
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel-mediated N–N bond formation and N2O liberation via nitrogen oxyanion reduction†
Daniel M.
Beagan
,
Alyssa C.
Cabelof
,
Maren
Pink
,
Veronica
Carta
,
Xinfeng
Gao
 and
Kenneth G.
Caulton
*
and
Kenneth G.
Caulton
*
Indiana University, Department of Chemistry, 800 E. Kirkwood Ave., Bloomington, IN 47401, USA. E-mail: caulton@indiana.edu
First published on 13th July 2021
Abstract
The syntheses of (DIM)Ni(NO3)2 and (DIM)Ni(NO2)2, where DIM is a 1,4-diazadiene bidentate donor, are reported to enable testing of bis boryl reduced N-heterocycles for their ability to carry out stepwise deoxygenation of coordinated nitrate and nitrite, forming O(Bpin)2. Single deoxygenation of (DIM)Ni(NO2)2 yields the tetrahedral complex (DIM)Ni(NO)(ONO), with a linear nitrosyl and κ1-ONO. Further deoxygenation of (DIM)Ni(NO)(ONO) results in the formation of dimeric [(DIM)Ni(NO)]2, where the dimer is linked through a Ni–Ni bond. The lost reduced nitrogen byproduct is shown to be N2O, indicating N–N bond formation in the course of the reaction. Isotopic labelling studies establish that the N–N bond of N2O is formed in a bimetallic Ni2 intermediate and that the two nitrogen atoms of (DIM)Ni(NO)(ONO) become symmetry equivalent prior to N–N bond formation. The [(DIM)Ni(NO)]2 dimer is susceptible to oxidation by AgX (X = NO3−, NO2−, and OTf−) as well as nitric oxide, the latter of which undergoes nitric oxide disproportionation to yield N2O and (DIM)Ni(NO)(ONO). We show that the first step in the deoxygenation of (DIM)Ni(NO)(ONO) to liberate N2O is outer sphere electron transfer, providing insight into the organic reductants employed for deoxygenation. Lastly, we show that at elevated temperatures, deoxygenation is accompanied by loss of DIM to form either pyrazine or bipyridine bridged polymers, with retention of a BpinO− bridging ligand.
Introduction
Reduction of CO2 is widely sought and has classically focused on deoxygenation to C1 products,1–3 with recent advances moving towards multi-carbon and value-added products which preserve terrestrial use of that carbon resource.4–9 There is a nitrogen analogue of this challenge, accomplished biologically through denitrification, which requires an N–N bond formation upon reduction of NOx− back to dinitrogen. It is widely recognized that the natural nitrogen cycle has been perturbed as an unintended consequence of inexpensive Haber–Bosch ammonia used to enhance food production.10–12 Soluble nitrate is abundant in runoff from agricultural fields, and accumulates in poorly flushed rivers, bays and seacoasts which ultimately leads to eutrophication and oxygen-poor “dead zones.”13–15The reduction of nitrogen oxidation states is biologically accomplished by a variety of enzymes16–23 and its synthetic conversion is essential to repurpose environmental pollutants into value-added compounds that might be derived from nitrogen oxyanions. An attractive route to nitrogen oxyanion reduction is through deoxygenation,24–32 and the oxygen can be captured by protons, metal electrophiles, or main group elements known for their oxophilicity. The same is true for CO2 reduction and both carbon and nitrogen reduction would be richer if they could form products with C–C and N–N bonds, respectively. Designing methods to encourage such element/element reductive coupling is a valuable pursuit.
One attractive strategy to promote N–N bond formation is to design complexes which pre-organize two nitrogen oxyanions at a single metal center and explore the deoxygenation chemistry of these systems. There are several reported examples of N–N bond formation at a single metal complex,33–36 and most terminal cis-hyponitrite complexes are reported with group 10 metals.37–41 The reagent bis-pinacolylboryl pyrazine (Scheme 1) has been shown to deoxygenate two nitrates coordinated to iron to yield a dinitrosyl iron complex.31 This reagent is electron rich (8π electron ring) and carries two electrophilic boron atoms making it a potent reductant with attractive kinetic reactivity, including polar B–N bonds.
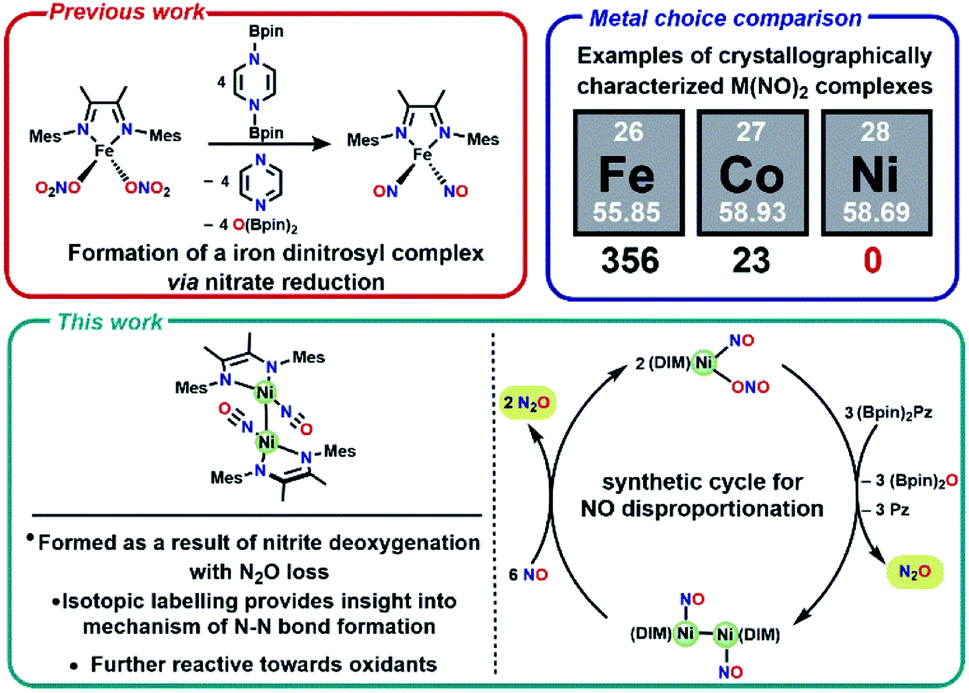 | ||
| Scheme 1 Previously reported nitrate reduction and motivation for utilizing Ni compounds (top), and summary of new reactivity presented (bottom). | ||
We chose a bidentate ancillary ligand with a steric profile designed to allow for multidentate binding of nitrogen oxyanions if needed. These 1,4-diazadienes (DIM, Scheme 1) ligands are established to be redox active and can assist in the stabilization of low valent nickel complexes.42–45 Herein we show stepwise deoxygenation of oxyanions coordinated to nickel, where the location of nickel late in the 3d series disfavors two coordinated nitrosyls in comparison to Fe or Co (Scheme 1), and instead deoxygenation leads to N–N bond formation and nitrous oxide liberation.
Results and discussion
Nickel nitrate and nitrite complexes
A precursor complex, (DIM)NiBr2, is synthesized from (DME)NiBr2 (DME = dimethoxyethane) and DIM in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio in DCM and workup affords a red/brown solid. 1H NMR spectroscopy (Fig. S1†) of (DIM)NiBr2 reveals a paramagnetic species with resonances ranging from −22 to 36 ppm and is fully consistent with a C2v symmetric product. Salt metathesis reactions of (DIM)NiBr2 with AgNO3 or AgNO2 in acetonitrile at room temperature quickly form the bis-nitrate and bis-nitrite complexes respectively with concurrent precipitation of AgBr (Scheme 2). The bis-nitrate and bis-nitrite complexes are also paramagnetic and twofold symmetric as judged by NMR spectroscopy (Fig. S3 and S5†) and their molecular structures (Fig. 1) exhibit bidentate binding of both nitrate and nitrite creating an octahedral coordination environment about the nickel metal centers.
:1 mole ratio in DCM and workup affords a red/brown solid. 1H NMR spectroscopy (Fig. S1†) of (DIM)NiBr2 reveals a paramagnetic species with resonances ranging from −22 to 36 ppm and is fully consistent with a C2v symmetric product. Salt metathesis reactions of (DIM)NiBr2 with AgNO3 or AgNO2 in acetonitrile at room temperature quickly form the bis-nitrate and bis-nitrite complexes respectively with concurrent precipitation of AgBr (Scheme 2). The bis-nitrate and bis-nitrite complexes are also paramagnetic and twofold symmetric as judged by NMR spectroscopy (Fig. S3 and S5†) and their molecular structures (Fig. 1) exhibit bidentate binding of both nitrate and nitrite creating an octahedral coordination environment about the nickel metal centers.
 | ||
| Scheme 2 Synthesis of (DIM)Ni(NOx)2 complexes, as well as the synthesis of (DIM)Ni(NO)(ONO) from (DIM)Ni(NO2)2. The bis-nitrate and bis-nitrite complexes are O-bound and bidentate in the solid-state molecular structures (Fig. 1), we have drawn them as monodentate here for simplicity. | ||
 | ||
| Fig. 1 (a) ORTEP representation of the molecular structure (50% probabilities) of the nonhydrogen atoms of (a) (DIM)Ni(NO2)3 and (b) (DIM)Ni(NO2)2 (right), showing selected atom labelling (unlabelled atoms are carbon). Selected structural parameters for (DIM)Ni(NO2)3 (Å): Ni1–O1, 2.110(4); Ni1–O2, 2.093(4); Ni1–O4, 2.050(4); Ni1–O5, 2.097(4); Ni1–N3, 2.009(5); Ni1–N4, 2.058(4); O1–N1, 1.265(6); O2–N1, 1.273(6); O3–N1, 1.230(6); O4–N2, 1.298(6); O5–N2, 1.271(6); O6–N2, 1.255(6); and (DIM)Ni(NO2)2 (Å): Ni1–O1, 2.094(6); Ni1–O2, 2.091(6); Ni1–O3, 2.099(6); Ni1–O4, 2.072(6); Ni1–N3, 2.035(7); Ni1–N4, 2.038(7); O1–N1, 1.287(9); O2–N1, 1.269(9); N2–O3, 1.257(10); N2–O4, 1.290(11). | ||
Single deoxygenation of (DIM)Ni(NO2)2
Reaction of (DIM)Ni(NO2)2 with equimolar (Bpin)2Pz occurs to completion within 12 h at room temperature in THF with a color change from brown to green (Scheme 2). Analysis by 1H NMR spectroscopy shows complete consumption of the boryl reagent, formation of equimolar pyrazine and (Bpin)2O and a diamagnetic metal complex with the expected 4:6:6:12 intensity 1H NMR resonances of a single DIM-containing product. Workup by pentane wash to remove organic materials yields a green solid, and the molecular structure (Fig. 2) confirms the presence of the singly deoxygenated species (DIM)Ni(NO)(ONO), assigned as {Ni(NO)}10 in the Enemark–Feltham notation. The Ni1–N3–O1 angle is linear (175.5°), and the complex has a strong nitrosyl stretching frequency at 1785 cm−1, similar to other LNi(NO)(X) complexes.46–49 The nitrito linkage in (DIM)Ni(NO)(ONO) contrasts the N-bound nitrite linkage in (Me2-ortho-phenanthroline)Ni(NO)(NO2).49
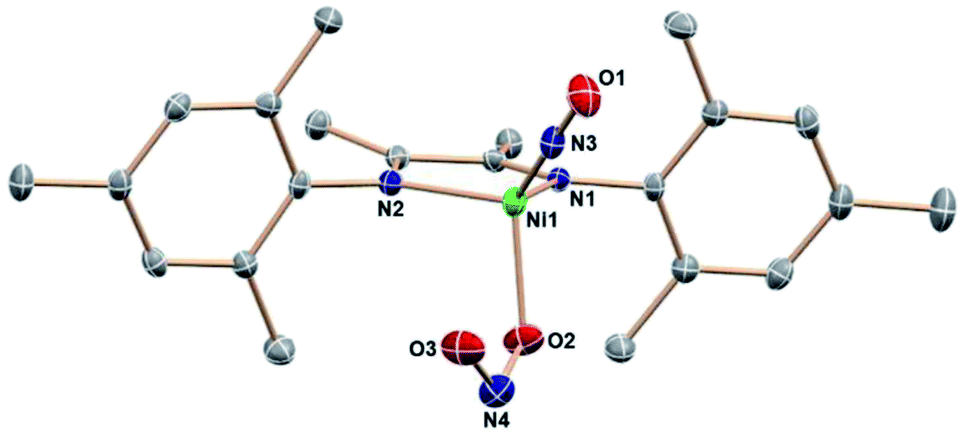 | ||
| Fig. 2 ORTEP representation of the molecular structure (50% probabilities) of the nonhydrogen atoms of (DIM)Ni(NO)(ONO) showing selected atom labelling (unlabelled atoms are carbon). Selected structural parameters: Ni1–N3, 1.637(2); Ni1–O2, 1.9952(19); Ni1–N2, 2.0075(19); Ni1–N1, 2.0204(19); O1–N3, 1.176(3), O2–N4, 1.280(3); O3–N4, 1.228(3); N4–O2–Ni1, 118.25(16); O1–N3–Ni1, 175.5(2). | ||
Deoxygenation of (DIM)Ni(NO)(ONO)
The tetrahedral nickel(0) (DIM)Ni(NO)(ONO) complex reacts with (Bpin)2Pz (1:1.5 mole ratio) at room temperature in THF. Monitoring the reaction by 1H NMR spectroscopy shows the formation of boryl ether and pyrazine, as well as the depletion of signals for (DIM)Ni(NO)(ONO). Crystalline precipitate forms during the course of the reaction. Executing this reaction in CD3CN shows complete consumption of starting materials in less than four hours at room temperature. All DIM-containing product is insoluble based on the lack of any ligand resonances, but the pyrazine and boryl ether byproducts are detected by 1H NMR spectroscopy (Fig. S19†) in the correct stoichiometric ratio in the colorless solution. This confirms that no free ligand is formed upon deoxygenation, nor any degradation products containing the DIM ligand. The reaction executed in CD2Cl2 shows the same result, including no detectable 1H NMR signals of the product nickel complex. This solvent gives slightly larger crystals, which were used for the better of two single crystal structure determinations. The molecular structure (Fig. 3) as determined by SCXRD reveals the product to be [(DIM)Ni(NO)]2, a dimer with a Ni–Ni bond.
 | ||
| Fig. 3 ORTEP representation of the molecular structure (50% probabilities) of the nonhydrogen atoms of centrosymmetric [(DIM)Ni(NO)]2, showing selected atom labelling (unlabelled atoms are carbon). Selected structural parameters: Ni1–N3, 1.960(4) Å; Ni1–N2, 1.893(4); Ni1–N1, 1.647(3); O1–N1, 1.193(4); O1–N1–Ni1 172.5(3)°. | ||
[(DIM)Ni(NO)]2 contains a crystallographic inversion center at the Ni–Ni midpoint and the Ni–Ni bond length (2.6103(10) Å) is short compared to other Ni–Ni bonds with no bridging ligands.50–53 There are no other examples of two Ni–NO units linked solely by a Ni–Ni bond. The Ni lies only 0.16 Å out of the three-nitrogen atom plane, and the Ni′/N1 distance is longer by 1.24 Å, indicating that the NO is not bridging. Analysis of the bond lengths in the DIM backbone (N–C–C–N) (Table S1†) reveals a long-short-long pattern (vs. the comparison compounds in Table S1†) consistent with increased electron density in the di-imine moiety. The C–C bond in particular is quite short (1.436 Å) compared to the other (DIM)Ni complexes, indicative of either a singly or doubly reduced ligand. The monomer (DIM)Ni(NO) is an odd-electron (radical) species which dimerizes to the crystallographically observed product. The ΔG° for dissociation of the dimer into two (DIM)Ni(NO) radical monomers is calculated to be 14.7 kcal mol−1, similar to reported values for dissociation of an iron carbonyl dimer.54 The MALDI-TOF spectrum (Fig. S18†) of [(DIM)Ni(NO)]2 has an m/z at 816.3, consistent with the intact dimer. The trans rotational conformation around the nickel–nickel bond is sterically favored. This conformation has the stabilizing effect of opposing the dipoles in each (DIM)Ni(NO) monomer and benefits from opposed NOδ+ to DIMδ− attraction between monomer units. Because the dimeric product is insoluble in a wide range of solvents of polarity up to acetonitrile and dichloromethane, a 1H NMR spectrum could not be obtained. The solid state IR spectrum (Fig. S15†) shows a strong NO absorption at 1664 cm−1, a low value consistent with greater back donation to the nitrosyl than in (DIM)Ni(NO)(ONO). This can be attributed to the increased donor ability of the reduced DIM ligand as well as better orbital overlap.
The reaction of (DIM)Ni(NO)(ONO) with equimolar (Bpin)2Pz shows complete consumption of borylated reagent along with incomplete consumption of nickel starting material. In order to establish the correct stoichiometry, (DIM)Ni(NO)(ONO) was reacted with excess (3:1 mole ratio) (Bpin)2Pz in the presence of naphthalene as an internal standard. From this experiment, pyrazine is produced in a 1:1.5 mole ratio with respect to the (DIM)Ni(NO)(ONO) starting material, indicating a 2:3 stoichiometry of nickel to borylating reagent. Labelling studies are employed here for characterization, and later for mechanistic insight. The nitrosyl stretch shifts to 1625 cm−1 (Fig. S16†) for isotopically labelled [(DIM)Ni(15NO)]2, which is consistent with the expected reduced mass calculation. The reduced nitrogen containing byproduct upon deoxygenation of (DIM)Ni(NO)(ONO) was confirmed to be N2O via vacuum transfer of the headspace volatiles to a gas IR cell, which clearly show the P and R bands of N2O at 2224 cm−1 (Fig. S30†). All of this allows us to write a balanced reaction for the formation of [(DIM)Ni(NO)]2 with concomitant loss of N2O (Scheme 3).
Deoxygenation of isotopically labelled (DIM)Ni(15NO)(O15NO) results in the formation of (15N)2O, evidenced by a band at 2158 cm−1via gas IR (Fig. S30†). The deoxygenating agent employed in Scheme 3 can be replaced by the reduced 4,4′-bipyridine analogue, (Bpin)2Bpy, which reacts completely within 2 hours. The faster deoxygenation using (Bpin)2Bpy vs. (Bpin)2Pz is a consequence of reduced bipyridine being a more potent reductant than reduced pyrazine.55
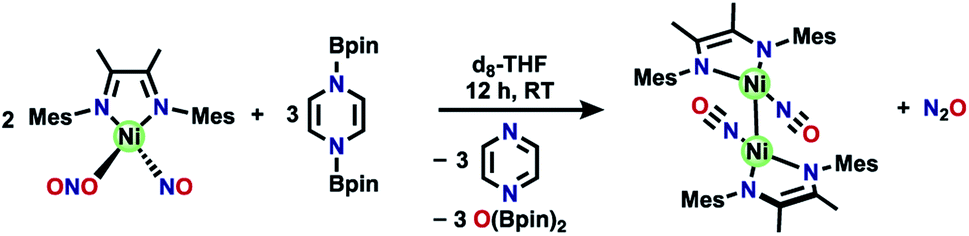 | ||
| Scheme 3 Synthesis of [(DIM)Ni(NO)]2 from (DIM)Ni(NO)(ONO) and (Bpin)2Pz. | ||
XPS of nickel complexes
XPS provides useful resolution of the chemical environment in these four molecules (see Fig. S44†). The nitrogen 1s region resolves nitrate, nitrite, nitrosyl and all the DIM nitrogen atoms, with progressively lower NOx binding energy along that series. The oxygen 1s intensity decreases progressively from nitrate to nitrite to nitrosyl when integrated with respect to the nitrogen. Compound (DIM)Ni(NO)(ONO) shows two oxygen 1s binding energies with relative intensities of 1:2, indicating resolution of two of the three types of oxygens. The oxygen 1s binding energy decreases progressively from (DIM)Ni(NO3)2 to (DIM)Ni(NO2)2 to [(DIM)Ni(NO)]2, consistent with a lower nitrogen oxidation state being less electron withdrawing towards the oxygen. Although linear nitrosyl and nitrite are both formally trivalent nitrogen, the lower binding energy of oxygen by 0.57 eV in [(DIM)Ni(NO)]2 compared to (DIM)Ni(NO2)2 can be attributed to the increased backdonation into the nitrosyl of the dimer, consistent with the observed low νNO stretching frequency. The nitrogen 1s spectrum of [(DIM)Ni(NO)]2 resolves the nitrosyl from imine in the intensity ratio of 1:2. The Ni 2p spectrum shows a lower metal binding energy for [(DIM)Ni(NO)]2 compared to the bis-nitrate and bis-nitrite complexes, consistent with it having a lower metal oxidation state.
Redox reactivity of [(DIM)Ni(NO)]2
Despite the insolubility of [(DIM)Ni(NO)]2, it reacts readily (Scheme 4) with oxidants to cleave the Ni–Ni bond. The reaction between a slurry of [(DIM)Ni(NO)]2 with AgNO2 or AgNO3 results in homogeneity of the solution and a color change to green in both cases. Isolation of the green solid from the reaction with AgNO2 reveals the product to be the previously characterized (DIM)Ni(NO)(ONO) by 1H and 13C NMR spectroscopy along with the diagnostic NO stretch at 1785 cm−1. The corresponding reaction of the dimer and AgNO3 results in the formation of the {Ni(NO)}10 complex (DIM)Ni(NO)(ONO2), indicating that the one electron oxidation product of [(DIM)Ni(NO)]2 is susceptible to nucleophilic attack even by weak nucleophiles such as nitrate. It is important here to control the Ag:Ni stoichiometry because both (DIM)Ni(NO)(ONO) and (DIM)Ni(NO)(ONO2) are subject to further oxidation by silver salts, forming the bis-nitrite or bis-nitrate complexes respectively. The oxidations of the mixed oxyanion complexes result in liberation of an equivalent of nitric oxide, which was detected via gas phase IR (Fig. S34†). There is precedence for nitric oxide liberation upon one electron oxidation of [Ni(NO)(bipy)][PF6] with either AgPF6 or [NO][PF6] as the oxidant.48
 | ||
| Scheme 4 Summary of oxidative reactivity with [(DIM)Ni(NO)]2. | ||
Even an oxidant with a more weakly nucleophilic triflate anion succeeds in this oxidative process. The reaction of [(DIM)Ni(NO)]2 with AgOTf in THF results in a color change to green and homogeneity of the solution. The 19F NMR spectrum (Fig. S21†) is indicative of one diamagnetic product, and the 1H NMR (Fig. S21†) spectrum indicates twofold symmetry. The solid state nitrosyl stretch appears at 1780 cm−1 (Fig. S22†) which is shifted to lower energy by 100 cm−1 compared to authentic 3-coordinate (bpy)Ni(NO)+ (bpy = bipyridine).49 The shift in νNO can be attributed to coordination of THF solvent to give the 4-coordinate species [(DIM)Ni(NO)(THF)]+. When the reaction of [(DIM)Ni(NO)]2 and AgOTf is repeated in the non-coordinating solvent DCM, the νNO is higher by 31 cm−1 in the solid state (Fig. S23†) and 45 cm−1 in a DCM solution (Fig. S24†). If the same material is redissolved in THF, the IR spectrum shows two nitrosyl stretches suggesting an equilibrium of THF binding to the nickel center. Crystals were grown via vapor diffusion of pentane into a concentrated THF solution and the solid state molecular structure (Fig. 4) shows the {Ni(NO)}10 complex (DIM)Ni(NO)(OTf), with triflate coordinated to nickel. However, based on spectroscopic data, the OTf is in equilibrium when dissolved in coordinating solvents.
 | ||
| Fig. 4 ORTEP representation of the molecular structure (50% probabilities) of the nonhydrogen atoms of (DIM)Ni(NO)(OTf), showing selected atom labeling (unlabelled atoms are carbon). Selected structural parameters (Å, °): Ni1–O2, 2.059(2); Ni1–N3, 1.994(2); Ni1–N2, 2.010(2); Ni1–N1, 1.630(2); O1–N1–Ni1, 173.1(3); N1–Ni1–O2, 124.22(11). | ||
With this weak nucleophile, the tetrahedron is distorted to flatten the (DIM)NiN(O) plane: the angle from the DIM N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N midpoint to the Ni center to N1 is 135.6°. Regardless of the abundance of THF as crystallization solvent, this neutral molecule is apparently the least soluble equilibrium participant.
C–N midpoint to the Ni center to N1 is 135.6°. Regardless of the abundance of THF as crystallization solvent, this neutral molecule is apparently the least soluble equilibrium participant.
Nitric oxide disproportionation
With the knowledge that [(DIM)Ni(NO)]2 reacts rapidly with silver oxidants, we were interested in its reactivity with NO gas. A suspension of [(DIM)Ni(NO)]2 in THF was exposed to excess NO and there was an immediate color change to a homogeneous brown/green solution. Workup of the solution indicates that the nickel-containing product of this reaction is (DIM)Ni(NO)(ONO), which is established by a variety of spectroscopic techniques along with an independent unit cell determination of the molecular structure via SCXRD. Vacuum transfer of the volatiles generated during the reaction into a gas IR cell show the formation of N2O (Fig. S31†). This reaction therefore accomplishes disproportionation of nitric oxide to N2O and NO2, but the latter undergoes one electron reduction by nickel to yield the coordinated nitrito product (Scheme 4).As evidenced by Scheme 5, two low valent nickel centers of [(DIM)Ni(NO)]2 mediate the disproportionation of 6 moles of NO resulting in the formation of two Ni-(ONO) complexes and two moles of N2O, a reaction which has precedence with first row transition metals.49,56–63 The reaction of [(DIM)Ni(NO)]2 with NO gas can be accomplished in the absence of solvent. Addition of excess nitric oxide to a solid sample of [(DIM)Ni(NO)]2 over the duration of 15 min, followed by extraction into THF indicates the formation of (DIM)Ni(NO)(ONO) via IR and 1H NMR spectroscopy. Attempts to detect an intermediate in this disproportionation reaction by solid state IR were investigated by exposing a solid sample of [(DIM)Ni(NO)]2 to 1–4 equivalents of nitric oxide. This resulted in up to 15% growth of (DIM)Ni(NO)(ONO) but no new intermediate terminal nitrosyl and no growth of other bands between 1000 and 1600 cm−1 (Fig. S37†). The fact that this reaction occurs cleanly in the solid state, where diffusive separation of intermediate nickel monomers is physically inhibited, makes it plausible that an intermediate is two identical (DIM)Ni(NO)2.
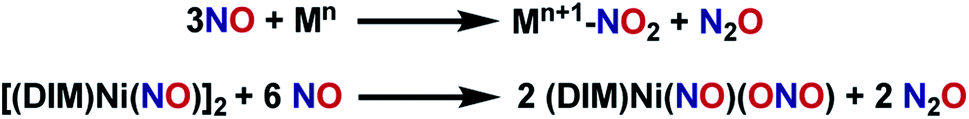 | ||
| Scheme 5 Metal-mediated nitric oxide disproportionation. | ||
Nitrogen isotope label studies
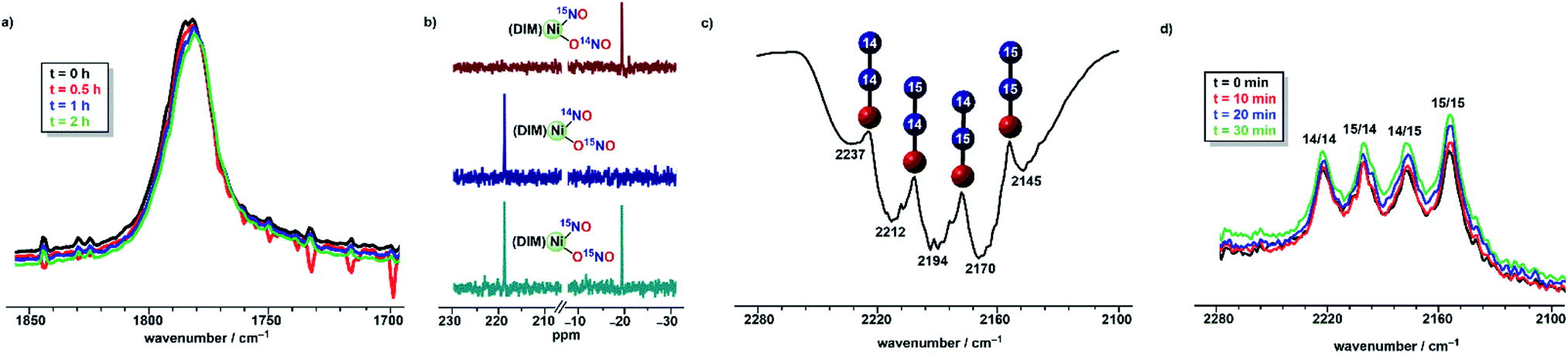
:1 mole ratio, where any detected 14N15NO or 15N14NO would indicate that the N–N bond formation occurs within a bimolecular Ni2 species. We isotopically labelled (DIM)Ni(15NO2)2 and found that it exhibits a broad 15N NMR chemical shift at 573.8 ppm that shows a T−1 dependence of chemical shift which is characteristic of paramagnetic complexes (Fig. S7†). Furthermore, the nitrite ligand stretches shift from 1295 cm−1 and 1210 cm−1 to 1276 cm−1 and 1180 cm−1 in the 15N isotopomer (Fig. S9†). Doubly labelled (DIM)Ni(15NO)(O15NO) can be synthesized by reductive borylation of the labelled (DIM)Ni(15NO2)2 starting material. Isotopically labelled (DIM)Ni(15NO)(O15NO) shows a diagnostic shift for both NO and nitrite IR signals (Fig. S13†). Curiously, 15N NMR of (DIM)Ni(15NO)(O15NO) in both THF and MeCN at room temperature shows only one signal at 3.6 ppm (Fig. S11†) despite chemically inequivalent nitrogen atoms. However, room temperature 15N NMR in DCM (Fig. S11†) shows the same signal at ∼3.6 ppm along with a broad signal at 206 ppm. Cooling this solution to −42 °C results in two sharp signals at −19.1 and 216.3 ppm in a 1:1 intensity ratio (Fig. 5). To confirm the assignment of the two signals, the isotopomers (DIM)Ni(14NO)(O15NO) and (DIM)Ni(15NO)(O14NO) were synthesized via the reaction of the appropriately labelled [(DIM)Ni(NO)]2 dimer with either AgNO2 or Ag15NO2 (see ESI† for isotopic details). At −42 °C in DCM, 15N NMR of (DIM)Ni(14NO)(O15NO) shows one sharp signal at 216.3 ppm, and (DIM)Ni(15NO)(O14NO) shows one sharp signal at −19.1 ppm (Fig. 5). The specific isotopomers confirm that the original unseen 15N NMR signal of (DIM)Ni(15NO)(O15NO) in THF and MeCN corresponds to the nitrito ligand, consistent with a dynamic process that exchanges the nitrito ligand between nickel complexes. This dynamic process is confirmed via mass spectrometry with a 1:1 THF solution of (DIM)Ni(15NO)(O15NO) and (DIM)Ni(14NO)(O14NO). The ESI+ spectrum is consistent with isotopic scrambling which shows a statistical mixture of the original doubly labelled and unlabelled compounds, together with equal amounts of (DIM)Ni(14NO)(O15NO) and (DIM)Ni(15NO)(O14NO) (Fig. S38†). The presence of the mixed labelled isotopomers is consistent with a dynamic process of intermolecular nitrite exchange between metal centers, and this dynamic process is supported by the line broadening 15N NMR observations above.
 | ||
| Fig. 5 (a) Solution IR of the nitrosyl stretch in (DIM)Ni(14NO)(O15NO) monitored over time (b) 15N NMR of (DIM)Ni(15NO)(O15NO) (bottom), (DIM)Ni(14NO)(O15NO) (middle), and (DIM)Ni(15NO)(O14NO) (top) all in CD2Cl2 at −42 °C (c) gas phase IR spectrum of volatiles from the reaction of (DIM)Ni(14NO)(O15NO) and 3 equivalents of (Bpin)2Pz and (d) THF solution IR spectrum of the reaction of (DIM)Ni(14NO)(O15NO) and 3 equivalents of (Bpin)2Bpy. | ||
:1 mixture of (DIM)Ni(15NO)(O15NO) and (DIM)Ni(14NO)(O14NO) precludes any mechanistic insight obtained from a mixture of these two complexes. Instead, we focused on the individual isotopomers (DIM)Ni(14NO)(O15NO) and (DIM)Ni(15NO)(O14NO) to obtain mechanistic information about the formation of N2O. Both (DIM)Ni(14NO)(O15NO) and (DIM)Ni(15NO)(O14NO) are isotopically pure as observed by the nitrosyl stretching frequencies and 15N NMR (Fig. 5), and monitoring a solution of either isotopomer in THF shows no isotopic scrambling as judged by 15N NMR and IR spectroscopy (Fig. 5) among the two NOx sites during the time scale relevant to the reactivity with (Bpin)2Pz. Focusing on one isotopomer, the reaction between (DIM)Ni(14NO)(O15NO) and (Bpin)2Pz in a 2:3 mole ratio provides insight into the mechanism based on the nature of the N2O produced. Vacuum transfer of the product volatiles to a gas IR cell reveals the presence of all four isotopomers, (15N)2O, (14N)2O, 14N15NO and 15N14NO (Fig. 5), indicating the N–N bond forming event occurs in a dinickel species. Moreover, the detection of both 14N15NO and 15N14NO demands an intermediate where both nitrogen atoms are chemically equivalent, which makes (DIM)Ni(NO)2 a plausible precursor.
Isotopomer (DIM)Ni(14NO)(O15NO) was then reacted with (Bpin)2Bpy which begins to react in the time of mixing. The solution infrared spectrum (Fig. 5) recorded within five minutes of combining the reagents showed all four isotopomers of nitrous oxide. This excludes any mechanism where nitrous oxide comes exclusively from (DIM)Ni(NO)(ONO) nitrite or exclusively from its nitrosyl nitrogen. The presence of 15N in both Nα and Nβ of nitrous oxide requires a transition state or intermediate where the two nitrogen atoms are symmetry-equivalent and thus equally likely to become Nα or Nβ in a subsequent N–N bond forming step. In addition, seeing both unlabelled and doubly 15N labelled N2O in this experiment indicates that the N–N bond forming event occurs after formation of a dinickel intermediate because N–N bond formation at a single nickel center would give only singly labelled N2O. Confirming the above, the solid state IR spectrum of [(DIM)Ni(NO)]2 formed from (DIM)Ni(14NO)(O15NO) and 3 equivalents of (Bpin)2Pz shows a diagnostic nitrosyl stretches for all three expected isotopomers, [(DIM)Ni(14NO)]2, [(DIM)Ni(15NO)]2, and (DIM)2Ni2(14NO)(15NO) (Fig. S17†), consistent with computationally predicted nitrosyl stretches for each isotopomer.
 | ||
| Fig. 6 (a) Solution IR of (DIM)Ni(NO)(ONO) and (Bpin)2Bpy in a 1:1 mole ratio showing complete conversion to the reduced [(DIM)Ni(NO)(ONO)]− species by the first spectrum collected (red trace), as well as the increase of the new nitrosyl stretch (solid black trace) after 30 minutes. The dotted black trace corresponds to the nitrosyl stretch of unreduced (DIM)Ni(NO)(ONO). (b) X-band EPR spectrum of [(DIM)Ni(NO)(ONO)]− produced from OSET using (Bpin)2Bpy. | ||
The EPR spectrum obtained after a one electron reduction of (DIM)Ni(15NO)(O15NO) is identical to the EPR spectrum of doubly 14N complex, indicating that the nitrosyl and nitrito nitrogen atoms are not involved in the coupling observed. The DIM ligand-based reduction is corroborated by density functional theory (DFT) electronic structure analysis of [(DIM)Ni(NO)(ONO)]− where the spin density and corresponding orbital diagram are consistent with a DIM ligand-based SOMO (Fig. S48†). Within 30 minutes of the reaction, there is a clear appearance of a second intermediate with νNO at 1795 cm−1 (Fig. 6). This intermediate is a result of downstream borylation, as this nitrosyl stretch at 1795 cm−1 does not appear following electron transfer from Cp*2Co.
:4) at 80 °C for two hours results in a color change from brown to blue. Monitoring by 1H NMR spectroscopy (Fig. S28†) reveals complete consumption of (Bpin)2Pz and the paramagnetic (DIM)Ni(NO3)2 complex, as well as the growth of O(BPin)2 and pyrazine. Upon cooling the NMR sample to room temperature, X-ray quality crystals are present in the J-Young tube. Single crystal XRD of these crystals show that the molecular structure is polymeric (Fig. 7), with a repeat unit Ni2(NO)2(OBpin)2(Pz). Bond lengths within this structure are consistent with unreduced (neutral) pyrazine and the linear Ni–N–O is best described as tetrahedral Ni(0). Each OBpin ligand derived from deoxygenation of nitrate bridges two nickel centers creating a Ni2(OBpin)2 diamond core. We postulate that a zero valent nickel nitrosyl complex is a thermodynamic sink and having excess pyrazine in solution facilitates the crystal growth of this polymer. The OBpin ligand shows that borylation using (Bpin)2Pz can be stepwise and it is not limited to concerted double boryl transfer to one oxygen. The stepwise borylation is supported by the ability of the borylated N-heterocycles to engage in outer sphere electron transfer. There is precedence for OBpin as a ligand including OBpin derived from deoxygenation of CO2.65–70 The solid state IR spectrum (Fig. S29†) of Ni2(NO)2(OBpin)2(Pz) shows a strong NO stretch at 1810 cm−1. We propose that loss of DIM occurs because its bite angle (∼81°) is not ideal for the tetrahedral needs of nickel in competition with pyrazine available as another suitable donor. Free DIM is observed by 1H NMR spectroscopy (Fig. S28†). Reacting (DIM)Ni(NO3)2 with 4 moles of (Bpin)2Bpy as the deoxygenating agent at 80 °C proceeds analogously, forming Ni2(NO)2(OBpin)2(Bpy) (Fig. 7). The τ4 values for the nickel in the pyrazine and bipyridine linked polymers vary slightly: 0.73 and 0.67, respectively. The smaller τ4 value for the bipyridine derivative is accompanied by a smaller nitrosyl angle, with Ni1–N1–O1 being 156.84°, compared to 176.06° for the pyrazine-bridged analogue.
 | ||
| Fig. 7 ORTEP representation of the molecular structures (50% probabilities) of the nonhydrogen atoms of (a) Ni2(NO)2(OBpin)2(Pz) and (b) Ni2(NO)2(OBpin)2(Bpy), showing selected atom labelling (unlabelled atoms are carbon). Selected structural parameters for Ni2(NO)2(OBpin)2(Pz) (Å): Ni1–N1, 1.634(3); Ni1–N2, 2.040(2); Ni1–O2, 1.988(2); N1–O1, 1.173(3); O1–N1–Ni1, 176.1(2); and Ni2(NO)2(OBpin)2(Bpy) (Å): Ni1–N1, 1.9914(17); Ni1–N2, 1.6386(19); Ni1–O1, 1.9960(16); N2–O2, 1.181(3); O2–N2–Ni1, 156.83(18). | ||
Much like in the synthesis of [(DIM)Ni(NO)]2, these polymers are deficient in nitrogen with respect to the bis-nitrate starting material. After reacting (DIM)Ni(NO3)2 with 4 moles of (Bpin)2Pz in THF at 80 °C for 2 hours, vacuum transfer of the headspace gas into a gas IR cell shows the characteristic doublet for N2O at 2224 cm−1 (Fig. S41†), indicating N–N bond formation. With N2O confirmed, as well as (Bpin)2O, pyrazine, and free DIM, a balanced reaction for this transformation is presented in Scheme 6. The formation of an N–N bond from the reaction of (DIM)Ni(NO)(ONO) with (Bpin)2Pz mirrors the N–N bond formation observed in Scheme 3; however, DIM now is replaced by bridging OBpin and bridging pyrazine or bipyridine ligands. From this result, we hypothesize that (DIM)Ni(NO)(ONO) is formed in the synthesis of polymeric Ni2(NO)2(OBpin)2(Pz), and DIM is lost as a result of heating after the nitrite/nitrosyl complex is formed. This is supported by the fact that (DIM)Ni(NO)(ONO) is thermally unstable, and heating in THF for 16 hours results in a color change from brown/green to blue. Monitoring this decomposition by 1H NMR shows the formation of many new DIM containing species, including free ligand, as judged by numerous new singlets in the aromatic region corresponding to the mesityl meta-H (Fig. S43†). Furthermore, these same polymeric structures can be obtained from the reactivity of (DIM)Ni(NO2)2 with either (Bpin)2Pz or (Bpin)2Bpy in a 1:2 mole ratio at 80 °C, once again showing elevated temperatures induce DIM loss and polymer formation.
 | ||
| Scheme 6 Synthesis of pyrazine linked polymer facilitated by N2O loss. | ||
Conclusions
We have shown that reductive borylation on both (DIM)Ni(NO2)2 and (DIM)Ni(NO3)2 at elevated temperatures leads to loss of DIM and nitrous oxide formation, followed by the formation of either pyrazine or 4,4′-bipyridine bridged polymers. With milder conditions, single deoxygenation of (DIM)Ni(NO2)2 is facile to yield the mixed oxyanion compound (DIM)Ni(NO)(ONO). The (DIM)Ni(NO)(ONO) complex undergoes further deoxygenation to form the dimeric [(DIM)Ni(NO)]2, while also liberating nitrous oxide. Isotopic labelling studies indicate that the N–N bond formation event happens bimolecularly, with a C2 symmetric intermediate. The dimer is susceptible to oxidation with AgX (X = NO3−, NO2−, or OTf−) to form (DIM)Ni(NO)(X) compounds, and is competent to facilitate nitric oxide disproportionation. While mechanistically complex, we have experimental evidence that the first step in the reaction to form [(DIM)Ni(NO)]2 is outer sphere electron transfer from the electron rich (Bpin)2Bpy reductant.Data availability
The data supporting the findings of this study are available within the paper and its ESI files. Crystallographic data for the crystal structures has been deposited at the CCDC under 2083107 for (DIM)Ni(NO3)2, 2083108 for (DIM)Ni(NO2)2, 2083111 for (DIM)Ni(NO)(ONO), 2083110 for [(DIM)Ni(NO)]2, 2083109 for (DIM)Ni(NO)(OTf), 2083112 for (OBpin)2Ni2(NO)2(Pz), and 2083113 for (OBpin)2Ni2(NO)2(Bpy), and can be obtained from https://ccdc.cam.ac.uk/structures/.Author contributions
D. M. B. designed and performed the experiments and computations, interpreted the results and assisted with the manuscript. A. C. C. helped synthesize and characterize the nitrate/nitrite starting materials and the polymers, and assisted with the manuscript. M. P. and V. C. collected and refined the X-ray data. X. G. assisted with the nitrogen NMR experiments. K. G. C. helped design experiments, interpret results, and assisted with the manuscript writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the IU Nanoscale Characterization Facility for access to instrumentation. This work was supported by the Indiana University Office of Vice President for Research and the National Science Foundation, Chemical Synthesis Program (SYN), by grant CHE-1955887.Notes and references
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2020, 6, eaay3111 CrossRef CAS PubMed.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- J. Resasco and A. T. Bell, Trends Chem., 2020, 2, 825–836 CrossRef CAS.
- Q. Zhu, X. Sun, D. Yang, J. Ma, X. Kang, L. Zheng, J. Zhang, Z. Wu and B. Han, Nat. Commun., 2019, 10, 3851 CrossRef PubMed.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- J. N. Galloway, J. D. Aber, J. W. Erisman, S. P. Seitzinger, R. W. Howarth, E. B. Cowling and B. J. Cosby, BioScience, 2003, 53, 341–356 CrossRef.
- D.-P. Häder, A. T. Banaszak, V. E. Villafañe, M. A. Narvarte, R. A. González and E. W. Helbling, Sci. Total Environ., 2020, 713, 136586 CrossRef PubMed.
- R. J. Diaz and R. Rosenberg, Science, 2008, 321, 926 CrossRef CAS PubMed.
- W. K. Dodds, W. W. Bouska, J. L. Eitzmann, T. J. Pilger, K. L. Pitts, A. J. Riley, J. T. Schloesser and D. J. Thornbrugh, Environ. Sci. Technol., 2009, 43, 12–19 CrossRef CAS PubMed.
- M. Duca and M. T. M. Koper, Energy Environ. Sci., 2012, 5, 9726–9742 RSC.
- P. Tavares, A. S. Pereira, J. J. G. Moura and I. Moura, J. Inorg. Biochem., 2006, 100, 2087–2100 CrossRef CAS PubMed.
- J. P. Collman, Y. Yang, A. Dey, R. A. Decréau, S. Ghosh, T. Ohta and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15660–15665 CrossRef CAS PubMed.
- T. Hino, Y. Matsumoto, S. Nagano, H. Sugimoto, Y. Fukumori, T. Murata, S. Iwata and Y. Shiro, Science, 2010, 330, 1666–1670 CrossRef CAS PubMed.
- C. J. Reed, Q. N. Lam, E. N. Mirts and Y. Lu, Chem. Soc. Rev., 2021, 50, 2486–2539 RSC.
- Y. Li, M. Hodak and J. Bernholc, Biochemistry, 2015, 54, 1233–1242 CrossRef CAS PubMed.
- R. Hille, J. Hall and P. Basu, Chem. Rev., 2014, 114, 3963–4038 CrossRef CAS PubMed.
- M. P. Schopfer, J. Wang and K. D. Karlin, Inorg. Chem., 2010, 49, 6267–6282 CrossRef CAS PubMed.
- S. Hematian, M. A. Siegler and K. D. Karlin, J. Biol. Inorg Chem., 2014, 19, 515–528 CrossRef CAS PubMed.
- M. K. Assefa, G. Wu and T. W. Hayton, Chem. Sci., 2017, 8, 7873–7878 RSC.
- P. L. Damon, G. Wu, N. Kaltsoyannis and T. W. Hayton, J. Am. Chem. Soc., 2016, 138, 12743–12746 CrossRef CAS PubMed.
- M. Delgado and J. D. Gilbertson, Chem. Commun., 2017, 53, 11249–11252 RSC.
- C. L. Ford, Y. J. Park, E. M. Matson, Z. Gordon and A. R. Fout, Science, 2016, 354, 741 CrossRef CAS PubMed.
- J. Gwak, S. Ahn, M.-H. Baik and Y. Lee, Chem. Sci., 2019, 10, 4767–4774 RSC.
- C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 12023–12031 CrossRef CAS PubMed.
- A. C. Cabelof, V. Carta and K. G. Caulton, Chem. Commun., 2021, 57, 2780–2783 RSC.
- D. M. Beagan, V. Carta and K. G. Caulton, Dalton Trans., 2020, 49, 1681–1687 RSC.
- C. M. Moore and N. K. Szymczak, Chem. Sci., 2015, 6, 3373–3377 RSC.
- S. Kundu, P. N. Phu, P. Ghosh, S. A. Kozimor, J. A. Bertke, S. C. E. Stieber and T. H. Warren, J. Am. Chem. Soc., 2019, 141, 1415–1419 CrossRef CAS PubMed.
- C. Van Stappen and N. Lehnert, Inorg. Chem., 2018, 57, 4252–4269 CrossRef CAS PubMed.
- E. G. Abucayon, D. R. Powell and G. B. Richter-Addo, J. Am. Chem. Soc., 2017, 139, 9495–9498 CrossRef CAS PubMed.
- C. Ferousi, S. H. Majer, I. M. DiMucci and K. M. Lancaster, Chem. Rev., 2020, 120, 5252–5307 CrossRef CAS PubMed.
- N. Arulsamy, D. S. Bohle, J. A. Imonigie and S. Levine, Angew. Chem., Int. Ed., 2002, 41, 2371–2373 CrossRef CAS PubMed.
- N. Arulsamy, D. S. Bohle, J. A. Imonigie and R. C. Moore, Polyhedron, 2007, 26, 4737–4745 CrossRef CAS.
- S. Bhaduri, B. F. G. Johnson, A. Pickard, P. R. Raithby, G. M. Sheldrick and C. I. Zuccaro, J. Chem. Soc., Chem. Commun., 1977, 10, 354–355 RSC.
- A. M. Wright, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 9930–9933 CrossRef CAS PubMed.
- T. Mayer, P. Mayer and H.-C. Böttcher, J. Organomet. Chem., 2012, 700, 41–47 CrossRef CAS.
- I. R. Corn, P. D. Astudillo-Sánchez, M. J. Zdilla, P. E. Fanwick, M. J. Shaw, J. T. Miller, D. H. Evans and M. M. Abu-Omar, Inorg. Chem., 2013, 52, 5457–5463 CrossRef CAS PubMed.
- S. J. Kraft, U. J. Williams, S. R. Daly, E. J. Schelter, S. A. Kozimor, K. S. Boland, J. M. Kikkawa, W. P. Forrest, C. N. Christensen, D. E. Schwarz, P. E. Fanwick, D. L. Clark, S. D. Conradson and S. C. Bart, Inorg. Chem., 2011, 50, 9838–9848 CrossRef CAS PubMed.
- H. Nishiyama, H. Hosoya, B. F. Parker, J. Arnold, H. Tsurugi and K. Mashima, Chem. Commun., 2019, 55, 7247–7250 RSC.
- C. Zarate, H. Yang, M. J. Bezdek, D. Hesk and P. J. Chirik, J. Am. Chem. Soc., 2019, 141, 5034–5044 CrossRef CAS PubMed.
- J. Kriege-Simondsen, G. Elbaze, M. Dartiguenave, R. D. Feltham and Y. Dartiguenave, Inorg. Chem., 1982, 21, 230–236 CrossRef CAS.
- S. Soma, C. Van Stappen, M. Kiss, R. K. Szilagyi, N. Lehnert and K. Fujisawa, J. Biol. Inorg Chem., 2016, 21, 757–775 CrossRef CAS PubMed.
- A. M. Wright, H. T. Zaman, G. Wu and T. W. Hayton, Inorg. Chem., 2013, 52, 3207–3216 CrossRef CAS PubMed.
- A. M. Wright, H. T. Zaman, G. Wu and T. W. Hayton, Inorg. Chem., 2014, 53, 3108–3116 CrossRef CAS PubMed.
- U. Denninger, J. J. Schneider, G. Wilke, R. Goddard and C. Krüger, Inorg. Chim. Acta, 1993, 213, 129–140 CrossRef CAS.
- D. Herebian, E. Bothe, F. Neese, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 2003, 125, 9116–9128 CrossRef CAS PubMed.
- S.-M. Peng and V. L. Goedken, J. Am. Chem. Soc., 1976, 98, 8500–8510 CrossRef CAS.
- L. Rigamonti, A. Forni, S. Righetto and A. Pasini, Dalton Trans., 2019, 48, 11217–11234 RSC.
- A. R. Cutler and M. Rosenblum, J. Organomet. Chem., 1976, 120, 87–96 CrossRef CAS.
- T. Ohmura, Y. Morimasa and M. Suginome, J. Am. Chem. Soc., 2015, 137, 2852–2855 CrossRef CAS PubMed.
- S. Ghosh, H. Deka, Y. B. Dangat, S. Saha, K. Gogoi, K. Vanka and B. Mondal, Dalton Trans., 2016, 45, 10200–10208 RSC.
- A. M. Wright, C. Sun and M. Dincă, J. Am. Chem. Soc., 2021, 143, 681–686 CrossRef CAS PubMed.
- C. K. Brozek, J. T. Miller, S. A. Stoian and M. Dincă, J. Am. Chem. Soc., 2015, 137, 7495–7501 CrossRef CAS PubMed.
- K. J. Franz and S. J. Lippard, J. Am. Chem. Soc., 1998, 120, 9034–9040 CrossRef CAS.
- K. J. Franz and S. J. Lippard, J. Am. Chem. Soc., 1999, 121, 10504–10512 CrossRef CAS.
- S. Metz, Inorg. Chem., 2017, 56, 3820–3833 CrossRef CAS PubMed.
- C. E. Ruggiero, S. M. Carrier and W. B. Tolman, Angew. Chem., Int. Ed., 1994, 33, 895–897 CrossRef.
- G. B. Wijeratne, S. Hematian, M. A. Siegler and K. D. Karlin, J. Am. Chem. Soc., 2017, 139, 13276–13279 CrossRef CAS PubMed.
- G. B. Wijeratne, M. Bhadra, M. A. Siegler and K. D. Karlin, J. Am. Chem. Soc., 2019, 141, 17962–17967 CrossRef CAS PubMed.
- M. Ahijado Salomon, T. Braun and A. Penner, Angew. Chem., Int. Ed., 2008, 47, 8867–8871 CrossRef PubMed.
- A. S. Batsanov, J. A. Cabeza, M. G. Crestani, M. R. Fructos, P. García-Álvarez, M. Gille, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2016, 55, 4707–4710 CrossRef CAS PubMed.
- A. C. Fernandes, J. A. Fernandes, F. A. Almeida Paz and C. C. Romão, Dalton Trans., 2008, 6686–6688 RSC.
- S. Hawkeswood and D. W. Stephan, Dalton Trans., 2005, 2182–2187 RSC.
- S. I. Kalläne, T. Braun, M. Teltewskoi, B. Braun, R. Herrmann and R. Laubenstein, Chem. Commun., 2015, 51, 14613–14616 RSC.
- D. S. Laitar, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196–17197 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2083107 (DIM)Ni(NO3)2, 2083108 (DIM)Ni(NO2)2, 2083111 (DIM)Ni(NO)(ONO), 2083110 [(DIM)Ni(NO)]2, 2083109 (DIM)Ni(NO)(OTf), 2083112 (OBpin)2Ni2(NO)2(Pz) and 2083113 (OBpin)2Ni2(NO)2(Bpy). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02846d |
| This journal is © The Royal Society of Chemistry 2021 |