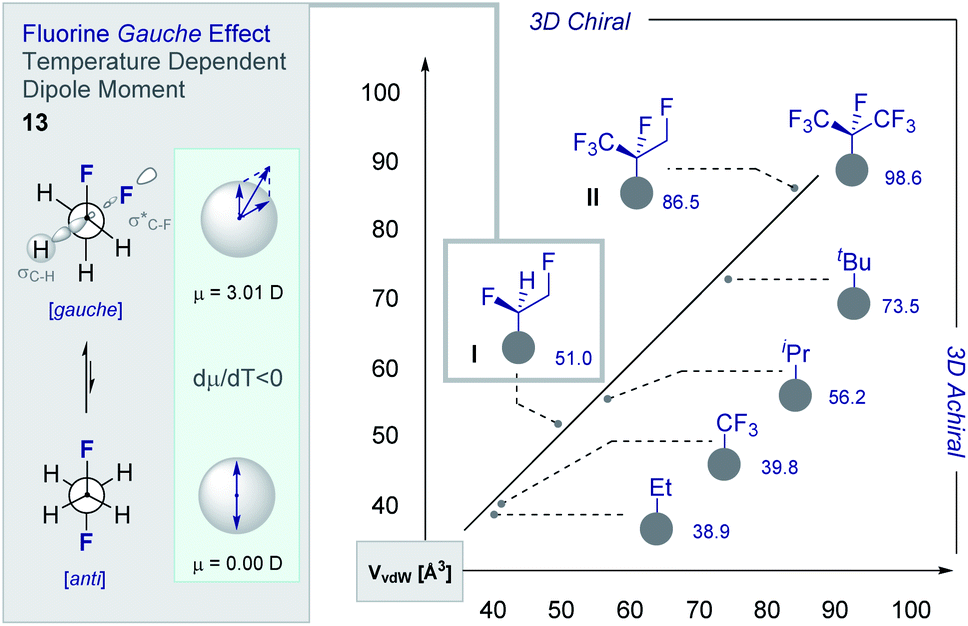 | ||
| Fig. 7 Calculated van der Waals radii [Å3] of short aliphatic and fluorinated aliphatic groups. Inset: the stereoelectronic gauche effect and the temperature-dependent dipole moment intrinsic to 1,2-difluoroethane. | ||
3.1 Catalysis-based vicinal difluorination of alkenes
Despite the popularity of fluorine bioisosterism in medicinal chemistry, and the notable advances in fluorination technologies that this has inspired,57 the catalytic, stereoselective vicinal fluorination of alkenes is comparatively under-developed.58 Direct fluorination using gaseous F2 in a carrier gas been reported by Rozen and Brand,59,60 but this approach presents safety and operational challenges for non-specialists that must be addressed (Fig. 8). As is evident from the conversion of coumarin 31 to product 32, the vicinal difluorination proceeds in a syn-selective fashion as was determined by coupling constant analysis (3JHF = 30 and 6 Hz). As a consequence, HF elimination occurs to generate the fluorinated coumarin 33. Tius has demonstrated that XeF2 enables the 1,2-difluorination of alkenes, thereby mitigating the safety concerns associated with handling strongly oxidising fluorine gas. Despite the operational simplicity of this approach, XeF2 is prohibitively expensive and translation to an enantioselective, catalysis-based platform would be challenging.61 In 1998, Hara, Yoneda and co-workers reported the direct difluorination of alkenes using stoichiometric p-TolIF2 (35) and Et3N·HF complex.62 This I(III)-reagent-based approach proceeds via a type II invertive mechanism (Type IIinv), resulting in a net syn-addition (34 → 36).58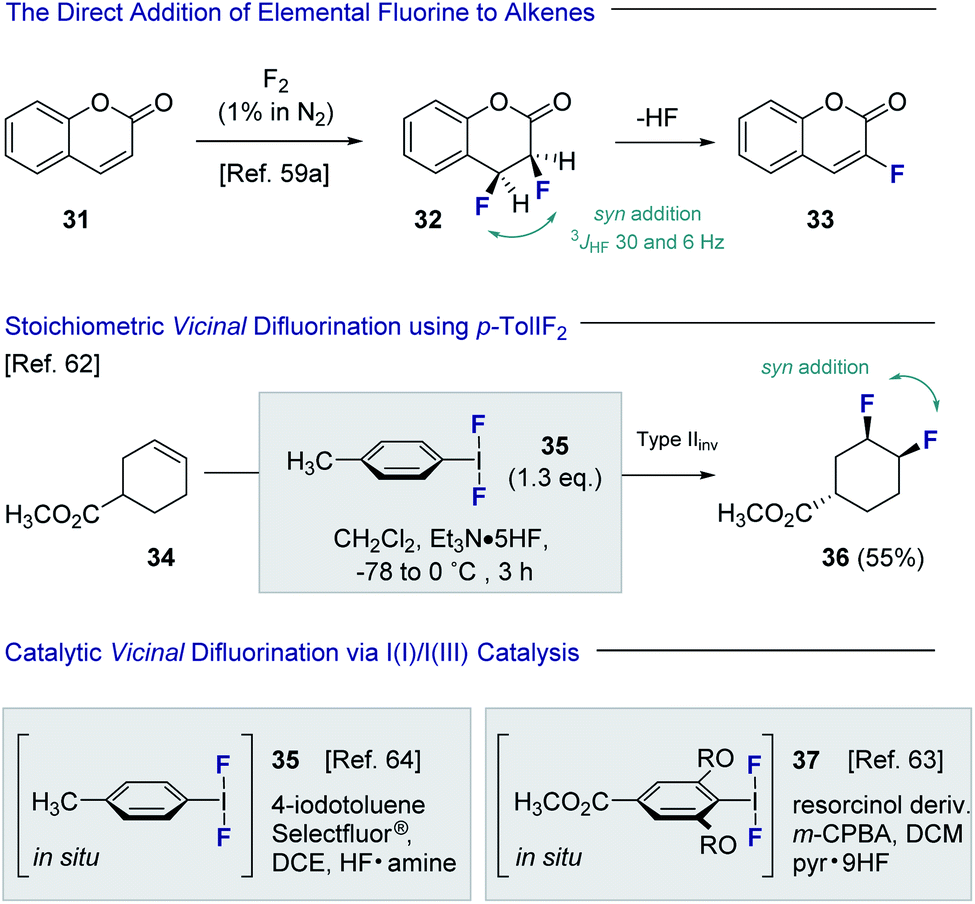 | ||
| Fig. 8 The direct vicinal difluorination of alkenes via I(I)/I(III) catalysis. | ||
Inspired by this seminal study, groups led by Jacobsen63 and Gilmour64 independently developed catalytic versions of this venerable transformation. Both strategies are predicated on the oxidation of simple aryl iodide organocatalysts, in the presence of an amine·HF complex, to generate the incipient ArIF2 species in situ.65,66 Whilst the Gilmour protocol employed Selectfluor® and various amine:HF ratios to generate 35in situ, the Jacobsen method employed m-CPBA as the terminal oxidant in conjunction with Olah's reagent to form the resorcinol derivative 37. Both groups disclosed preliminary validation of enantioselectivity, and this has since been expanded further to enable the generation of chiral motifs with broad functional group tolerance (vide infra). A scalable, electrochemical variant of the vicinal difluorination of alkenes mediated by p-TolIF2 has also been reported by Lennox and co-workers.67
In 2018, Gilmour and co-workers reported an enantioselective, catalytic vicinal difluorination of electron deficient styrenes (e.g.38) using a chiral resorcinol-derived aryl iodide (39, Fig. 9).68 This study revealed the importance of Brønsted acidity in biasing regioselectivity (vicinal versus geminal, 40 and 41, respectively) as a function of the amine:HF ratio. Varying amine:HF ratios are achieved by mixing commercially available amine·HF complexes, such as NEt3·3HF and Olah's reagent (Pyr·9HF). It is pertinent to note that the importance of Brønsted acid activators was reported by Cotter et al.69 in the activation of iodobenzene dichloride23c,70 by trifluoroacetic acid.
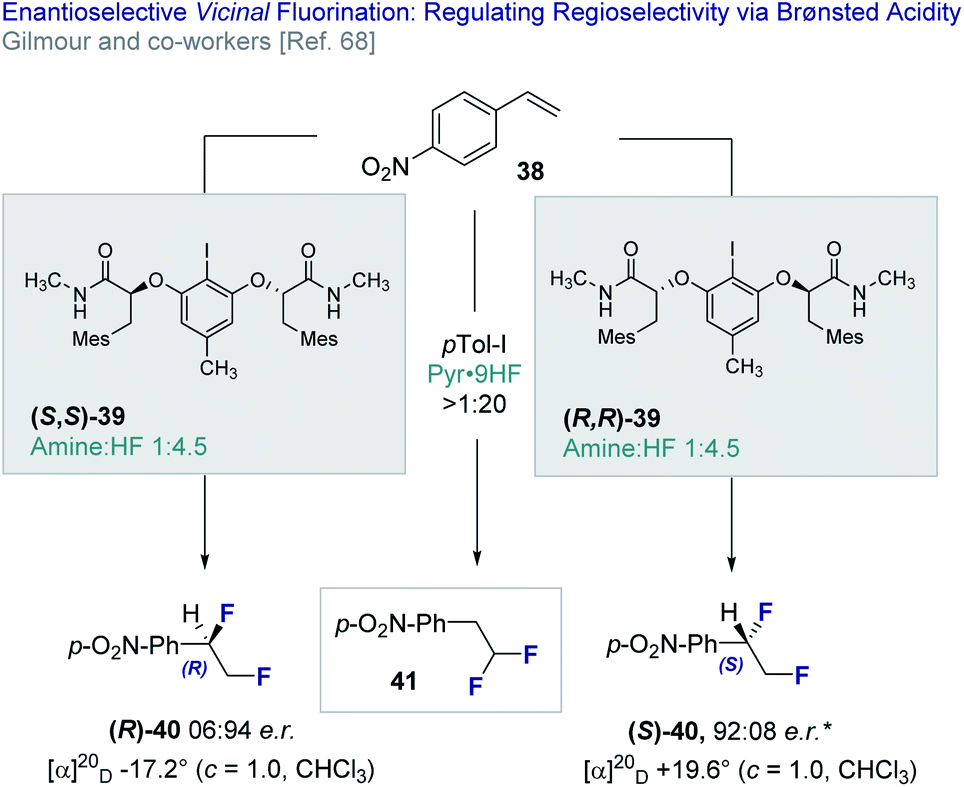 | ||
| Fig. 9 The enantioselective, catalytic vicinal difluorination of electron deficient styrenes. * 98:2 e.r. after recrystallisation from CH2Cl2/n-pentane. | ||
Jacobsen and co-workers have reported an enantio- and diastereo-selective vicinal difluorination of cinnamamides (42 → 44) using a chiral resorcinol-based aryl iodide (43).71 Regioselectivity is regulated through the anchimeric assistance of a N-tert-butyl amide substituent thereby suppressing phenonium ion rearrangement to deliver the geminal product (vide infra). This elegant solution enables the target difluorides to be generated in up to 98% ee (Fig. 10).
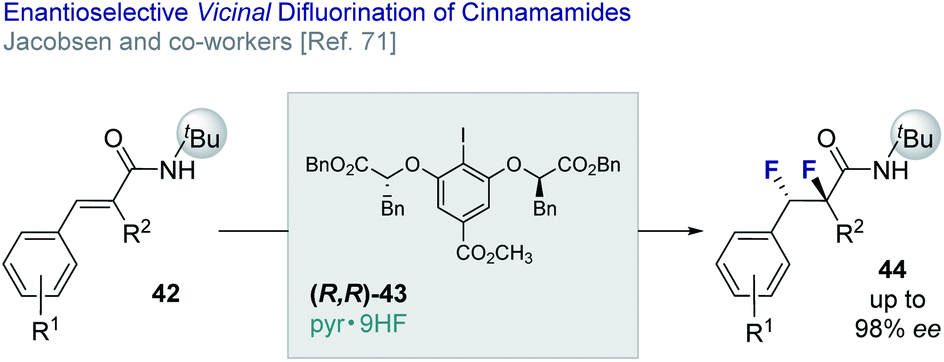 | ||
| Fig. 10 The enantio- and diastereoselective vicinal difluorination of cinnamamides. | ||
To date, this methodology64 has been leveraged to validate the 1,2-difluoromethylene motif as a chiral hybrid bioisostere of trifluoromethyl and ethyl (BITE group)8b in several small molecule drug candidates (Fig. 11). Examples from this laboratory include the synthesis of a series of Vorinostat (Zolinza®) derivatives (45) containing a pendant chain capped with a vicinal difluoro motif.72 The HDAC inhibitory behaviour of this compound set was evaluated relative to the non-fluorinated systems.73 In all cases, the FDA approved Vorinostat (Zolinza®) was used as a control.74 Several of the compounds containing the 1,2-difluoroethylene unit showed greater in vitro potency than the clinically approved drug itself against HDAC1. This trend was found to be general with the BITE-modified HDAC inhibitors performing significantly better than the ethyl derivatives.
 | ||
| Fig. 11 Small molecule drugs modified with the BITE group. | ||
BITE-modified analogues of the multiple sclerosis drug Fingolimod (Gilenya®) (46) have also been reported.75 Through detailed physicochemical analyses, it was possible to demonstrate that introduction of the BITE group is accompanied by a significant reduction in lipophilicity compared to the ethyl and trifluoromethyl systems. Most recently, the BITE group has been validated as a hybrid bioisostere of the trifluoromethyl and ethyl groups using matrix metalloproteases as structural probes.76 To that end, a series of modified barbiturate inhibitors (47) were evaluated as inhibitors of MMPs 2, 8, 9 and 13.77 The IC50 values of the BITE-modified inhibitors were found to intersect those of the corresponding Et and CF3 derivatives.55
The vicinal difluorination of alkenes has recently been extended to α-trifluoromethyl styrenes to generate fluorinated analogues of the isopropyl group (Fig. 12). Although the heptafluoroisopropyl group has become a privileged motif in agrochemical research3,7 and currently features in drug candidates78 and organocatalysts,79 routes to generate a chiral analogue remained conspicuously absent. Exposing simple α-trifluoromethyl styrenes (48) to fluorination conditions (various amine·HF complexes, Selectfluor®) in the presence of a chiral resorcinol catalyst ((R,R)-49),80 it was possible to generate chiral products efficiently (50) and with good levels of enantioselectivity.81 An interesting conformational feature of this motif is that the C(sp3)–CF3 bond is orthogonal to the plane of the aryl ring, thereby enabling stabilising hyperconjugative interactions,82 whilst mitigating 1,3-allylic strain.83 Moreover, the stereoelectronic gauche effect manifests itself as was determined by single crystal X-ray analysis of several derivatives. In an extension of this methodology, the vicinal difluorination of α-trifluoromethyl-β-difluoro-styrenes (51 → 52) was achieved through in situ generation of p-TolIF2 (35) by treatment of p-TolI with Selectfluor® in the presence of pyr·9HF complex.84 In line with the previous analysis, the structure displayed a degree of pre-organisation with one of the C(sp3)–CF3 bonds aligned with the π-system of the adjacent aryl ring. Curiously, a phthalimide derivative was found to display orthogonal C–F…C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O interactions with a neighbouring molecule in the solid state. This may prove to be useful given the increasing prominence of these interactions in medicinal chemistry.11,85
O interactions with a neighbouring molecule in the solid state. This may prove to be useful given the increasing prominence of these interactions in medicinal chemistry.11,85
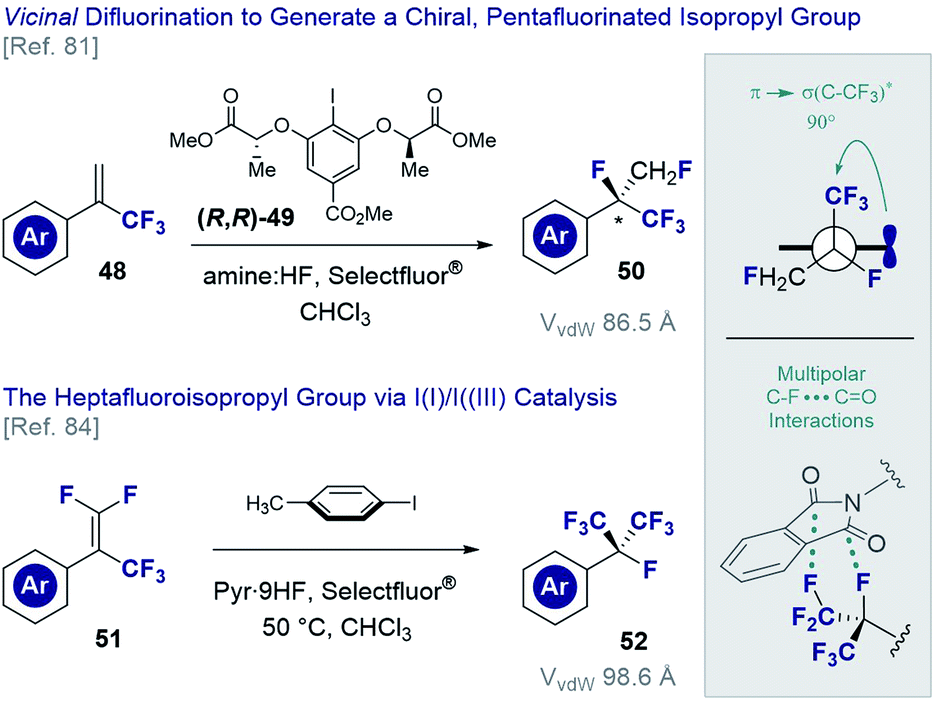 | ||
| Fig. 12 Generating fluorinated surrogates of the isopropyl group via I(I)/I(III) catalysis. | ||
3.2 Catalysis-based geminal difluorination of alkenes
Hypervalent iodine platforms have a venerable history in halogenation chemistry,86 and have also been successfully harnessed to generate geminal difluorination patterns (Fig. 13). Seminal examples include Hara and Yoneda's use of stoichiometric quantities of p-TolIF2 (35) to enable a difluorinative ring contraction of alkenes.87 The antipodal ring expansion has recently been reported by this laboratory to generate conformationally biased fluorinated tetralins.52 A silver-mediated geminal difluorination of styrenes has been developed by Szabó and co-workers using a fluoroiodoxazole reagent.88 Moreover, Murphy and co-workers have disclosed the geminal difluorination of phenylallenes using stoichiometric p-TolIF2via Lewis acid activation.89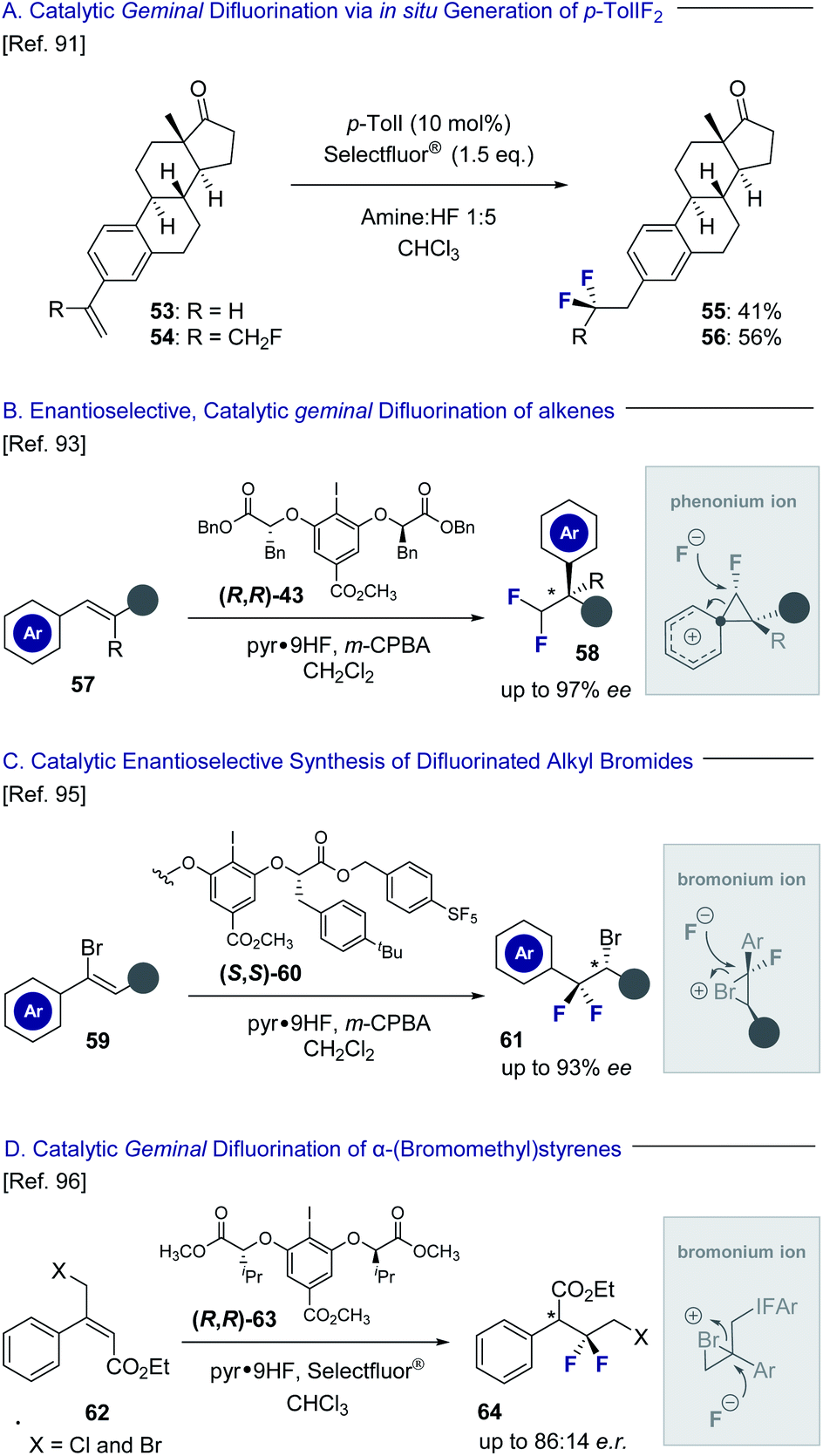 | ||
| Fig. 13 Geminal difluorination of alkenes to generate difluoromethylated stereocenters. | ||
Catalysis-based platforms have been developed to complement these reagent-based approaches and include Kitamura and co-workers protocol to generate 2,2-difluoroethylarenes from simple styrenes using p-TolI as the catalyst with m-CPBA as the oxidant.90 This laboratory has also reported the geminal difluorination of styrenes and extended it to include α-substituted styrenes bearing fluorine-containing groups (Fig. 13A; 53 → 55 and 54 → 56).91 The difluorination of alkenyl N-methyliminodiacetyl boronates has been reported by Fan and co-workers to generate synthetically useful building blocks for subsequent diversification.92 Particularly relevant to this Perspective dedicated to short, chiral fluorine-containing groups is the development of an enantioselective, catalytic 1,1-difluorination of alkenes (57) to construct difluoromethylated stereocenters (58) by Jacobsen and co-workers (Fig. 13B).93 Key to the success of this transformation is a stereospecific phenonium ion rearrangement94 to deliver highly versatile building blocks with excellent levels of enantioselectivity. The same laboratory has also leveraged a conceptually related reaction design, proceeding via bromonium ion formation, to process simple vinyl bromides to optically active difluorinated alkyl bromides (Fig. 13C, 59 → 61).95 Bromonium ion formation is a feature in the geminal difluorination of α-(bromomethyl)-styrenes reported by this laboratory to generate electrophilic linchpins (Fig. 13D, 62 → 64).96 Although the transformations discussed in Section 3.2 do not generate a stereogenic centre at the fluorine bearing carbon atom, their inclusion in this Perspective is instructive. Collectively, I(III) species have been central to the development of catalysis-based methods to enable the 1,1- and 1,2-difluorination alkenes, whilst also facilitating access to 1,3-difluoro motifs.97–99
4. Conclusions
Short, alkyl groups are prominent in the natural product repertoire and are a logical consequence of the iterative biosynthesis algorithms that underpin their genesis. The importance of these seemingly inconspicuous motifs in biology is reflected in the development of many synthetic bioactive small molecules in which the “magic methyl” effect manifests itself. Chiral antipodes of these structural units have a venerable history in mechanistic enzymology and would augment the existing drug module portfolio. However, with the exception of branched hydrocarbons, this requires the impractical introduction of deuterium and tritium. Hydrogen to fluorine (bio)isosterism, to generate multivicinal fluoroalkanes, proves an alternative to address this challenge and develop materials with unique properties. In what may be considered a conceptual merger of two units that are prevalent in nature; namely short alkyl groups and (vicinal) oxidation patterns, a plethora of selective processes have been reported that leverage I(I)/I(III) catalysis to expand organofluorine chemical space into chiral regions. Integrating these fluorine-containing fragments in focussed drug and agrochemical discovery libraries will fully reveal the physicochemical potential of these materials which will, in turn, provide an impetus for further innovation in the field. In recent years, the seemingly innocent replacement of H/OH by F in stereochemically complex biomolecules has led to striking changes in orientation when bound by the target enzyme: this has broad implications for molecular recognition and chemical biology in a more general sense.100,101 Expanding organofluorine chemical space has an important role to play in the design of molecular function and main group catalysis is currently centre stage.Author contributions
The manuscript was conceived by all authors and written by RG with input from SM and JH.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge generous financial support from the European Commission (ERC Consolidator Grant “RECON” 818949), the DFG SFB 858 and Excellence Cluster EXC 1003 “Cells in Motion”, and the WWU Münster.Notes and references
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC.
- (a) B. E. Smart, J. Fluorine Chem., 2001, 109, 3 CrossRef CAS; (b) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (d) J. Wang, M. Sánchez-Roselló, J. L. Acenaña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (e) J. Han, A. M. Remete, L. S. Dobson, L. Kiss, K. Izawa, H. Moriwaki, V. A. Soloshonok and D. O'Hagan, J. Fluorine Chem., 2020, 239, 109639 CrossRef CAS.
- (a) P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS; (b) T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16 CrossRef CAS; (c) S. Pazenok and F. R. Leroux in Frontiers of Organofluorine Chemistry, ed. I. Ojima, World Scientific, London, 2020, p. 695 Search PubMed; (d) Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
- (a) M. Pagliaro and R. Ciriminna, J. Mater. Chem., 2005, 15, 4981 RSC; (b) F. Babudri, G. M. Farinola, F. Naso and R. Ragni, Chem. Commun., 2007, 1003 RSC; (c) R. Ragni, A. Punzi, F. Babudri and G. M. Farinola, Eur. J. Org. Chem., 2018, 3500 CrossRef CAS.
- (a) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS; (b) D. O'Hagan and H. Deng, Chem. Rev., 2015, 115, 634 CrossRef PubMed; (c) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed.
- (a) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS; (b) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS; (c) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822 CrossRef CAS.
- M. E. Qacemi, S. Rendine and P. Mainfisch, in Fluorine in Life Sciences: Pharmaceuticals, Medicinal Diagnostics, and Agrochemicals, Copyright, Elsevier Inc, 2019, pp. 607–623 Search PubMed.
- (a) Q. A. Huchet, B. Kuhn, B. Wagner, N. A. Kratochwil, H. Fischer, M. Kansy, D. Zimmerli, E. M. Carreira and K. Müller, J. Med. Chem., 2015, 58, 9041 CrossRef CAS PubMed; (b) I. G. Molnár, C. Thiehoff, M. C. Holland and R. Gilmour, ACS Catal., 2016, 6, 7167 CrossRef.
- (a) A. L. Hopkins and G. R. Bickerton, Nat. Chem. Biol., 2010, 6, 482 CrossRef CAS PubMed; (b) J.-L. Reymond, R. van Deursen, L. C. Blum and L. Ruddigkeit, MedChemComm, 2010, 1, 30 RSC.
- (a) J. J. Mullins, J. Chem. Educ., 2012, 89, 834–836 CrossRef CAS; (b) L. E. Zimmer, C. Sparr and R. Gilmour, Angew. Chem., Int. Ed., 2011, 50, 11860 CrossRef CAS; (c) C. Thiehoff, Y. P. Rey and R. Gilmour, Isr. J. Chem., 2017, 57, 92 CrossRef CAS; (d) M. Aufiero and R. Gilmour, Acc. Chem. Res., 2018, 51, 1701 CrossRef CAS PubMed.
- (a) J. A. Olsen, D. W. Banner, P. Seiler, U. O. Sander, A. D'Arcy, M. Stihle, K. Müller and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 2507 CrossRef CAS PubMed; (b) F. Hof, D. M. Scofield, W. B. Schweizer and F. Diederich, Angew. Chem., Int. Ed., 2004, 43, 5056 CrossRef CAS PubMed.
- J. Han, L. Kiss, H. Mei, A. M. Remete, M. Ponikvar-Svet, D. M. Sedgwick, R. Roman, S. Fustero, H. Moriwaki and V. A. Soloshonok, Chem. Rev., 2021, 121, 4678 CrossRef CAS.
- D. O'Hagan and D. B. Harper, J. Fluorine Chem., 1999, 100, 127 CrossRef.
- (a) D. O'Hagan, C. Schaffrath, S. L. Cobb, J. T. G. Hamilton and C. D. Murphy, Nature, 2002, 416, 279 CrossRef PubMed; (b) C. Dong, F. Huang, H. Deng, C. Schaffrath, J. B. Spencer, D. O'Hagan and J. H. Naismith, Nature, 2004, 427, 561 CrossRef CAS PubMed; (c) X. Zhu, D. A. Robinson, A. R. McEwan, D. O'Hagan and J. H. Naismith, J. Am. Chem. Soc., 2007, 129, 14597 CrossRef CAS PubMed; (d) K. K. J. Chan and D. O'Hagan, Methods Enzymol., 2012, 516, 219 CAS; (e) H. Aldemir, S. V. Kohlhepp, T. Gulder and T. A. M. Gulder, J. Nat. Prod., 2014, 77, 2331 CrossRef CAS.
- F. Lovering, MedChemComm, 2013, 4, 515 RSC.
- (a) C. D. Murphy, Biotechnol. Lett., 2010, 32, 351 CrossRef CAS; (b) W. Tong, Q. Huang, M. Li and J.-b. Wang, Bioresour. Bioprocess., 2019, 6, 46 CrossRef.
- (a) J. Lüthy, J. Rétey and D. Arigoni, Nature, 1969, 221, 1213 CrossRef; (b) C. A. Townsend, T. Scholl and D. Arigoni, J. Chem. Soc., Chem. Commun., 1975, 921 RSC; (c) H. G. Floss and S. Lee, Acc. Chem. Res., 1993, 26, 116 CrossRef CAS.
- T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, J. Med. Chem., 2019, 62, 5276 CrossRef CAS PubMed.
- C. Schmidt, Nat. Biotechnol., 2017, 35, 493 CrossRef CAS PubMed.
- A. Eschenmoser, in ‘Chemical Synthesis, Gnosis to Prognosis’ed. C. Chatgilialoglu and V. Snieckus, NATO ASI, Kluwer Academic Publications, Dordrecht, 1994, pp. 231–232 Search PubMed.
- L. Nowack, C. S. Teschers, S. Albrecht and R. Gilmour, Nat. Prod. Rep., 2021, 38, 890–904 RSC.
- C. S. Teschers, C. G. Daniliuc, G. Kehr and R. Gilmour, J. Fluorine Chem., 2018, 210, 1–5 CrossRef CAS.
- (a) W.-J. Chung and C. D. Vanderwal, Angew. Chem., Int. Ed., 2016, 55, 4396 CrossRef CAS; (b) M. L. Landry and N. Z. Burns, Acc. Chem. Res., 2018, 51, 1260 CrossRef CAS; (c) J. C. Sarie, J. Neufeld, C. G. Daniliuc and R. Gilmour, ACS Catal., 2019, 9, 7232 CrossRef CAS.
- S. Fischer, N. Huwyler, S. Wolfrum and E. M. Carreira, Angew. Chem., Int. Ed., 2016, 55, 2555 CrossRef CAS.
- E. Zerhouni, Chem. Eng. News, 2003, 84, 12–17 Search PubMed.
- D. Seebach, Angew. Chem., Int. Ed., 1990, 29, 1320 CrossRef.
- (a) L. Hunter and D. O'Hagan, Org. Biomol. Chem., 2008, 6, 2843 RSC; (b) D. O'Hagan, J. Org. Chem., 2012, 77, 3689 CrossRef PubMed.
- F. Scheidt, P. Selter, N. Santschi, M. C. Holland, D. V. Dudenko, C. Daniliuc, C. Mück-Lichtenfeld, M. R. Hansen and R. Gilmour, Chem. – Eur. J., 2017, 23, 6142 CrossRef CAS PubMed.
- D. Wu, A. Tian and H. Sun, J. Phys. Chem. A, 1998, 102, 9901 CrossRef CAS.
- N. J. Leonard, Acc. Chem. Res., 1979, 12, 423 CrossRef CAS.
- G. Deniau, A. M. Z. Slawin, T. Lebl, F. Chorki, J. P. Issberner, T. van Mourik, J. M. Heygate, J. J. Lambert, L.-A. Etherington, K. T. Sillar and D. O'Hagan, ChemBioChem, 2007, 8, 2265 CrossRef CAS PubMed.
- I. Yamamoto, M. J. T. Jordan, N. Gavande, M. R. Doddareddy, M. Chebib and L. Hunter, Chem. Commun., 2012, 48, 829 RSC.
- D. O'Hagan, H. S. Rzepa, M. Schüler and A. M. Z. Slawin, Beilstein J. Org. Chem., 2006, 2, 19 Search PubMed.
- L. Hunter, Beilstein J. Org. Chem., 2010, 6, 38 Search PubMed.
- M. Schüler, D. O'Hagan and A. M. Z. Slawin, Chem. Commun., 2005, 4324 RSC.
- L. Hunter, K. A. Jolliffe, M. J. T. Jordan, P. Jensen and R. B. Macquart, Chem. – Eur. J., 2011, 17, 2340 CrossRef CAS PubMed.
- L. Hunter, S. Butler and S. B. Ludbrook, Org. Biomol. Chem., 2012, 10, 8911 RSC.
- M. Nicoletti, D. O'Hagan and A. M. Z. Slawin, J. Am. Chem. Soc., 2005, 127, 482 CrossRef CAS PubMed.
- V. A. Brunet, A. M. Z. Slawin and D. O'Hagan, Beilstein J. Org. Chem., 2009, 5, 61 Search PubMed.
- R. Cheerlavancha, A. Lawer, M. Cagnes, M. Bhadbhade and L. Hunter, Org. Lett., 2013, 15, 5562 CrossRef CAS PubMed.
- M. Nicoletti, M. Bremer, P. Kirsch and D. O'Hagan, Chem. Commun., 2007, 5075 RSC.
- S. Bresciani, T. Lebl, A. M. Z. Slawin and D. O'Hagan, Chem. Commun., 2010, 46, 5434 RSC.
- P. Bentler, N. Erdeljac, K. Bussmann, M. Ahlqvist, L. Knerr, K. Bergander, C. G. Daniliuc and R. Gilmour, Org. Lett., 2019, 21, 7741 CrossRef CAS PubMed.
- L. Hunter, D. O'Hagan and A. M. Z. Slawin, J. Am. Chem. Soc., 2006, 128, 16422 CrossRef CAS.
- L. Hunter, A. M. Z. Slawin, P. Kirsch and D. O'Hagan, Angew. Chem., Int. Ed., 2007, 46, 7887 CrossRef CAS PubMed.
- L. Hunter, P. Kirsch, J. T. G. Hamilton and D. O'Hagan, Org. Biomol. Chem., 2008, 6, 3105 RSC.
- D. Farran, A. M. Z. Slawin, P. Kirsch and D. O'Hagan, J. Org. Chem., 2009, 74, 7168 CrossRef CAS PubMed.
- L. Hunter, P. Kirsch, A. M. Z. Slawin and D. O'Hagan, Angew. Chem., Int. Ed., 2009, 48, 5457 CrossRef CAS PubMed.
- Z. Fang, D. B. Cordes, A. M. Z. Slawin and D. O'Hagan, Chem. Commun., 2019, 55, 10539 RSC.
- A. Rodil, S. Bosisio, M. S. Ayoup, L. Quinn, D. B. Cordes, A. M. Z. Slawin, C. D. Murphy, J. Michel and D. O'Hagan, Chem. Sci., 2018, 9, 3023 RSC.
- (a) N. S. Keddie, A. M. Z. Slawin, T. Lebl, D. Philp and D. O'Hagan, Nat. Chem., 2015, 7, 483 CrossRef CAS PubMed; (b) N. Santschi and R. Gilmour, Nat. Chem., 2015, 7, 467 CrossRef CAS PubMed; (c) B. E. Ziegler, M. Lecours, R. A. Marta, J. Featherstone, E. Fillion, W. S. Hopkins, V. Steinmetz, N. S. Keddie, D. O'Hagan and T. B. McMahon, J. Am. Chem. Soc., 2016, 138, 7460 CrossRef CAS PubMed; (d) For a synthetic study, see M. P. Wiesenfeldt, Z. Nairoukh, W. Li and F. Glorius, Science, 2017, 357, 908 CrossRef CAS PubMed.
- J. Neufeld, T. Stünkel, C. Mück-Lichtenfeld, C. G. Daniliuc and R. Gilmour, Angew. Chem., Int. Ed., 2021, 60, 13647–13651 CrossRef CAS PubMed.
- R. Mondal, M. Agbaria and Z. Nairoukh, Chem. – Eur. J., 2021, 27, 7193 CrossRef CAS PubMed.
- The Van der Waals volumes were calculated according to: Y. H. Zhao, M. H. Abraham and A. M. Zissimos, J. Org. Chem., 2003, 68, 7368 CrossRef CAS PubMed Please also see: A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef.
- M. Jagodzinska, F. Huguenot, G. Candiani and M. Zanda, ChemMedChem, 2009, 4, 49 CrossRef CAS PubMed.
- F. Akkerman, J. Buschmann, D. Lentz, P. Luger and E. Rödel, J. Chem. Crystallogr., 2003, 33, 969 CrossRef CAS.
- P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity and Applications, Wiley-VCH, Weinheim, 2013 Search PubMed.
- A. J. Cresswell, S. T.-C. Eey and S. E. Denmark, Angew. Chem., Int. Ed., 2015, 54, 15642 CrossRef CAS PubMed.
- (a) S. Rozen and M. Brand, J. Org. Chem., 1986, 51, 3607 CrossRef CAS; (b) I. Vints and S. Rozen, J. Org. Chem., 2014, 79, 7261 CrossRef CAS PubMed; (c) I. Vints and S. Rozen, Tetrahedron, 2016, 72, 632 CrossRef CAS.
- S. T. Purrington, B. S. Kagan and T. B. Patrick, Chem. Rev., 1986, 86, 997 CrossRef CAS.
- M. A. Tius, Tetrahedron, 1995, 51, 6605 CrossRef CAS.
- S. Hara, J. Nakahigashi, K. Ishi-I, H. Sakai, M. Sawaguchi, T. Fukuhara and N. Yoneda, Synlett, 1998, 495 CrossRef CAS.
- S. M. Banik, J. W. Medley and E. N. Jacobsen, J. Am. Chem. Soc., 2016, 138, 5000 CrossRef CAS PubMed.
- I. G. Molnár and R. Gilmour, J. Am. Chem. Soc., 2016, 138, 5004 CrossRef PubMed.
- C. Ye, B. Twamley and J. M. Shreeve, Org. Lett., 2005, 7, 3961 CrossRef CAS PubMed.
- (a) R. F. Weinland and W. Stille, Ber. Dtsch. Chem. Ges., 1901, 34, 2631 CrossRef CAS; (b) W. Carpenter, J. Org. Chem., 1966, 31, 2688 CrossRef CAS; (c) T. B. Patrick, J. J. Scheibel, W. E. Hall and Y. H. Lee, J. Org. Chem., 1980, 45, 4492 CrossRef CAS; (d) M. Zupan and A. Pollak, J. Chem. Soc., Chem. Commun., 1975, 17, 715 RSC; (e) J. C. Sarie, C. Thiehoff, R. J. Mudd, C. G. Daniliuc, G. Kehr and R. Gilmour, J. Org. Chem., 2017, 82, 11792 CrossRef CAS PubMed.
- (a) S. Doobary, A. T. Sedikides, H. P. Caldora, D. L. Poole and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 1155 CrossRef CAS PubMed; (b) S. Doobary and A. J. J. Lennox, Synlett, 2020, 31, 1333 CrossRef CAS; (c) D. M. Heard, S. Doobary and A. J. J. Lennox, ChemElectroChem, 2021, 8, 2070–2074 CrossRef CAS.
- F. Scheidt, M. Schäfer, J. C. Sarie, C. G. Daniliuc, J. J. Molloy and R. Gilmour, Angew. Chem., Int. Ed., 2018, 57, 16431 CrossRef CAS PubMed.
- J. L. Cotter, L. J. Andrews and R. M. Keefer, J. Am. Chem. Soc., 1962, 84, 793 CrossRef CAS.
- J. C. Sarie, J. Neufeld, C. G. Daniliuc and R. Gilmour, Synthesis, 2019, 51, 4408 CrossRef CAS.
- M. K. Haj, S. M. Banik and E. N. Jacobsen, Org. Lett., 2019, 21, 4919 CrossRef CAS PubMed.
- N. Erdeljac, K. Bussmann, A. Schöler, F. K. Hansen and R. Gilmour, ACS Med. Chem. Lett., 2019, 10, 1336 CrossRef CAS PubMed.
- J. W. Walton, J. M. Cross, T. Riedel and P. J. Dyson, Org. Biomol. Chem., 2017, 15, 9186 RSC.
- N. Reißing, V. Marquardt, C. G. W. Gertzen, A. Schöler, A. Schramm, T. Kurz, H. Gohlke, A. Aigner, M. Remke and F. K. Hansen, MedChemComm, 2019, 10, 1109 RSC.
- N. Erdeljac, G. Kehr, M. Ahlqvist, L. Knerr and R. Gilmour, Chem. Commun., 2018, 54, 12002 RSC.
- N. Erdeljac, C. Thiehoff, R. P. Jumde, C. G. Daniliuc, S. Höppner, A. Faust, A. K. H. Hirsch and R. Gilmour, J. Med. Chem., 2020, 63, 6225 CrossRef CAS PubMed.
- B. Fingleton, Curr. Pharm. Des., 2007, 13, 333 CrossRef CAS PubMed.
- A. Karmakar, R. Y. Nimje, A. Silamkoti, M. Pitchai, M. Basha, C. Singarayer, D. Ramasamay, G. T. V. Babu, R. Samikannu, S. Subramaniam, P. Anjanappa, M. Vetrichelvan, H. Kumar, A. G. Dikundwar, A. Gupta, A. K. Gupta, R. Rampulla, T. G. M. Dhar and A. Mathur, Org. Process Res. Dev., 2021, 25, 1001 CrossRef CAS.
- J. Guin, C. Rabalakos and B. List, Angew. Chem., Int. Ed., 2012, 51, 8859 CrossRef CAS PubMed.
- For selected examples, see (a) M. Fujita, Y. Yoshida, K. Miyata, A. Wakisaka and T. Sugimura, Angew. Chem., Int. Ed., 2010, 49, 7068 CrossRef CAS PubMed; (b) S. Haubenreisser, T. H. Wöste, C. Martínez, K. Ishihara and K. Muñiz, Angew. Chem., Int. Ed., 2016, 55, 413 CrossRef CAS PubMed.
- S. Meyer, J. Häfliger, M. Schäfer, J. J. Molloy, C. G. Daniliuc and R. Gilmour, Angew. Chem., Int. Ed., 2021, 60, 6430 CrossRef CAS PubMed.
- T. Schaefer, R. W. Schurko, R. Sebastian and F. E. Hruska, Can. J. Chem., 1995, 73, 816 CrossRef CAS.
- R. W. Hoffmann, Chem. Rev., 1989, 89, 1841 CrossRef CAS.
- V. Martín-Heras, C. G. Daniliuc and R. Gilmour, Synthesis, 2021 DOI:10.1055/a-1485-4916.
- (a) R. Paulini, K. Müller and F. Diederich, Angew. Chem., Int. Ed., 2005, 44, 1788 CrossRef CAS PubMed; (b) J. Pollock, D. Borkin, G. Lund, T. Purohit, E. Dyguda-Kazimierowicz, J. Grembecka and T. Cierpicki, J. Med. Chem., 2015, 58, 7465 CrossRef CAS PubMed.
- (a) N. Yoneda, J. Fluorine Chem., 2004, 125, 7 CrossRef CAS; (b) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS; (c) S. V. Kohlhepp and T. Gulder, Chem. Soc. Rev., 2016, 45, 6270 RSC; (d) A. M. Arnold, A. Ulmer and T. Gulder, Chem. – Eur. J., 2016, 22, 8728 CrossRef CAS PubMed.
- S. Hara, J. Nakahigashi, K. Ishi-i, T. Fukuhara and N. Yoneda, Tetrahedron Lett., 1998, 39, 2589 CrossRef CAS.
- (a) N. O. Ilchenko, B. O. A. Tasch and K. J. Szabó, Angew. Chem., Int. Ed., 2014, 53, 12897 CrossRef CAS; (b) N. O. Ilchenko and K. J. Szabó, J. Fluorine Chem., 2017, 203, 104 CrossRef CAS.
- Z. Zhao, L. Racicot and G. K. Murphy, Angew. Chem., Int. Ed., 2017, 56, 11620 CrossRef CAS PubMed Also see: Z. Zhao, A. J. To and G. K. Murphy, Chem. Commun., 2019, 55, 14821 RSC.
- (a) T. Kitamura, K. Muta and J. Oyamada, J. Org. Chem., 2015, 80, 10431 CrossRef CAS; (b) T. Kitamura, K. Yoshida, S. Mizuno, A. Miyake and J. Oyamada, J. Org. Chem., 2018, 83, 14853 CrossRef CAS PubMed.
- F. Scheidt, J. Neufeld, M. Schäfer, C. Thiehoff and R. Gilmour, Org. Lett., 2018, 20, 8073 CrossRef CAS PubMed.
- W.-X. Lv, Q. Li, J.-L. Li, Z. Li, E. Lin, D.-H. Tan, Y.-H. Cai, W.-X. Fan and H. Wang, Angew. Chem., Int. Ed., 2018, 57, 16544 CrossRef CAS PubMed.
- S. M. Banik, J. W. Medley and E. N. Jacobsen, Science, 2016, 353, 51 CrossRef CAS PubMed.
- T. Bykova, N. Al-Maharik, A. M. Z. Slawin and D. O'Hagan, J. Fluorine Chem., 2015, 179, 188 CrossRef CAS.
- M. D. Levin, J. M. Ovian, J. A. Read, M. S. Sigman and E. N. Jacobsen, J. Am. Chem. Soc., 2020, 142, 14831 CrossRef CAS.
- J. Häfliger, K. Livingstone, C. G. Daniliuc and R. Gilmour, Chem. Sci., 2021, 12, 6148 RSC.
- N. O. Ilchenko, M. Hedberg and K. J. Szabó, Chem. Sci., 2017, 8, 1056 RSC.
- S. M. Banik, K. M. Mennie and E. N. Jacobsen, J. Am. Chem. Soc., 2017, 139, 9152 CrossRef CAS.
- K. M. Mennie, S. M. Banik, E. C. Reichert and E. N. Jacobsen, J. Am. Chem. Soc., 2018, 140, 4797 CrossRef CAS PubMed.
- P. Bentler, K. Bergander, C. G. Daniliuc, C. Mück-Lichtenfeld, R. P. Jumde, A. K. H. Hirsch and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 10990 CrossRef CAS.
- A. Axer, R. P. Jumde, S. Adam, A. Faust, M. Schäfers, M. Fobker, J. Koehnke, A. K. H. Hirsch and R. Gilmour, Chem. Sci., 2021, 12, 1286 RSC.
Footnote |
| † This manuscript is dedicated with much respect to Prof. Dr David O'Hagan FRSE for his inspiration and example in organofluorine chemistry. |
| This journal is © The Royal Society of Chemistry 2021 |