
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amide-bridged conjugated organic polymers: efficient metal-free catalysts for visible-light-driven CO2 reduction with H2O to CO†
Fan
Wen‡
ac,
Fengtao
Zhang‡
ab,
Zhen
Wang
ab,
Xiaoxiao
Yu
ab,
Guipeng
Ji
a,
Dongyang
Li
ac,
Shengrui
Tong
a,
Yingbin
Wang
c,
Buxing
Han
 ab and
Zhimin
Liu
*ab
ab and
Zhimin
Liu
*ab
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Colloid and Interface and Thermodynamics, CAS, Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, 100190 Beijing, P. R. China. E-mail: liuzm@iccas.ac.cn
bSchool of Chemistry, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
cSchool of Science, China University of Geosciences, Beijing, 100083, P. R. China
First published on 13th July 2021
Abstract
The visible-light-driven photoreduction of CO2 to value-added chemicals over metal-free photocatalysts without sacrificial reagents is very interesting, but challenging. Herein, we present amide-bridged conjugated organic polymers (amide-COPs) prepared via self-condensation of amino nitriles in combination with hydrolysis, for the photoreduction of CO2 with H2O without any photosensitizers or sacrificial reagents under visible light irradiation. These catalysts can afford CO as the sole carbonaceous product without H2 generation. Especially, amide-DAMN derived from diaminomaleonitrile exhibited the highest activity for the photoreduction of CO2 to CO with a generation rate of 20.6 μmol g−1 h−1. Experiments and DFT calculations confirmed cyano/amide groups as active sites for CO2 reduction and second amine groups for H2O oxidation, and suggested that superior selectivity towards CO may be attributed to the adjacent redox sites. This work presents a new insight into designing photocatalysts for artificial photosynthesis.
Introduction
The massive consumption of fossil fuels has caused excessive CO2 emissions, and the CO2 issue has attracted worldwide attention.1 The photocatalytic reduction of CO2 with H2O to value-added chemicals is a promising and ideal way to realize CO2 transformation and has received much interest.2–4 To date, various kinds of photocatalysts including metal oxide/sulfide semiconductors,5,6 perovskite materials,7 metal–organic frameworks,8,9 and conjugated organic polymers (COPs)10,11 have been developed for the photocatalytic reduction of CO2. COPs as a class of metal-free materials with wide application have attracted continuous interest because of their high designability.12,13 Especially, COPs with CO2-philic functional groups (e.g. triazine, azo, amine and cyano groups) have been reported to show good performances in CO2 capture and transformation,14,15 and some heteroatom (e.g., N, P and S) doped COPs can realize CO2 photoreduction under visible light irradiation.16,17 For example, hexachlorocyclo-triphosphazene-derived COPs showed high CO2 absorption capacity and could photocatalyze CO2 reduction to CH4 under visible light irradiation in the presence of triethanolamine.18 So far, only a few photocatalysts can achieve the reduction of CO2 with H2O,10,19,20 and sacrificial reagents are required in most cases. Therefore, it is very demanding to efficiently couple the photoreduction of CO2 with the photooxidation of H2O in one photocatalytic system, especially under visible light irradiation. The natural photosynthesis of CO2 with H2O produces biomass with O2 release and no H2 generation. However, in the artificial photocatalytic process of CO2 with H2O the generation of side reaction products and H2 is hardly avoided. If the CO2 reduction could couple with the H2O oxidation perfectly, the H2 generation may be suppressed. To achieve this goal, a photocatalyst is the key, but such a photocatalyst is seldom reported.11Herein, we report amide-bridged COPs (amide-COPs, including amide-DAMN, amide-DAEN and amide-34AB) with –C–NH–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–C—as the structural unit and rich in the –CN group, for the photoreduction of CO2 with H2O, which were prepared via self-condensation of amino nitriles (i.e., diaminomaleonitrile, DAMN; 2,3-diaminobut-2-ene-1,4-dinitrile, DAEN; 3,4-diaminobenzonitrile, 34AB) in combination with subsequent hydrolysis,21,22 as illustrated in Scheme 1. These COPs could absorb visible light efficiently and exhibit high CO2 adsorption capacities. Importantly, they could realize the photoreduction of CO2 with H2O without any photosensitizer or sacrificial reagent under visible light irradiation, affording CO as the sole carbonaceous product without H2 generation. Especially, amide-DAMN with a suitable energy band structure (Eg = 2.19 eV, CB = −0.75 eV, and VB = 1.44 eV) displayed the highest activity for the photoreduction of CO2, affording CO with a production rate of 20.6 μmol g−1 h−1, much better than most reported metal-free catalysts. Density functional theory (DFT) calculations indicate that the adjacent redox sites of this kind of photocatalyst make the CO2 photoreduction couple well with H2O photooxidation, resulting in no H2 generation.
O)–C—as the structural unit and rich in the –CN group, for the photoreduction of CO2 with H2O, which were prepared via self-condensation of amino nitriles (i.e., diaminomaleonitrile, DAMN; 2,3-diaminobut-2-ene-1,4-dinitrile, DAEN; 3,4-diaminobenzonitrile, 34AB) in combination with subsequent hydrolysis,21,22 as illustrated in Scheme 1. These COPs could absorb visible light efficiently and exhibit high CO2 adsorption capacities. Importantly, they could realize the photoreduction of CO2 with H2O without any photosensitizer or sacrificial reagent under visible light irradiation, affording CO as the sole carbonaceous product without H2 generation. Especially, amide-DAMN with a suitable energy band structure (Eg = 2.19 eV, CB = −0.75 eV, and VB = 1.44 eV) displayed the highest activity for the photoreduction of CO2, affording CO with a production rate of 20.6 μmol g−1 h−1, much better than most reported metal-free catalysts. Density functional theory (DFT) calculations indicate that the adjacent redox sites of this kind of photocatalyst make the CO2 photoreduction couple well with H2O photooxidation, resulting in no H2 generation.
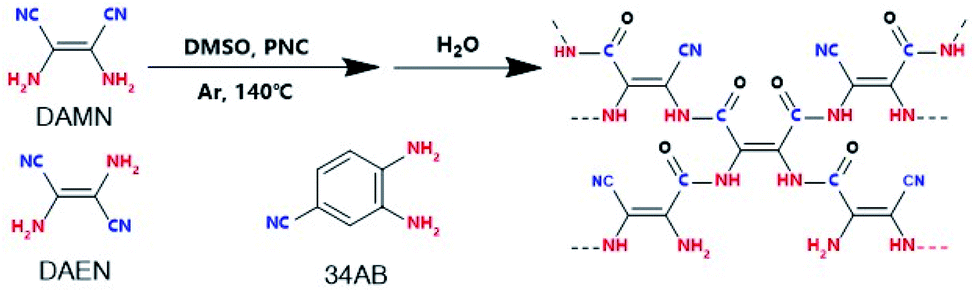 | ||
| Scheme 1 Synthetic procedure for amide-COP production. | ||
Results and discussion
The formation of amide-COPs was confirmed by Fourier-transform infrared (FT-IR), cross-polarization magic-angle spinning (CP/MAS) 13C NMR and X-ray photoelectron spectroscopy (XPS) analyses. In the FT-IR spectra (Fig. 1a and S1†), the cyano signal at around 2228 cm−1 of each polymer was significantly weakened as compared with that of the corresponding monomer, and new peaks appeared at 1672 and 1501 cm−1 corresponding to the stretching vibration of the CO bond and the deformation vibration of N–H in the samples.23,24 In CP/MAS 13C NMR spectra (Fig. S2†), the sp2 hybridized vinyl carbon and aromatic carbon species in the benzene rings are located in the range of 105 to 158 ppm. The well-defined signals at 160–165 ppm and 116–119 ppm can be attributed to the carbon in the amide groups and cyano groups, respectively. The signals of the aliphatic area may be due to the residual DMSO in the material and the partially added sp2 hybridized carbon.9,25 In the C 1s XPS spectrum of amide-DAMN (Fig. 1b), four peaks with the binding energies (BEs) at 288.5, 286.8, 286.2, and 284.7 eV are deconvoluted, ascribing to the CO, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, C–N and C–C/CC bonds, respectively.26 The deconvolution of the N 1s XPS spectrum (Fig. 1c) exhibits two peaks attributed to amino groups (N–Hx, x = 1, 2) at 400.0 eV and to the CN group at 399.1 eV.27,28 Correspondingly, amide-DAEN and amide-34AB showed similar XPS spectra (Fig. S3†).
N, C–N and C–C/CC bonds, respectively.26 The deconvolution of the N 1s XPS spectrum (Fig. 1c) exhibits two peaks attributed to amino groups (N–Hx, x = 1, 2) at 400.0 eV and to the CN group at 399.1 eV.27,28 Correspondingly, amide-DAEN and amide-34AB showed similar XPS spectra (Fig. S3†).
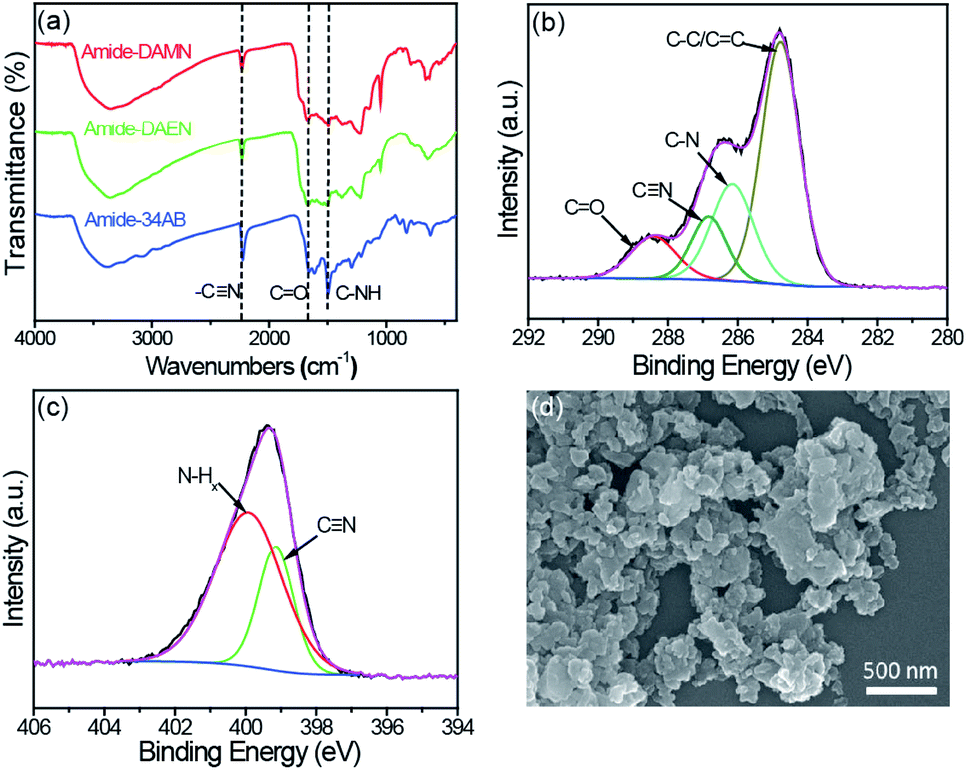 | ||
| Fig. 1 (a) FT-IR spectra of amide-COPs. (b) High resolution XPS C 1s spectrum of amide-DAMN. (c) High resolution XPS N 1s spectrum of amide-DAMN. (d) SEM image of amide-DAMN. | ||
The powder X-ray diffraction (XRD) analysis revealed that amide-COPs were amorphous (Fig. S4†), and field-emission scanning electron microscopy (SEM) and transmission electron microscopy (TEM) (Fig. 1d and Fig. S5†) observations showed that the polymers were an assembly of nanoparticles with irregular shapes. Their N2 adsorption–desorption isotherms represent typical type IV isotherms with definite type H3 hysteresis loops (0.6 < P/P0 < 1) (Fig. S6†), indicating the coexistence of mesopores and macropores in the polymeric matrices, consistent with Barrett–Joyner–Halenda (BJH) pore size distribution. The Brunauer–Emmett–Teller (BET) surface areas of amide-DAMN, amide-DAEN and amide-34AB were determined to be 47, 60 and 31 m2 g−1, respectively (Fig. S7†). The CO2 adsorption capacities of amide-DAMN, amide-DAEN and amide-34AB reached 23.6, 14.5 and 32.7 mg g−1 at 1 bar at 273 K, respectively (Fig. S8†). The porous structures and high CO2 adsorption capacities of amide-COPs may be favorable to the adsorption and activation of H2O and CO2 molecules.
The photophysical properties of amide-COPs were measured by UV-vis diffuse reflectance spectroscopy (DRS). As illustrated in Fig. 2a, amide-COPs have strong capability to absorb visible light. Based on the Tauc plots, the optical band gaps of amide-COPs were estimated to be 2.19, 2.09 and 1.74 eV for amide-DAMN, amide-DAEN and amide-34AB, respectively (Fig. S9†). Applying Mott–Schottky analysis, the conduction band (CB) positions were determined to be located at −0.75, −0.78 and −0.79 eV (vs. Ag/AgCl, pH = 6.8) for amide-DAMN, amide-DAEN and amide-34AB, respectively (Fig. S10†), and the positive slopes of the plots demonstrated their typical characters of n-type semiconductors.29 Accordingly, the valence band (VB) positions of amide-DAMN, amide-DAEN and amide-34AB were calculated to be at 1.44, 1.31 and 0.95 eV (vs. Ag/AgCl, pH = 6.8), respectively. From their band gap structures (Fig. 2b), it seems that all amide-COP samples may be capable of powering the reduction of CO2 to CO (−0.69 eV vs. Ag/AgCl, pH = 6.8) and oxidation of H2O to O2 (0.64 eV vs. Ag/AgCl, pH = 6.8). This implies that amide-COPs may have the capability to catalyze the photoreduction of CO2 with H2O.
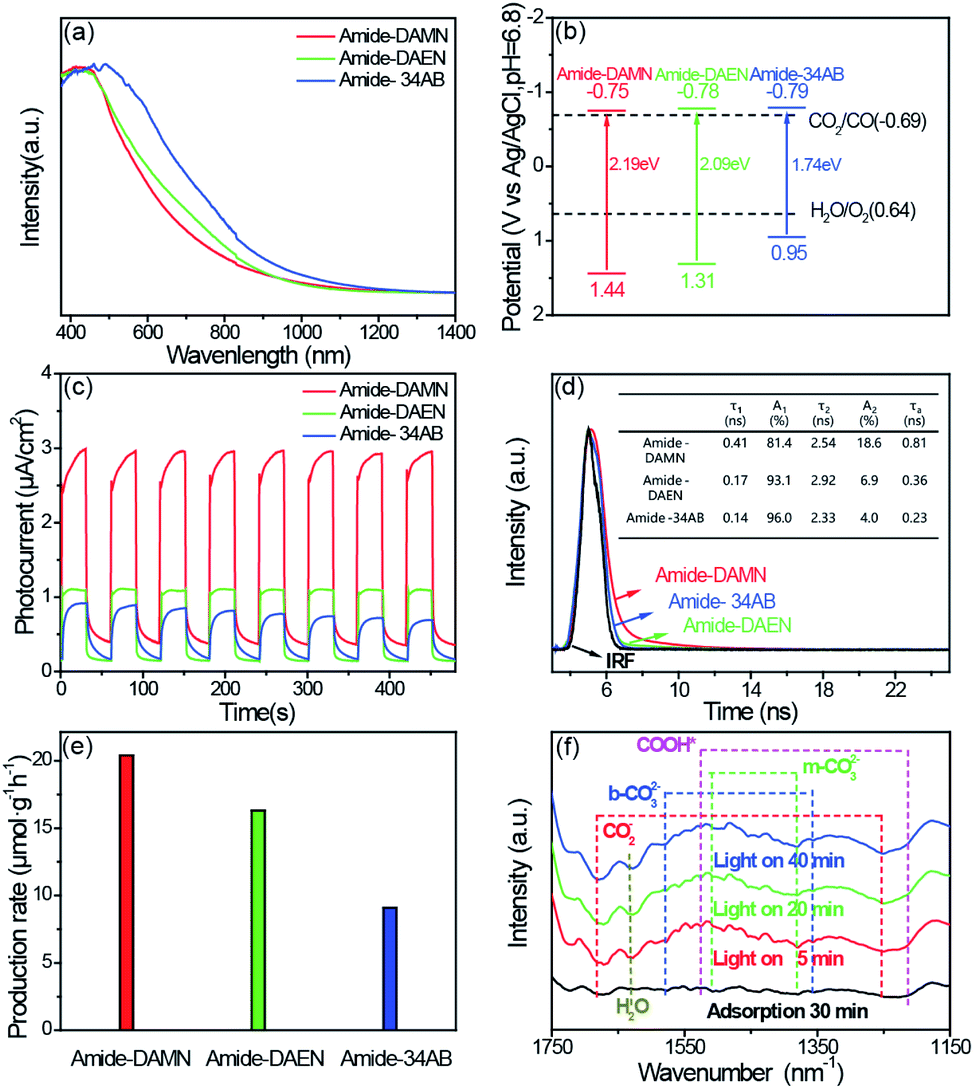 | ||
| Fig. 2 (a) UV/Vis light absorption spectra of amide-COPs. (b) Schematic band structure of amide-COPs. (c) Transient photocurrent responses of amide-COPs under visible light irradiation (λ > 420 nm). (d) Time-resolved PL spectra of amide-COPs. (e) Average product evolution rate over amide-COPs under visible light irradiation (λ > 420 nm). (f) In situ FT-IR spectra of amide-DAMN. | ||
The separation efficiency of photogenerated electrons and holes and their migration rate are related to the activity of the photocatalyst. Electrochemical characterization including transient photo-current, electrochemical impedance spectroscopy (EIS) and steady-state and time-resolved photoluminescence (PL and TRPL) was performed to determine the abilities of charge separation and transfer in amide-COPs. As depicted in Fig. 2c, fast and consistent photocurrent responses were revealed by turning lights on and off at the same time interval. Obviously, the photocurrent intensity of amide-DAMN (2.99 μA cm−2) was much higher than those of amide-DAEN (1.12 μA cm−2) and amide-34AB (0.93 μA cm−2), indicating that it is the most efficient for electron–hole pair separation. After 8 cycles, the peak photocurrents did not attenuate, which can infer that the catalysts can continuously and stably output electrons and holes during visible light irradiation. EIS spectra were recorded to evaluate the ability to transport charge carriers to the reactive sites.30 Amide-DAMN shows the smallest radius of the semicircular Nyquist plot (Fig. S11†), reflecting the lowest charge transfer resistance. The separation and recombination of photogenerated electron–hole pairs can be measured by steady-state PL analysis, and the low intensity of the emission peak indicates that the recombination of the photogenerated electron–hole pairs is efficaciously inhibited. The order of the emission peak positions of amide-COPs is consistent with the UV-Vis absorption spectra, and amide-DAMN shows the lowest photo-luminescence intensity (Fig. S12†), indicating that it has the highest separation efficiency of carriers.31 As shown in Fig. 2d, amide-DAMN shows the longest average photoluminescence lifetime of 0.81 ns, which is 2.3 and 3.5 times higher than those of amide-DAEN and amide-34AB, respectively. This result means that amide-DAMN could offer more opportunities for free charges to participate in the surface photoreaction, thus exhibiting higher photocatalytic activity.
The photocatalytic CO2 reduction performances of amide-COPs were tested under visible-light irradiation (λ > 420 nm) in a CO2 atmosphere at room temperature. As illustrated in Fig. 2e, each amide-COP was effective for the photoreduction of CO2 with H2O, affording CO as the only carbonaceous product, and no H2 was detectable by GC (Fig. S13†). Amide-DAMN afforded the highest CO production rate of 20.6 μmol h−1 g−1, showing much better performance than most reported metal-free photocatalysts, and even better than many metal-containing photocatalytic systems (Table S1†). Notably, these metal-free catalysts realized the visible-light-driven photoreduction of CO2 at very low overpotentials that were not found in a literature survey. This may be ascribed to their unique structures.
In contrast, amide-DAMN and amide-DAEN showed different photocatalytic activities though DAMN and DAEN are cis–trans isomers. This may be attributed to the differences in the structures and band gaps of the resultant polymers, and amide-DAMN was more conducive to the photo-generated charges to reach reactive sites.
To explore the origin of CO, a control experiment was performed using amide-DAMN as the catalyst in an Ar atmosphere under visible light irradiation, and no carbonaceous product was detected. The isotope labelling experiment of 13CO2 photoreduction with H2O afforded 13CO (m/z = 29) detected by GC-MS (Fig. S14a†), indicating that CO resulted from CO2 reduction. Furthermore, using a mixture of H218O (1 mL) and H2O (9 mL) as the reaction medium to perform the reaction, 16O18O (m/z = 34) and 18O2 (m/z = 36) were detected by GC-MS analysis, confirming that the photooxidation of H2O generated O2 (Fig. S14b†). The above results indicate that amide-COPs can realize the photocatalytic reduction of CO2 with H2O to CO and O2 under visible light irradiation. The fact that no H2 was detectable implies that the photoreduction of CO2 with H2O might follow a new mechanism different from those reported previously. In addition, amide-DAMN still retained high activity with only a slight decrease after being reused five times (Fig. S15†). TEM observation and FT-IR analysis (Fig. S16†) of the amide-DAMN sample reused 5 times indicated that no obvious morphology and structure changes were observed, unveiling its good robustness and durability in the reaction process.
To reveal the reaction mechanism for CO2 photoreduction with H2O over amide-DAMN, in situ FT-IR spectra were recorded after the photocatalyst was exposed to CO2 and water vapor for 30 min and subsequently irradiated with visible light. As shown in Fig. 2f, new peaks appeared and their intensity changed with the extension of irradiation time from 5 to 40 min. Obviously, the CO2 and H2O co-adsorbed on amide-DAMN gave rise to the signals of bidentate carbonate (b-CO32− at 1580 and 1358 cm−1),32 monodentate carbonate (m-CO32− at 1510 and 1380 cm−1)33,34 and physically adsorbed H2O (wide band at 1632 cm−1).35 The bands at 1684 and 1253 cm−1 are attributed to the vibration frequency of CO2−.32 It can be rationally inferred that the cyano groups and amide groups could capture CO2 and H2O and activate them. Importantly, new peaks at 1526 and 1214 cm−1 gradually strengthened with the prolonged irradiation time and match well with the COOH* species, giving information on the key intermediate for CO2 reduction to CO.33,34
To explore the photo-induced electron and charge carrier transfer in the catalyst, DFT calculations were performed taking the typical structural unit of amide-DAMN, named S-1 as shown in Fig. 3a and b, as a model compound. The natural bond orbital (NBO) analysis of the highest occupied molecular orbital (HOMO) (Fig. 3c) and the lowest unoccupied molecular orbital (LUMO) (Fig. 3d) were used to reveal the transformation of the photoexcited charge carrier in S-1. It was indicated that the HOMO of S-1 is mainly composed of the 2pz orbitals of 1(C), 6(N) and 21(C) centers (Table S2†). These atoms can serve as light-absorption sites and produce holes to realize the oxidation of water. The LUMO of S-1 mainly consists of the 2pz orbitals of 2(C), 3(O) and 12(N) centers (Table S3†), indicating that the photogenerated electrons could accumulate at the carbonyl/cyano groups for CO2 reduction. Consistent with the above results, the secondary amine group and the carbonyl/cyano groups are at the extreme points of the highest and lowest electrostatic potentials (ESPs), respectively, which are considered as potential redox active sites (Fig. 3e).
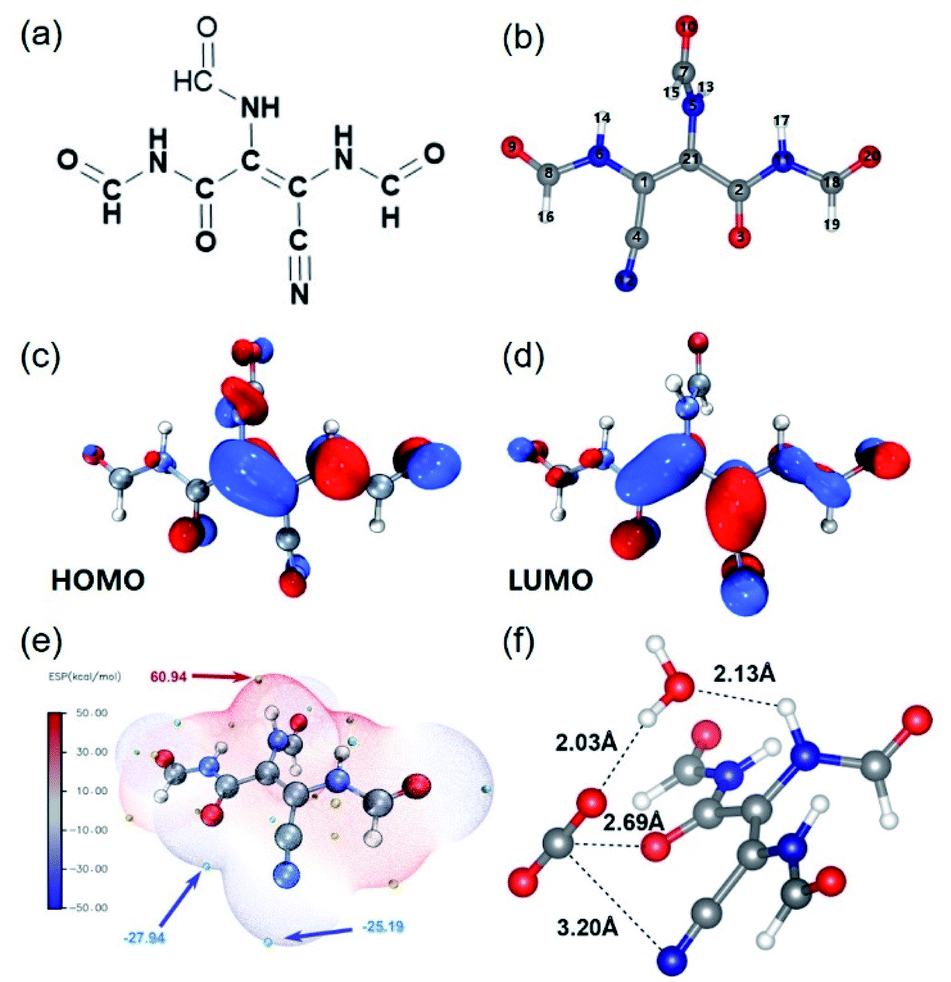 | ||
| Fig. 3 (a) Chemical structure of S-1. (b) Optimized structure of S-1, obtained by the NBO method. Blue, red, gray, and white spheres denote the positions of nitrogen, oxygen, carbon, and hydrogen atoms, respectively. (c) HOMO of S-1. (d) LUMO of S-1. (e) Electrostatic potential mapped of S-1. Color coding for ESP is from blue (negative) to red (positive). (f) The optimized structure of the intermediate in photocatalytic reactions. | ||
To further determine the reaction pathway, the interaction sites to attract CO2 on S-1 were explored by DFT calculations. As shown in Fig. S17,† both –CN and –CO groups in different positions could adsorb CO2. Both –CN and –CO groups that are adjacent to CC have higher binding energies than –CO that is far from CC, indicating that the –CN and –CO groups along the side of CC have more electron cloud density due to the inductive effect (Fig. S17a–c†). In addition, it is possible for both –CN and –CO to co-adsorb CO2 in a way as shown in Fig. S17d.† It can be inferred that CO2 is adsorbed and activated by carbonyl/cyano groups, at which it accepts photogenerated electrons (e−) from the LUMO of the catalyst to form CO2−. Meanwhile H2O is adsorbed and activated by the secondary amine group, at which it accepts photogenerated holes (h+) to realize oxidation, generating protons and releasing O2. From the optimized structure of the intermediate of CO2 reduction with H2O over S-1 (Fig. 3e), it is clear that one H2O molecule can be attracted by –NH– via forming a H-bond with a bond length of 2.13 Å, while one CO2 molecule can be co-captured by –CO and CN groups with bond lengths of 2.69 and 3.20 Å, respectively. Importantly, the captured H2O can form a H-bond with the captured CO2 with a bond length of 2.03 Å. From these results, it can be deduced that the proton generated from H2O oxidation may directly transfer to CO2− and involve in its reduction. This may explain why no H2 is generated in the reaction process.
Based on the above experimental results and analysis, a rational mechanism of photoreduction of CO2 with H2O over amide-DAMN is proposed (Fig. S18†). Upon visible-light irradiation, the photogenerated electrons accumulate at the cyano/carbonyl sites and transfer to the adsorbed CO2; meanwhile the photogenerated holes accumulate at the secondary amine sites and oxidize the adsorbed H2O, generating protons and releasing O2. Since the oxidation sites and the reduction sites match well, the proton generated from H2O oxidation may directly transfer to CO2− at CB, thereby producing the COOH* species, which is further reduced to CO.
Conclusions
In summary, amide-bridged COPs have been developed for the photoreduction of CO2 with H2O under visible-light irradiation without sacrificial agents and additives, which can afford CO as the sole carbonaceous product without H2 generation. Especially, amide-DAMN shows the highest CO production rate of 20.6 μmol g−1 h−1, much better than most reported metal-free catalysts. DFT calculations suggest that the reaction sites for CO2 reduction match perfectly with the sites for H2O oxidation, which makes the CO2 reduction couple well with the H2O oxidation, inhibiting the generation of H2. This work presents a new insight into designing COP photocatalysts for artificial photosynthesis.Experimental
Chemicals
All reagents and solvents were purchased from commercial sources and were used without further purification, unless indicated otherwise. Diaminomaleonitrile (≥98.5%, DAMN), 2,3-diaminobut-2-ene-1,4-dinitrile (≥95%, DAEN) and dimethyl sulfoxide (≥99.9%, DMSO) was purchased from J&K China Chemical Ltd. 3,4-Diaminobenzonitrile (≥97%, 34AB) and phosphonitrilic chloride trimer (≥98%, PNC) were purchased from Innochem China Chemical Ltd.Catalyst preparation
Typically, 1 mmol of phosphonitrilic chloride trimer (PNC) and 6 mmol of diaminomaleonitrile (DAMN) were, respectively, dissolved in 10 mL of ultra-dry DMSO. At 140 °C, under an argon atmosphere, the DMSO solution of DAMN was added dropwise into the DMSO solution of PNC. The mixture was stirred for 18 h at 140 °C. After being cooled to room temperature, 20 mL of water was added to the reaction system, and then the solid was collected via filtration, followed by washing with distilled water, tetrahydrofuran and ethanol several times. After being extracted using a Soxhlet extractor with ethanol, H2O and THF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) for 48 h, the final sample was obtained after being dried at 80 °C in a vacuum for 12 h, and named amide-DAMN.
:1:1) for 48 h, the final sample was obtained after being dried at 80 °C in a vacuum for 12 h, and named amide-DAMN.
Similarly, using 2,3-diaminobut-2-ene-1,4-dinitrile (DAEN) and 3,4-diaminobenzonitrile (34AB) as the monomers, the corresponding polymers, named amide-DAEN and amide-34AB, were obtained.
Photocatalytic carbon dioxide reduction experiment
In a typical photocatalytic experiment, 5 mg of polymer catalyst dispersed in 1 mL of ethanol was immobilized onto a glass sheet of 3 cm × 3 cm, and ethanol was then evaporated using an infrared lamp. The glass sheet with the catalyst was moved into a 250 mL photoreactor with 10 mL of H2O and suspended on the top of the reactor. Prior to illumination, the reactor was vacuumed and was subsequently backfilled with ultra-pure CO2 (99.999%) for about 1 h to reach the adsorption/desorption equilibrium of CO2 on the surface of the polymer catalyst. The pressure of the reactor was 1 atm and the temperature was kept at 25 °C using circulating water. Then the reactor was illuminated for a desired time (e.g., 10 h) with a 300 W Xe lamp (Aulight CEL-HX, Beijing) equipped with a 420 nm cut-off filter positioned 5 cm above the reactor. After irradiating for 10 h, 2 mL of the gaseous mixture was periodically sampled from the glass reactor at a given time interval, and analyzed by using a gas chromatograph equipped with a thermal conductivity detector (4890, Agilent Technology) with Ar as the carrier gas. The isotope-labelling experiment was performed using 13CO2 instead of 12CO2, H218O (1 mL) and H216O (9 mL) and the gaseous products were analyzed using gas chromatography-mass spectrometry.Data availability
All characterization, computational methods and data, copies of FT-IR, CP/MAS 13C NMR, XPS, XRD, EIS, photoluminescence spectra, TEM/SEM images, N2 sorption isotherms and pore size distribution, CO2 adsorption capacity isotherms, Tauc plots together with the bandgaps, and Motto–Schottky plots of the photocatalysts, GC spectrum of CO2 photoreduction products, GC-MS spectrum of 13CO2 photoreduction, calculation data, related to this study, can be found in the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (21773266 and 21890761).Notes and references
- S. J. Davis, K. Caldeira and H. D. Matthews, Science, 2010, 329, 1330–1333 CrossRef CAS.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS.
- T. A. Faunce, W. Lubitz, A. W. Rutherford, D. MacFarlane, G. F. Moore, P. Yang, D. G. Nocera, T. A. Moore, D. H. Gregory, S. Fukuzumi, K. B. Yoon, F. A. Armstrong, M. R. Wasielewski and S. Styring, Energy Environ. Sci., 2013, 6, 695–698 RSC.
- S.-H. Yu, Acta Phys.–Chim. Sin., 2020, 137, 2004010 Search PubMed.
- S. Neaţu, J. A. Maciá-Agullo, P. Concepción and H. Garcia, J. Am. Chem. Soc., 2014, 136, 15969–15976 CrossRef PubMed.
- L.-Z. Wu, Acta Phys.–Chim. Sin., 2020, 48, 2004005 Search PubMed.
- X.-D. Wang, Y.-H. Huang, J.-F. Liao, Y. Jiang, L. Zhou, X.-Y. Zhang, H.-Y. Chen and D.-B. Kuang, J. Am. Chem. Soc., 2019, 141, 13434–13441 CrossRef CAS.
- L. Zeng, X. Guo, C. He and C. Duan, ACS Catal., 2016, 6, 7935–7947 CrossRef CAS.
- Y. Benseghir, A. Lemarchand, M. Duguet, P. Mialane, M. Gomez-Mingot, C. Roch-Marchal, T. Pino, M.-H. Ha-Thi, M. Haouas, M. Fontecave, A. Dolbecq, C. Sassoye and C. Mellot-Draznieks, J. Am. Chem. Soc., 2020, 142, 9428–9438 CrossRef CAS PubMed.
- X. Yu, Z. Yang, B. Qiu, S. Guo, P. Yang, B. Yu, H. Zhang, Y. Zhao, X. Yang, B. Han and Z. Liu, Angew. Chem., Int. Ed., 2019, 58, 632–636 CrossRef CAS.
- M. Lu, J. Liu, Q. Li, M. Zhang, M. Liu, J.-L. Wang, D.-Q. Yuan and Y.-Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 12392–12397 CrossRef CAS PubMed.
- C. Dai and B. Liu, Energy Environ. Sci., 2020, 13, 24–52 RSC.
- Z. A. Lan, G. Zhang, X. Chen, Y. Zhang, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 10236–10240 CrossRef CAS.
- X. Zhu, C. Tian, G. M. Veith, C. W. Abney, J. Dehaudt and S. Dai, J. Am. Chem. Soc., 2016, 138, 11497–11500 CrossRef CAS.
- Y. Zeng, R. Zou and Y. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS PubMed.
- S. Guo, Z. Deng, M. Li, B. Jiang, C. Tian, Q. Pan and H. Fu, Angew. Chem., Int. Ed., 2016, 55, 1830–1834 CrossRef CAS PubMed.
- G. Liu, P. Niu, C. Sun, S. C. Smith, Z. Chen, G. Q. Lu and H. M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642–11648 CrossRef CAS PubMed.
- S. Guo, H. Zhang, Y. Chen, Z. Liu, B. Yu, Y. Zhao, Z. Yang, B. Han and Z. Liu, ACS Catal., 2018, 8, 4576–4581 CrossRef CAS.
- K. Lei, D. Wang, L. Ye, M. Kou, Y. Deng, Z. Ma, L. Wang and Y. Kong, ChemSusChem, 2020, 13, 1725–1729 CrossRef CAS.
- C. Bie, B. Zhu, F. Xu, L. Zhang and J. Yu, Adv. Mater., 2019, 1902868 CrossRef CAS.
- S. Song, X. Li, J. Wei, W. Wang, Y. Zhang, L. Ai, Y. Zhu, X. Shi, X. Zhang and N. Jiao, Nat. Catal., 2020, 3, 107–115 CrossRef CAS.
- C. L. Perrin and O. Nuñez, J. Am. Chem. Soc., 1987, 109, 522–527 CrossRef CAS.
- J. Zimmermann, M. C. Thielges, Y. J. Seo, P. E. Dawson and F. E. Romesberg, Angew. Chem., Int. Ed., 2011, 50, 8333–8337 CrossRef CAS.
- K. Gottschling, G. Savasci, H. Vignolo-González, S. Schmidt, P. Mauker, T. Banerjee, P. Rovó, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2020, 142, 12146–12156 CrossRef CAS PubMed.
- C. Mo, M. Yang, F. Sun, J. Jian, L. Zhong, Z. Fang, J. Feng and D. Yu, Adv. Sci., 2020, 7, 1902988 CrossRef CAS.
- B. A. Sexton and N. R. Avery, Surf. Sci., 1983, 129, 21–36 CrossRef CAS.
- T. Takahagi, I. Shimada, M. Fukuhara, K. Morita and A. Ishitani, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 3101–3107 CrossRef CAS.
- Q. Han, Z. Cheng, B. Wang, H. Zhang and L. Qu, ACS Nano, 2018, 12, 5221–5227 CrossRef CAS.
- Z. Zhang, J. Long, L. Yang, W. Chen, W. Dai, X. Fu and X. Wang, Chem. Sci., 2011, 2, 1826–1830 RSC.
- J. Ran, T. Y. Ma, G. Gao, X. W. Du and S. Z. Qiao, Energy Environ. Sci., 2015, 8, 3708–3717 RSC.
- I. V. Lightcap and P. V. Kamat, J. Am. Chem. Soc., 2012, 134, 7109–7116 CrossRef CAS PubMed.
- J. Tang, R. Guo, W. Pan, W. Zhou and C. Huang, Appl. Surf. Sci., 2019, 467–468, 206–212 CrossRef CAS.
- J. Wu, X. Li, W. Shi, P. Ling, Y. Sun, X. Jiao, S. Gao, L. Liang, J. Xu, W. Yan, C. Wang and Y. Xie, Angew. Chem., Int. Ed., 2018, 57, 8719–8723 CrossRef CAS.
- X. Jiao, X. Li, X. Jin, Y. Sun, J. Xu, L. Liang, H. Ju, J. Zhu, Y. Pan, W. Yan, Y. Lin and Y. Xie, J. Am. Chem. Soc., 2017, 139, 18044–18051 CrossRef CAS PubMed.
- L. Liu, H. Zhao, J. M. Andino and Y. Li, ACS Catal., 2012, 2, 1817–1828 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02499j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |