
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidation of ustilaginoidin biosynthesis reveals a previously unrecognised class of ene-reductases†
Dan
Xu
a,
Ruya
Yin
a,
Zhiyao
Zhou
a,
Gan
Gu
a,
Siji
Zhao
a,
Jin-Rong
Xu
b,
Junfeng
Liu
a,
You-Liang
Peng
a,
Daowan
Lai
 *a and
Ligang
Zhou
*a
*a and
Ligang
Zhou
*a
aDepartment of Plant Pathology and MOA Key Lab of Pest Monitoring and Green Management, College of Plant Protection, China Agricultural University, Beijing 100193, China. E-mail: dwlai@cau.edu.cn; lgzhou@cau.edu.cn
bDepartment of Plant Pathology, College of Plant Protection, Northwest A&F University, Yangling 712100, China
First published on 3rd November 2021
Abstract
Ustilaginoidins are a type of mycotoxin featuring a dimeric naphtho-γ-pyrone skeleton, produced by the rice false smut pathogen Ustilaginoidea virens. Here we used gene disruption, heterologous expression in Aspergillus oryzae, feeding experiments, and in vitro experiments to fully elucidate the biosynthesis of ustilaginoidins. A new route to dimeric 2,3-unsaturated naphtho-γ-pyrones via dimerization of YWA1 (and 3-methyl YWA1) followed by dehydration was discovered. Intriguingly, the reduction of the 2,3-double bond of the pyrenone ring was catalyzed by a phospholipid methyltransferase-like enzyme (UsgR). The reductase was specific for reduction of monomeric, linear naphtho-γ-pyrenones, but not for the dimers. Atroposelective coupling of various monomers by the laccase (UsgL) led to diverse ustilaginoidins. Moreover, 3-epimerism of the 3-methyl-2,3-dihydro-naphtho-γ-pyrones adds additional complexity to the biosynthesis.
Introduction
Ustilaginoidins (usg) are polyketides produced by the rice false smut (RFS) pathogen Ustilaginoidea virens.1 Structurally, they are 9,9′-linked, dimeric naphtho-γ-pyrones of aR configuration. So far 27 members have been reported from U. virens.1–4 They showed teratogenicity towards mouse embryo limb bud and midbrain cells,5 cytotoxicity against cancer cells,3,4,6,7 and phytotoxicity against the elongation of the radicle or plumule of rice seeds,3,4 as well as antibacterial activity.4,8Due to the atropisomeric structure and interesting bioactivity, it was attractive to study the biosynthesis of ustilaginoidins. The putative biosynthetic gene cluster (BGC) of ustilaginoidins was predicted by genome analysis of U. virens,9 and later linked to the biosynthesis of ustilaginoidins by three independent studies almost simultaneously.10–12 It was confirmed that the laccase was responsible for the coupling of two monomeric naphtho-γ-pyrones.10–12 However, our understanding of the biosynthesis of ustilaginoidins is incomplete, especially the biosynthesis of the monomers, for which different biosynthetic pathways were proposed.11,12
Herein, we report the complete biosynthesis of ustilaginoidins using gene disruption, heterologous expression in Aspergillus oryzae, feeding experiments, and in vitro experiments. Intriguingly, the stereoselective reduction of the 2,3-double bond of 4H-benzo[g]chromen-4-one was catalyzed by a distinct class of ene-reductases not found previously. The biosynthesis of ustilaginoidins was reconstructed in A. oryzae and in vitro by using various monomers with the laccase.
Results
Deletion of various genes in the usg BGC
The usg BGC comprises genes encoding a non-reducing polyketide synthase (nrPKS, uvpks1), a putative dehydrase (usgD), a C-methyltransferase (C-MeT, usgM), a laccase (usgL), a putative flavin-dependent oxidoreductase (usgO), and several proteins of unknown function (Fig. 1A). Interestingly, UsgM has a ψ-ACP-MT didomain, as multiple sequence alignment revealed that the phosphopantetheine modification site in this ACP domain is DPL, which differs from the classic DSL sequence (Fig. 1C and ESI Fig. S1†).13 Such a didomain was also found in the trans-acting C-MeT (TlnC) that was involved in the biosynthesis of tricholignan A,13 in which ψ-ACP was crucial for methylation.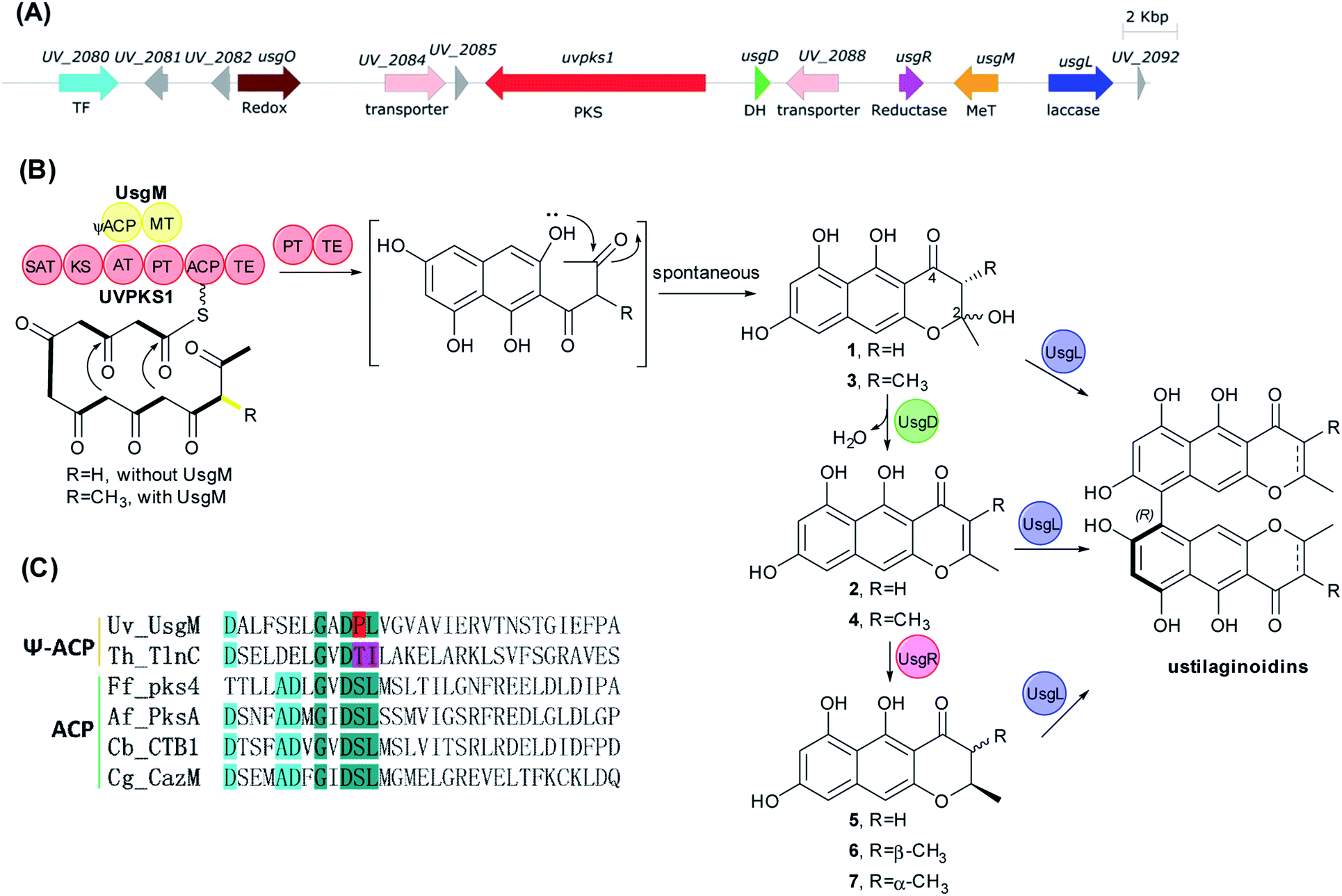 | ||
| Fig. 1 The biosynthesis of ustilaginoidins. (A) Biosynthetic gene cluster; (B) the proposed biosynthetic pathway. TF: transcription factor; Redox: oxidoreductase; PKS: polyketide synthase; DH: dehydratase; MeT/MT: methyltransferase; SAT: starter ACP transacylase; KS: β-ketoacyl synthase, AT: acyl transferase, PT: product template; ACP: acyl carrier protein; ΨACP: pseudo ACP; TE: thioesterase; (C) active site motifs of nrPKS ACP. | ||
Using the CRISPR/Cas9-mediated gene knock-out method,10 we successively deleted various genes in the usg BGC. Deletion of uvpks1 completely abolished the production of ustilaginoidins (Fig. 2(i)), while deletion of usgM, encoding the trans-acting C-methyltransferase, led to the accumulation of usg F (18), G (11), and A (10) (Fig. 2(ii)), all without a 3-methyl substitution. Deletion of the laccase-encoding gene (usgL) resulted in the production of semi-ustilaginoidins, i.e., hemiustilaginoidin F (5), epihemiustilaginoidin D (6), and hemiustilaginoidin D (7) (Fig. 2(iii)). The chemical profiles of these deleted mutants were in good agreement with those in previous reports,10,11 suggesting the role of these genes in the biosynthesis of ustilaginoidins. Heterologous expression of the nrPKS in A. aculeatus revealed that YWA1 (1) was the first metabolite in usg biosynthesis;12 however, the dehydration from YWA1 or its derivative to form the Δ2-derivatives, and subsequent reduction to yield the 2,3-dihydrogenated derivatives were unclear yet. Thus, we set forth to delete other putative genes in the BGC.
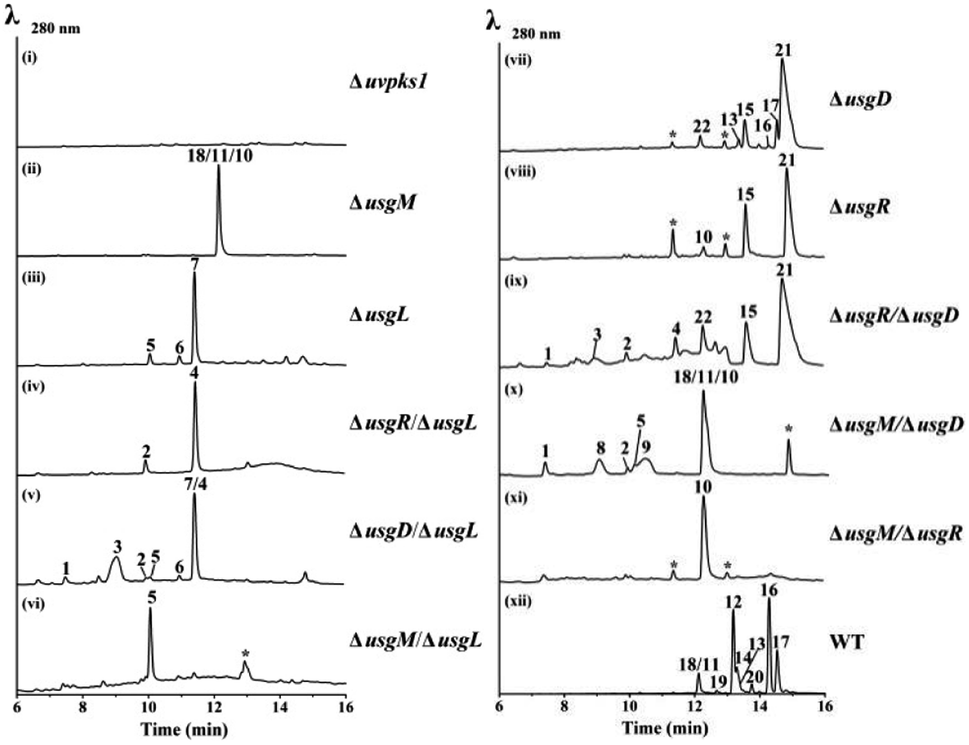 | ||
| Fig. 2 HPLC-MS profiles (UV 280 nm) of the gene-deletion mutants. For traces (ii) and (x), 18, 11, and 10 were resolved by using a different column (Fig. S15 and S17†). For trace (v), 7 and 4 were resolved likewise (Fig. S16†). * – unrelated peaks. WT: wild type. | ||
Deletion of the putative dehydratase gene (usgD) led to the accumulation of usg K (21), L (15), and M (17), in addition to the minor congeners including usg N (13) and D (16), and an unknown metabolite (22, Rt 12.2 min, MW 560, vide infra) (Fig. 2(vii)). In contrast, usg D (16) and E (12), the 2,3-reduced congeners of 21 and 15, respectively, are present as the two dominant metabolites in the wild type (WT) (Fig. 2(xii)). This was unexpected, and it seemed that usgD played a role in Δ2-reduction.
Meanwhile, deletion of usgR, which encodes a protein of unknown function (belonging to the phospholipid methyltransferase superfamily, Fig. S2†), resulted in the accumulation of the Δ2-type of ustilaginoidins (i.e., 10, 15, and 21) (Fig. 2(viii)). Interestingly, when both usgR and usgL were deleted, the ΔusgR/ΔusgL mutant accumulated only 2,3-unsaturated, monomeric naphtho-γ-pyrones (2 and 4) (Fig. 2(iv)). Their structures were elucidated as norrubrofusarin (2) and 3-methyl-norrubrofusarin (4) by UV, MS, and NMR analysis (Fig. S30–S33 and S41–S44; Tables S11 and S13†). Thus, UsgR was deduced to be the Δ2-reductase. Consistent with this, further deletion of usgR on the ΔusgM background led to the production of 10 as the sole ustilaginoidin metabolite (Fig. 2(xi)). In addition, the reduction was quite efficient, as only the reduced monomers can be seen in the ΔusgL (Fig. 2(iii)) or ΔusgM/ΔusgL mutant (Fig. 2(vi)), and the reduced dimers were dominant in the WT (Fig. 2(xii)).
As an apparent reductive role for usgD could not be ruled out, several double deletion experiments involving this gene were conducted (Fig. 2(v), (ix) and (x)). Interestingly, when the laccase encoding gene was deleted, the double-deletion mutant (ΔusgD/ΔusgL) further produced 3-methyl-YWA1 (3) as a major compound, and three minor peaks of YWA1 (1), norrubrofusarin (2), and 3-methyl-norrubrofusarin (4) were observed (Fig. 2(v)), compared to that of the ΔusgL (Fig. 2(iii)). The structures of 1 and 3 were unambiguously elucidated by UV, HRMS, and NMR analysis, and the absolute configuration of C-3 in 3 was determined to be 3S by conversion to its methyl ketal (3c) followed by ECD calculations (Fig. S26–S29 and S34–S40, Scheme S1, and Tables S10 and S12†), while 2 and 4 were identified by LC-MS. This revealed that UsgD was indeed a dehydratase; however, the co-existence of the 2,3-saturated monomers (i.e., 5–7) in ΔusgD/ΔusgL indicated that dehydration of 1 and 3 did happen even without usgD to give 2 and 4, which were then reduced by usgR to yield 5–7. Meanwhile, the ΔusgM/ΔusgD mutant produced five additional metabolites (Fig. 2(x)), including the known monomers (1, 2, and 5) and two unknown metabolites (8 and 9), compared to that of ΔusgM (Fig. 2(ii)). The molecular mass of 8 and 9 corresponded to the homodimer of 1 (m/z 549.09 [M–H]− and 551.10 [M + H]+) and heterodimers of 1 and 2 (m/z 531.08 [M–H]− and 533.09 [M + H]+), respectively. Compound 9 was purified and characterized by NMR as a 1/2-heterodimer (Fig. S53–S55 and Table S14†). However, an attempt to isolate 8 for NMR analysis failed, as it was quickly converted to the dehydrated products (9 and 10) after workup. Based on the HRMS and UV data, and its relationship to 10, the structure of 8 was proposed (Scheme 1). Similarly, we found that the minor metabolite (22) in ΔusgD (Fig. 2(vii)) was quickly converted to usg K (21) upon isolation. Based on this relationship, together with the HRMS data, the structure of 22 was characterized as a 3/4-heterodimer. This suggested that the laccase can dimerize YWA1 and its 3-methyl congener, and dehydration could happen in the dimeric YWA1 or its analogue without usgD (Scheme 1). The third double mutant (ΔusgR/ΔusgD) produced four monomeric naphtho-γ-pyrones (1–4) (Fig. 2(ix)), albeit in minor amounts, and dimer 22, compared to that of ΔusgR (Fig. 2(viii)). It seems that the dimerization was not 100% completed without both usgD and usgR, as there were no monomeric congeners found in either the ΔusgD or ΔusgR strain (Fig. 2(vii) and (viii)). Similarly, monomers were uncovered in the ΔusgM/ΔusgD mutant (Fig. 2(x)), but not in the ΔusgM/ΔusgR mutant (Fig. 2(xi)).
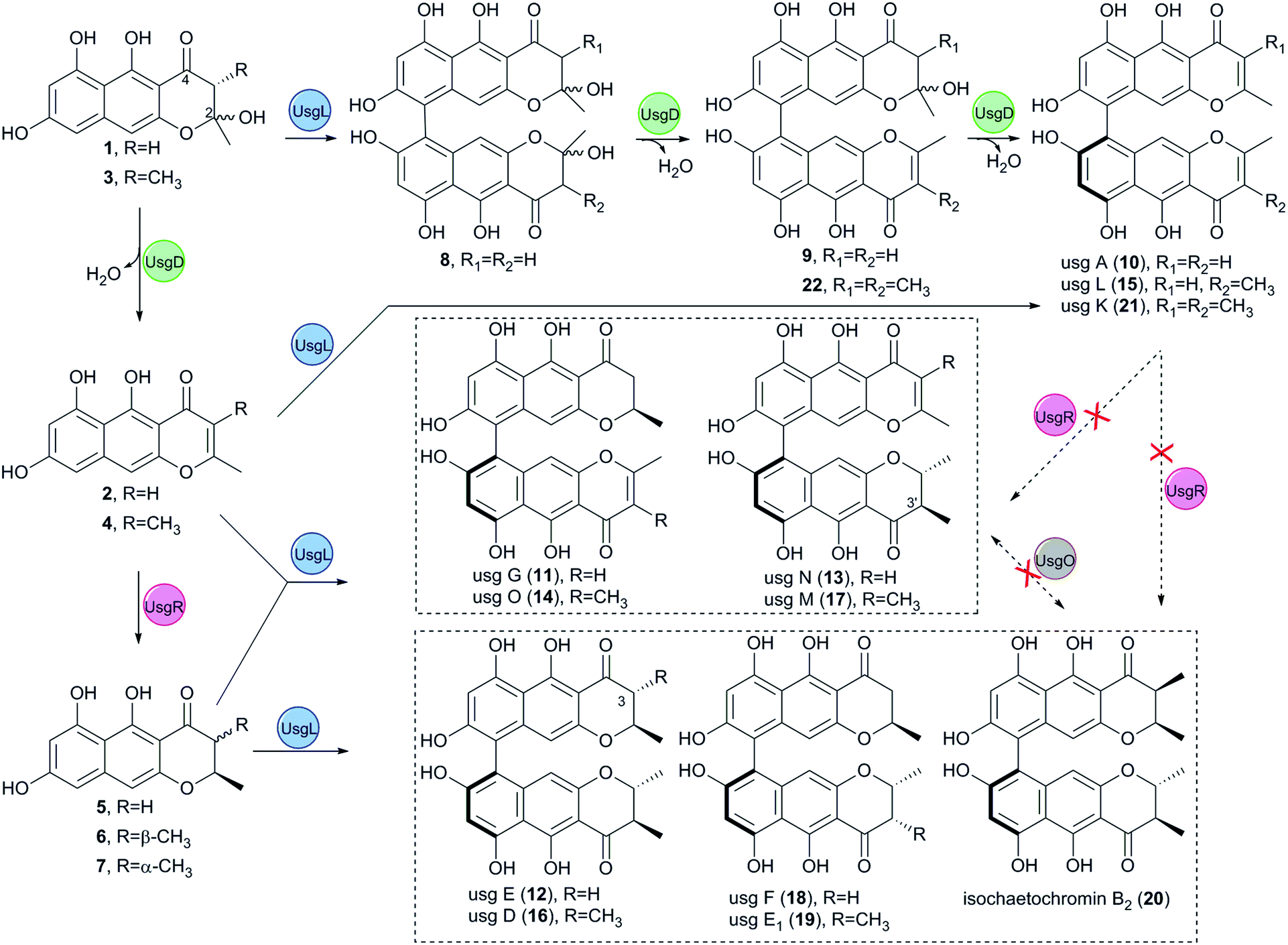 | ||
| Scheme 1 Biosynthesis of the dimeric naphtho-γ-pyrones revealed by gene-deletion experiments and heterologous expression in A. oryzae. | ||
When comparing the profiles of ΔusgD/ΔusgL and ΔusgD (Fig. 2(v) and (vii)), one can see that the abundant peaks in the former, except 3, were for the reduced monomer (7), while in the latter, the abundant one was a dimer of Δ2–7 (i.e., usg K (21)). In contrast, in the WT (Fig. 2(xii)), the 2,3-reduced dimers, i.e., usg E (12) and usg D (16), are dominant. This suggested that the reduction for Δ2-monomers was quite efficient (Fig. 2(v)), but not for the Δ2-dimers (Fig. 2(vii)). The formation of 2,3-unsaturated metabolites indicates that dehydration happens without usgD, albeit slower. The accumulation of Δ2-dimers in ΔusgD (Fig. 2(vii)) indicates an alternative route to the Δ2-dimers starting from dimeric YWA1 and its 3-methyl derivatives via sequential dehydration (Scheme 1, top), as otherwise, this mutant should enrich reduced dimers if it follows the route shown in the left side of Scheme 1.
Interestingly, deletion of the putative FAD dependent oxidoreductase encoding gene usgO did not change the metabolite profile compared to the WT (Fig. S14†). This was in contrast to the previous report,11 in which the ΔusgO mutant still produced all the ustilaginoidins compared to the WT, but the ratio of semi-reduced usg vs. the fully reduced counterparts (i.e., usg N vs. E; usg M vs. D) increased, so a reductive role of usgO was proposed. The discrepancy might result from the different medium used (rice and PSB media in this study vs. PSA in the literature11). The heterologous expression in A. oryzae and feeding experiments (vide infra) has unambiguously excluded the catalytic role of usgO in ustilaginoidin biosynthesis.
Heterologous reconstruction of the ustilaginoidin biosynthesis in A. oryzae NSAR1
To verify the function of the usg genes, we carried out a heterologous expression of combinations of the usg genes in A. oryzae NSAR1 (Table 1 and Fig. 3). All gene fragments were amplified from cDNA and cloned into fungal expression vectors (see ESI Fig. S8†). Expression of the uvpks1 alone (Table 1, entry 1) resulted in the production of YWA1 (1) as the major compound, as well as its dehydrated derivative norrubrofusarin (2) in a smaller amount (Fig. 3, EXP1). It is unknown whether the dehydration was catalyzed by the dehydratase of the host, or non-enzymatically. Further expression of the dehydratase (usgD) in A. oryzae (Table 1, entry 2) led to an almost complete turnover to norrubrofusarin (2) (Fig. 3, EXP2), revealing that this DH was highly efficient. Co-expressing uvpks1 and C-MeT usgM in A. oryzae (Table 1, entry 4) resulted in the observation of two new metabolites (3 and 4) compared to that of EXP1. LC-MS analysis revealed that they were 3-methyl derivatives of 1 and 2, respectively (Fig. 3, EXP4). Likewisely, adding the DH to the previous set of genes in A. oryzae (Table 1, entry 8) converted the hemiketals (1 and 3) to the dehydrated congeners (2 and 4) almost quantitatively (Fig. 3, EXP8).| EXP | Uvpks1 | UsgD | UsgR | UsgM | UsgL | UsgO | Product |
|---|---|---|---|---|---|---|---|
| PKS | DH | Red | MeT | Laccase | Redox | ||
| a Trace amount. | |||||||
| 1 | √ | — | — | — | — | — | 1, 2 |
| 2 | √ | √ | — | — | — | — | 2 |
| 3 | √ | — | √ | — | — | — | 1, 2, 5 |
| 4 | √ | — | — | √ | — | — | 1–4 |
| 5 | √ | — | — | — | √ | — | 1, 2, 8–10 |
| 6 | √ | — | — | — | — | √ | 1, 2 |
| 7 | √ | √ | √ | — | — | — | 2, 5 |
| 8 | √ | √ | — | √ | — | — | 2, 4 |
| 9 | √ | √ | — | — | √ | — | 2, 10 |
| 10 | √ | — | √ | √ | — | — | 1–5, 6,a7 |
| 11 | √ | — | — | — | √ | √ | 1, 2, 8–10 |
| 12 | √ | √ | √ | √ | — | — | 2, 4, 5, 6,a7 |
| 13 | √ | √ | √ | — | √ | — | 2, 5, 9–11, 18 |
| 14 | √ | √ | — | √ | √ | — | 2, 4, 10, 15, 21 |
| 15 | √ | √ | √ | √ | — | √ | 2, 4, 5, 6,a7 |
| 16 | √ | √ | √ | √ | √ | — | 2, 4, 5, 7, 10–18 |
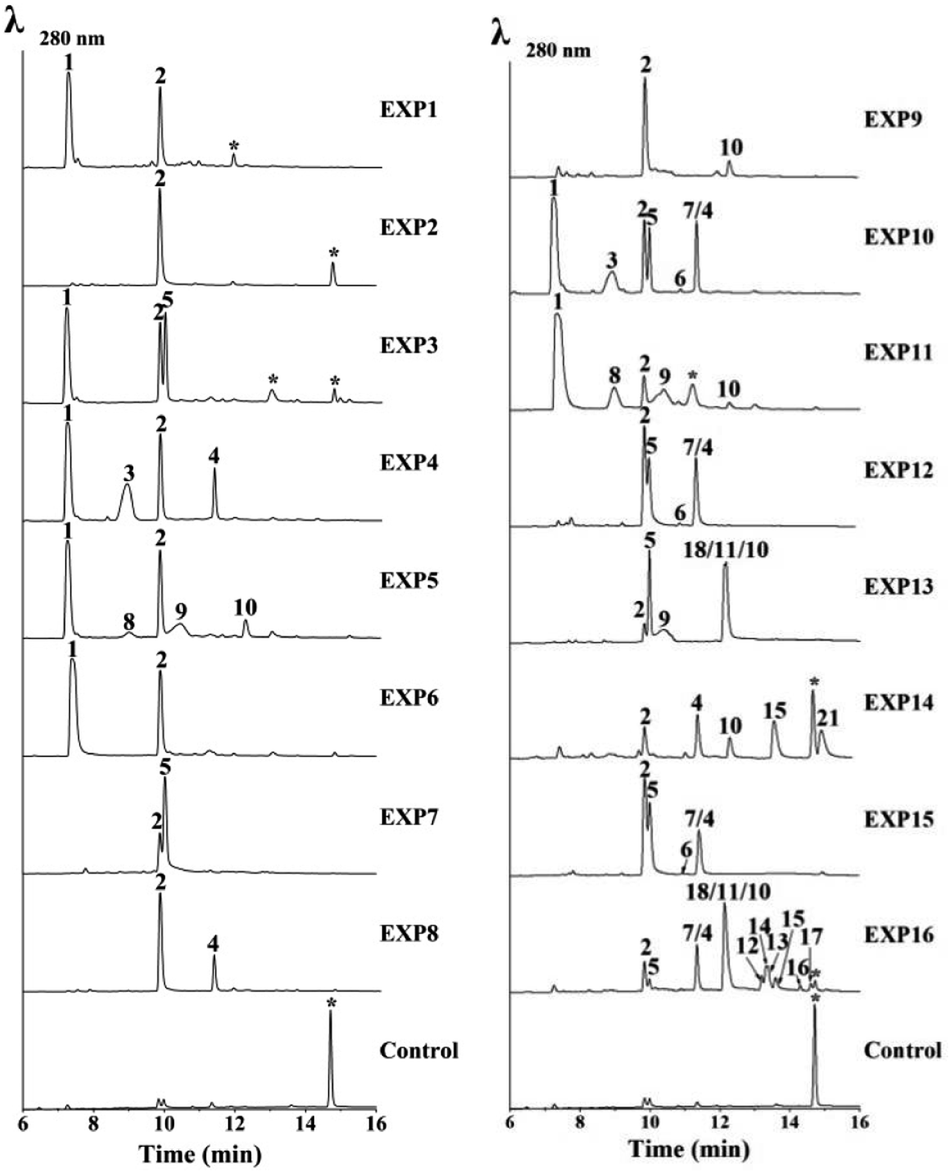 | ||
| Fig. 3 HPLC-MS analysis of the expression of usg genes in A. oryzae. EXP1–16: UV (λ = 280 nm) traces of extracts of A. oryzae transformants. EXP refers to the gene combinations in Table 1. * – unrelated peaks. For EXP10, EXP12 and EXP15, 4 and 7 were resolved by using a different column (Fig. S18†). For EXP13, peaks of 18, 11, and 10 were resolved likewise (Fig. S19†). | ||
Co-expressing uvpks1 and usgR (EXP3, Table 1) resulted in the production of one additional metabolite when compared with EXP1. This metabolite was identified to be the 2,3-dihydrogenated derivative of 2, i.e. hemiustilaginoidin F (5). Further adding usgM to the combination (EXP10; Fig. 3) led to the production of four additional 3-methyl naphtho-γ-pyrones (3, 4, 6, and 7) compared to that of EXP3, among which 6 and 7 were the 2,3-reduced congeners of 4. Similarly, adding the DH to the combinations (EXP7 and EXP12, Table 1) greatly diminished 1 and 3, while the dehydrated products (2 and 4) and the reduced metabolites (5 and 7) were increased, compared to EXP3 and EXP10, respectively. These results confirmed that UsgR was a reductase. Intriguingly, the reduction was highly stereoselective. The absolute configurations of 5–7 were determined previously,10 and all have a 2R configuration. Regarding the 3-methyl substrate, this enzyme predominantly gave a 2,3-trans-dimethyl product (7), in addition to a minor amount of 2,3-cis product (6).
Although there was neither a YWA1 (1) nor 3-methyl-YWA1 (3) moiety found in the reported ustilaginoidins, we hypothesized that they could be dimerized by the laccase. Indeed, when both uvpks1 and usgL were co-expressed in A. oryzae (Table 1, entry 5), we observed three dimers including the 1/1-homodimer (8), 1/2-heterodimer (9), and 2/2-homodimer (10), in addition to the monomers (1 and 2) (Fig. 3, EXP5). Again, adding the DH to the above combination (EXP9, Fig. 3) led to the enrichment of the dehydrated monomer (2) and dimer (10), as expected.
In EXP13, combining the PKS, DH, Red, and laccase resulted in the production of the monomers (2 and 5), and the dimeric metabolites (9–11 and 18) building from 2, 5, and 1 (Fig. 3). Meanwhile, expressing all the usg genes except the reductase usgR (EXP14) led to the almost exclusive production of Δ2-metabolites, including the monomers (2 and 4), and the 2/2-homodimer (10), 2/4-heterodimer (15), and 4/4-homodimer (21) (Fig. 3).
Finally, heterologous expression of all five genes (Table 1, entry 16) led to the observation of a variety of ustilaginoidins (10–18), as well as the monomers 2, 4, 5, and 7. The identity of the ustilaginoidins was confirmed by co-chromatography with the standards isolated previously by our group.3,4
The putative FAD-dependent oxidoreductase, usgO, was also co-expressed with key usg genes (Table 1, EXP6, 11, and 15) in A. oryzae to confirm our deduction from the knockout experiments. Indeed, UsgO does not function as a tailoring enzyme, and as can be seen from Fig. 3 (EXP6, 11, and 15), the metabolite profiles did not change compared to those of EXP1, EXP5, and EXP12, respectively. However, when usgO was present, the ratio of the dehydrated product (2) to its precursor (1) was significantly decreased, compared to those without usgO (Fig. 3, EXP6 vs. EXP1, and EXP11 vs. EXP5, all lacking a DH in the combinations), especially in EXP11, where the 1/1-homodimer (8) increased, but the 2/2-homodimer (10) decreased, probably resulting from the difference in availability of 1 and 2. In EXP15, however, the metabolite profile was the same as that of EXP12. It seems that UsgO might have inhibited side dehydration when the DH was absent.
Meanwhile, we fed 2 and 4 to A. oryzae expressing usgO, respectively, and no conversion was found (Fig. S12†). This again indicated that UsgO was not a reductase. Furthermore, the semi-reduced ustilaginoidins, i.e., usg M (17) and O (14), were also fed to this host; however, no turnover was detected by LC-MS (Fig. S12†). This excludes a possible role of usgO in adjusting the equilibrium of fully reduced and semi-reduced ustilaginoidins proposed previously.11
In vitro biosynthesis of ustilaginoidins with the laccase
Since the laccase was shown to dimerize norrubrofusarin (2) in vitro,12 we further tested all the possible monomers found in the reported ustilaginoidins, together with their precursors (YWA1 (1) and 3-methyl-YWA1 (3)) in various combinations. The results are shown in Fig. 4, S9,† and Table 2.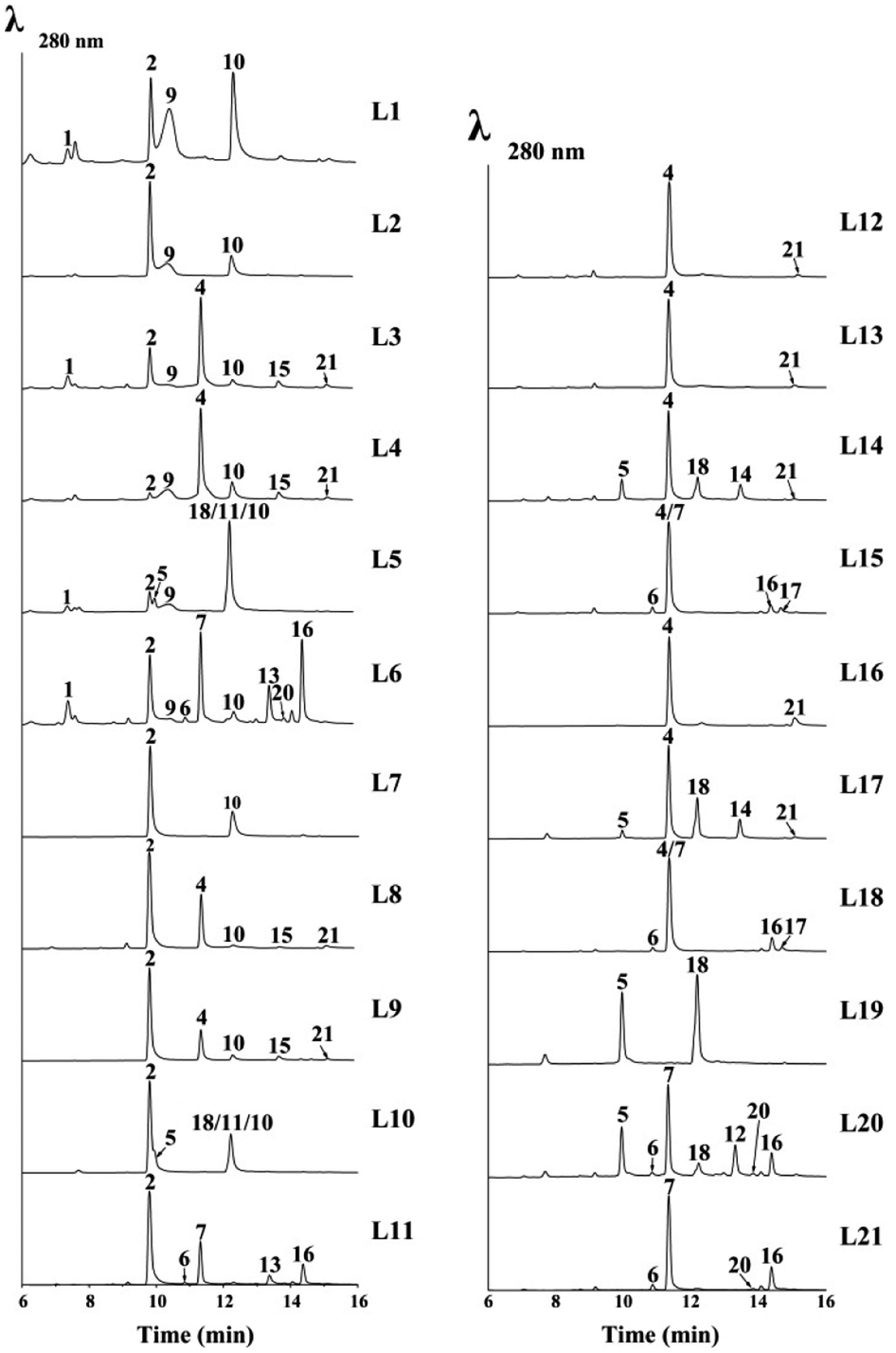 | ||
| Fig. 4 LC-MS analysis of various combinations of monomers in the cell-free extract of UsgL (24 h). L1–L21 refer to the EXPs in Table 2. For L5 and L10, peaks of 18, 11, and 10 were resolved by using a different column (Fig. S20†). | ||
| EXP | Monomera | Product | |||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 7 | ||
| a 6 was not tested due to its limited amount. b Trace amount. | |||||||
| L1 | √ | — | — | — | — | — | 2, 9, 10 |
| L2 | √ | √ | — | — | — | — | 9, 10 |
| L3 | √ | — | √ | — | — | — | 2, 4, 9, 10, 15, 21 |
| L4 | √ | — | — | √ | — | — | 2, 9, 10, 15, 21 |
| L5 | √ | — | — | — | √ | — | 2, 9–11, 18 |
| L6 | √ | — | — | — | — | √ | 2, 6, 9, 10, 13, 16, 20 |
| L7 | — | √ | — | — | — | — | 10 |
| L8 | — | √ | √ | — | — | — |
4, 10,b15,b21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
| L9 | — | √ | — | √ | — | — |
10, 15, 21b |
| L10 | — | √ | — | — | √ | — | 10, 11, 18 |
| L11 | — | √ | — | — | — | √ | 6,b13, 16 |
| L12 | — | — | √ | — | — | — |
4, 21b |
| L13 | — | — | √ | √ | — | — |
21b |
| L14 | — | — | √ | — | √ | — |
4, 14, 18, 21b |
| L15 | — | — | √ | — | — | √ | 4, 6, 16, 17 |
| L16 | — | — | — | √ | — | — | 21 |
| L17 | — | — | — | √ | √ | — |
14, 18, 21b |
| L18 | — | — | — | √ | — | √ | 6, 16, 17 |
| L19 | — | — | — | — | √ | — | 18 |
| L20 | — | — | — | — | √ | √ | 6, 12, 16, 18, 20 |
| L21 | — | — | — | — | — | √ | 6, 16, 20 |
The cell-free extract (CFE) of A. oryzae expressing the laccase (usgL) in citrate buffer (pH 6.0) was able to dimerize norrubrofusarin (2) (entry L7, Table 2) to produce usg A (10) (Fig. 4, L7), which was consistent with a previous report,12 while no dimerization was found in Tris buffer (pH 8.0), indicating that the protonation of the phenolic hydroxyl group is essential for the activity. The CFE was constructed thereafter in citrate buffer for all the experiments (Table 2). The results are shown in Fig. 4 (for full data, see Fig. S9†).
When using YWA1 (1) as the substrate, the expected dimer (8) could not be detected. Instead, a partial dehydration product (9) and the fully dehydrated congener (10) were found to be the major products, together with the dehydrated monomer 2 (entry L1, Table 2, and Fig. 4). In the control experiment, 1 was treated with boiled CFE, and ca. 2/3 of 1 was found to be converted to 2 (Fig. S9,† trace EXP L1). It is possible that the dimerization surpassed the dehydration in the case of 1, which first gave rise to 8, and subsequent dehydration to 9, and finally to 10, as the yield of 10 in EXP L1 was significantly higher than that in EXP L7 (Fig. 4). Interestingly, when 1 and 2 were co-fed to the medium, the yield of 9 and 10 was significantly lower, while 1 was completely used up (EXP L2). The above data indicated that YWA1 (1) might be a preferred substrate compared to its dehydrated congener (2) by the laccase.
In contrast, the 3-methyl derivative of 2, i.e., 3-methyl-norrubrofusarin (4), was only partially (ca. 6%) converted to the dimer usg K (21) (entry L16, Fig. 4). However, when reacting with 3-methyl-YWA1 (3) (entry L12), a major peak of the dehydrated monomer (4), together with a trace amount of usg K (21), was observed (Fig. 4). In comparison, compound 3 was fully dehydrated in the control experiment (Fig. S9†). The even lower yield of 21 in L12 than that in L16 suggested a relatively better reactivity for 4 than 3, under the tested conditions. It seems that the dehydration was faster than dimerization by the laccase in the case of 3, as neither the dimerized product of 3 nor its partially dehydrated product was seen, in analogy to that of 1 (EXP L1). A similar profile was found when 3 and 4 were mixed (EXP L13). Interestingly, the co-reaction of 1 and 3 (EXP L3) led to the production of the dehydrated monomers 2 and 4 as the major products, while only a minor amount of the dimers usg A (10), L (15), and K (21), together with a trace amount of 9, could be detected. A similar profile was found in EXP L4 when 1 and 4 were added together; however, the yield of 9 and usg A (10) was increased, while that of 2 was decreased, compared to EXP L3.
In EXP L5, 1 and 5 were the reactants, which caused the production of a 5/5-homodimer (usg F, 18) and a 2/5-heterodimer (usg G, 11), together with 2, 9 and 10 as found in EXP L1. In EXP L6, co-incubation of 1 and 7 resulted in a 7/7-homodimer (usg D, 16) as the major product, a 2/7-heterodimer (usg N, 13) as the second major product, and a 6/7-heterodimer (isochaetochromin B2, 20), 2/2-homodimer (10), and 1/2-heterodimer (9) as minor metabolites. These dimers arose from the combination of four available monomers: 1 and its dehydrated product 2, and 7 and its 3-epimer 6, as all of them were detected in the control experiment (Fig. S9).† Similarly, when 7 was fed alone to the CFE (EXP L21), the dimeric products 16/20 were produced, as well as 6.
When comparing EXP L5 vs. L2, and L6 vs. L4, one can see that the 2,3-reduced monomer was more efficiently converted by the laccase than its Δ2-analogue. And judging from the peak area of the residual starting material, the 3-methyl substitution has a negative impact on the dimerization (Fig. 4, L6 vs. L5; L3 vs. L1; L7 vs. L16).
Interestingly, using 2 instead of 1 in various combinations (entries L7–L11, Table 2) led to a similar profile of the dimerized products (except in the case of 9) to that found with 1 (entries L1 and L3–L6, respectively); however, the yield was significantly lower. This also revealed that the laccase preferred 1 over 2.
As mentioned previously, 3-methyl-YWA1 (3) was quickly dehydrated under the tested conditions, leading to a trace product of 4/4-homodimer (21) (EXP L12 and L13). However, adding a 2,3-dihydrogenated monomer (5) to the reaction mixture (EXP L14) significantly increased the yield of the dimerized product, including a 5/5-homodimer (18) and 5/4-heterodimer (usg O, 14), though 21 was still obtained in a trace amount. In EXP L15, upon mixing 3 with one other 2,3-reduced monomer (7), the dimeric products were minor; however, they included the 7/7-homodimer (16) and 4/7-heterodimer (usg M, 17), respectively, in a descending order of yield.
Meanwhile, using 3-methyl-norrubrofusarin (4) instead of 3 in the combination (EXP L16–L18, Table 2) resulted in a noticeable increase of the dimeric products; however, a similar profile was found (Fig. 4, L16–L18 vs. L13–L15). This suggested that 4 was preferred by UsgL over 3, which was in contrast to 1vs.2.
In EXP L19, the 5/5-homodimer (usg F, 18) was generated in a good yield using 5 as the substrate. When mixing 5 and 7 in EXP L20, the 5/7-heterodimer (usg E, 12) was the major product, which was followed by the 7/7-homodimer (usg D, 16) and 5/5-homodimer (usg F, 18) as the 2nd and 3rd major products, respectively, together with a minor amount of 20. When comparing with the control (Fig. S9†), one can find that 5 was consumed more than 7, suggesting that 5 might be a better substrate for the laccase.
Obermaier et al.12 reported that the atropselectivity of the laccase was dependent on the protein concentration. In the current study, the atropselectivity of UsgL was investigated at different concentrations using 5 as the substrate, which was chosen due to its better reactivity; diastereomeric products were formed if the atropselectivity varied, and thus there was no need to use a chiral column. Indeed, the P/M-selectivity varied with the change of the protein concentration, with a higher concentration causing a higher M-selectivity (Fig. S10†). These results were consistent with a previous study using 2 as the substrate.12
UsgR is a distinct class of ene-reductases
Bioinformatics analysis indicated that UsgR contained a conserved domain belonging to the phospholipid methyltransferase superfamily, similar to DUF1295 (a family of unknown function), and the C-terminal domain of the steroid 5-alpha reductase family (Fig. S2†). UsgR was predicted to be a membrane-bound protein (Fig. S3†). An attempt to express it in Escherichia coli failed despite various trials. Then, two truncated versions of UsgR were designed and tested: UsgR76-273 and UsgR160-264. The former was designed based on the alignment to Δ14-sterol reductase in SWISS-MODEL, while the latter contained only the predicted conserved domain of UsgR (Fig. S21†). We found that UsgR76-273 was not expressed in E. coli despite testing various expression vectors and strains, while UsgR160-264 was expressed in three vectors (pETM10, pMBP_1a, and pGEX6p-1) upon addition of IPTG (Table S9†). However, further analysis of the expression pattern revealed that UsgR160-264 was insoluble and found only in the cell debris (Fig. S22†).We expressed usgR in A. oryzae, and the CFE showed no reducing ability towards the ene-substrates (2 and 4), not surprisingly. Then, we fed the substrates to the A. oryzae strain expressing usgR, in DPY medium and citrate buffer, respectively (Fig. 5 and S11†). The results showed that norrubrofusarin (2) was fully converted to its dihydrogenated product 5 (Fig. 5(i) and (ii)). Meanwhile, feeding 3-methyl-norrubrofusarin (4) led to the production of two reduced epimers (6 and 7), with the 2,3-trans-methylated one (7) as the predominant product (approx. ratio of 6 to 7, 9:91 and 7:93, Fig. 5(iii) and (iv), respectively). On the other hand, we fed 7 and 5 to the same strain, respectively, and no conversion back to 4 and 2 was found after 48 h. This was in contrast to the steroid 5α-reductase (EC 1.3.1.22), which catalyzed the conversion of 3-oxo-Δ4 steroids into their corresponding 5α form, and vice versa. By the same token, we fed the ene-substrates to the E. coli and A. oryzae recombinant strains expressing usgR160-264, and no products were found in neither strain (Fig. S23 and S24†). This indicated that this truncated protein was nonfunctional.
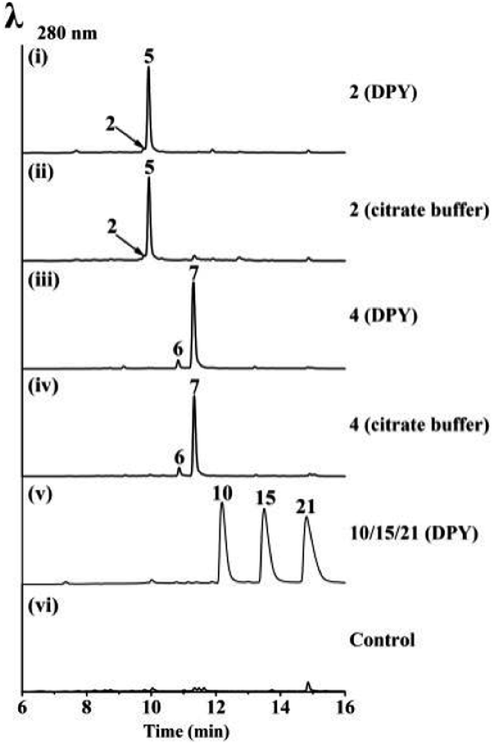 | ||
| Fig. 5 LC-MS analysis of the feeding experiments in A. oryzae-usgR. The feeding substances (media) are indicated on the right side of each trace. | ||
The heterologous expression of usgR in yeasts Pichia pastoris GS115 and Saccharomyces cerevisiae BY4741 succeeded as well, as confirmed by feeding experiments (Fig. S25A and B†), albeit less efficient than in A. oryzae (Fig. S25C†). However, the microsomal fractions from these recombinant strains failed to reduce 2 despite various trials (Fig. S25†).
Interestingly, when we fed usg A (10), L (15), and K (21), three dimeric Δ2-naphtho-γ-pyrones, to the A. oryzae-usgR, no product was found after 48 h (Fig. 5(v) and S11†).
For the 2,3-dimethyl precursor, the reduction gave a predominant 2,3-trans product (7), with a minor amount of 3-epimer (6). It is probable that the 3-epimerism is non-enzymatic, as the keto–enol tautomerism could destroy the stereocenter at C-3 (Fig. 6). The formation of 7 could be thermodynamically favored because it is more stable when both methyl groups are oriented towards a different face. In order to test these hypotheses, 6 or 7 was incubated with various tested buffers, media or solvent (Table 3). The results showed that 7 was stable in the citrate buffer, DPY and PSB media, and in MeOH, for 2 d (the same as the feeding experiment), while the cis-isomer 6 was partially (∼20%) converted to the trans-isomer (7) in the citrate buffer (pH = 6); however, it was very stable in the tested media and in MeOH. This indicated that 7 was more stable than 6. However, ∼2% of 7 was converted to 6, after about 1 month at 4 °C, while a ∼6% conversion rate was found in DMSO even when it was stored at −20 °C. Interestingly, when 7 was incubated with the boiled CFE of A. oryzae-usgL, 8% of 6 was formed. This ratio is close to that found in the feeding experiment (4 + A. oryzae-usgR) and is also similar to that in the knockout mutant (ΔusgL). These data suggested that the cellular components of the host promoted the epimerism, but non-enzymatically.
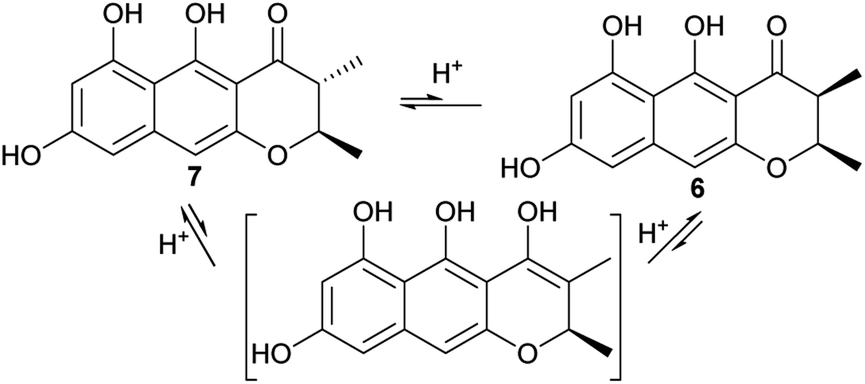 | ||
| Fig. 6 Keto–enol tautomerism led to epimerism at C-3. | ||
| Tested conditions | Substrate | Ratio of 6/7a |
|---|---|---|
| a The ratio was estimated from the peak area at 280 nm. The feeding experiments and CFE tests were performed at 28 °C for 2 days and 1 day, respectively. CFE: cell-free extract constructed in citrate buffer. | ||
| Citrate buffer | 7 | <1:99 (2 d, 28 °C) |
| 2:98 (30 d, 4 °C) |
||
| 6 | 81:19 (2 d, 28 °C) |
|
| 80:20 (30 d, 4 °C) |
||
| DPY | 7 | <1:99 (2 d, 28 °C) |
| 6 | >99:1 (2 d, 28 °C) |
|
| PSB | 7 | <1:99 (2 d, 28 °C) |
| 2:98 (30 d, 4 °C) |
||
| 6 | 96:4 (2 d, 28 °C) |
|
| 94:6 (30 d, 4 °C) |
||
| MeOH | 7 | <1:99 (2 d, 28 °C) |
| 2:98 (30 d, 4 °C) |
||
| 6 | 97:3 (2 d, 28 °C) |
|
| 96:4 (30 d, 4 °C) |
||
| DMSO | 7 | 6:94 (30 d, −20 °C) |
| Boiled CFE of A. oryzae-usgL | 7 | 8:92 (1 d, 28 °C) |
| A. oryzae-usgR, citrate buffer | 4 | 7:93 (2 d, 28 °C) |
| A. oryzae-usgR, DPY | 4 | 9:91 (2 d, 28 °C) |
| ΔusgL | — | 8:92–7:93 |
Although a large number of ene-reductases have been discovered for asymmetric reduction of activated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds,14 no examples have been reported for the reduction of naphtho-γ-pyrenones, to the best of our knowledge. Moreover, only a few enzymatic reductions of chromen-4-one-containing substrates, including flavones15 and isoflavones,16 have been reported. In this study, we also tested some representatives containing such a moiety, including one flavone (23), one isoflavone (24), four chromen-4-ones (25–28), and one benzo[h]chromen-4-one (29) (Fig. 7); however, no reduction was found after feeding them to A. oryzae-usgR for 2 d (data not shown). This suggested that the reductase was specific for reduction of linear naphtho-γ-pyrenones (i.e., 4H-benzo[g]chromen-4-one) (e.g.2 and 4).
C bonds,14 no examples have been reported for the reduction of naphtho-γ-pyrenones, to the best of our knowledge. Moreover, only a few enzymatic reductions of chromen-4-one-containing substrates, including flavones15 and isoflavones,16 have been reported. In this study, we also tested some representatives containing such a moiety, including one flavone (23), one isoflavone (24), four chromen-4-ones (25–28), and one benzo[h]chromen-4-one (29) (Fig. 7); however, no reduction was found after feeding them to A. oryzae-usgR for 2 d (data not shown). This suggested that the reductase was specific for reduction of linear naphtho-γ-pyrenones (i.e., 4H-benzo[g]chromen-4-one) (e.g.2 and 4).
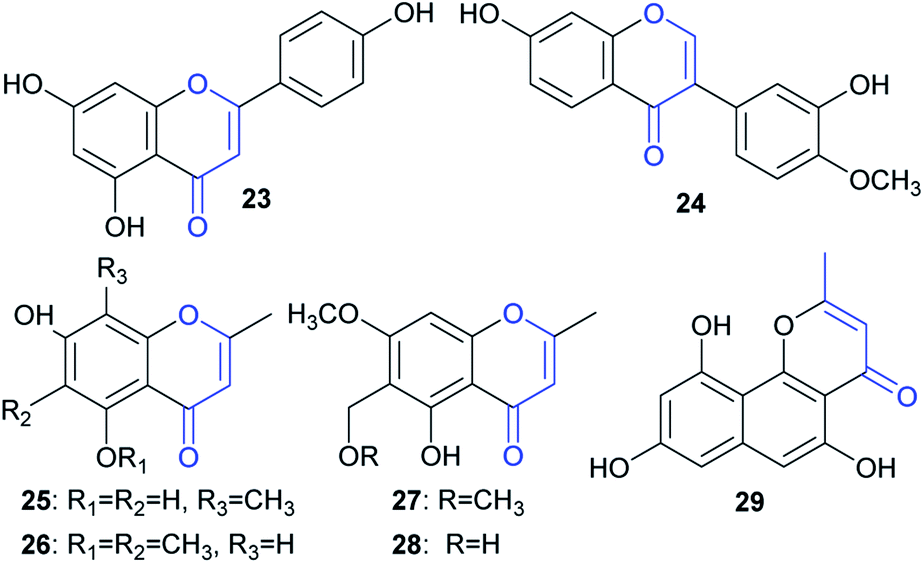 | ||
| Fig. 7 Tested compounds (23–29) containing a chromen-4-one moiety. | ||
Discussion
Bis-naphtho-γ-pyrones are widely distributed in filamentous fungi, and displayed a broad range of biological activities.17 Among them, ustilaginoidins and chaetochromins are mycotoxins, sharing a 9/9′-linked bis-naphtho-γ-pyrone skeleton but differing in the axial configuration (M and P, respectively).As can be seen from the structures of ustilaginoidins, they vary at the constituted monomers due to having different saturations at C-2/C-3, and methylation at C-3 (Fig. 1). Different biosynthetic schemes have been proposed for these monomers,11,12 but the key steps (methylation, dehydration, and reduction) have not been validated by biochemical or genetic studies. Moreover, it is unclear whether some modifications on the dimers could happen or not during the biosynthesis.
In this study, the complete biosynthetic pathway of ustilaginoidins was revealed, as shown in Fig. 1B and Scheme 1. UVPKS1 incorporated one acetyl CoA and six malonyl CoA to form an ACP-bound heptaketide, and was further processed by the product template (PT) and thioesterase (TE) domain before releasing the phenolic ketone, which was spontaneously ketalized to produce YWA1 (1). UsgM contains a ψ-ACP-MT didomain, similar to TlnC,13 in which ψ-ACP was essential for the C-methylation of tricholignan A. The ψ-ACP of TlnC was found to inhibit the KS (β-ketoacyl synthase) catalyzed chain elongation to facilitate methylation. It is likely that UsgM can work in a similar manner. It should work on the ACP-bound growing ketides before chain release to form the 3-methyl derivative (3). This can explain why feeding YWA1 (1) to A. oryzae-usgM or its CFE did not result in any 3-methylated product (Fig. S13†). Moreover, the methylation was so efficient that the 3-methyl derivatives are predominant (Fig. 2, xii; especially iii and iv), and judging from ΔusgR/ΔusgL (Fig. 2(iv)), the ratio of non-methylation vs. methylation is ca. 1 to 7.
The hemi-ketals, YWA1 (1) and its derivative (3), were dehydrated by UsgD to form the Δ2-derivatives (2 and 4). These metabolites were prone to dehydration without UsgD, albeit it was slower (Fig. 3, EXP1 and EXP4; Fig. 2(v)).
The reduction of the 2,3-double bond by UsgR afforded the 2,3-dihydro-naphtho-γ-pyrones (5–7). The reduction was stereo-selective, yielding the 2R substrates. With regard to the 2,3-dimethyl precursor, the major product was the trans-isomer (7), in addition to a minor amount of cis-isomer (6). The production of the 3-epimer is probably non-enzymatic under the culture conditions.
Atroposelective dimerization of these monomers by the laccase UsgL gives rise to the diverse ustilaginoidins. In vitro experiments revealed that the reactivity of the monomers followed the order 3-nonmethyl > 3-methyl; 2,3-saturated > 2,3-unsaturated; surprisingly, YWA1 (1) > its dehydrated counterpart (2); however, the trend is reversed for 3-methyl YWA1 (3). To compensate for the relatively low reactivity, the fungus produced a significant excess of 3-methyl metabolites to nonmethylated ones as mentioned above. All these together contribute to the observed diversity of ustilaginoidins (Scheme 1 and Fig. 2(xii)).
Meanwhile, the gene-deletions and heterologous expression experiments revealed complex interactions between the DH (usgD), C-MeT (usgM), reductase (usgR), and laccase (usgL). For instance, gene-deletion of usgD did not result in the enrichment of 2-hydroxyl derivatives (Fig. 2(vii)), while further deletion of either usgL, usgR, or usgM did (Fig. 2(v), (ix) and (x)). Moreover, disrupting usgD alone did not lead to the accumulation of monomers (Fig. 2(vii)), but upon further deletion of either the upstream usgM or the downstream usgR, the monomers appeared (Fig. 2(ix) and (x)). This indicated two different ways to biosynthesize the Δ2-dimers (i.e., 10, 15, and 21), either through dimerization of the Δ2-monomers, or dimerization of the 2-hydroxyl intermediates followed by dehydration (Scheme 1). Further reduction of these Δ2-dimers by usgR or the putative oxidoreductase (usgO) was not observed (Scheme 1). Thus, the fully reduced dimers (12, 16, and 18–20) and the semi-reduced ustilaginoidins (11, 13, 14, and 17) had to be formed by dimerization of two corresponding monomers by the laccase.
The non-enzymatic epimerism at C-3 was found between the monomers (6 and 7) as stated previously (Table 3). Likewisely, the epimerism could be possible in the dimers containing either 6 or 7. Indeed, we observed a partial conversion (6%) from usg D (16) to its 3-epimer 20, from usg M (17) to its 3′-epimer usg M1,2 and from usg N (13) to its 3′-epimer usg P,10 in the frozen samples of 16, 17, and 13, respectively, which had been stored at −20 °C for 4 years. This suggested one alternative route to generate the 6-containing dimers (such as 19 and 20) via 3-epimerism of the corresponding dimers (e.g.12 → 19, 16 → 20), in addition to the laccase-catalyzed dimerization of the respective monomers (Scheme 1).
UsgR is a previously unknown reductase that specifically catalyzed the reduction of 4H-benzo[g]chromen-4-one. A BLASTP search using the usgR sequence as a query identified 97 putative enzymes (amino acid sequence identity ≥30%, coverage ≥80%, e-value ≤1e − 10) from over 73 different species of ascomycetes (Table S1†). Phylogenetic analysis revealed that it is closely related to Metarhizium spp. and Thermothelomyces sp. (Fig. S4†). Further searching for similar PKSs and DHs in the genome of these fungi resulted in 12 species that could be possible producers of 2,3-reduced naphtho-γ-pyrone (Table S2†), including the species in the genera of Metarhizium (three species), Thermothelomyces, Rhizodiscina, Lindgomyces, Melanomma, Corynespora, Glonium, Cenococcum, Pseudogymnoascus, and Chaetomium. Interestingly, the DH was found to cluster with the PKS in all these species, among which nine species contained the reductase in the same cluster (Fig. S5†). As shown in Fig. S5,† high synteny was found for U. virens, C. olivicolor, T. thermophiles, M. robertsii, M. brunneum, and M. anisopliae, indicating that they could produce similar metabolites. Among them, Chaetomium spp.18 are well-known producers of chaetochromins, though not reported from C. olivicolor. M. anisopliae is reported to produce monomeric (indigotides G, H, and B) and dimeric 2,3-reduced naphtho-γ-pyrones (i.e., 16 and 20),8 in which 2,3-trans-dimethylated products were found together with the 2,3-cis products; however, the stereochemistry (C-2, C-3) of the pyrone ring was assigned reversely between the monomers and the dimers. Comparing the CD profiles of these monomers with those of 5–7 (ref. 10) revealed that the stereochemistry was probably incorrectly assigned, and should be revised to 2R. Hence, the reductases in these species showed the same stereoselectivity.
In summary, the biosynthesis of ustilaginoidins was revealed by gene-deletions and reconstructed by heterologous expression in A. oryzae, and in vitro experiments. The dehydration step was cryptic at first, but was unveiled by further deletion of one other gene in the ΔusgD background, and confirmed by heterologous expression. A new route to 2,3-unsaturated dimeric naphtho-γ-pyrones was discovered via dimerization of YWA1 (and 3-methyl YWA1) followed by dehydration. The reduction of C2C3 was catalysed by UsgR, which was distinct from any characterized ene-redcutases. The reduction was stereo-controlled, and worked only on monomeric, linear naphtho-γ-pyrenones. In addition, the epimerism at C-3 was non-enzymatic. Finally, the laccase atroposelectively coupled the various building blocks to generate ustilaginoidins, in which different substrate preferences were found. It is interesting to note that ustilaginoidins with a 2-hydroxymethyl substituent were found in the RFS balls,3 but not in the culture of U. virens;4 hence, the oxygenase-encoding gene must locate outside the usg BGC.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Conceptualisation, supervision and writing – L. Z., and D. L.; investigation and methodology –D. X., R. Y., Z. Z., G. G., S. Z., J.-R. X., J. L., Y.-L. P., D. L.; all authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 31471729), the National Key R&D Program of China (No. 2017YFD0200501 and 2017YFC1600905), and the Chinese Universities Scientific Fund (No. 2019TC058). We thank Professor Russell Cox (Leibniz University of Hannover) for providing the fungal heterologous expression host (A. oryzae NSAR1) and plasmids (pTYGSarg and pTYGSmet).Notes and references
- K. Koyama and S. Natori, Chem. Pharm. Bull., 1988, 36, 146–152 CrossRef CAS.
- J. Meng, S. Zhao, P. Dang, Z. Zhou, D. Lai and L. Zhou, Nat. Prod. Res., 2021, 35, 1555–1560 CrossRef CAS PubMed.
- W. Sun, A. Wang, D. Xu, W. Wang, J. Meng, J. Dai, Y. Liu, D. Lai and L. Zhou, J. Agric. Food Chem., 2017, 65, 5151–5160 CrossRef CAS PubMed.
- S. Lu, W. Sun, J. Meng, A. Wang, X. Wang, J. Tian, X. Fu, J. Dai, Y. Liu, D. Lai and L. Zhou, J. Agric. Food Chem., 2015, 63, 3501–3508 CrossRef CAS PubMed.
- T. Tsuchiya, S. Sekita, K. Koyama, S. Natori and A. Takahashi, Congenital Anomalies, 1987, 27, 245–250 CrossRef CAS.
- K. Koyama, K. Ominato, S. Natori, T. Tashiro and T. Tsuruo, J. Pharmacobio-Dyn., 1988, 11, 630–635 CrossRef CAS PubMed.
- K. Kawai, K. Hisada, S. Mori, Y. Nozawa, K. Koyama and S. Natori, Proc. Jpn. Assoc. Mycotoxicol., 1991, 1991, 31–35 CrossRef.
- X. Kong, X. Ma, Y. Xie, S. Cai, T. Zhu, Q. Gu and D. Li, Arch. Pharmacal Res., 2013, 36, 739–744 CrossRef CAS PubMed.
- Y. Zhang, K. Zhang, A. Fang, Y. Han, J. Yang, M. Xue, J. Bao, D. Hu, B. Zhou, X. Sun, S. Li, M. Wen, N. Yao, L.-J. Ma, Y. Liu, M. Zhang, F. Huang, C. Luo, L. Zhou, J. Li, Z. Chen, J. Miao, S. Wang, J. Lai, J.-R. Xu, T. Hsiang, Y.-L. Peng and W. Sun, Nat. Commun., 2014, 5, 3849 CrossRef CAS PubMed.
- D. Lai, J. Meng, D. Xu, X. Zhang, Y. Liang, Y. Han, C. Jiang, H. Liu, C. Wang, L. Zhou and J.-R. Xu, Sci. Rep., 2019, 9, 1855 CrossRef PubMed.
- Y. Li, M. Wang, Z. Liu, K. Zhang, F. Cui and W. Sun, Environ. Microbiol., 2019, 21, 2629–2643 CrossRef CAS PubMed.
- S. Obermaier, W. Thiele, L. Fürtges and M. Müller, Angew. Chem., Int. Ed., 2019, 58, 9125–9128 CrossRef CAS PubMed.
- M. Chen, Q. Liu, S.-S. Gao, A. E. Young, S. E. Jacobsen and Y. Tang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 5499–5504 CrossRef CAS PubMed.
- H. S. Toogood and N. S. Scrutton, ACS Catal., 2018, 8, 3532–3549 CrossRef CAS PubMed.
- G. Yang, S. Hong, P. Yang, Y. Sun, Y. Wang, P. Zhang, W. Jiang and Y. Gu, Nat. Commun., 2021, 12, 790 CrossRef CAS PubMed.
- N. L. Paiva, R. Edwards, Y. Sun, G. Hrazdina and R. A. Dixon, Plant Mol. Biol., 1991, 17, 653–667 CrossRef CAS PubMed.
- S. Lu, J. Tian, W. Sun, J. Meng, X. Wang, X. Fu, A. Wang, D. Lai, Y. Liu and L. Zhou, Molecules, 2014, 19, 7169–7188 CrossRef PubMed.
- S. Sekita, K. Yoshihira, S. Natori, S. Udagawa, T. Muroi, Y. Sugiyama, H. Kurata and M. Umeda, Can. J. Microbiol., 1981, 27, 766–772 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: including all experimental and spectroscopic details. See DOI: 10.1039/d1sc02666f |
| This journal is © The Royal Society of Chemistry 2021 |