
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoinduced 1,2-dicarbofunctionalization of alkenes with organotrifluoroborate nucleophiles via radical/polar crossover†
María Jesús
Cabrera-Afonso‡
 a,
Anasheh
Sookezian‡
a,
Shorouk O.
Badir
a,
Mirna
El Khatib
b and
Gary A.
Molander
*a
a,
Anasheh
Sookezian‡
a,
Shorouk O.
Badir
a,
Mirna
El Khatib
b and
Gary A.
Molander
*a
aRoy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, Pennsylvania 19104-6323, USA. E-mail: gmolandr@sas.upenn.edu
bDepartment of Biochemistry and Biophysics, Perelman School of Medicine, University of Pennsylvania, Stellar-Chance Building, 422 Curie Boulevard, Philadelphia, Pennsylvania 19104-6059, USA
First published on 7th June 2021
Abstract
Alkene 1,2-dicarbofunctionalizations are highly sought-after transformations as they enable a rapid increase of molecular complexity in one synthetic step. Traditionally, these conjunctive couplings proceed through the intermediacy of alkylmetal species susceptible to deleterious pathways including β-hydride elimination and protodemetalation. Herein, an intermolecular 1,2-dicarbofunctionalization using alkyl N-(acyloxy)phthalimide redox-active esters as radical progenitors and organotrifluoroborates as carbon-centered nucleophiles is reported. This redox-neutral, multicomponent reaction is postulated to proceed through photochemical radical/polar crossover to afford a key carbocation species that undergoes subsequent trapping with organoboron nucleophiles to accomplish the carboallylation, carboalkenylation, carboalkynylation, and carboarylation of alkenes with regio- and chemoselective control. The mechanistic intricacies of this difunctionalization were elucidated through Stern–Volmer quenching studies, photochemical quantum yield measurements, and trapping experiments of radical and ionic intermediates.
Introduction
The vicinal difunctionalization of alkenes has emerged as an enabling technology in organic synthesis to access diverse structural skeletons from readily available building blocks.1,2 In particular, intermolecular 1,2-dicarbofunctionalization (DCF) reactions represent a powerful method to install two carbon subunits across an unsaturated system in one step with an accompanying increase in molecular complexity.3 Traditionally, DCF efforts have been largely limited to transition-metal-mediated processes, with1 or without2 the aid of a photocatalyst (Fig. 1A). Palladium,4 nickel,5 copper,6 and other metals7 have all been utilized to afford 1,2-substituted products. Although these DCF advancements represented milestones in their own right, the necessity of a metal catalyst can lead to numerous deleterious pathways, such as β-hydride elimination, homocoupling, isomerization, or proto-demetalation.1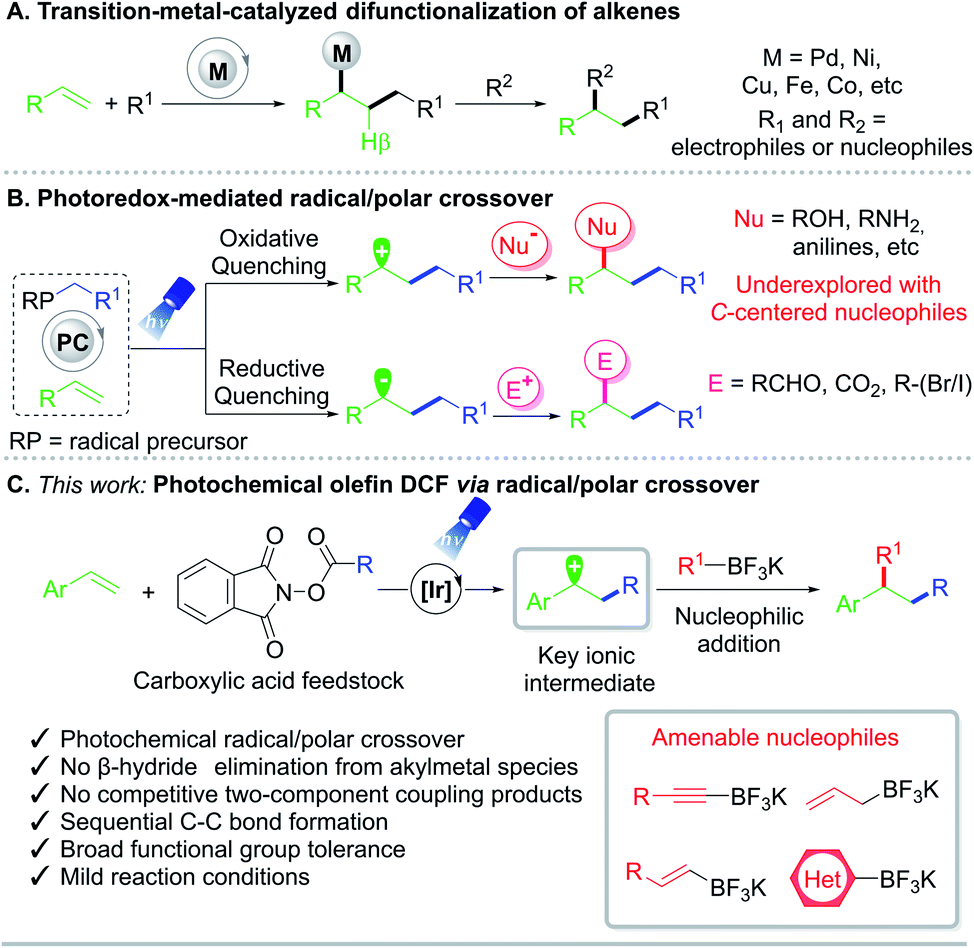 | ||
| Fig. 1 Strategies for olefin 1,2-difunctionalization. (A) Transition-metal-catalyzed difunctionalizations. (B) Overview of photochemical radical/polar crossover. (C) Developed vicinal dicarbofunctionalization using organotrifluoroborate nucleophiles. | ||
Radical/polar crossover (RPC) has recently been enlisted to assemble challenging structural motifs under mild reaction conditions (Fig. 1B).8,9 Under RPC paradigms, odd-electron intermediates are generated through single-electron transfer (SET) and then engage in further transformations. The resulting radical species can subsequently undergo single-electron reduction10,11 or oxidation12,13 to enter the two-electron reaction domain for further diversification. Although photo- and electrochemical RPC-mediated intermolecular 1,2-DCFs that proceed with in situ formed carbanions have been reported, these efforts are largely limited to carbonyl alkylation8d,10 or carbocarboxylation.8d,11
Methods to incorporate carbon-centered nucleophiles typically rely on the use of electrophilic radicals including perfluoroalkyl feedstocks as well as strongly nucleophilic, electron-rich systems in conjunction with Lewis acids or peroxides as additives.13 In this vein, the development of a general RPC route employing two carbon-based coupling partners for DCF remains elusive. Importantly, the implementation of a unified approach toward the carboallylation, carboalkenylation, carboalkynylation, and carboarylation of alkenes presents a formidable, yet highly powerful scenario to rapidly assemble molecular complexity from commodity chemicals.
Discussion
As part of a program centered on the development of catalytic tools for alkene functionalization, a photochemical intermolecular 1,2-dicarbofunctionalization of olefins with alkyl N-(acyloxy)phthalimide redox-active esters (RAEs) as radical progenitors has been developed (Fig. 1C). RAEs are bench-stable solids readily accessible from carboxylic acids with an established propensity to undergo decarboxylative fragmentation upon single-electron reduction.14 We envisioned that radical addition to vinyl arenes would generate key radical and carbocation intermediates that could be harnessed in sequential bond formation through RPC. With these goals in mind, the interrogation of potassium organotrifluoroborates as nucleophiles to construct C–C linkages under photoredox catalysis was considered, as they have been shown to engage in addition reactions under Brønsted and Lewis acid catalysis.15 Furthermore, alkynyltrifluoroborates have been enlisted as alternative nucleophilic partners in Suzuki–Miyaura cross-couplings, with the Bsp3–Csp bond being adequately polarized to engender a direct transmetalation event.16 From a synthetic standpoint and through the same RPC reactivity mode, the proposed strategy would facilitate carboallylation, carboalkenylation, carboalkynylation, and carboarylation from commodity chemicals with regio- and chemoselective control. Notably, this net-neutral photochemical RPC process is driven by SET events that occur predominantly between the photocatalyst and substrates/intermediates, bypassing the requirement for stoichiometric external reductants or oxidants.To examine the feasibility of the proposed reaction design, 4-acetoxystyrene 1a, aliphatic RAE 2a, and potassium (2-phenylethynyl)trifluoroborate 3a were employed (Table 1). A more detailed optimization of the equivalences of reaction components, catalyst loading, solvent, and reaction concentration is provided in the ESI.† Here, the most relevant findings are highlighted, namely the critical performance of photoredox catalysts on the DCF outcome.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Deviation from std. conditions | % Yieldb | Entry | Deviation from std. conditions | % Yieldb |
| a Reaction conditions: styrene 1a (0.1 or 0.2 mmol), RAE 2a (1.5 equiv.), potassium organotrifluoroborate salt 3a (2 equiv.), Ir(ppy)3 (3 mol%) in MeCN (0.1 M), 24 h irradiation with blue LEDs (λmax = 456 nm). b Yields were determined by 1H NMR analysis using trimethoxybenzene as internal standard. Abbreviations: std, standard; nr, no reaction. | |||||
| 1 | None | 89 | 10 | PC10 instead PC1 | nr |
| 2 | PC2 instead PC1 | 81 | 11 | PC11 instead PC1 | Trace |
| 3 | PC3 instead PC1 | 67 | 12 | PC12 instead PC1 | nr |
| 4 | PC4 instead PC1 | 91 | 13 | PC13 instead PC1 | nr |
| 5 | PC5 instead PC1 | 89 | 14 | 20 mol% Cu(OTf)2 | 32 |
| 6 | PC6 instead PC1 | 52 | 15 | 20 mol% Yb(OTf)3 | 36 |
| 7 | PC7 instead PC1 | 58 | 16 | No PC | nr |
| 8 | PC8 instead PC1 | 68 | 17 | No light | nr |
| 9 | PC9 instead PC1 | 81 | |||

Notably, the crux of this net-neutral RPC approach is a series of well-orchestrated, single-electron oxidation and reduction steps. To achieve chemo- and regio-selectivity, the following criteria must be considered: (i) the aliphatic RAE should be more susceptible to reduction than the alkene or the resulting benzylic radical formed upon addition to the olefin. (ii) The alkyl radical should react with alkene 1 at a rate faster than its single-electron oxidation to a carbocation intermediate or radical dimerization. (iii) The rate of benzylic radical oxidation must be competitive with its addition to another equivalent of the styrene. (iv) The rate of single-electron oxidation of the benzylic radical must be faster than that of the radical intermediate generated from RAE reduction. (v) The rate of nucleophilic addition of the potassium organotrifluoroborate to the benzylic carbocation should take place preferentially over single-electron oxidation of the organoboron reagent under photoredox conditions. (vi) This rate must also be competitive with the nucleophilic addition of phthalimide anions generated upon decarboxylative fragmentation of the RAE. In this vein, the choice of photocatalyst significantly impacts product distributions. Given the propensity of RAEs to undergo SET (Ered1/2 = −1.26 V vs. SCE for 1-methylcyclohexyl-N-hydroxyphthalimide-ester17), a palette of catalysts in acetonitrile (MeCN) was surveyed (entries 1–13). Reducing iridium-based (Ir) photocatalysts in combination with 2-phenylpyridine (ppy) derived-ligands (entries 1–9) exhibited optimal reactivity, affording 4a in excellent yield. Importantly, in the absence of a highly oxidizing photocatalyst, the corresponding organotrifluoroborates would not undergo SET.18 As expected, weaker reductants, PC10 (IrIV/*IrIIIE1/2 = −1.00 V vs. SCE19) and organic dye PC11 (PC˙+/PC* E1/2 = −1.12 V vs. SCE19), showed little (entry 11) to no conversion (entry 10). Notably, iron-based catalyst PC12 (FeIII/*FeIIE1/2 = −1.65 V vs. SCE19) and organic sensitizer PC13 (PC·+/PC* E1/2 = −2.1 V vs. SCE19) resulted in full recovery of the styrene derivative (entries 12–13), presumably because they possess a short excited-state lifetime (for PC13, τ = ∼0.8–2.3 ns (ref. 19)). Inspired by precedents on the activation of RAEs with Lewis acids,20 the influence of these additives on the outcome of the three-component photochemical paradigm was studied. Surprisingly, the addition of Cu(OTf)2 or Yb(OTf)3 led to a decrease in yield (entries 14–15). Control experiments omitting light as well as photocatalyst validated the necessity of all reaction components to facilitate sequential bond formation (entries 16–17).
With suitable conditions established, the scope of RAEs with nucleophile 3a was evaluated (Scheme 1). In general, the reaction is amenable to an array of unactivated secondary and tertiary radical architectures. The method further benefits from broad substrate tolerance, facilitating the incorporation of a strained cyclobutane subunit (4b), a Boc-protected amine (4d), a bridged bicycle (4e), acyclic moieties, as well as biologically relevant scaffolds including lipid-lowering agent gemfibrozil (4h). In addition, efficient product formation occurs in the presence of an internal olefin (4c), showcasing the chemoselectivity of this protocol toward styrenyl-type systems. Notably, RAE bearing a chloride handle (4g, 4k, 4y, 4ah, 4ai) can be introduced without compromising yields, delivering linchpins that can drive molecular complexity through subsequent transition metal-mediated functionalization.
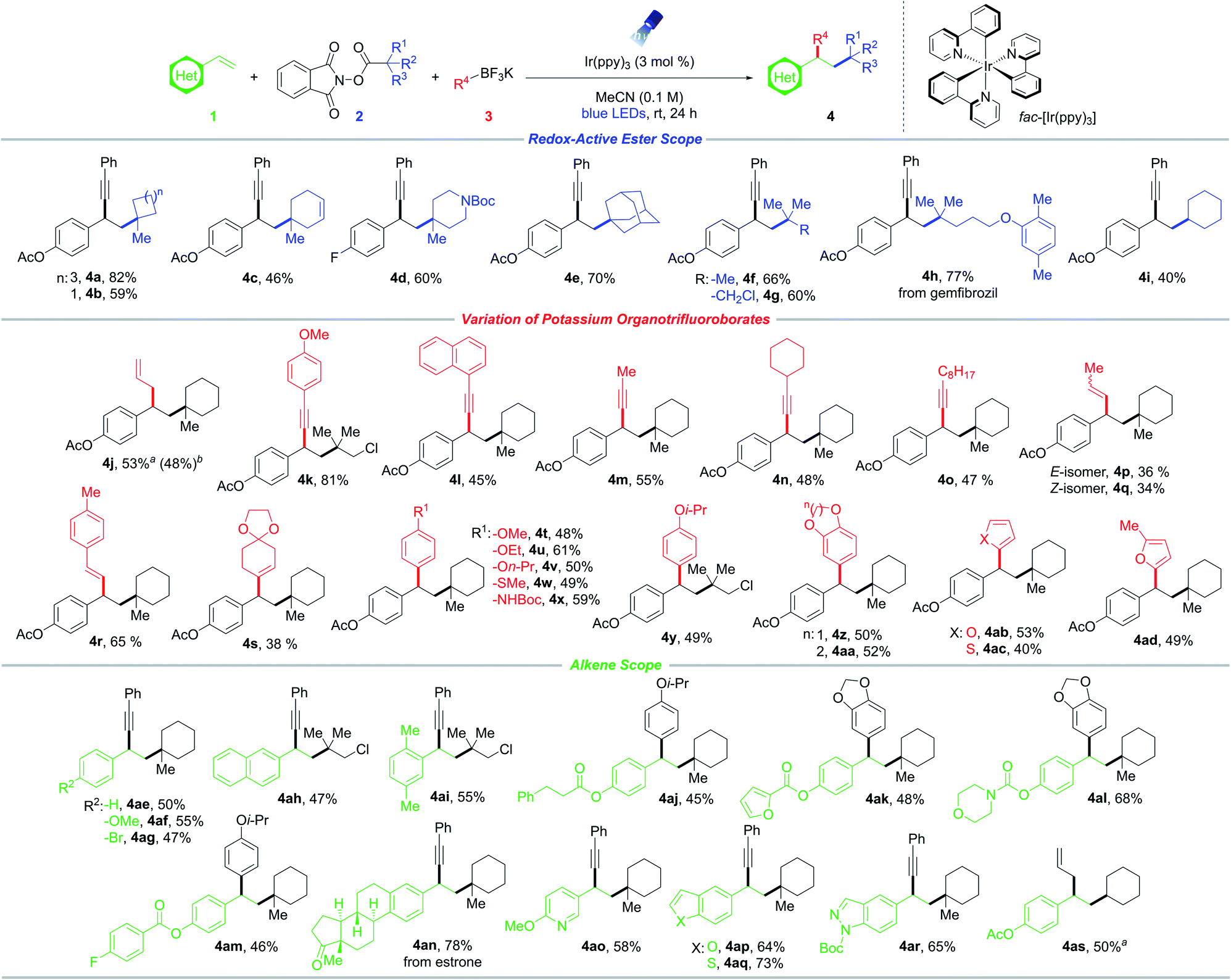 | ||
| Scheme 1 Evaluation of substrate scope. Reaction conditions: styrene 1 (0.3 mmol), RAE 2 (0.45 mmol, 1.5 equiv.), potassium organotrifluoroborate salt 3 (0.6 mmol, 2 equiv.), Ir(ppy)3 (3 mol%) in MeCN (3.0 mL, 0.1 M), 24 h irradiation with blue LEDs (λmax = 456 nm). a[Ir(dtbbpy)(ppy)2]PF6 (3 mol%) was used instead Ir(ppy)3. bGram scale reaction: styrene 1 (6.2 mmol). | ||
Next, the reactivity of various potassium organotrifluoroborates using 4-acetoxystyrene 1a was evaluated (Scheme 1). Carboallylation proved feasible, affording the difunctionalized product (4j) in good yield, while simultaneously incorporating an olefinic moiety that can engage in diverse downstream alkene transformations. Although progress has been made in conjunctive cross-couplings employing transition-metal catalysts with alkynyl, alkenyl, alkyl, and aryl electrophiles, the use of allyl counterparts remains scarce.21,22 In addition to β-hydride elimination associated with alkylmetal species, these processes are further complicated by the generation of undesired two-component allylation products. Importantly, the allyl handle might be susceptible to additional insertion events resulting in oligomerization.21,22 In this vein, the utility of the developed three-component allylation is partially driven by its ability to deliver two C(sp3)–C(sp3) linkages selectively from readily available building blocks. Similarly, potassium arylethynyltrifluoroborates bearing electron-donating (para-methoxy, 4k) or electron-neutral (naphthyl, 4l) substituents serve as effective nucleophiles. The phenyl moiety was successfully replaced by primary short- (4m) and long-chain (4o) alkyl groups as well as more sterically hindered carbocycles (4n). The scope was further extended to alkenyltrifluoroborates (4p–4s), with aryl-substituted derivatives performing slightly better under the reaction conditions. Notably, both the (E)- and (Z)-isomers of 1-propenyltrifluoroborate resulted in product formation with retention of stereochemistry about the olefin (4p, 4q).
Remarkably, aryltrifluoroborates function as competent nucleophiles, facilitating intermolecular 1,2-alkylarylation (Scheme 1). Substitution at the para- and meta-positions of the aryl scaffolds was explored, whereby efficient photocoupling took place. Specifically, alkoxy derivatives, a methyl thioether, and a Boc-protected amine were successfully harnessed to afford difunctionalized synthetic frameworks (4t–4aa). The amenability of aryltrifluoroborates provides a facile, unique approach toward the synthesis of 1,1-diaryl compounds of significance in drug discovery efforts.23 Furthermore, medicinally relevant heterocycles such as furan (4ab, 4ad) and thiophene (4ac) moieties exhibited good reactivity. Notably, this photoredox-mediated RPC proceeds exceptionally well with electron-rich aryl systems that suffer from lower coupling efficiency in certain transition-metal-catalyzed cross-couplings. The mild reaction conditions (room temperature, additive-free, and near-neutral pH) serve to suppress side reactivity stemming from an otherwise competitive hydrodeboration of the trifluoroborate starting material.
Finally, the scope of olefins was investigated (Scheme 1). In general, styrenes bearing no substitution, electron-donating, and electron-withdrawing groups at the ortho-, meta-, and para-positions exhibited comparable reactivity (4ae–4ai). Of note, aryl bromide 4ag proved to be a suitable substrate with complete retention of the halide handle, providing a clear advantage in terms of scope over traditional transition-metal-catalyzed DCFs. A broad array of functional groups is tolerated, including esters, ketones, Boc-protected amines, and carbamates (4aj–4ar). Additionally, a substrate derived from estrone was examined, generating steroid derivative 4an in excellent yield. Heterocyclic compounds including pyridine, benzofuran, benzothiophene, and indazole systems were readily incorporated under the developed conditions (4ao–4ar). In particular, these Lewis basic moieties are traditionally challenging structures in cross-couplings because of their ability to bind and poison the catalyst.
To confirm this RPC protocol was unique to potassium organotrifluoroborates, boronic acid 3xa, pinacol boronate 3xb, and MIDA boronate 3xc were tested as partner nucleophiles but proved ineffectual (Scheme 2A). These results are in accordance with the N-parameters reported by Mayr,24 a solvent-dependent nucleophilicity scale, which identified potassium organotrifluoroborates as one of the most reactive nucleophilic organoboron sources.
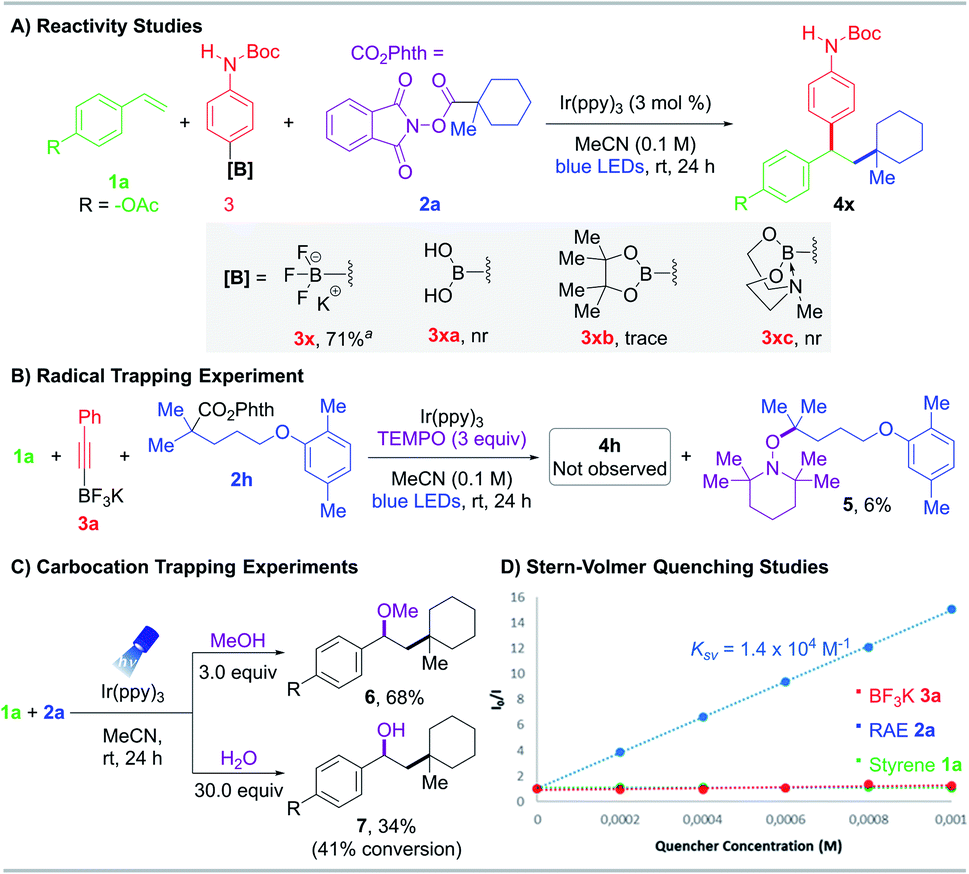 | ||
| Scheme 2 (A) Reactivity studies of organoboron compounds. (B) Radical trapping experiments. (C) Carbocation trapping experiments. (D) Stern–Volmer quenching. Reaction conditions: styrene 1 (0.2 mmol), RAE 2 (0.3 mmol, 1.5 equiv.), potassium organotrifluoroborate salt 3 (0.4 mmol, 2 equiv.), Ir(ppy)3 (3 mol%) in MeCN (2.0 mL, 0.1 M), 24 h irradiation with blue LEDs (λmax = 456 nm). aYield was determined by 1H NMR analysis using trimethoxybenzene as internal standard. Abbreviations: nr, no reaction. | ||
To investigate the reaction mechanism, radical and carbocation trapping studies were performed under standard conditions (Scheme 2). Addition of TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) inhibited product generation. Specifically, recovery of 4-acetoxystyrene 1a was observed, and the corresponding TEMPO adduct 5 was isolated and confirmed via NMR and HRMS analysis (Scheme 2B). To probe the intermediacy of carbocation species, nucleophilic trapping experiments were conducted using O-centered nucleophiles with slight modifications in the loading of these reagents (3.0 equiv. of MeOH or 30.0 equiv. of H2O). The corresponding ether 6 and alcohol 7 were successfully isolated and characterized, providing further credence to the existence of an ionic pathway (Scheme 2C).
Stern–Volmer luminescence studies of individual reaction components established that the excited state photocatalyst was quenched most effectively by the aliphatic RAE with an observed constant KSV of 1.4 × 104 M−1 (Scheme 2D, see the ESI†). Furthermore, the photochemical quantum yield Φ of this reaction is 0.26, indicating that a radical chain mechanism is unlikely or inefficient (see the ESI†).25
Based on these findings, a mechanistic scenario is postulated (Scheme 3) whereby excitation of Ir(ppy)3 under blue light irradiation generates a potent excited state *[Ir]III complex (E1/2 [IrIV/Ir*III] = −1.88 V vs. SCE19). Single-electron transfer (SET) to RAE 2 (Ered1/2 = −1.26 V vs. SCE for 1-methylcyclohexyl-N-hydroxyphthalimide ester17) induces formation of C(sp3)-hybridized radical A followed by extrusion of carbon dioxide. Subsequent addition of this reactive intermediate to vinyl arene 1 furnishes a relatively stabilized 2° benzylic radical B (Eox1/2 = 0.37 V vs. SCE26). Single-electron oxidation of this species by [Ir]IV (E1/2 [IrIV/IrIII] = 0.77 V vs. SCE19) yields the corresponding carbocation C, restoring the ground-state photocatalyst. At this critical juncture, ionic intermediate C is intercepted by the organotrifluoroborate nucleophile 3 to furnish the desired 1,2-dicarbofunctionalized product 4.
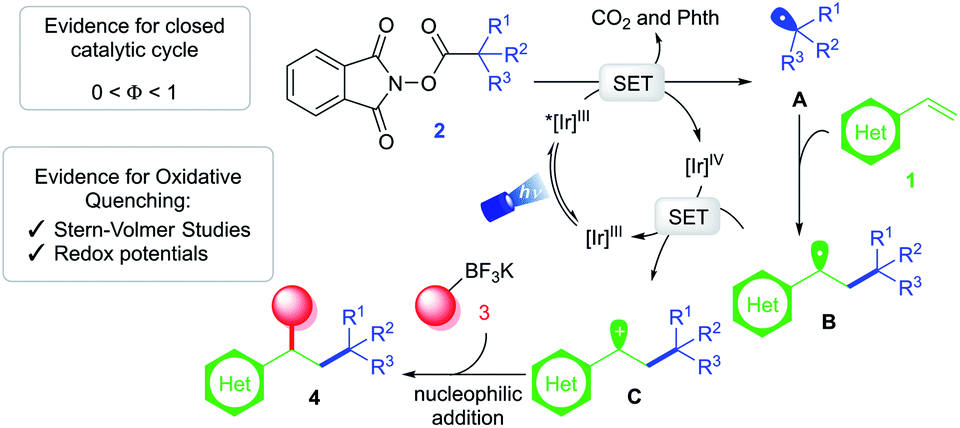 | ||
| Scheme 3 Proposed mechanism of photoinduced olefin dicarbofunctionalization via RPC. | ||
Conclusions
In summary, a unified protocol to achieve the carboallylation, carboalkenylation, carboalkynylation, and carboarylation of olefins with regio- and chemoselective control has been developed. Through an oxidative quenching pathway, the photoreduction of aliphatic RAEs has been enlisted to generate reactive alkyl radical intermediates that react with alkene feedstocks in a regulated fashion. The resulting C-centered radical undergoes single-electron oxidation to afford a key carbocation intermediate that is intercepted by organotrifluoroborate nucleophiles to facilitate sequential C–C bond formation under mild reaction conditions. Mechanistic studies, including Stern–Volmer quenching studies, photochemical quantum yield measurements, and trapping experiments of radical and ionic intermediates, emphasize that a RPC-mediated mechanism is likely operational. Most importantly, this report provides a general blueprint toward 1,2-dicarbofunctionalizations in the absence of organometal species.Author contributions
The project was conceived by Shorouk O. Badir. Dr María Jesús Cabrera-Afonso and Shorouk O. Badir performed Stern–Volmer quenching studies. Dr Mirna El Khatib conducted quantum yield experiments. Dr María Jesús Cabrera-Afonso and Anasheh Sookezian contributed equally. Dr María Jesús Cabrera-Afonso, Anasheh Sookezian, and Shorouk O. Badir performed experiments with input from Professor Gary A. Molander. Shorouk O. Badir, Dr María Jesús Cabrera-Afonso, and Anasheh Sookezian wrote the manuscript with input from Professor Gary A. Molander.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support provided by NIGMS (R35 GM 131680 to G. M.). Shorouk O. Badir is supported by the Bristol-Myers Squibb Graduate Fellowship for Synthetic Organic Chemistry. Dr María Jesús Cabrera-Afonso acknowledges the Fundación Ramón Areces for a Postgraduate Fellowship in Life and Matter Sciences. Anasheh Sookezian thanks Janssen Pharmaceuticals for financial support. Dr Mirna El Khatib is thankful for financial support provided by NHLBI (K25 HL145092-01). The NSF Major Research Instrumentation Program (award NSF CHE-1827457), the NIH supplement awards 3R01GM118510-03S1 and 3R01GM087605-06S1, as well as the Vagelos Institute for Energy Science and Technology supported the purchase of the NMRs used in this study. We thank Dr Sergei Vinogradov (UPenn) for his input in the photochemical quantum yield measurements. Stern–Volmer measurements were conducted in the Facility for Spectroscopic Excellence (UPenn) through a collaboration with JASCO Corporation. We thank Dr Charles W. Ross, III (UPenn) for mass spectral data. Johnson Matthey is acknowledged for the donation of iridium(III) chloride, Kessil for the donation of lamps, and Frontier Scientific for the donation of organotrifluoroborates.Notes and references
- For selected reviews in the presence of a photocatalyst, see: (a) J. Li, Y. Luo, H. W. Cheo, Y. Lan and J. Wu, Chem, 2019, 5, 192 Search PubMed; (b) S. O. Badir and G. A. Molander, Chem, 2020, 6, 1327 Search PubMed; (c) C. Zhu, H. Yue, L. Chu and M. Rueping, Chem. Sci., 2020, 11, 4051 Search PubMed.
- For selected reviews on DCF in the absence of a photocatalyst, see: (a) R. K. Dhungana, S. Kc, P. Basnet and R. Giri, Chem. Rec., 2018, 18, 1314 Search PubMed; (b) R. Giri and S. KC, J. Org. Chem., 2018, 83, 3013 Search PubMed; (c) J. Lin, R.-J. Song, M. Hu and J.-H. Li, Chem. Rec., 2019, 19, 440 Search PubMed.
- For representative synthesis of bioactive compounds, see: (a) S. KC, P. Basnet, S. Thapa, B. Shrestha and R. Giri, J. Org. Chem., 2018, 83, 2920 Search PubMed; (b) S. KC, R. K. Dhungana, V. Aryal and R. Giri, Org. Process Res. Dev., 2019, 23, 1686 Search PubMed; (c) T. Yang, X. Chen, W. Rao and M. J. Koh, Chem, 2020, 6, 738 Search PubMed.
- For representative examples using palladium, see: (a) M. Koy, P. Bellotti, F. Katzenburg, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 2375 Search PubMed; (b) M. Catellani and G. P. Chiusoli, Tetrahedron Lett., 1982, 23, 4517 Search PubMed; (c) E. W. Werner, K. B. Urkalan and M. S. Sigman, Org. Lett., 2010, 12, 2848 Search PubMed; (d) L. Liao, R. Jana, K. B. Urkalan and M. S. Sigman, J. Am. Chem. Soc., 2011, 133, 5784 Search PubMed; (e) M. Orlandi, M. J. Hilton, E. Yamamoto, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 12688 Search PubMed.
- For representative examples using nickel, see: (a) X. Qi and T. Diao, ACS Catal., 2020, 10, 8542 Search PubMed; (b) J. Derosa, O. Apolinar, T. Kang, V. T. Tran and K. M. Engle, Chem. Sci., 2020, 11, 4287 Search PubMed; (c) S. KC, R. K. Dhungana, B. Shrestha, S. Thapa, N. Khanal, P. Basnet, R. W. Lebrun and R. Giri, J. Am. Chem. Soc., 2018, 140, 9801 Search PubMed; (d) Q. Lin and T. Diao, J. Am. Chem. Soc., 2019, 141, 17937 Search PubMed; (e) D. Anthony, Q. Lin, J. Baudet and T. Diao, Angew. Chem., Int. Ed., 2019, 58, 3198 Search PubMed; (f) W. Shu, A. García-Domínguez, M. T. Quirós, R. Mondal, D. J. Cárdenas and C. Nevado, J. Am. Chem. Soc., 2019, 141, 13812 Search PubMed; (g) L. Guo, H.-Y. Tu, S. Zhu and L. Chu, Org. Lett., 2019, 21, 4771 Search PubMed; (h) S. -Z. Sun, Y. Duan, R. S. Mega, R. J. Somerville and R. Martin, Angew. Chem., Int. Ed., 2020, 59, 4370 Search PubMed; (i) S. KC, R. K. Dhungana, N. Khanal and R. Giri, Angew. Chem., Int. Ed., 2020, 59, 8047 Search PubMed; (j) R. K. Dhungana, R. R. Sapkota, L. M. Wickham, D. Niroula and R. Giri, J. Am. Chem. Soc., 2020, 142, 20930 Search PubMed; (k) X. Wei, W. Shu, A. García-Domínguez, E. Merino and C. Nevado, J. Am. Chem. Soc., 2020, 142, 13515 Search PubMed; (l) D. Anthony and T. Diao, Synlett, 2020, 31, 1443 Search PubMed; (m) L. Guo, M. Yuan, Y. Zhang, F. Wang, S. Zhu, O. Gutierrez and L. Chu, J. Am. Chem. Soc., 2020, 142, 20390 Search PubMed.
- For representative examples using copper, see: (a) Z. L. Li, G. C. Fang, Q. S. Gu and X. Y. Liu, Chem. Soc. Rev., 2020, 49, 32 Search PubMed; (b) L. Wu, F. Wang, X. Wan, D. Wang, P. Chen and G. Liu, J. Am. Chem. Soc., 2017, 139, 2904 Search PubMed; (c) L. Fu, S. Zhou, X. Wan, P. Chen and G. Liu, J. Am. Chem. Soc., 2018, 140, 10965 Search PubMed; (d) W. Sha, L. Deng, S. Ni, H. Mei, J. Han and Y. Pan, ACS Catal., 2018, 8, 7489 Search PubMed; (e) L. Wu, F. Wang, P. Chen and G. Liu, J. Am. Chem. Soc., 2019, 141, 1887 Search PubMed; (f) P.-Z. Wang, Y. Gao, J. Chen, X.-D. Huan, W.-J. Xiao and J.-R. Chen, Nat. Commun., 2021, 12, 1815 Search PubMed.
- For representative examples using other metals, see: (a) L. Liu, W. Lee, M. Y. Youshaw, M. B. Geherty, P. Y. Zavalij and O. Gutierrez, Chem. Sci., 2020, 11, 8301 Search PubMed; (b) J. Terao, K. Saito, S. Nii, N. Kambe and N. Sonoda, J. Am. Chem. Soc., 1998, 120, 11822 Search PubMed; (c) K. Mizutani, H. Shinokubo and K. Oshima, Org. Lett., 2003, 5, 3959 Search PubMed; (d) X.-H. Ouyang, R.-J. Song, M. Hu, Y. Yang and J.-H. Li, Angew. Chem., Int. Ed., 2016, 55, 3187 Search PubMed.
- For representative electrochemical DCF methods, see: (a) G. S. Sauer and S. Lin, ACS Catal., 2018, 8, 5175 Search PubMed; (b) H. Mei, Z. Yin, J. Liu, H. Sun and J. Han, Chin. J. Chem., 2019, 37, 292 Search PubMed; (c) J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547 Search PubMed; (d) W. Zhang and S. Lin, J. Am. Chem. Soc., 2020, 142, 20661 Search PubMed.
- For selected examples on photoredox-mediated RPC, see: (a) R. J. Wiles and G. A. Molander, Isr. J. Chem., 2020, 60, 281 Search PubMed; (b) L. Pitzer, J. L. Schwarz and F. Glorius, Chem. Sci., 2019, 10, 8285 Search PubMed.
- Q.-F. Bao, Y. Xia, M. Li, Y.-Z. Wang and Y.-M. Liang, Org. Lett., 2020, 22, 7757 Search PubMed.
- For representative RPC-mediated carboxylation reactions, see: (a) V. R. Yatham, Y. Shen and R. Martin, Angew. Chem., Int. Ed., 2017, 56, 10915 Search PubMed; (b) J. Hou, A. Ee, H. Cao, H.-W. Ong, J.-H. Xu and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 17220 Search PubMed; (c) B. Zhang, Y. Yi, Z.-Q. Wu, C. Chen and C. Xi, Green Chem., 2020, 22, 5961 Search PubMed; (d) H. Wang, Y. Gao, C. Zhou and G. Li, J. Am. Chem. Soc., 2020, 142, 8122 Search PubMed.
- For selected examples using heteroatom-based nucleophiles, see: (a) L. Li, H. Chen, M. Meia and L. Zhou, Chem. Commun., 2017, 53, 11544 Search PubMed; (b) X.-H. Ouyang, Y. Li, R.-J. Song and J.-H. Li, Org. Lett., 2018, 20, 6659 Search PubMed; (c) T. Patra, P. Bellotti, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 3172 Search PubMed; (d) E. W. Webb, J. B. Park, E. L. Cole, D. J. Donnelly, S. J. Bonacorsi, W. R. Ewing and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 9493 Search PubMed; (e) S. Shibutani, K. Nagao and H. Ohmiya, Org. Lett., 2021, 23, 1798 Search PubMed; (f) G. Fumagalli, S. Boyd and M. F. Greaney, Org. Lett., 2013, 15, 4398 Search PubMed.
- For selected RPC-mediated protocols using C-nucleophiles, see: (a) A. Carboni, G. Dagousset, E. Magnier and G. Masson, Chem. Commun., 2014, 50, 14197 Search PubMed; (b) Y. Duan, W. Li, P. Xu, M. Zhang, Y. Cheng and C. Zhu, Org. Chem. Front., 2016, 3, 1443 Search PubMed; (c) X. Wang, Y.-F. Han, X.-H. Ouyang, R.-J. Song and J.-H. Li, Chem. Commun., 2019, 55, 14637 Search PubMed; (d) M. Lux and M. Klussmann, Org. Lett., 2020, 22, 3697 Search PubMed; (e) F.-D. Lu, L.-Q. Lu, G.-F. He, J.-C. Bai and W.-J. Xiao, J. Am. Chem. Soc., 2021, 143, 4168 Search PubMed.
- S. Murarka, Adv. Synth. Catal., 2018, 360, 1735 Search PubMed.
- For selected examples, see: (a) S. Lee and D. W. C. MacMillan, J. Am. Chem. Soc., 2007, 129, 15438 Search PubMed; (b) C.-V. T. Vo, T. A. Mitchell and J. W. Bode, J. Am. Chem. Soc., 2011, 133, 14082 Search PubMed; (c) A. S. Vieira, P. F. Fiorante, T. L. S. Hough, F. P. Ferreira, D. S. Lüdtke and H. A. Stefani, Org. Lett., 2008, 10, 5215 Search PubMed; (d) S. Roscales and A. Csákÿ, Chem. Commun., 2014, 50, 454 Search PubMed; (e) T. N. Nguyen, T. S. Nguyen and J. A. May, Org. Lett., 2016, 18, 3786 Search PubMed; (f) R. William, S. Wang, A. Mallick and X.-W. Liu, Org. Lett., 2016, 18, 4458 Search PubMed; (g) S. Roscales, V. Ortega and A. G. Csákÿ, J. Org. Chem., 2018, 83, 11425 Search PubMed; (h) T. N. Nguyen and J. A. May, Org. Lett., 2018, 20, 3618 Search PubMed; (i) S. Sundstrom, T. S. Nguyen and J. A. May, Org. Lett., 2020, 22, 1355 Search PubMed.
- For selected reviews, see: (a) G. A. Molander, J. Org. Chem., 2015, 80, 7837 Search PubMed; (b) S. Roscales and A. Csákÿ, Chem. Soc. Rev., 2014, 43, 8215 Search PubMed.
- G. L. Lackner, K. W. Quasdorf, G. Pratsch and L. E. Overman, J. Org. Chem., 2015, 80, 6012 Search PubMed.
- For selected examples using organotrifluoroborates as radical precursors, see: (a) J. K. Matsui, D. N. Primer and G. A. Molander, Chem. Sci., 2017, 8, 3512 Search PubMed; (b) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2019, 58, 6152 Search PubMed; (c) J. Yi, S. O. Badir, R. Alam and G. A. Molander, Org. Lett., 2019, 21, 4853 Search PubMed.
- Y. Wu, D. Kim and T. S. Teets, Synlett, 2021, 32, A Search PubMed.
- S. P. Pitre, T. K. Allred and L. E. Overman, Org. Lett., 2021, 23, 1103 Search PubMed.
- For selected Tsuji–Trost allylation reactions, see: (a) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 Search PubMed; (b) J. T. Mohr and B. M. Stoltz, Chem.–Asian J., 2007, 2, 1476 Search PubMed; (c) J. D. Weaver, A. Recio III, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846 Search PubMed; (d) B. M. Trost, Tetrahedron, 2015, 71, 5708 Search PubMed; (e) N. A. Butt and W. Zhang, Chem. Soc. Rev., 2015, 44, 7929 Search PubMed.
- For selected allylation reactions, see: (a) A. Yanagisawa, N. Nomura, S. Habaue and H. Yamamoto, Tetrahedron Lett., 1989, 30, 6409 Search PubMed; (b) N. Nomura and T. V. RajanBabu, Tetrahedron Lett., 1997, 38, 1713 Search PubMed; (c) K. Lee, J. Lee and P. H. Lee, J. Org. Chem., 2002, 67, 8265 Search PubMed; (d) S. Son and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 2756 Search PubMed; (e) Y. Sumida, S. Hayashi, K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10, 1629 Search PubMed; (f) B. J. Stokes, S. M. Opra and M. S. Sigman, J. Am. Chem. Soc., 2012, 134, 11408 Search PubMed; (g) X. Cui, S. Wang, Y. Zhang, W. Deng, Q. Qian and H. Gong, Org. Biomol. Chem., 2013, 11, 3094 Search PubMed; (h) H. D. Srinivas, Q. Zhou and M. P. Watson, Org. Lett., 2014, 16, 3596 Search PubMed; (i) K. Hojoh, Y. Shido, K. Nagao, S. Mori, H. Ohmiya and M. Sawamura, Tetrahedron, 2015, 71, 6519 Search PubMed; (j) B. Yang and Z.-X. Wang, J. Org. Chem., 2017, 82, 4542 Search PubMed; (k) H. Chen, X. Jia, Y. Yu, Q. Qian and H. Gong, Angew. Chem., Int. Ed., 2017, 56, 13103 Search PubMed; (l) J. L. Hofstra, A. H. Cherney, C. M. Ordner and S. E. Reisman, J. Am. Chem. Soc., 2018, 140, 139 Search PubMed; (m) X.-G. Jia, P. Guo, J. Duan and X.-Z. Shu, Chem. Sci., 2018, 9, 640 Search PubMed; (n) K. Semba, N. Ohta, Y. Yano and Y. Nakao, Chem. Commun., 2018, 54, 11463 Search PubMed; (o) K. Semba, N. Ohta and Y. Nakao, Org. Lett., 2019, 21, 4407 Search PubMed; (p) H. Li, M. Zhang, H. Mehfooz, D. Zhu, J. Zhao and Q. Zhang, Org. Chem. Front., 2019, 6, 3387 Search PubMed; (q) V. T. Tran, Z. Q. Li, T. J. Gallagher, J. Derosa, P. Liu and K. M. Engle, Angew. Chem., Int. Ed., 2020, 59, 7029 Search PubMed.
- For selective examples, see: (a) D. Ameen and T. J. Snape, MedChemComm, 2013, 4, 893 Search PubMed; (b) X. Liu, Y. Xiao, J.-Q. Li, B. Fu and Z. Qin, Mol. Diversity, 2019, 23, 809 Search PubMed.
- G. Berionni, B. Maji, P. Knochel and H. Mayr, Chem. Sci., 2012, 3, 878 Search PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 Search PubMed.
- D. D. M. Wayner, D. J. McPhee and D. Griller, J. Am. Chem. Soc., 1988, 110, 132 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and mechanistic studies details, as well as spectral data. See DOI: 10.1039/d1sc02547c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |