
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-molecule FRET and conformational analysis of beta-arrestin-1 through genetic code expansion and a Se-click reaction†
Ming-Jie
Han‡
a,
Qing-tao
He‡
bde,
Mengyi
Yang‡
c,
Chao
Chen
af,
Yirong
Yao
c,
Xiaohong
Liu
e,
Yuchuan
Wang
g,
Zhong-liang
Zhu
h,
Kong-kai
Zhu
i,
Changxiu
Qu
b,
Fan
Yang
b,
Cheng
Hu
e,
Xuzhen
Guo
e,
Dawei
Zhang
a,
Chunlai
Chen
*c,
Jin-peng
Sun
*bd and
Jiangyun
Wang
 *efg
*efg
aTianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin Airport Economic Area, Tianjin, 300308, China
bKey Laboratory Experimental Teratology of the Ministry of Education and Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, 44 Wenhua Xi Road, Jinan, 250012, Shandong, China. E-mail: sunjinpeng@sdu.edu.cn
cSchool of Life Sciences, Tsinghua-Peking Joint Center for Life Sciences, Beijing Advanced Innovation Center for Structural Biology, Tsinghua University, Haidian District, Beijing, 100084, China. E-mail: chunlai@tsinghua.edu.cn
dDepartment of Physiology and Pathophysiology, School of Basic Medical Sciences, Peking University, Key Laboratory of Molecular Cardiovascular Science, Ministry of Education, Haidian District, Beijing, 100191, China
eInstitute of Biophysics, Chinese Academy of Sciences, Chaoyang District, Beijing, 100101, China
fUniversity of the Chinese Academy of Sciences (UCAS), Shijingshan District, Beijing, 100049, China
gShenzhen Institute of Transfusion Medicine, Shenzhen Blood Center, Futian District, Shenzhen, 518052, China
hSchool of Life Sciences, University of Science and Technology of China, Baohe District, Anhui 230026, China
iSchool of Biological Science and Technology, University of Jinan, Jinan, Shandong 250022, China
First published on 31st May 2021
Abstract
Single-molecule Förster resonance energy transfer (smFRET) is a powerful tool for investigating the dynamic properties of biomacromolecules. However, the success of protein smFRET relies on the precise and efficient labeling of two or more fluorophores on the protein of interest (POI), which has remained highly challenging, particularly for large membrane protein complexes. Here, we demonstrate the site-selective incorporation of a novel unnatural amino acid (2-amino-3-(4-hydroselenophenyl) propanoic acid, SeF) through genetic expansion followed by a Se-click reaction to conjugate the Bodipy593 fluorophore on calmodulin (CaM) and β-arrestin-1 (βarr1). Using this strategy, we monitored the subtle but functionally important conformational change of βarr1 upon activation by the G-protein coupled receptor (GPCR) through smFRET for the first time. Our new method has broad applications for the site-specific labeling and smFRET measurement of membrane protein complexes, and the elucidation of their dynamic properties such as transducer protein selection.
Introduction
Single-molecule Förster resonance energy transfer (smFRET) is a pivotal tool for investigating the dynamic properties of biomacromolecules,1,2 providing important insight into numerous fundamental biological processes including transcription,3 translation,4 protein folding,5 signal transduction, and enzyme mechanisms.6 Compared with other techniques such as nuclear magnetic resonance (NMR), X-ray crystallography, and cryo-electron microscopy, smFRET has the advantage of capturing multiple and minor populated conformational states of macromolecules under close-to-physiological conditions without the need of ensemble averaging.2,7 Precise protein labeling with two or more fluorophores at desirable sites is an essential prerequisite of smFRET measurement.8,9 An ideal protein labeling strategy to enable smFRET requires that (1) unique functional groups should be introduced site-specifically into proteins, without introducing extra mutations and causing loss of functions; (2) the site-specifically labeled protein should react at high rate and selectively with fluorescent dyes under physiological conditions.Currently, the cysteine–maleimide reaction is the most popular protein bio-conjugation approach for smFRET study, which has enabled the unraveling of the dynamic mechanisms of membrane proteins, including G protein coupled receptors (GPCRs).10 Enzymatic labeling strategies with SNAP/CLIP tags, sulfatase, and biotin ligase11 have been applied to the detection of HIV-1 (ref. 12) and mGluR2 receptor conformational change using smFRET.13 These investigations have significantly advanced our understanding of transmembrane protein signaling.14 As the receptor family with the largest number of drug targets, GPCRs convert extracellular signals to intracellular signaling cascades through both G protein and arrestins. Importantly, G-protein or arrestin biased GPCR ligands may exhibit higher potency and lower side-effects as pharmacological agents.15 Recent X-ray diffraction or cryo-electron microscopy (Cryo-EM) studies of GPCR–arrestin complexes have provided important structural information on how receptors engage with arrestins, and how a biased ligand induces specific receptor conformational changes.16,17 However, the single-molecule perspective of receptor–arrestin signaling by smFRET has not been established.
Notably, the seven surface-exposed cysteine residues on arrestin represent a major challenge for the study of receptor–arrestin signaling using the smFRET method with the cysteine–maleimide reaction, since mutations of all seven cysteines in arrestin may significantly affect its function. Moreover, the large molecular weight of SNAP/CLIP tags may significantly perturb target protein function18 (Table S1†).
In contrast to these methods, unnatural amino acid incorporation through genetic code expansion19,20 has the unique advantage of introducing site-specific protein modifications without introducing extra mutations.21 Therefore, this method has a unique advantage for labeling membrane protein complexes, such as GPCR–arrestins, and exploring their dynamic properties.22 Moreover, bioorthogonal reactions have been developed extensively, including the Cu(I) mediated azide–alkyne click reaction,23 strain-promoted azide–alkyne click reaction,24 Diels–Alder reactions,25,26 inverse electron-demand Diels–Alder cycloaddition,25,27 ketone/aldehyde and hydrazine/hydroxylamine reaction,28 “π-clamp” reaction,29 methionine–oxaziridine ReACT reaction,30 palladium catalyzed bioorthogonal reaction,31 photo-click reaction32 and others (Table S2†). Although these bioorthogonal reactions are extremely valuable, a more user-friendly protein labeling method generating homogenous products with high reaction rate and without the use of acidic pH and metal catalysis will further advance the application of smFRET in investigating membrane protein complexes. In addition, a fairly long hydrophobic linker (typically with more than 10 covalent bonds) usually lies between the fluorophore and the protein backbone. The hydrophobic linker and fluorophore may interact with the lipid membrane,33,34 which complicates data analysis. Therefore, a labeling method with a short linker will enrich the labeling toolbox and may benefit smFRET measurement under certain circumstances.35
In this work, we demonstrate the highly efficient and specific genetic incorporation of a novel unnatural amino acid, 2-amino-3-(4-hydroselenophenyl) propanoic acid or seleno-phenylalanine (SeF) (Fig. 2G), through genetic code expansion, followed by a Se-click reaction to conjugate the Bodipy593 fluorophore on proteins (Fig. 1A and S2†). Compared with other protein conjugation methods, the Se-click reaction possesses several advantages that make it uniquely suitable for smFRET: (1) SeF differs only by a single atom from tyrosine (Fig. 2G) and, therefore, it introduces minimal perturbation in the target protein; (2) the second-order rate constant of the Se-click reaction is 26.3 ± 0.4 M−1 s−1, 3500-fold higher than that of the N-acetyl cysteine/Bodipy593 reaction rate, enabling rapid and selective protein labeling in the presence of multiple surface-exposed cysteine residues; (3) after the Se-click reaction, Bodipy593 undergoes a 47 nm bathochromic shift, which significantly reduces the background fluorescence signal from unreacted fluorescent dye; and (4) SeF-Bodipy593 has a short linker between the protein backbone and chromophore, which may reduce interactions between the fluorophore and lipid bilayers and benefit smFRET measurements of membrane proteins. We demonstrate the utility of the Se-click reaction in smFRET measurement by labeling SeF-Bodipy593 and FlAsH as a FRET pair to βarr1. smFRET measurement using this precisely doubly-labeled βarr1 mutant reveals that the JNK3 binding site of βarr1, the β-1 strand, underwent conformational changes related to the C-edge domain, in response to active phospho-β2AR engagement.
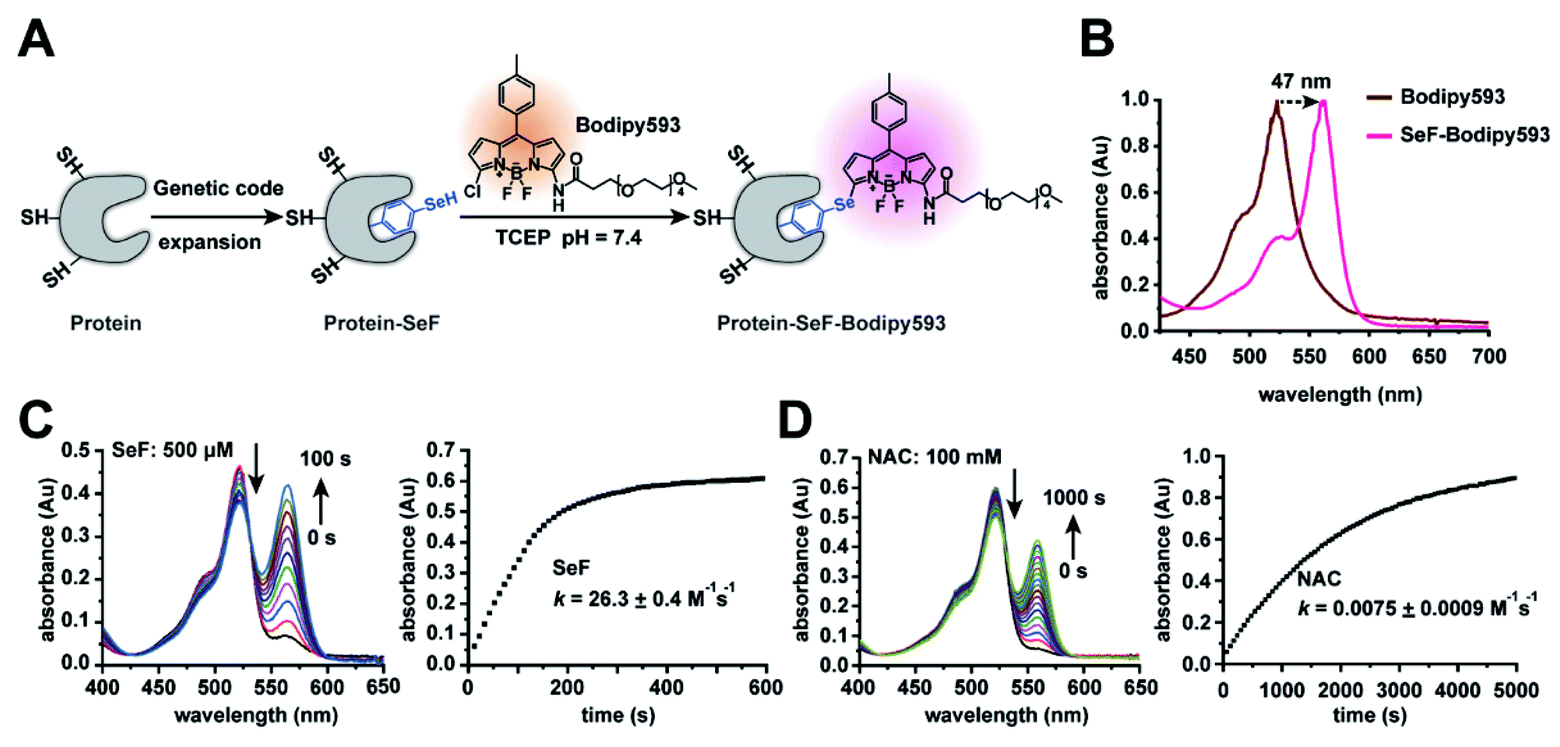 | ||
| Fig. 1 Characterization of the Se-click reaction. (A) Se-click reaction applied to site-specific protein labeling. (B) The absorbance of Bodipy593 exhibits a 47 nm bathochromic shift after the Se-click reaction. (C) Kinetic measurement of the reaction rate between SeF and Bodipy593. During the reaction, absorption at 518 nm decreased accompanied by an increase at 565 nm. The extinction coefficient of SeF-Bodipy593 is larger than that of Bodipy593, so the absorbance increase at 565 nm is larger than the decrease at 518 nm. The second-order rate constant was 26.3 ± 0.4 M−1 s−1, generated by fitting the absorbance at 570 nm. Reaction conditions: 20 μM Bodipy593 and 500 μM SeF (reduced by 1 mM TCEP). (D) Kinetic measurement of the reaction rate between N-acetyl-cysteine (NAC) and Bodipy593. The rate constant was 0.0075 ± 0.0009 M−1 s−1 generated by fitting the absorbance at 560 nm (reaction conditions: 20 μM Bodipy593 and 100 mM NAC). All experiments were performed in HEPES buffer (pH = 7.4, 200 mM) with 10% CH3CN as the co-solvent at room temperature. Data are shown as mean ± s.e.m. (C) and (D) show representative data of three independent experiments. | ||
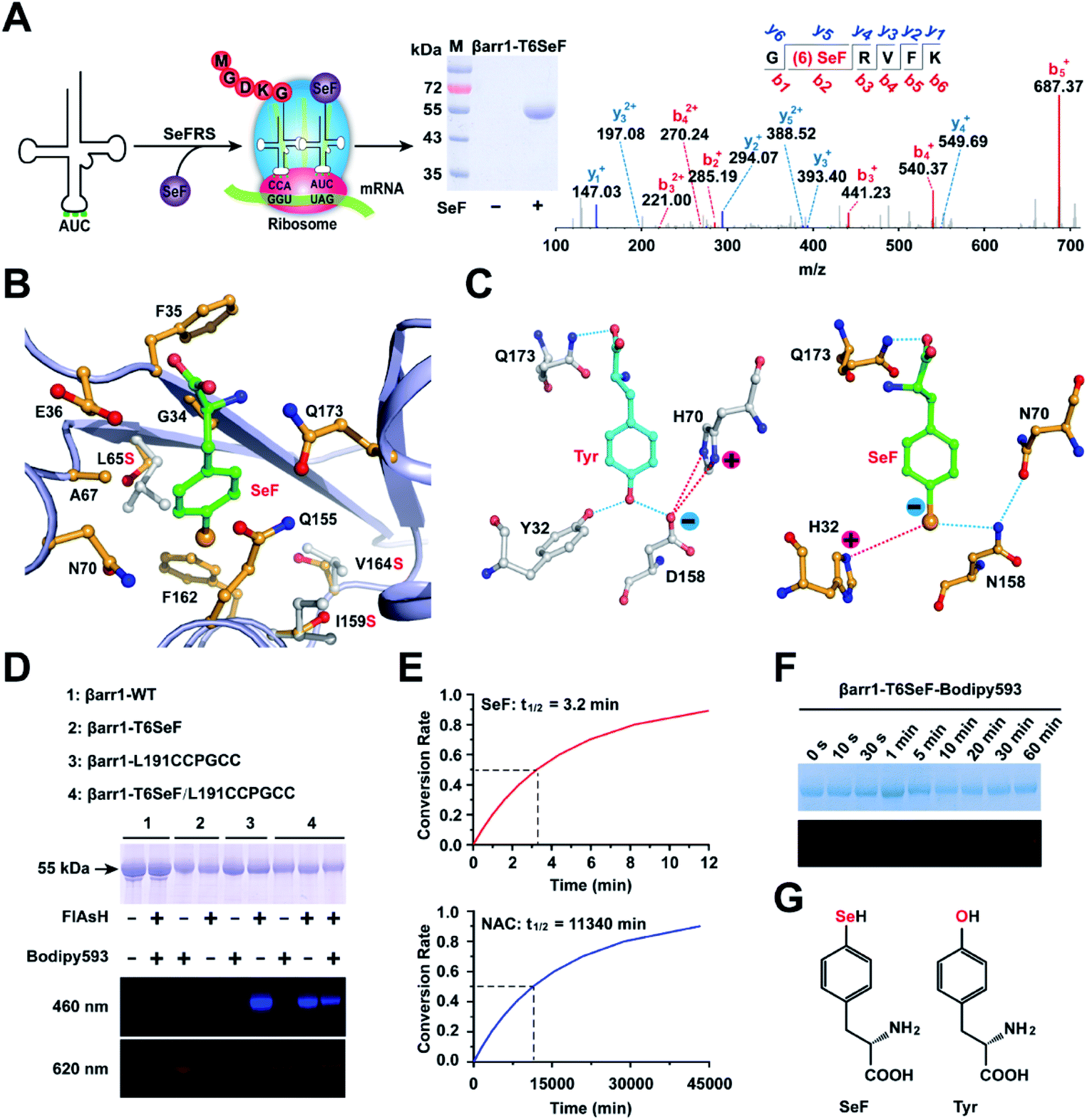 | ||
| Fig. 2 Site-specific incorporation of SeF in protein. (A) Schematic for the site-specific incorporation of SeF into βarr1 through genetic code expansion. The purified βarr1-T6SeF protein was analyzed by SDS-PAGE and LC-MS/MS spectroscopy (b-ions refer to the N-terminal fragments of the peptide, and y-ions represent the C-terminal fragments). (B) X-ray diffraction reveals the substrate binding pocket in SeFRS for SeF. The hydrophobic residues are shown as yellow sticks. Three key mutations (Leu65Ser, Ile159Ser, and Val164Ser) are indicated using grey sticks. (C) Structural determinants for the selective recognition of the Se anions by the SeFRS pocket residues. The corresponding interactions within the Tyr-aaRs are shown for comparison. Charge–charge interactions and hydrogen bonds formed between H32 and N158 in the SeFRS with Se anions are highlighted with blue and red dotted lines. (D) The selectivity of the Se-click reaction on protein conjugation. Bodipy593 only reacted with SeF, and FlAsH only reacted with the CCPGCC motif. (E) Half-life of 150 μM Bodipy593 in the presence of βarr1-T6SeF (top) and βarr1-T6Cys (bottom) (pH = 7.4). (F) In-gel fluorescent image (bottom) and SDS-PAGE (top) of βarr1 T6SeF after incubation with 150 μM Bodipy593 in Tris buffer (50 mM Tris, 150 mM NaCl, pH = 7.4) for 0–60 min. (G) Chemical structures of the unnatural amino acid SeF and tyrosine. | ||
Results and discussion
Characterization of the Se-click reaction
We chose to synthesize and genetically incorporate 2-amino-3-(4-hydroseleno-phenyl) propanoic acid (SeF), due to the significantly lowered pKa (5.9) and enhanced nucleophilicity of the benzeneselenol sidechain36 in comparison to the phenol (pKa = 10.2) sidechain in tyrosine. In addition, selenium can be used to determine protein structures through the multi-wavelength anomalous diffraction phasing method.37 We then examined the second-order reaction rate constant between SeF and Bodipy593 in 200 mM HEPES buffer (pH = 7.4) with 10% CH3CN as the co-solvent at room temperature, using liquid chromatography-mass spectrometry (LC-MS) and UV-Vis spectroscopy. To our delight, the reaction proceeded rapidly and generated an instantaneous color change through selenoate substitution of chlorine in Bodipy593 (Fig. 1B). The nucleophilic substitution reaction between mono-chlorinated Bodipy derivatives and nucleophiles relied on the electron-donating ability of the substituent groups. We designed the probe Bodipy593 by carefully choosing the amide moiety, whose electron-donating ability decreases the electrophilicity of Bodipy593 such that aliphatic thiols react with Bodipy593 at a very slow rate. However, due to the much stronger nucleophilicity of the phenylselenol group and its lower pKa (5.9) than cysteine (8.5), SeF reacts with Bodipy 3500-fold faster than N-acetyl-cysteine under physiological conditions (Fig. 1).The next step was to compare the reaction rate between SeF and N-acetyl cysteine. SeF reacted with Bodipy593 very rapidly, with a second-order rate constant of 26.3 ± 0.4 M−1 s−1 (Fig. 1C) which is comparable with that of the Cu(I) catalyzed azide–alkyne click reaction (Table S2†). In contrast, N-acetyl cysteine reacted with Bodipy593 very slowly (reaction rate is 0.0075 ± 0.0009 M−1 s−1, Fig. 1D), 3500 times slower than SeF. The lower pKa of selenol and the much higher nucleophilicity of selenoate versus thiol under neutral conditions38 may account for this drastic reaction rate advantage. These results indicate that if the protein labeling reaction is performed in pH 7.4 buffer where the SeF-labeled protein of interest (POI) and the Bodipy593 dye are present in 30 μM and 150 μM concentrations, respectively, more than 90% of the SeF-labeled POI is labeled within 15 min, while less than 0.1% of the cysteine residue is labeled (Fig. 2F). Since this reaction is carried out in neutral pH without requiring any catalyst and both the protein and fluorescent dye are only present in low concentrations, this bioconjugation reaction can be conveniently applied for the site-specific labeling of diverse protein complexes, without concern of either protein denaturation/precipitation in acidic buffer or the presence of copper/palladium catalysts. Importantly, the excitation and emission maxima of SeF-Bodipy593 were determined to be 565 nm and 585 nm, respectively, giving an approximately 50 nm bathochromic shift compared with Bodipy593 (Fig. 1B and S3†). Since a fluorescence background generated by the unconjugated fluorophore (which is usually hydrophobic and sticks to the hydrophobic surface of proteins or membranes) is often a big issue for fluorescence imaging and FRET measurement,7 this dramatic bathochromic shift constitutes an effective approach for the discrimination of the free Bodipy593 dye and labeled protein.
Incorporation of SeF into protein through genetic code expansion
To selectively incorporate SeF at defined sites in proteins in E. coli, a mutant Methanococcus jannaschii tyrosyl amber suppressor tRNA (MjtRNATyrCUA)/tyrosyl-tRNA synthetase (MjTyrRS) pair was evolved, which which uniquely specifies unnatural amino acid SeF in response to the TAG condon, but not any natural amino acid in response to the TAG codon. An MjTyrRS library, pBK-lib-jw1, was used to screen for active and selective mutants for SeF as previously reported.22 One MjTyrRS clone emerged after three rounds of positive selections and two rounds of negative selections, which grew by 120 μg mL−1 of chloramphenicol in the presence of 1 mM SeF, but by only 20 μg mL−1 of chloramphenicol in its absence.39 This clone was named SeFRS and sequencing revealed the following mutations: Tyr32His, Leu65Ser, His70Asn, and Asp158Asn. The structure of SeFRS was solved at 1.7 Å resolution to elucidate the molecular mechanism of the selective recognition of SeF by SeFRS (Fig. 2B and Table S3†). The complex structure of SeF/SeFRS was modeled using GLIDE 5.5 (grid-based ligand docking with energetics) program40 (Fig. S4†). Compared to the natural amino acid Tyr, the Se atom in SeF has a larger atomic radius and bears a negative charge in neutral pH. To accommodate the increased volume of SeF, three hydrophobic residues (L65, I159, and V164) in the binding pocket of the wild-type Mj-Tyr-aaRS were replaced by serines in SeFRS, whose smaller side-chain provides sufficient space to accommodate SeF (Fig. 2B and E). The D158N and Y32H mutations favored interactions between the positively charged amino acid sidechain and the anionic sidechain in SeF (Fig. 2C). These observations provided the structural basis for the specific and efficient incorporation of SeF in SeFRS. The selective recognition of SeF by SeFRS is a remarkable demonstration of the power of directed evolution, since SeF is an isostere of tyrosine, with single selenium atom replacement of the oxygen atom of tyrosine (Fig. 2C).Dual labeling of βarr1 with SeF-Bodipy593 and FlAsH
We next used the evolved SeFRS to incorporate SeF into βarr1. In the presence of SeFRS, the full-length βarr1-T6SeF protein was obtained after His-tag affinity column purification in 1 mg L−1 yield. By contrast, wild-type βarr1 was obtained in 2 mg L−1 yield (Fig. 2A and D). Mass spectrometric analysis showed the successful incorporation of SeF at the desired position T6 in βarr1 with approximately 100% efficiency after purification (Fig. 2A). We then evaluated the Se-click reaction for the site-specific modification of proteins. To 30 μM βarr1-T6SeF in reaction buffer (50 mM Tris, 150 mM NaCl pH = 7.4) 150 μM Bodipy593 was added, and then the mixture was incubated at room temperature for 30 min in the dark. The Se-click modification yield could then be monitored by in-gel fluorescence imaging (Fig. 2D). To achieve specific incorporation of the FRET donor for the βarr1-T6SeF-Bodipy593, we used FlAsH technology to introduce the fluorescent dye FlAsH-EDT2 into a CCPGCC motif41 inserted between the βarr1 Leu191/Met192 sites. The incorporation of FlAsH and Biodipy593 was confirmed by fluorescence emission at 460 and 620 nm, respectively (Fig. 2D). In the absence of the CCPGCC motif, no 460 nm emission was observed, and in the absence of SeF incorporation, no 620 nm emission was observed. Only in the presence of both the CCPGCC motif and SeF incorporation were both 460 and 620 nm emissions observed. These results indicate that FlAsH-EDT2 binds selectively to the CCPGCC site and Biodipy593 reacts selectively to SeF, demonstrating the mutual orthogonality of the FlAsH-EDT2/CCPGCC and Biodipy593/SeF protein labeling system. Since the FlAsH-EDT2/CCPGCC labeling method is already well-established and has been broadly applied to cellular imaging studies,41 the combination of the FlAsH-EDT2/CCPGCC and Biodipy593/SeF labeling system should provide a broadly applicable toolkit for smFRET studies.Characterization of SeF-Bodipy593 for smFRET
We then compared the photo-chemical properties of SeF-Bodipy593 with those of Cy3, which is one of the most popular fluorescent dyes used in smFRET. Our results show that SeF-Bodipy593 and Cy3 have similar absorption and emission spectra and photostability (Fig. 3A, Table S4†). Although SeF-Bodipy593 has a much shorter linker (3.4 Å, Fig. S5†) than Cy3 (15 Å),42 fluorescence anisotropy indicated that the orientational freedom of SeF-Bodipy593 and FlAsH is similar to that of Cy3 and Cy5 when they label proteins (Tables S5 and S6†). Together, we show that SeF-Bodipy593 is suitable for smFRET applications.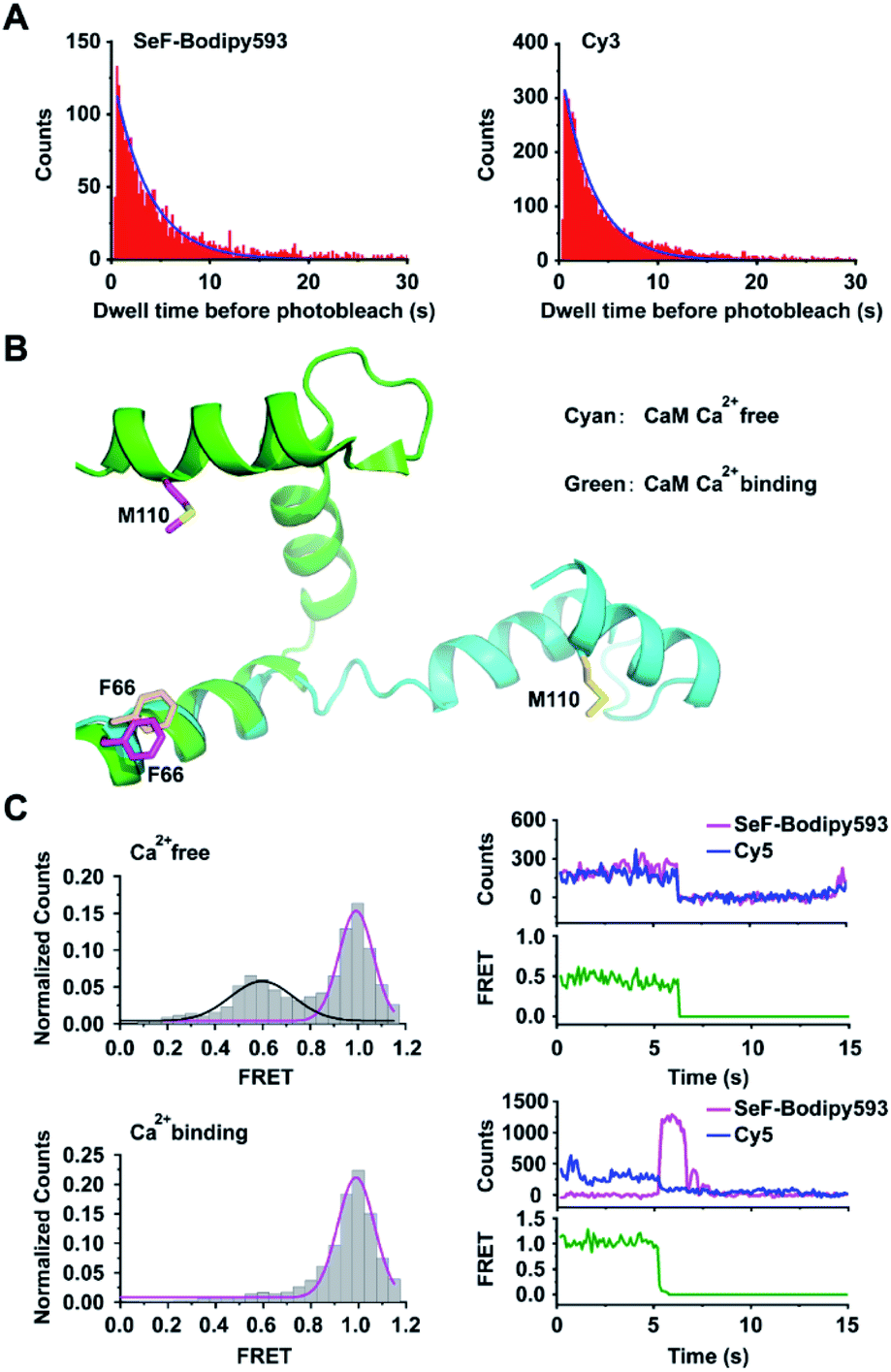 | ||
| Fig. 3 Application of Se-click based smFRET to capture conformational changes of calmodulin. (A) Comparison of the photobleaching rate between SeF-Bodipy593 and Cy3. Bodipy593 was labeled at the F66SeF position of CaM, and Cy3 was labeled at the ribosomal L11 protein.42 (B) Superimposition of the Ca2+-free CaM structure (cyan, extended conformation, PDB code 1cfd) and the Ca2+-bound CaM structure (green, compact conformation, PDB code 1prw). (C) FRET distribution of Ca2+-free CaM (top, FRET trace number: 662) and Ca2+-bound CaM (bottom, FRET trace number: 1022). The FRET efficiency of Ca2+-bound CaM is 0.99 ± 0.01, but the Ca2+-free CaM has two FRET distributions: E1 = 0.59 ± 0.02 and E2 = 0.99 ± 0.01. In the upper right panel, the donor photobleached at ∼7 s, leading to the loss of both donor and acceptor signals. In the lower right panel, the acceptor photobleached at ∼6 s, leading to the loss of FRET signal and a corresponding increase in donor signal. Both transition events are signatures to confirm that FRET is occurring. | ||
We then chose calmodulin (CaM),43 whose conformation is modulated by Ca2+ binding, to evaluate the application of the Se-click reaction in smFRET. CaM was labeled by expressing the CaM-F66SeF/M110C double mutant, followed by the Se-click reaction and cysteine–maleimide reaction, to generate the doubly labeled CaM-66SeF-Bodipy593/110C-Cy5 protein44 (Fig. 3B). Since there is a substantial overlap between the SeF-Bodipy593 fluorescence emission spectrum and Cy5 absorption spectrum, SeF-Bodipy593 and Cy5 form a good FRET pair with an R0 value of 43 Å (Fig. S6†). In the smFRET experiment, CaM-66SeF-Bodipy593/110C-Cy5 shows a single high-FRET population (FRET efficiency: 0.99 ± 0.01) in the presence of 0.1 mM Ca2+, but a low-FRET population (FRET efficiency: 0.59 ± 0.02) appeared when Ca2+ was removed through EDTA addition (Fig. 3C). These results agree with previous observations,45 and demonstrate the utility of Se-click in smFRET measurements.
Monitoring conformational changes of βarr1 activated by V2Rpp through smFRET
We then genetically incorporated SeF into functionally important structural motifs of βarr1, an important signaling component downstream of most G protein coupled receptor activations.46,47 Although recent crystallographic and NMR studies have unveiled critical knowledge about the arrestin structure and function,16,48,49 a single-molecule perspective of arrestin activation has not been established. One technical difficulty in studying arrestin by smFRET is the multiple cysteines harbored in native arrestins (7 for βarr1 and 8 for β-arrestin-2). Mutations of these cysteines to Ala would facilitate site-specific chemical labeling but also significantly perturb the structural and functional properties of arrestin. Therefore, to facilitate smFRET study of βarr1 activation, we exploited the Se-click strategy to introduce the SeF-Bodipy593 at a specific site of βarr1 as a FRET acceptor and a unique binding motif (CCPGCC) for the fluorescein arsenical hairpin (FlAsH-EDT2) at βarr1 in the C-terminal (R418) as a FRET donor50 (Fig. 4A). We selected two functionally important sites for SeF incorporation. One site was K157SeF, which is the proposed NFκB binding site located in the N-terminal half (N-domain) of βarr1 and mediates the regulatory role of arrestin in inflammatory functions (Fig. 4A). The other site was the T6SeF, which is the proposed JNK3 binding site at the N-domain and plays important roles in many physiological and pathological processes such as cell cycle, reproduction, apoptosis, and cell stress (Fig. 4A).51 The fidelity of SeF incorporation was confirmed by SDS-PAGE, and Bodipy593 labeling was easily detected by gel imaging (Fig. 2D). In order to validate that genetic incorporation of SeF or labeled dyes in βarr1 did not affect the functional integrity of βarr1, we conducted functional testing of clathrin recruitment. Clathrin is the key signaling molecule for receptor internalization and recognizes the active arrestin conformation.52,53 We used the fully phosphorylated 29 amino-acid carboxy-terminal peptide derived from the human V2 vasopressin receptor (V2Rpp),52 a general and widely used tool to stimulate the active arrestin conformation, to promote the interaction between clathrin and arrestin wild type or mutants. The results indicated that mutations or labeled dyes in βarr1 did not affect the V2Rpp induced βarr1 activation in the clathrin binding assay, thus indicating its functional integrity (Fig. S7†).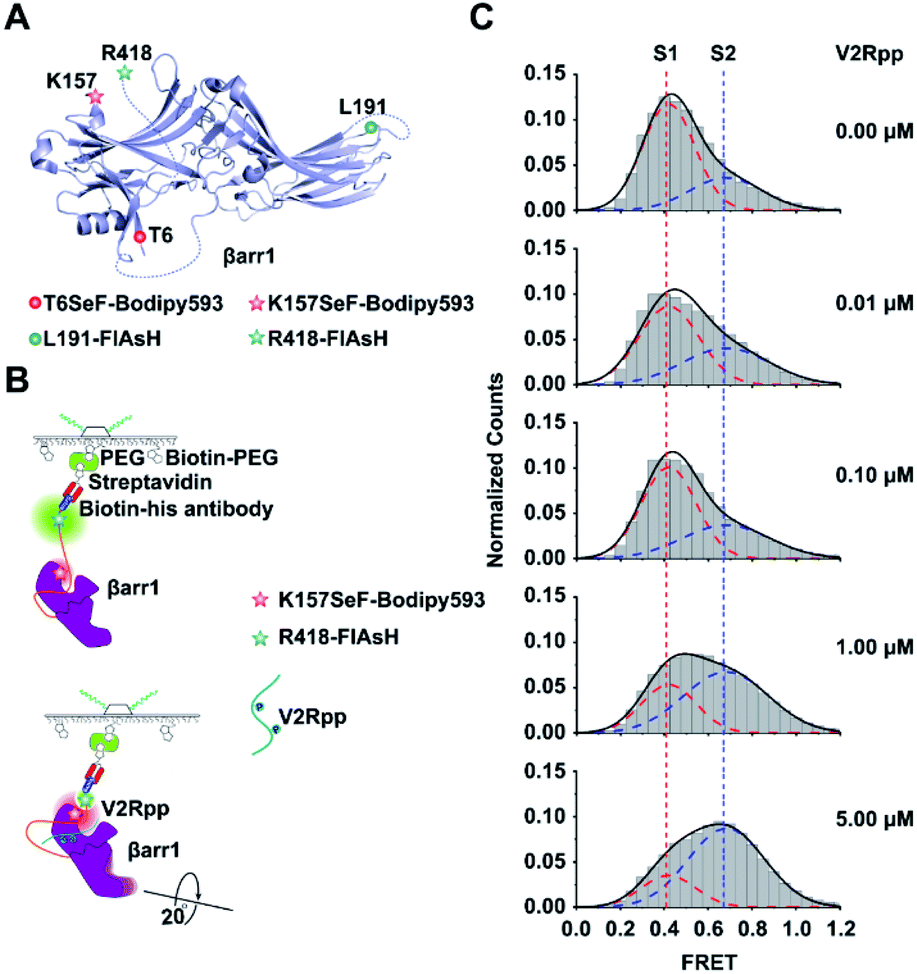 | ||
| Fig. 4 Application of the Se-click reaction in monitoring conformational changes of βarr1 activated by V2Rpp. (A) Schematic of labeling sites on βarr1. FlAsH is the FRET donor and Se-Bodipy593 is the FRET acceptor. The double mutant K157SeF-Bodipy593/R418-FlAsH examines conformational changes between the N-domain and C tail. The double mutant T6SeF-Bodipy593/L191-FlAsH examines conformational changes between the N- and C-domains. (B) Schematics of the smFRET analysis assay of βarr1 activated by V2Rpp. (C) Conformational population shift upon V2Rpp addition revealed by smFRET. The FRET trace numbers for smFRET efficiency histograms from top to bottom are 909, 949, 1188, 1273 and 1167. | ||
Two major sets of intramolecular interactions including the three-element (β-strand I, α-helix I and the C terminus of arrestin) interaction and the polar-core (five interacting charged residues: D26, R169, D290, D297 and R393) interaction have been proposed to constrain arrestin in an inactive conformation. A significant conformational rearrangement occurred once V2Rpp binds to βarr1. In V2Rpp-bound βarr1 structures, the N and C domains of βarr1 undergo a substantial twist relative to one another with a 20° rotation around a central axis, accompanied by conformation changes of the finger loop, middle loop and lariat loop exhibiting exhibiting the active state of βarr1.17,52 The V2Rpp-induced arrestin activation has been observed in both crystal structures and cryo-EM structures.48 In order to establish a single-molecule perspective of arrestin activation, arrestin activation by V2Rpp was then examined by imaging βarr1-K157SeF-Bodipy593/R418-FlAsH immobilized with biotin-conjugated His-antibodies (Fig. 4B). The activated βarr1 was generated by incubating βarr1 and V2Rpp at room temperature for 30 min and the sample was analyzed via smFRET (Fig. S8A and B†).17 In the absence of V2Rpp, two populations of βarr1 were identified by smFRET, which included a low-FRET state S1 (population 70%) centered at 0.42 ± 0.01 and a high-FRET state S2 (population 30%) centered at 0.68 ± 0.01 (Fig. 4C). Upon increasing the concentration of V2Rpp, the peak volume of S1 gradually decreased, and the peak volume of S2 increased (Fig. 4C). In order to better stabilize the S2 state and because of the difficulty in capturing the active conformation of βarr1 due to the significant conformational flexibility of active arrestin, the active arrestin conformation selective antibody Fab30 was used. Fab30 is extensively used in functional and structural study of arrestins.17 The formation of the active V2Rpp–arrestin–Fab30 complex was confirmed by fluorescence size exclusion chromatography (FSEC) (Fig. S9†). The addition of Fab30 resulted in the further population increase of the high-FRET state centered at 0.68 ± 0.01 (Fig. S10†). Together, these results indicate that the high FRET state S2 is the active arrestin state, whereas the low FRET state S1 is the inactive arrestin state.
Monitoring subtle conformational changes of the βarr1/GPCR complex
Our recent results have suggested that the β-1 strand is one of the functional motifs in mediating JNK3 activation downstream of β-arrestins.51 We therefore selected the T6 in the β-1 strand of arrestin and incorporated Bodipy593 at this site to examine how receptor binding triggers arrestin conformational change. We selected two labeling sites for smFRET, the T6 site and the L191 position, whose relative distance undergoes a 7 Å change (from 63 Å to 70 Å) (Table S7†) during the activation process of arrestin.17 To make a direct comparison between our SeF-Bodipy593/FlAsH FRET pair and the widely-used Cy3/Cy5 pair, two doubly-labeled proteins, βarr1-T6SeF-Bodipy593/L191FlAsH and βarr1-T6C/L191C-(Cy3/Cy5), were generated and subjected to smFRET measurement in the presence and absence of activated GPCRs. In order to monitor subtle conformational changes of the βarr1/GPCR complex,54 we exploited the model of pp-β2V2R, which is a chimaeric receptor generated by sortase-based ligation of a synthetic phosphorylated peptide (V2Rpp) onto the carboxyl terminus of β2AR, named “pp-β2V2R” in previously published studies.55 β2V2R maintains pharmacological properties identical to those of β2AR, but it binds βarr1 with higher affinity than wild-type β2AR. The phosphorylated V2R C-terminal tail is necessary to promote the interaction and improve the affinity between pp-β2V2R and βarr1 for structural and functional studies. Recent Cryo-EM studies exploiting these chimeric receptors with the conjugated ppV2R tail have provided important structural insights into the active conformations of βarr1.16,48 The labeled arrestin (1 nM) was incubated with pp-β2V2R (1 μM) and Fab30 (20 nM), stimulated by ISO (isopreterenol, 100 μM) at room temperature for 30 min. The sample was then analyzed via smFRET.Interestingly, structural changes could not be detected for βarr1-T6C/L191C-(Cy3/Cy5) before or after ISO/pp-β2V2R/Fab30 addition (Fig. S8C and D†), which in both cases only displayed a single conformational state (FRET efficiency E = 0.34 ± 0.02, Fig. 5B) corresponding to a 67 ± 2 Å Cy3/Cy5 distance. By contrast, while βarr1-T6SeF-Bodipy593/L191FlAsH presented a single high-FRET population in its inactive state, upon ISO/pp-β2V2R/Fab30 addition, two FRET populations emerged, including a high-FRET S1 state centered at 0.66 ± 0.02 (54 ± 1 Å between SeF-Bodipy593 and FlAsH) with a 51% population ratio, and full-width at half-maximum height values of 0.59 ± 0.05. Importantly, we observed a new low-FRET state S2 centered at 0.25 ± 0.01 (72 ± 1 Å between SeF-Bodipy593 and FlAsH), with a population ratio of 49%, and full-width at half-maximum height (FWHM) values of 0.19 ± 0.01 (Fig. 5C and S8E–G†). Thus, these results show that approximately half of the JNK3 binding site of βarr1, the β-1 strand, underwent conformational changes with respect to the C-edge domain (L191 position) in response to active phospho-β2AR engagement. Our results demonstrate that the SeF-Bodipy593/FlAsH FRET pair has significant advantages over the popular Cy3/Cy5 pair. All seven surface-exposed cysteines must be mutated for Cy3/Cy5 pair labeling in βarr1-T6C/L191C-(Cy3/Cy5), which might introduce significant perturbation to βarr1 and impair its ability to undergo conformation change. Due to the intrinsic limitation of the cysteine–maleimide chemistry, βarr1-T6C/L191C-(Cy3/Cy5) is a mixture of four components: βarr1-T6C-Cy3/L191C-Cy5, βarr1-T6C-Cy5/L191C-Cy3, βarr1-T6C-Cy3/L191C-Cy3, and βarr1-T6C-Cy5/L191C-Cy5. By contrast, no endogenous cysteine residue needs to be mutated to obtain the doubly-labeled βarr1-T6SeF-Bodpy593/L191-FlAsH, and each fluorescent dye was labeled uniquely at a specific position. In addition, the short linker between the protein backbone and SeF-Bodipy593/FlAsH dye is likely to decrease their AVs (Fig. 5A and Table S8†) and narrow the distributions of their relative distances,56 which reduces their spatial uncertainties and makes them more accurate reporters to indicate small distance changes in βarr1 than the Cy3/Cy5 dye pair. In addition, the short linker may minimize interactions between the fluorophore and detergent molecules in solution. Together, these properties may explain our observation that only the double mutant βarr1-T6SeF-Bodpy593/L191-FlAsH can successfully monitor the subtle conformational change of βarr1 upon activation. Together, we demonstrate the power of the Se-click reaction for precise and efficient protein labeling and its application in smFRET measurements for capturing subtle conformation changes.
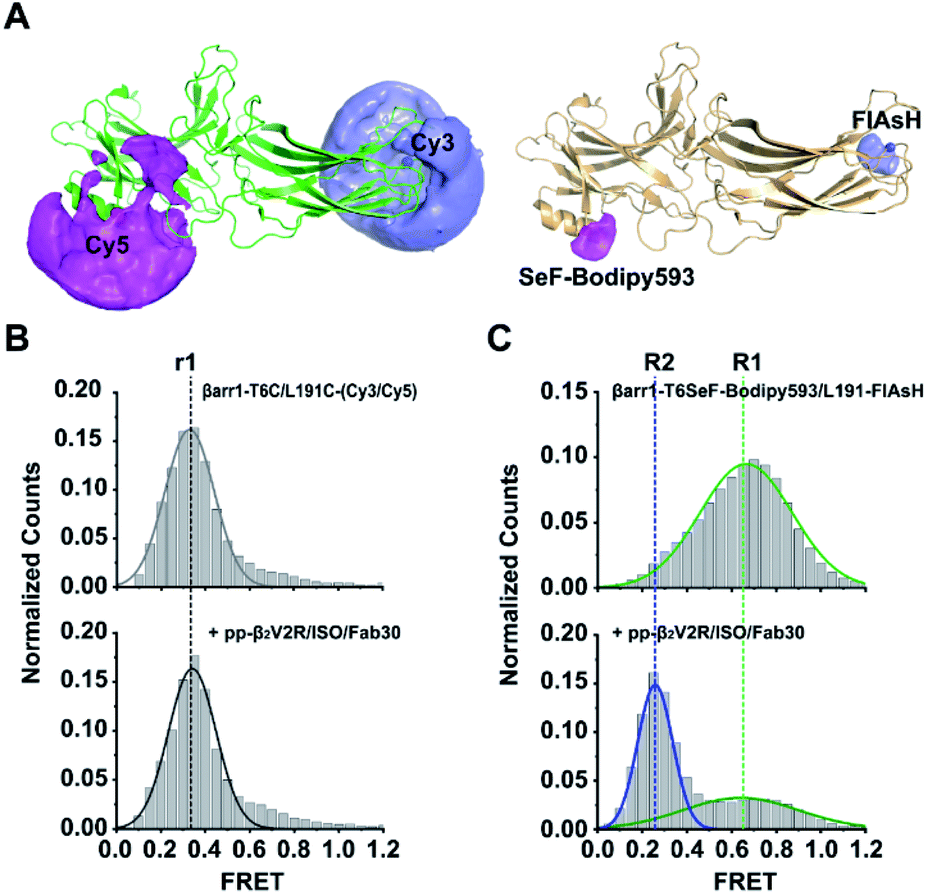 | ||
| Fig. 5 Subtle conformational changes captured using the SeF-Bodipy593/FlAsH FRET pair. (A) Accessible volumes (AVs) of various dyes (Cy3, Cy5, SeF-Bodipy593, and FlAsH)56 generated using software for FRET-restrained positioning and screening (FPS). (B) Conformation distributions of βarr1-T6C/L191C-(Cy3/Cy5), dashed lines r1 (black) indicate the mean FRET values. (C) βarr1-T6SeF-Bodpy593/L191-FlAsH before (top panel) and after (bottom panel) pp-β2V2R addition, measured by smFRET. Dashed lines highlight the mean FRET values of βarr1-T6SeF-Bodpy593/L191-FlAsH before (R1, green) and after (R2, blue) pp-β2V2R activation. R0 of Cy3/Cy5 is 60 Å, and R0 of SeF-Bodipy593/FlAsH is 60 Å. Data are shown as mean ± s.e.m. (B) and (C) show the representative data of three independent experiments. The FRET trace numbers for smFRET efficiency histograms: B (top): 1487, B (bottom): 1187, C (top): 3018, C (bottom): 2920. | ||
Conclusion
In conclusion, we have developed a facile Se-click reaction for site-specific protein labeling, which is particularly suitable for smFRET studies. The genetically encoded unnatural amino acid SeF is different from tyrosine by only one atom and should have minimal influence on the target protein. In comparison to other bioorthogonal reactions (Table S2†), the Se-click reaction generates a homogeneously labeled protein particularly important for smFRET measurement without requiring a catalyst, acidic pH or light irradiation. Nevertheless, if other suitable dyes were to be used, the electrophilicity of the dyes should be fine-tuned if the protein of interest contains cysteines, to ensure site-specific protein modification. After the reaction, SeF-Bodipy593 undergoes a 47 nm bathochromic shift compared with unreacted Bodipy593, which is advantageous for the separation of unreacted fluorescent dye signals. Compared with the popular fluorescent dye Cy3, SeF-Bodipy593 possesses similar absorption/emission spectra and comparable photophysical characteristics. Together, the Se-click reaction serves as a user-friendly site-specific protein labeling method, which will greatly facilitate application of smFRET to cysteine-rich proteins.Using the Se-click reaction, we investigated how a specific functional related conformation of arrestin was distributed during the engagement of an active phosphorylated GPCR. It is worth noting that arrestins and G proteins are two important downstream effectors of the GPCR superfamily, and account for more than 34% of direct targets of clinically used drugs.57 Although recent studies have used smFRET to provide the dynamics of GPCRs and their interactions with G proteins,10,58 the conformational perspective of arrestin engagement with GPCRs has not been achieved. Upon interaction with a GPCR harboring a phosphorylated C-tail and a conformation selective antibody, Fab30, only half of the JNK3 binding site of arrestin assumed an active conformation. It is likely that this small portion of the active arrestin conformation at this specific site was sufficient to direct the specific downstream signaling of a particular GPCR, considering the relatively high intracellular concentration of arrestin compared to other signaling proteins in cells. These observations are also consistent with the multi-functional and fast dynamic characteristics of arrestin.10,58 Taken together, our research suggests that smFRET measurement facilitated by the Se-click reaction and genetic code expansion may represent a general and facile approach for investigating the dynamic conformation change of membrane protein signaling complexes. Here, we provided a glimpse at the single-molecule level of the activation of a particular functional site of arrestin, the JNK3 interaction motif, in response to a phospho-receptor engagement. More information on the kinetics of the receptor–arrestin interaction, the conformational distributions of the receptor–arrestin complexes, and other important functional sites within the arrestin will be explored in the future using Se-click reaction assisted smFRET measurements.
Data availability
The datasets supporting this article have been uploaded as part of the supplementary information. Crystallographic data for SeFRS has been deposited at the PBD under 7C5C.Author contributions
J.-Y. W. designed the Se-click reaction and its application in smFRET. J.-Y. W., J.-P. S. and C.-L. C. conceived all the smFRET measurements. M.-J. H. performed experiments including synthesis of SeF and Boidpy593; characterization of the dynamics of the Se-click reaction and photophysical properties of the fluorophore, purification and conjugation of CaM and the βarr1 mutant; purification of pp-β2V2R; smFRET measurement; and data treatment. Q.-T. H. performed the SeF RNA synthase crystallization and X-ray diffraction, βarr1 mutant, pp-β2V2R and Fab30 purification, western blot detection, and smFRET measurement. C. C. and A.-P. H. synthesized the SeF compound. C.-L. C. and M.-Y. Y. performed the smFRET measurement of arrestin157SeF-Bodipy593/418FlAsH activation by V2Rpp and CaM data treatment. C.-L. C. and Y.-R. Y. performed the data treatment of the βarr1 mutant activation by pp-β2V2R. X.-H. L. and F. Y. participated in the experiment design. Z.-L. Z. and K.-K. Z. performed the SeFRS crystal analysis and docking. C. H. and X.-Z. G. participated in the dynamic data analysis and R0 calculation. J.-Y. W., J.-P. S. and C.-L. C. supervised all project design and execution. J.-Y. W., J.-P. S., C.-L. C., M.-J. H. and Q.-T. H. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank S. S. Zang and X.-H. L. for help with NMR spectra determination; X. Ding and Z. Xie for protein mass spectrometry measurement; and J.-H. L. for the anisotropy measurement. We thank the staff of BL19U1 beamlines at the National Center for Protein Sciences Shanghai and Shanghai Synchrotron Radiation Facility for assistance during data collection. We are grateful for the financial support from the National Key Research and Development Program of China (2016YFA0501502, 2019YFA0904200), the National Natural Science Foundation of China (21750003 to J. Y. W., 21503268 to M.-J. H., 21922704 and 21877069 to C. C., 31800716), the Sanming Project of Medicine in Shenzhen (No. SZSM201811092), the National Science Fund for Distinguished Young Scholars Grant (81825022 to J.-P. S.), the Beijing Natural Science Foundation (Z200019 to J.-P. S.), and the Shandong Provincial Natural Science Foundation (ZR2020ZD39 to J.-P. S.).References
- F. R. Hill, E. Monachino and A. M. van Oijen, Biochem. Soc. Trans., 2017, 45, 759–769 Search PubMed.
- T. Ha, T. Enderle, D. F. Ogletree, D. S. Chemla, P. R. Selvin and S. Weiss, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 6264–6268 Search PubMed.
- A. Robinson and A. M. van Oijen, Nat. Rev. Microbiol., 2013, 11, 303–315 Search PubMed.
- J. B. Munro, A. Vaiana, K. Y. Sanbonmatsu and S. C. Blanchard, Biopolymers, 2008, 89, 565–577 Search PubMed.
- Y. Gambin and A. A. Deniz, Mol. BioSyst., 2010, 6, 1540–1547 Search PubMed.
- X. Zhuang, L. E. Bartley, H. P. Babcock, R. Russell, T. Ha, D. Herschlag and S. Chu, Science, 2000, 288, 2048–2051 Search PubMed.
- E. Lerner, T. Cordes, A. Ingargiola, Y. Alhadid, S. Chung, X. Michalet and S. Weiss, Science, 2018, 359, 288 Search PubMed.
- W. R. Algar, N. Hildebrandt, S. S. Vogel and I. L. Medintz, Nat. Methods, 2019, 16, 815–829 Search PubMed.
- B. Hellenkamp, S. Schmid, O. Doroshenko, O. Opanasyuk, R. Kuhnemuth, S. R. Adariani, B. Ambrose, M. Aznauryan, A. Barth, V. Birkedal, M. E. Bowen, H. T. Chen, T. Cordes, T. Eilert, C. Fijen, C. Gebhardt, M. Gotz, G. Gouridis, E. Gratton, T. Ha, P. Y. Hao, C. A. Hanke, A. Hartmann, J. Hendrix, L. L. Hildebrandt, V. Hirschfeld, J. Hohlbein, B. Y. Hua, C. G. Hubner, E. Kallis, A. N. Kapanidis, J. Y. Kim, G. Krainer, D. C. Lamb, N. K. Lee, E. A. Lemke, B. Levesque, M. Levitus, J. J. McCann, N. Naredi-Rainer, D. Nettels, T. Ngo, R. Y. Qiu, N. C. Robb, C. Rocker, H. Sanabria, M. Schlierf, T. Schroder, B. Schuler, H. Seidel, L. Streit, J. Thurn, P. Tinnefeld, S. Tyagi, N. Vandenberk, A. M. Vera, K. R. Weninger, B. Wunsch, I. S. Yanez-Orozco, J. Michaelis, C. A. M. Seidel, T. D. Craggs and T. Hugel, Nat. Methods, 2018, 15, 984 Search PubMed.
- G. G. Gregorio, M. Masureel, D. Hilger, D. S. Terry, M. Juette, H. Zhao, Z. Zhou, J. M. Perez-Aguilar, M. Hauge, S. Mathiasen, J. A. Javitch, H. Weinstein, B. K. Kobilka and S. C. Blanchard, Nature, 2017, 547, 68–73 Search PubMed.
- I. S. Carrico, B. L. Carlson and C. R. Bertozzi, Nat. Chem. Biol., 2007, 3, 321–322 Search PubMed.
- M. Lu, X. Ma, L. R. Castillo-Menendez, J. Gorman, N. Alsahafi, U. Ermel, D. S. Terry, M. Chambers, D. Peng, B. Zhang, T. Zhou, N. Reichard, K. Wang, J. R. Grover, B. P. Carman, M. R. Gardner, I. Nikić-Spiegel, A. Sugawara, J. Arthos, E. A. Lemke, A. B. Smith, M. Farzan, C. Abrams, J. B. Munro, A. B. McDermott, A. Finzi, P. D. Kwong, S. C. Blanchard, J. G. Sodroski and W. Mothes, Nature, 2019, 568, 415–419 Search PubMed.
- H. Tian, A. Furstenberg and T. Huber, Chem. Rev., 2017, 117, 186–245 Search PubMed.
- R. Lamichhane, J. J. Liu, G. Pljevaljcic, K. L. White, E. van der Schans, V. Katritch, R. C. Stevens, K. Wuthrich and D. P. Millar, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14254–14259 Search PubMed.
- L. M. Wingler, M. Elgeti, D. Hilger, N. R. Latorraca, M. T. Lerch, D. P. Staus, R. O. Dror, B. K. Kobilka, W. L. Hubbell and R. J. Lefkowitz, Cell, 2019, 176, 468–478 Search PubMed.
- W. Huang, M. Masureel, Q. Qianhui, J. Janetzko, A. Inoue, H. E. Kato, M. J. Robertson, K. C. Nguyen, J. S. Glenn, G. Skiniotis and B. K. Kobilka, Nature, 2020, 579, 303–308 Search PubMed.
- A. K. Shukla, A. Manglik, A. C. Kruse, K. H. Xiao, R. I. Reis, W. C. Tseng, D. P. Staus, D. Hilger, S. Uysal, L. Y. Huang, M. Paduch, P. Tripathi-Shukla, A. Koide, S. Koide, W. I. Weis, A. A. Kossiakoff, B. K. Kobilka and R. J. Lefkowitz, Nature, 2013, 497, 137–141 Search PubMed.
- E. M. Milczek, Chem. Rev., 2018, 118, 119–141 Search PubMed.
- S. Milles, D. Mercadante, I. V. Aramburu, M. R. Jensen, N. Banterle, C. Koehler, S. Tyagi, J. Clarke, S. L. Shammas, M. Blackledge, F. Grater and E. A. Lemke, Cell, 2015, 163, 734–745 Search PubMed.
- M. G. Romei, C. Y. Lin, I. I. Mathews and S. G. Boxer, Science, 2020, 367, 76–79 Search PubMed.
- J. Fredens, K. Wang, D. de la Torre, L. F. H. Funke, W. E. Robertson, Y. Christova, T. Chia, W. H. Schmied, D. L. Dunkelmann, V. Beranek, C. Uttamapinant, A. G. Llamazares, T. S. Elliott and J. W. Chin, Nature, 2019, 569, 514–518 Search PubMed.
- C. C. Liu and P. G. Schultz, Annu. Rev. Biochem., 2010, 79, 413–444 Search PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 Search PubMed.
- B. Gold, G. B. Dudley and I. V. Alabugin, J. Am. Chem. Soc., 2013, 135, 1558–1569 Search PubMed.
- K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317–10320 Search PubMed.
- T. Plass, S. Milles, C. Koehler, J. Szymanski, R. Mueller, M. Wiessler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2012, 51, 4166–4170 Search PubMed.
- M. Baalmann, L. Neises, S. Bitsch, H. Schneider, L. Deweid, P. Werther, N. Ilkenhans, M. Wolfring, M. J. Ziegler, J. Wilhelm, H. Kolmar and R. Wombacher, Angew. Chem., Int. Ed., 2020, 59, 12885–12893 Search PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 Search PubMed.
- C. Zhang, M. Welborn, T. Zhu, N. J. Yang, M. S. Santos, T. Van Voorhis and B. L. Pentelute, Nat. Chem., 2016, 8, 120–128 Search PubMed.
- S. X. Lin, X. Y. Yang, S. Jia, A. M. Weeks, M. Hornsby, P. S. Lee, R. V. Nichiporuk, A. T. Iavarone, J. A. Wells, F. D. Toste and C. J. Chang, Science, 2017, 355, 597–602 Search PubMed.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 Search PubMed.
- R. K. V. Lim and Q. Lin, Acc. Chem. Res., 2011, 44, 828–839 Search PubMed.
- L. D. Hughes, R. J. Rawle and S. G. Boxer, PLoS One, 2014, 9, e87649 Search PubMed.
- Z. F. Zhang, D. Yomo and C. Gradinaru, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 1242–1253 Search PubMed.
- J. Liu, R. Cheng, N. Van Eps, N. Wang, T. Morizumi, W. L. Ou, P. C. Klauser, S. Rozovsky, O. P. Ernst and L. Wang, J. Am. Chem. Soc., 2020, 142, 17057–17068 Search PubMed.
- H. J. Reich and M. L. Cohen, J. Org. Chem., 1979, 44, 3148–3151 Search PubMed.
- W. A. Hendrickson, Science, 1991, 254, 51–58 Search PubMed.
- R. Mousa, R. N. Dardashti and N. Metanis, Angew. Chem., Int. Ed., 2017, 56, 15818–15827 Search PubMed.
- B. Yang, N. Wang, P. D. Schnier, F. Zheng, H. Zhu, N. F. Polizzi, A. Ittuveetil, V. Saikam, W. F. DeGrado, Q. Wang, P. G. Wang and L. Wang, J. Am. Chem. Soc., 2019, 141, 7698–7703 Search PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 Search PubMed.
- C. Hoffmann, G. Gaietta, M. Bunemann, S. R. Adams, S. Oberdorff-Maass, B. Behr, J. P. Vilardaga, R. Y. Tsien, M. H. Eisman and M. J. Lohse, Nat. Methods, 2005, 2, 171–176 Search PubMed.
- C. Chen, B. Stevens, J. Kaur, D. Cabral, H. Liu, Y. Wang, H. Zhang, G. Rosenblum, Z. Smilansky, Y. E. Goldman and B. S. Cooperman, Mol. Cell, 2011, 42, 367–377 Search PubMed.
- M. S. DeVore, A. Braimah, D. R. Benson and C. K. Johnson, J. Phys. Chem. B, 2016, 120, 4357–4364 Search PubMed.
- A. Yildiz, J. N. Forkey, S. A. McKinney, T. Ha, Y. E. Goldman and P. R. Selvin, Science, 2003, 300, 2061–2065 Search PubMed.
- C. K. Johnson, Biochemistry, 2006, 45, 14233–14246 Search PubMed.
- D. L. Zhang, Y. J. Sun, M. L. Ma, Y. J. Wang, H. Lin, R. R. Li, Z. L. Liang, Y. Gao, Z. Yang, D. F. He, A. Lin, H. Mo, Y. J. Lu, M. J. Li, W. Kong, K. Y. Chung, F. Yi, J. Y. Li, Y. Y. Qin, J. X. Li, A. R. B. Thomsen, A. W. Kahsai, Z. J. Chen, Z. G. Xu, M. Y. Liu, D. L. Li, X. Yu and J. P. Sun, Elife, 2018, 7, e33432 Search PubMed.
- C. H. Liu, Z. Gong, Z. L. Liang, Z. X. Liu, F. Yang, Y. J. Sun, M. L. Ma, Y. J. Wang, C. R. Ji, Y. H. Wang, M. J. Wang, F. A. Cui, A. Lin, W. S. Zheng, D. F. He, C. X. Qu, P. Xiao, C. Y. Liu, A. R. B. Thomsen, T. J. Cahill, A. W. Kahsai, F. Yi, K. H. Xiao, T. Xue, Z. A. Zhou, X. Yu and J. P. Sun, Nat. Commun., 2017, 8, 14335 Search PubMed.
- D. P. Staus, H. Hu, M. J. Robertson, A. L. W. Kleinhenz, L. M. Wingler, W. D. Capel, N. R. Latorraca, R. J. Lefkowitz and G. Skiniotis, Nature, 2020, 579, 297–302 Search PubMed.
- T. Weinert, P. Skopintsev, D. James, F. Dworkowski, E. Panepucci, D. Kekilli, A. Furrer, S. Brunle, S. Mous, D. Ozerov, P. Nogly, M. Wang and J. Standfuss, Science, 2019, 365, 61–65 Search PubMed.
- S. R. Adams and R. Y. Tsien, Nat. Protoc., 2008, 3, 1527–1534 Search PubMed.
- J. Y. Park, C. X. Qu, R. R. Li, F. Yang, X. Yu, Z. M. Tian, Y. M. Shen, B. Y. Cai, Y. Yun, J. P. Sun and K. Y. Chung, Structure, 2019, 27, 1162–1170 Search PubMed.
- F. Yang, X. Yu, C. Liu, C. X. Qu, Z. Gong, H. D. Liu, F. H. Li, H. M. Wang, D. F. He, F. Yi, C. Song, C. L. Tian, K. H. Xiao, J. Y. Wang and J. P. Sun, Nat. Commun., 2015, 6, 8202 Search PubMed.
- N. R. Latorraca, M. Masureel, S. A. Hollingsworth, F. M. Heydenreich, C. M. Suomivuori, C. Brinton, R. J. L. Townshend, M. Bouvier, B. K. Kobilka and R. O. Dror, Cell, 2020, 183, 1813–1825 Search PubMed.
- D. P. Staus, L. M. Wingler, M. J. Choi, B. Pani, A. Manglik, A. C. Kruse and R. J. Lefkowitz, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3834–3839 Search PubMed.
- F. Yang, P. Xiao, C. X. Qu, Q. Liu, L. Y. Wang, Z. X. Liu, Q. T. He, C. Liu, J. Y. Xu, R. R. Li, M. J. Li, Q. Li, X. Z. Guo, Z. Y. Yang, D. F. He, F. Yi, K. Ruan, Y. M. Shen, X. Yu, J. P. Sun and J. Y. Wang, Nat. Chem. Biol., 2018, 14, 876–886 Search PubMed.
- S. Sindbert, S. Kalinin, H. Nguyen, A. Kienzler, L. Clima, W. Bannwarth, B. Appel, S. Muller and C. A. Seidel, J. Am. Chem. Soc., 2011, 133, 2463–2480 Search PubMed.
- C. M. Suomivuori, N. R. Latorraca, L. M. Wingler, S. Eismann, M. C. King, A. L. W. Kleinhenz, M. A. Skiba, D. P. Staus, A. C. Kruse, R. J. Lefkowitz and R. O. Dror, Science, 2020, 367, 881–887 Search PubMed.
- T. Sungkaworn, M. L. Jobin, K. Burnecki, A. Weron, M. J. Lohse and D. Calebiro, Nature, 2017, 550, 543–547 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02653d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |