
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransfer of polyantimony units†
Veronika
Heinl
,
Andreas E.
Seitz
,
Gábor
Balázs
,
Michael
Seidl
and
Manfred
Scheer
 *
*
Institut für Anorganische Chemie, Universität Regensburg, 93040 Regensburg, Germany. E-mail: Manfred.Scheer@ur.de; Web: https://www.uni-regensburg.de/chemie-pharmazie/anorganische-chemie-scheer/
First published on 17th June 2021
Abstract
Transfer reagents are useful tools in chemistry to access metastable compounds. The reaction of [Cp′′2ZrCl2] with KSb(SiMe3)2 leads to the formation of the novel polyantimony triple decker complex [(Cp′′Zr)2(μ,η1:1:1:1:1:1-Sb6)] (1, Cp′′ = 1,3-di-tertbutyl-cyclopentadienyl), containing a chair-like Sb66− ligand. Compound 1 represents a valuable transfer reagent to form novel antimony ligand complexes. Thus, the reaction of 1 with CpR-substituted transition metal halides of nickel, cobalt and iron leads to the formation of a variety of novel Sbn ligand complexes, such as the cubane-like compounds [(Cp′′′Ni)4(μ3-Sb)4] (2) and [(Cp′′′Co)4(μ3-Sb4)] (3a) or the complexes [(CpBnCo)3(μ3-Sb)2] (4) and [(Cp′′′Fe)3(μ3-Sb)2] (5), representing a trigonal bipyramidal structure. Moreover, beside the transfer of Sb1 units, also the complete entity can be transferred as seen in the iron complex [(Cp′′′Fe)3(μ3,η4:4:4-Sb6)] (6). DFT calculations shed light on the bonding situation of the products.
Introduction
The reactivity of polypnictogen ligand complexes is an active field of chemical research, which resulted in numerous novel En complexes (E = P, As, Sb).1,2 Especially for phosphorus and arsenic, a large variety of examples have been synthesized so far, usually by employing white phosphorus or yellow arsenic in the reaction with transition metal or main group compounds.3–6 In contrast, the number of polyantimony ligand complexes is very limited and, therefore, their chemistry was far less investigated.7 Reasons for this can be attributed to various challenges such as the sensitivity of antimony-containing compounds towards light, the weak Sb–Sb bonding8 and especially the lack of suitable antimony sources. Since Sb4 is only known in the gas phase or under special conditions, e.g. trapped in a solid neon matrix,9 grey antimony, Zintl ions,10 organo-substituted Sb precursors11 and quite simply SbCl3 (ref. 12–14) and its derivatives were used as starting materials. In the 1970s, the first Sbn ligand complex [{Co(CO)3}4(μ3-Sb)4] was published by Dahl et al. by the reaction of Co(OAc)2·H2O with SbCl3 as antimony source (I, Fig. 1).12 Even in the case of the complexes [Sb2{W(CO)5}3]13 (II) and [{LGa(NMe2)}2(μ,η1:1-Sb4)]14 (III), SbCl3 and Sb(NMe2)3 are used as precursors (Fig. 1, L = HC[C(Me)N(2,6-C6H3iPr2)]2).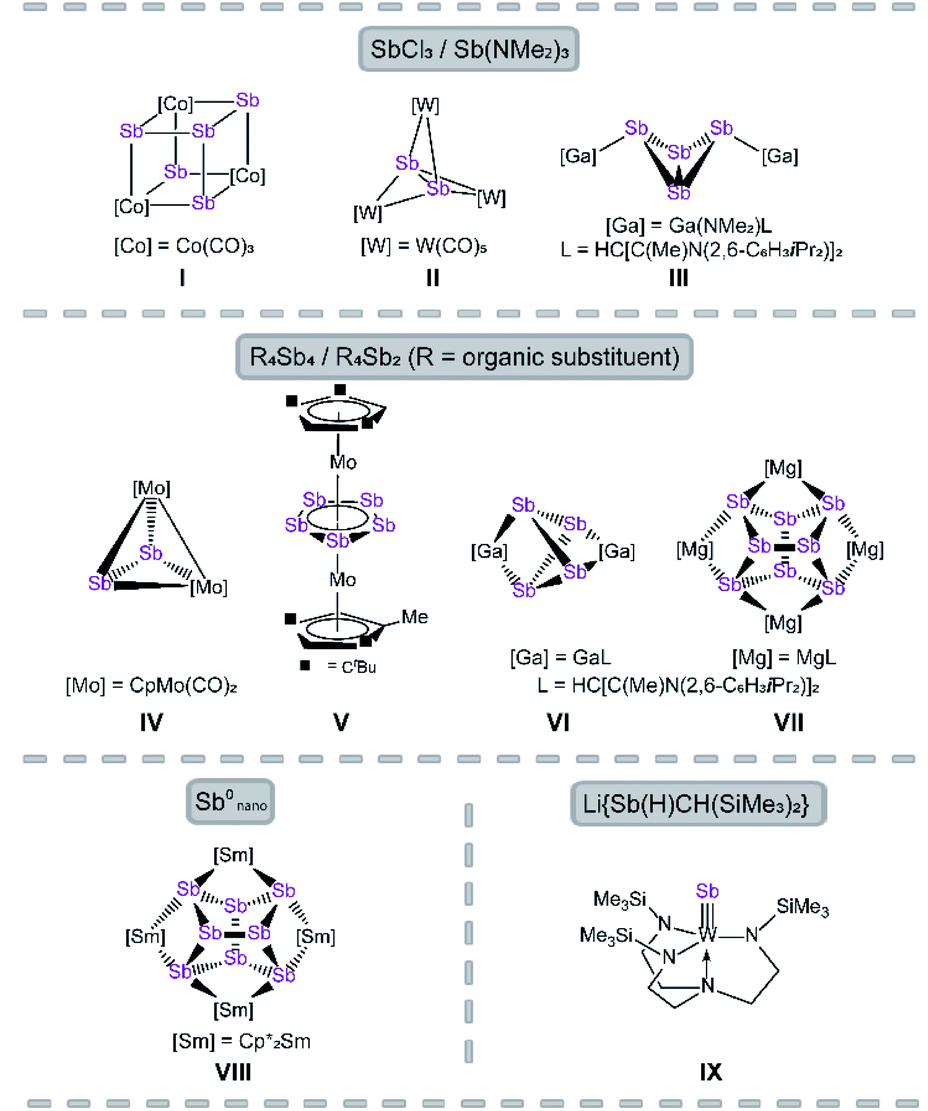 | ||
| Fig. 1 Selected examples of Sbn ligand complexes. | ||
The most commonly applied antimony sources, to build polyantimony ligand complexes or main group compounds, are neutral organo-substituted antimony units such as R4Sb4 (R = Cp*, tBu; Cp* = η1-C5Me5)15–17 and R′4Sb2 (R′ = Me, Et).18 Rösler et al. succeeded in the synthesis of the molybdenum complexes [{CpMo(CO)2}2(μ,η2-Sb2)] (IV) and [Cp′′′Mo(μ,η5:5-Sb5)MoCpR] (V, Fig. 1; Cp′′′ = η5-C5H2tBu3, CpR = η5-C5H2tBu2Me) by thermolysis of tBu4Sb4 with [CpMo(CO)3]2 and [Cp′′′Mo(CO)3CH3], respectively.15,16V represents the first triple-decker sandwich complex containing a cyclo-Sb5 middle deck. Interestingly, one tBu group of the Cp′′′ ligand is degraded to a methyl group during the thermolytic reaction.16
Recently, the chemistry of the polyantimony main group compounds has experienced a revival5,19 especially by the introduction of the nacnac ligand in combination with Mg and Ga (i.e. LMg and LGa) in this field. In addition to the aforementioned compound III, [(LGa)2(μ,η2:2-Sb4)]17 (VI) and [(LMg)4(μ4,η2:2:2:2-Sb8)]18 (VII) are interesting representatives made of organo-substituted antimony precursors. VI is formed by the reaction of LGa with Cp*4Sb4via reductive elimination of decamethyl-dihydrofulvalene Cp*2 and oxidative addition of LGa to the Sb4 subunit.17 To form VII, the reducing agent [(LMg)2] was used to initiate the cleavage of the Sb–C bonds in R′4Sb2.18 Another possibility to build polyantimony ligand complexes was shown by P. Roesky et al. by the usage of Sb0 nanoparticles in the thermolysis reaction with [Cp*2Sm], leading to the formation of the f-element polystibide [(Cp*2Sm)(μ4,η2:2:2:2-Sb8)] (VIII), similar to VII.20 Also, our group is interested in the generation of Sbn ligand complexes with unprecedented structural motifs. For instance, we succeeded in the synthesis of [(N3N)WSb] (VI, N3N = tren = N(CH2CH2NSiMe3)3) which possesses a first tungsten antimony triple bond compound.21 Here, a lithium antimonide salt was used as Sb1 source. Due to the lack of suitable antimony precursors, the access to polyantimony compounds is strongly limited (vide supra), in principle due to their instability. The classical synthetic procedures, i.e. thermolysis or photolysis, lead in most of the cases to gray antimony.
Besides the usual synthetic pathways such as thermolysis, photolysis or reactions with unsaturated fragments, the transfer of En units constitutes a promising procedure for the synthesis of metastable En ligand complexes, since such reactions proceed under mild conditions and enable the formation of compounds that are not accessible by other means.4,6,22 A prominent product of such a transfer reaction is the synthesis of AsP3, reported by Cummins et al., where a P33− unit was transferred from a niobium complex to AsCl3.23 Asn units were also transferred from the zirconium complexes [Cp′′2Zr(μ,η1:1-E4)] (E = P, As) to LSi, leading to the formation of the inorganic benzene derivatives L3Si3E3 (E = P, As; L = PhC(NtBu)2).24 Moreover, this approach was extended to transition metals.25 Besides the transfer of the entire En unit, its degradation as well as aggregation were also observed. Therefore, the question arises whether it would be possible to synthesize a related zirconium complex containing a polyantimony unit. Such derivative could be used to transfer the antimony entity to transition metals or main group compounds to build metastable polyantimony compounds. Herein we report on the successful synthesis of [(Cp′′Zr)2(μ,η1:1:1:1:1:1-Sb6)] (1) and its unprecedented use as transfer reagent for Sbn species. Nevertheless, in comparison to Pn and Asn transfer reagents, one should keep in mind the lesser stability of Sb–Sb bonds during the transfer, due to the much lower E–E single bond energy.
Results and discussion
To discover a suitable starting material for transfer reactions, we investigated the reaction of [Cp′′2ZrCl2] with KSb(SiMe3)2, surprisingly leading to the formation of the polyantimony triple decker complex [(Cp′′Zr)2(μ,η1:1:1:1:1:1-Sb6)] (1) as the only isolable and identified product (Scheme 1). Compound 1 is well soluble in solvents such as n-pentane, toluene or THF, and is highly air- and light-sensitive. It decomposes rapidly, especially in solution, even at low temperatures (−80 °C) after few days. Therefore, the recorded 1H NMR spectrum of 1 at r.t. reveals the expected signals for the Cp′′ ligands, but also unidentified decomposition products. Also, in the mass spectrum of 1, the molecular ion peak with a correct isotopic pattern was detected at m/z = 1267.61, beside some other peaks, corresponding to decomposition products.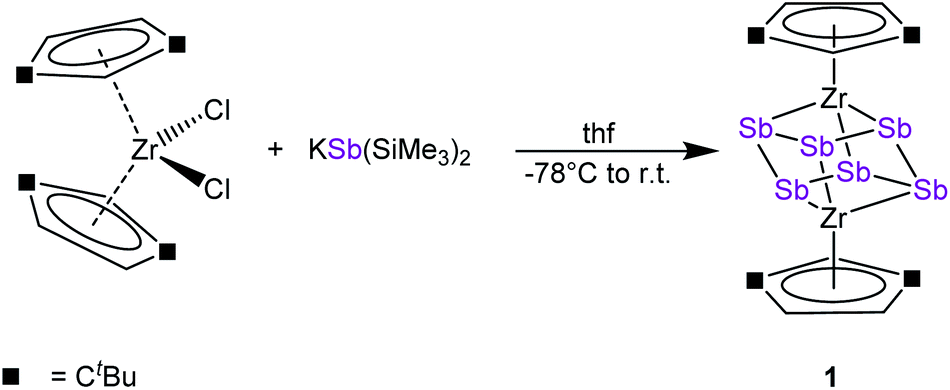 | ||
| Scheme 1 Synthesis of 1. | ||
Crystals of 1 suitable for single crystal X-ray diffraction analysis were obtained by storing a concentrated solution in dichloromethane or n-pentane at −30 °C. The molecular structure of 1 is depicted in Fig. 2.
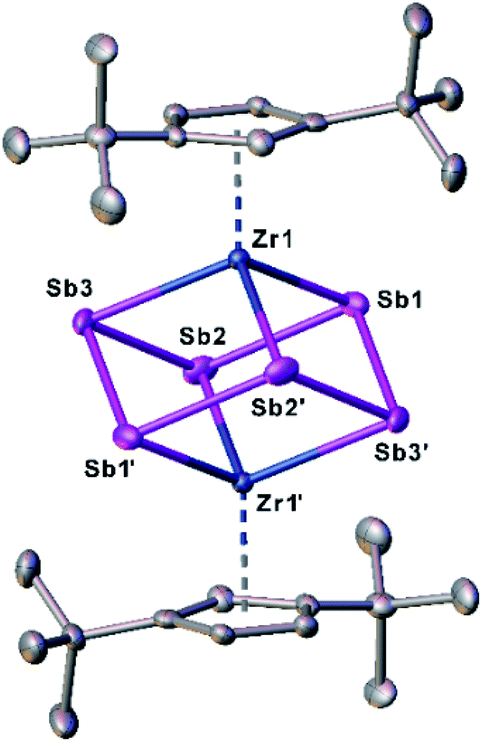 | ||
| Fig. 2 Molecular structure of 1 in the solid state with thermal ellipsoids at 50% probability level. H atoms are omitted for clarity. | ||
The central structural core represents a chair-type Sb6 unit attached to two [Cp′′Zr] fragments in an η1:1:1:1:1:1 fashion, leading to a cube-like Zr2Sb6 core. The Sb–Sb distances of 2.8381(7) Å to 2.8783(6) Å are in the range of a single bond and in good agreement with other polyantimony ligand complexes.14,16–18,26 Complexes containing chair-like Sb6 ligands are not known. The most related compounds are the boat-like cyclo-Sb64− species in the Zintl ion [(Cp*Ru)2(μ,η4:2:2-Sb6)]2− reported by B. Eichhorn et al.10c and the phosphine ligand stabilized compound [(R3P)4Sb6]4+ containing a bicyclic Sb6 moiety.10d Otherwise, only organo-antimony compounds bearing organic substituents ((RSb)6, R = C6H5, C7H7) were published.26,27 Complexes containing chair-like or skew boat-like E6 (E = P, Bi) moieties have also been reported.28 The most related complex to 1, [(Cp*Ti)2(μ,η1:1:1:1:1:1-P6)], was synthesized by the co-thermolysis of [Cp*Ti(CO)2] with white phosphorus.28a This compound can be formally described as a lighter congener of 1. Only a few examples of compounds containing antimony and zirconium are known so far, but organic substituents are attached to the antimony atoms.29
DFT calculations were performed at the B3LYP/def2-TZVP level of theory to elucidate the electronic structure and the nature of the Sb6 ligand in 1. The Wiberg Bond Indices (WBI) of the Zr–Sb bonds of 0.97 (in average) indicate a covalent nature of these bonds. This applies also to the Sb–Sb bonds with a slightly lower value of 0.84 (average), confirming the presence of single bonds within the Zr2Sb4 unit. The natural charge distribution shows a positive charge concentration on the cyclo-Sb6 ligand (+0.48) and Zr (+0.11 each), while the negative charge is located on the Cp′′ ligand (−0.35 each).
With compound 1 in hand, we investigated its suitability to transfer Sbn units under mild conditions to late transition metals. For this purpose, transition metal halides of the type [Cp′′′MX]2 of nickel, cobalt and iron were used. The formation of the thermodynamically favored zirconium halogen bond represents the driving force of these reactions. Due to the very limited stability of 1, in situ generated solutions were used under strict exclusion of light. All mentioned reactions were performed in THF at r.t. and a subsequent column chromatographic workup was needed for purifications, significantly reducing the yield of the formed products (cf.Scheme 2). The reaction of 1 with [Cp′′′NiBr]2 leads to [(Cp′′′Ni)4(μ3-Sb)4] (2), containing a cubane-like Ni4Sb4 core and the co-crystallization of nickelocene [Cp′′′2Ni] (2′). Under similar reaction conditions, 1 reacts with [Cp′′′CoCl]2 exclusively to the heterocubane derivative [(Cp′′′Co)4(μ3-Sb)4] (3a). [CpBnCoCl]2, containing the sterically less encumbered CpBn substituent, reacts with 1 to a mixture of [(CpBnCo)4(μ3-Sb)4] (3b) and [(CpBnCo)3(μ3-Sb)2] (4).30 Despite numerous attempts, 3b and 4 could not be separated to be obtained as analytically pure compounds. Furthermore, the reaction of 1 with [Cp′′′FeBr]2 leads to two products after chromatographic workup, [(Cp′′′Fe)3(μ3-Sb)2] (5) and [(Cp′′′Fe)3(μ3,η4:4:4-Sb6)] (6). Whereas in the afore-reported products 2, 3a, 3b, 4 and 5 only Sb1 units are incorporated, in 6, the complete Sb6 entity was transferred. The 1H and 13C{1H} NMR spectra of 2/2′ reveal the expected signals for 2. Due to the paramagnetic nature of 2′, no signals are detected in the diamagnetic region. 1H NMR measurements of the mixture of 3b and 4 show two sets of signals, which can be attributed to 3b and 4, respectively. Although 3b is paramagnetic, the 1H NMR signals occur in the diamagnetic region probably due to the strong localization of the spin density on the cobalt centers. Thus, the ratio between 3b and 4 of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 could be identified. In the 1H NMR spectra of 3a, 5 and 6, only broad signals for the tBu groups are observed, due to their paramagnetic character. The molecular ion peaks with the correct isotope pattern were detected for all mentioned compounds by mass spectrometry.31
:1 could be identified. In the 1H NMR spectra of 3a, 5 and 6, only broad signals for the tBu groups are observed, due to their paramagnetic character. The molecular ion peaks with the correct isotope pattern were detected for all mentioned compounds by mass spectrometry.31
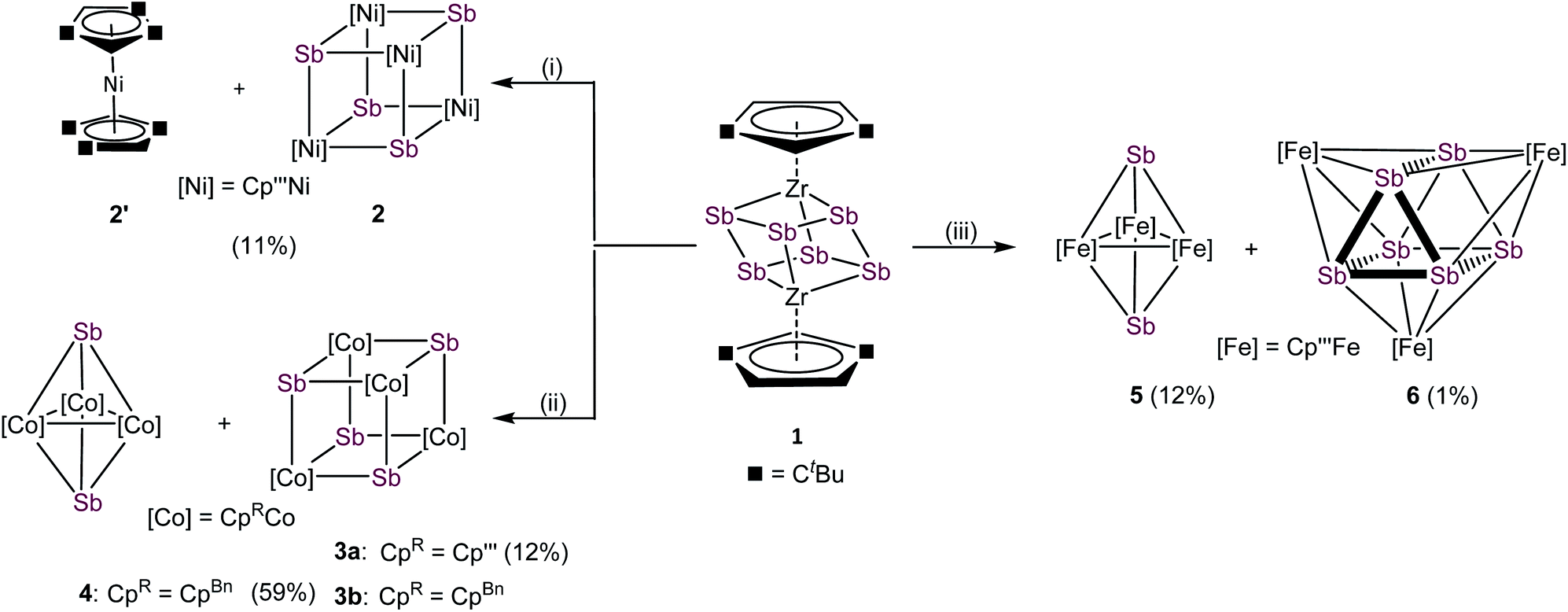 | ||
| Scheme 2 Reaction of in situ prepared [(Cp′′Zr)2(μ,η1:1:1:1:1:1-Sb6)] (1) with transition metal halides at r.t. in THF. (i): 1 with [Cp′′′NiBr]2; (ii): 1 with [CpRCoCl]2 (CpR = CpBn, Cp′′′); (iii): 1 with in situ prepared [Cp′′′FeBr]2: isolated yields are given in parentheses. | ||
Single crystals of 2/2′ and 3a suitable for crystal X-ray diffraction analysis were obtained by layering a toluene solution with acetonitrile (2/2′) or by storing a concentrated n-hexane solution at −30 °C (3a) (Fig. 3). Surprisingly, 2 co-crystallizes with 2′ in a ratio of 2:1 (ESI: Fig. S2†). The central structural core of 2 and 3a consists of a distorted hetero-cubane unit built up by four nickel or cobalt and four antimony atoms (Fig. 3). The square faces of the cubanes show a kite-like distortion in which the Sb atoms approach each other. All nickel and cobalt atoms are coordinatively saturated by the coordination of a Cp′′′ ligand. The Ni–Sb bond lengths in 2 (2.5202(7) Å to 2.5602(7) Å)32,33 and the Co–Sb bond lengths in 3a (2.5023(19) Å to 2.6305(14) Å)12,34–37 are in the range of corresponding single bonds. While, in 2, the Ni–Ni distances are quite long (3.9352(9) Å to 3.962(1) Å) indicating no bonding interaction, in 3a, there are two short Co⋯Co distances of 3.071(4) Å and two long Co⋯Co distances of 4.034(3) Å. Similarly, the Sb–Sb distances in 2 are reasonably similar (3.0767(4) Å to 3.1613(4) Å), while, in 3a, there are two very short (2.9453(13) Å) and two longer (3.1763(13) Å) Sb–Sb distances. The former lies in the range of an elongated single bond.38 The distortion of the M4Sb4 core is more accentuated in 3a than in 2, probably due to the Co⋯Co and Sb⋯Sb interactions. The rhombic distortion of the cubanes [(Cp*M)4E4] (M = Cr, Mo, E = O, S) was attributed to ferromagnetic as well as antiferromagnetic couplings of the metal-based electrons.39
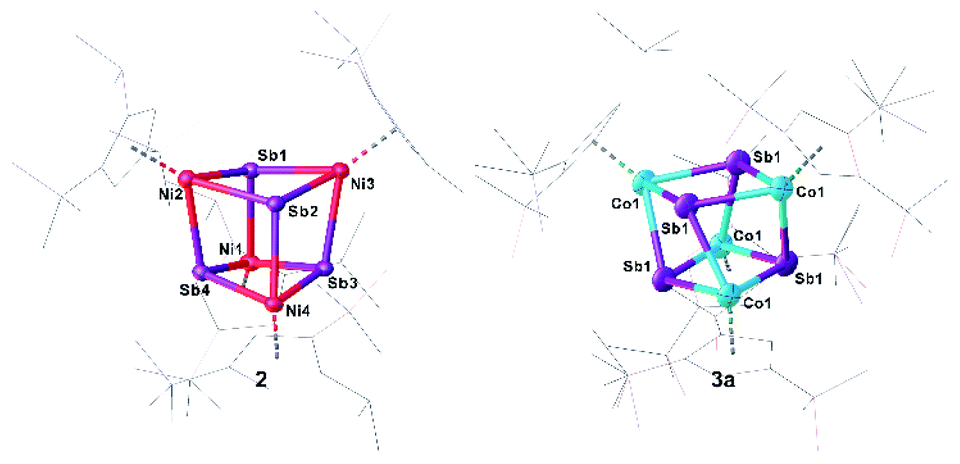 | ||
| Fig. 3 Molecular structures of 2 (left) and 3a (right) in the solid state with thermal ellipsoids at 50% probability level. H atoms are omitted and the Cp′′′ ligands drawn in the wire frame model for clarity. | ||
DFT calculations reproduce well the geometric parameters of the model compound [(Cp*Co)3(μ-Sb)2] (4m; vide infra), while the short Co–Co distances in 3a are slightly longer in the optimized geometry, irrespective of a singlet or a triplet spin state. The short Sb–Sb distances in 3a are well reproduced. The presence of Sb⋯Sb interactions in 2 and 3a are substantiated by the Mayer bond orders, which vary from 0.25 to 0.35 and from 0.27 to 0.52, respectively correlate with the corresponding distances. Based on the analysis of the Intrinsic Bonding Orbitals,40 the Co⋯Co interaction in 3m is realized via the Co–Sb σ-bond to which contributions of roughly 10% from the other two Co atoms are mixed. Additionally, on each Co center, there are three non-bonding d orbitals which do not overlap to give Co–Co bonds. The Mayer bond order for the Co–Co interactions in 4m is roughly 0.48 while for the Co–Sb bonds vary from 0.92 to 0.98. To investigate the electronic structure of 3a in more detail, EPR and Evans NMR investigations were performed. 3a is EPR-silent and has an effective magnetic moment of μeff = 2.34 μB, corresponding roughly to two unpaired electrons as determined by the Evans method.41
Compounds 4 and 5 crystallize as brown and violet blocks, respectively, by layering a toluene solution with acetonitrile. Their central structural motif consists of an [M3Sb2] trigonal bipyramide, built up by two antimony and three metal atoms (M = Co (4), Fe (5), Fig. 4). The cobalt and iron atoms are located in the equatorial plane and coordinated by a CpR ligand, the antimony atoms occupy the apical positions. While the Fe–Sb distances of 2.4895(6) Å to 2.55342(5) Å are in good agreement with reported single bonds,1,42,43 the Co–Sb distances of 4 (2.4362(4) Å to 2.4547(4) Å) are shortened.12,34–37 A remarkable difference between the geometry of 4 and 5 is represented by the M–M distances. While, in 4, the Co–Co distances are all very similar (2.7284(6) Å to 2.7332(5) Å) and lie in the same range of elongated single bonds,33,43 in 5, there are one short (2.4489(6) Å and 2.4895(6) Å) and two longer Fe–Fe distances (2.8076(6) Å to 2.9230(6) Å).44 Interestingly, the nitrogen congener of 5, [(Cp′′′Fe)3(μ3-N)2], possesses uniform Fe–Fe distances varying from 2.4727(4) Å to 2.4734(4) Å,45 while the phosphorus and arsenic derivatives [(Cp*Fe)3(μ3-E)2] (E = P, As)46 show a distortion of the Fe3 fragment that is similar to 5, but less accentuated. The structure of 4 represents a new structural motif and can be regarded as a closo-type cluster with 12 skeletal electrons according to the Wade-Mingos rule. Similar complexes of iron and antimony are known only for antimony atoms coordinating to Lewis acidic metal fragments as in [Fe3(CO)9(μ3-SbMLn)2] (MLn = CpMn(CO)2, Cr(CO)5, W(CO)5, Mo(CO)5).1,42
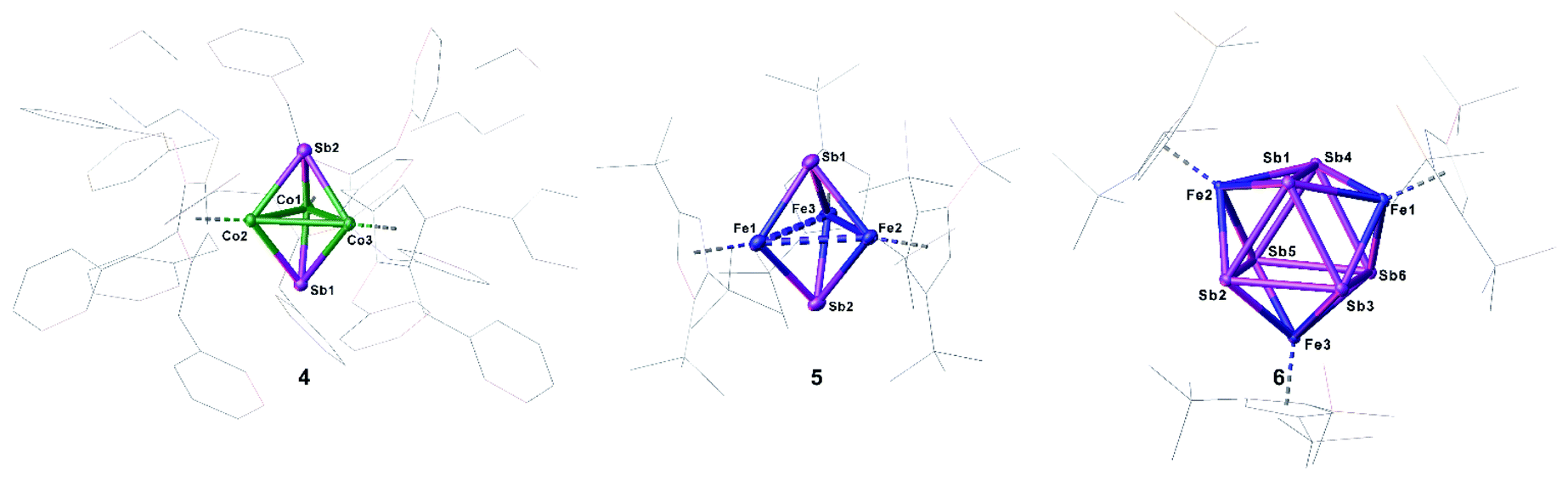 | ||
| Fig. 4 Molecular structures of 3b (left), 5 (middle) and 6 (right) in the solid state with thermal ellipsoids at 50% probability level. H atoms are omitted and the Cp′′′ ligands drawn in the wire frame model for clarity. | ||
To explore the electronic structure of 5, EPR and Evans NMR investigations were performed. The X-band EPR spectrum of 5 at 77 K in frozen toluene shows an axial signal (gx = gy = 2.177 and gz = 2.443 (giso = 2.266)) with no hyperfine splitting (Fig. 5). The doublet spin state of 5 is further supported by Evans NMR measurements, resulting in μeff = 1.65 μB, corresponding roughly to one unpaired electron.41
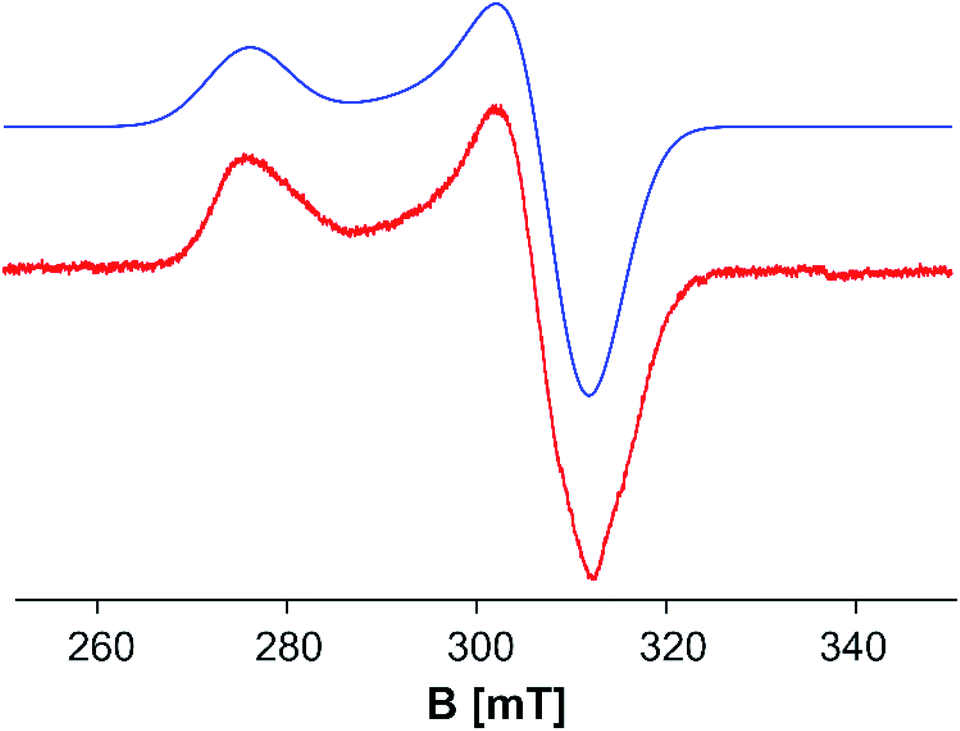 | ||
| Fig. 5 X-band EPR spectrum of 5 in toluene at 77 K: gx = gy = 2.177 and gz = 2.443 (giso = 2.266). Red: experimental; blue: simulated.47 | ||
In order to elucidate the electronic structure of 5, DFT calculations were performed. The geometry of 5 in different spin states (S = 1/2 to 4) was optimized by using different functionals. The results show that only the OPBE functional in a doublet spin state reproduces the experimental geometry of 5 found in the solid state. Hybrid functionals lead to optimized geometries with considerably longer Fe–Fe distances, while a similar trend is observed for the OPBE functional in higher spin states (cf. ESI†). Single point calculations with both functionals OPBE and B3LYP (OPBE, doublet spin state optimized geometry) predict the doublet spin state as the energetically lowest state, while the other spin states lie energetically higher (cf. ESI†). Therefore, the ground state of 5 can be viewed as being a doublet spin state, which is in accordance with the EPR and Evans NMR data (vide supra). For the nitrogen analog of 5, [(Cp′′′Fe)3(μ3-N)2], the presence of three low-spin iron(III) (S = 1/2) centers and a total spin of Stot = 1/2 was reported, due to complex antiferromagnetic coupling.45
Single crystals of 6 suitable for X-ray diffractions were obtained after chromatographic workup by layering a toluene solution with acetonitrile. The central structural motif of 6 consists of a Sb6 prism with its rectangular faces being capped by [Cp′′′Fe] fragments (Fig. 4, right). Within the triangular faces, the Sb–Sb distances (2.9029(4) Å to 2.9365(4) Å) are slightly shorter than the Sb–Sb distances between them (3.0283(4) Å to 3.0646(4) Å). This variation of the Sb–Sb distances nicely correlates with the corresponding Mayer bond orders (OPBE/def2-SVP, doublet spin state) of 0.53 and 0.33, respectively. The Mayer bond orders for the Fe–Sb bonds vary only slightly between 0.69 to 0.73. Although the Sb–Sb distances are slightly longer than usual Sb–Sb single bonds, they lie in the range of elongated single bonds.14,16,18,48 DFT calculations at the OPBE/def2-SVP level predict the doublet spin state for 6 as ground state, with the spin density being evenly distributed over the three Fe centers, while the other spin states lie energetically higher (for example the quartet spin state is with 53 kJ mol−1 higher in energy; cf. ESI†). To the best of our knowledge, 6 is the first complex containing a Sb6 prism. Comparable complexes are only known for other group 15 homologous.49
Conclusions
In summary, we herein report the synthesis of the first zirconium antimony ligand complex [(Cp′′Zr)2(μ,η1:1:1:1:1:1-Sb6)] (1), which contains an unprecedented chair-like Sb6 ligand coordinated to two Cp′′Zr fragments. Complex 1 can be used as an effective antimony transfer reagent towards transition metal halides of nickel, cobalt and iron. That way, a variety of novel Sbn ligand complexes such as the cubane-like compounds [(Cp′′′Ni)4(μ3-Sb)4]/[Cp′′′2Ni] (2/2′) and [(Cp′′′Co)4(μ3-Sb4)] (3a) or the complexes [(CpBnCo)3(μ3-Sb)2] (4) and [(Cp′′′Fe)3(μ3-Sb)2] (5) containing a trigonal bipyramidal structure were obtained and fully characterized. Beside the partial transfer of the Sb1 units, also the complete transfer of a Sb6 unit was achieved. Here, the iron complex [(Cp′′′Fe)(μ,η4:4:4-Sb6)] (6) is formed which shows the potential of 1 as transfer reagent for polyantimony units. In principle, an initial transfer of larger Sbn units from 1 should have been possible, however, the low stability of such complexes (especially during column chromatographic work-up) might be the reason that mostly only Sb1-unit-containing complexes were isolated. Future work in this area is dedicated to developing smoother reaction pathways.Data availability
Crystallographic data for 1–6 has been deposited at the CCDC/ICSD under 2079755 (1), 2079756 (2/2′), 2079757 (3a), 2079758 (4·C7H8), 2079759 (5) and 2079760 (6).Author contributions
Veronika Heinl, synthesis and characterization of compounds 2, 3a, 5 and 6 and writing the paper. Dr Andreas E. Seitz, synthesis and characterization of compounds 1, 3b and 4. Dr Michael Seidl, recalculating the X-ray structures. Dr Gábor Balázs, computational details. Manfred Scheer, supervising the whole research work, fund raising and writing the paper. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Deutsche Forschungsgemeinschaft within the project Sche 384/32-2 supported this work. V. Heinl is grateful to the Fonds der Chemischen Industrie for a PhD fellowship.Notes and references
- B. E. Collins, Y. Koide, C. K. Schauer and P. S. White, Inorg. Chem., 1997, 36, 6172–6183 CrossRef CAS.
- C. M. Hoidn, D. J. Scott and R. Wolf, Chem.–Eur. J., 2021, 27, 1886–1920 CrossRef CAS PubMed.
- (a) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS; (b) F. Scalambra, M. Peruzzini and A. Romerosa, Adv. Organomet. Chem., 2019, 173–222 CrossRef CAS; (c) N. A. Giffin and J. D. Masuda, Coord. Chem. Rev., 2011, 255, 1342–1359 CrossRef CAS; (d) O. J. Scherer, Acc. Chem. Res., 1999, 32, 751–762 CrossRef CAS.
- B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164–4177 CrossRef CAS PubMed.
- L. Qiao, C. Zhang, X. W. Zhang, Z. C. Wang, H. Yin and Z. M. Sun, Chin. J. Chem., 2020, 38, 295–304 CrossRef CAS.
- M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 CrossRef CAS PubMed.
- (a) O. J. Scherer, Angew. Chem., Int. Ed. Engl., 1985, 24, 924–943 CrossRef; (b) O. J. Scherer, Angew. Chem., Int. Ed. Engl., 1990, 29, 1104–1122 CrossRef.
- Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, 2007 Search PubMed.
- (a) J. Kordis and K. A. Gingerich, J. Chem. Phys., 1973, 58, 5141–5149 CrossRef CAS; (b) J. Mühlbach, P. Pfau, E. Recknagel and K. Sattler, Surf. Sci., 1981, 106, 18–26 CrossRef; (c) V. E. Bondybey, G. P. Schwartz and J. E. Griffiths, J. Mol. Spectrosc., 1981, 89, 328–332 CrossRef CAS; (d) R. Prasad, V. Venugopal, Z. Singh and D. D. Sood, J. Chem. Thermodyn., 1979, 11, 963–970 CrossRef CAS; (e) H. Zhang and K. Balasubramanian, J. Chem. Phys., 1992, 97, 3437–3444 CrossRef CAS.
- (a) S. Scharfe, F. Kraus, S. Stegmaier, A. Schier and T. F. Fässler, Angew. Chem., Int. Ed., 2011, 50, 3630–3670 CrossRef CAS PubMed; (b) R. S. P. Turbervill and J. M. Goicoechea, Chem. Rev., 2014, 114, 10807–10828 CrossRef CAS PubMed; for the use of K3Sb cf.: (c) Y. Wang, P. Zavalij and B. Eichhorn, Chem. Commun., 2018, 54, 11917–11920 RSC; (d) S. S. Chitnis, N. Burford, J. J. Weigand and R. McDonald, Angew. Chem., Int. Ed., 2015, 54, 7828–7832 CrossRef CAS.
- H. J. Breunig, Organic Arsenic, Antimony and Bismuth Compounds 1994, John Wiley & Sons, pp. 563–577 Search PubMed.
- L. F. Dahl and A. S. Foust, J. Am. Chem. Soc., 1970, 92, 7337–7341 CrossRef CAS.
- G. Huttner, U. Weber, B. Sigwarth and O. Schneidsteger, Angew. Chem., Int. Ed. Engl., 1982, 21, 215–216 CrossRef.
- L. Tuscher, C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, Angew. Chem., Int. Ed., 2015, 54, 10657–10661 CrossRef CAS PubMed.
- H. J. Breunig, R. Rösler and E. Lork, Angew. Chem., Int. Ed. Engl., 1997, 36, 2819–2821 CrossRef CAS.
- H. J. Breunig, N. Burford and R. Rösler, Angew. Chem., Int. Ed., 2000, 39, 4148–4150 CrossRef CAS PubMed.
- C. Ganesamoorthy, J. Krüger, C. Wölper, A. S. Nizovtsev and S. Schulz, Chem.–Eur. J., 2017, 23, 2461–2468 CrossRef CAS PubMed.
- C. Ganesamoorthy, C. Wölper, A. S. Nizovtsev and S. Schulz, Angew. Chem., Int. Ed., 2016, 55, 4204–4209 CrossRef CAS PubMed.
- (a) Y. Wang, P. Zavalij and B. Eichhorn, Chem. Commun., 2018, 54, 11917–11920 RSC; (b) C. Ganesamoorthy, C. Helling, C. Wölpe, W. Frank, E. Bill, G. E. Cutsail and S. Schulz, Nat. Commun., 2018, 9, 87 CrossRef PubMed; (c) C. Schoo, S. Bestgen, A. Egeberg, S. Klementyeva, C. Feldmann, S. N. Konchenko and P. W. Roesky, Angew. Chem., Int. Ed., 2018, 57, 5912–5916 CrossRef CAS PubMed; (d) C. Helling, C. Wölper and S. Schulz, Dalton Trans., 2020, 49, 11835–11842 RSC; (e) C. Helling, G. E. Cutsail, H. Weinert, C. Wölper and S. Schulz, Angew. Chem., Int. Ed., 2020, 59, 7561–7568 CrossRef CAS PubMed.
- C. Schoo, S. Bestgen, A. Egeberg, S. Klementyeva, C. Feldmann, S. N. Konchenko and P. W. Roesky, Angew. Chem., Int. Ed., 2018, 57, 5912–5916 CrossRef CAS.
- G. Balázs, M. Sierka and M. Scheer, Angew. Chem., Int. Ed., 2005, 44, 4920–4924 CrossRef.
- T. Wetting, B. Geissler, R. Schneider, S. Barth, P. Binger and M. Regitz, Angew. Chem., Int. Ed. Engl., 1992, 31, 758–759 CrossRef.
- B. M. Cossairt, M.-C. Diawara and C. C. Cummins, Science, 2009, 323, 602 CrossRef CAS PubMed.
- A. E. Seitz, M. Eckhardt, A. Erlebach, E. V. Peresypkina, M. Sierka and M. Scheer, J. Am. Chem. Soc., 2016, 138, 10433–10436 CrossRef CAS PubMed.
- M. Schmidt, A. E. Seitz, M. Eckhardt, G. Balázs, E. V. Peresypkina, A. V. Virovets, F. Riedlberger, M. Bodensteiner, E. M. Zolnhofer, K. Meyer and M. Scheer, J. Am. Chem. Soc., 2017, 139, 13981–13984 CrossRef CAS PubMed.
- H. J. Breunig, K. Häberle, M. Dräger and T. Severengiz, Angew. Chem., Int. Ed. Engl., 1985, 24, 73 CrossRef.
- H. J. Breunig, K. H. Ebert, S. Gülec and J. Probst, Chem. Ber., 1995, 128, 599–603 CrossRef CAS.
- (a) O. J. Scherer, H. Swarowsky, G. Wolmershäuser, W. Kaim and S. Kohlmann, Angew. Chem., Int. Ed. Engl., 1987, 26, 1153–1155 CrossRef; (b) J. M. Goicoechea, M. W. Hull and S. C. Sevov, J. Am. Chem. Soc., 2007, 129, 7885–7893 CrossRef CAS; (c) H.-G. von Schnering, M. Wittmann and R. Nesper, J. Less Common Met., 1980, 76, 213–226 CrossRef CAS.
- (a) S. R. Wade, M. G. H. Wallbridge and G. R. Willey, J. Organomet. Chem., 1984, 267, 271–276 CrossRef CAS; (b) R. Zitz, J. Baumgartner and C. Marschner, Organometallics, 2015, 34, 1431–1439 CrossRef CAS PubMed.
- Despite several attempts, it was not possible to structurally identify 3a. Its composition and structure are assigned based on mass spectrometric data and are presumably isostructural to the cubanes 2 and 3b.
- Note that the peak for 3b is relatively weak presumably due to its high mass (2785 Da).
- (a) P. D. Mlynek and L. F. Dahl, Organometallics, 1997, 16, 1641–1654 CrossRef CAS; (b) S. Charles, B. W. Eichhorn and S. G. Bott, J. Am. Chem. Soc., 1993, 115, 5837–5838 CrossRef CAS.
- U. Vogel, G. Baum and M. Scheer, Z. Anorg. Allg. Chem., 2000, 626, 444–449 CrossRef CAS.
- T. A. Albright, K. A. Yee, J. Y. Saillard, S. Kahlal, J. F. Halet, J. S. Leigh and K. H. Whitmire, Inorg. Chem., 1991, 30, 1179–1190 CrossRef CAS.
- N. C. Norman, P. M. Webster and L. J. Farrugia, J. Organomet. Chem., 1992, 430, 205–219 CrossRef CAS.
- J. S. Leigh, K. H. Whitmire, K. A. Yee and T. A. Albright, J. Am. Chem. Soc., 1989, 111, 2726–2727 CrossRef CAS.
- S. N. Konchenko, A. V. Virovets, S. A. Apenina and S. V. Tkachev, Inorg. Chem., 1999, 555–557 CAS.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186–197 CrossRef.
- J. E. McGrady, J. Chem. Soc., Dalton Trans., 1999, 1393–1399 RSC.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS.
- Due to the high molecular weight and bad solubility, the results of the Evans NMR should be treated with care.
- H. Lang, G. Huttner, L. Zsolnai, G. Mohr, B. Sigwarth, U. Weber, O. Orama and I. Jibril, J. Organomet. Chem., 1986, 304, 157–179 CrossRef CAS.
- T. Gröer, T. Palm and M. Scheer, Eur. J. Inorg. Chem., 2000, 2591–2595 CrossRef.
- There are two different bond distances due to two molecules in the asymmetric unit.
- M. Reiners, D. Baabe, K. Münster, M.-K. Zaretzke, M. Freytag, P. G. Jones, Y. Coppel, S. Bontemps, I. D. Rosal, L. Maron and M. D. Walter, Nat. Chem., 2020, 12, 740–746 CrossRef PubMed.
- S. Reisinger, Organometallic Pnictogen Chemistry – Three Aspects, PhD thesis, University of Regensburg, 2014.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- H. J. Breunig, R. Rösler and E. Lork, Angew. Chem., Int. Ed. Engl., 1997, 36, 2819–2821 CrossRef CAS.
- (a) S. Heinl, A. Y. Timoshkin, J. Müller and M. Scheer, Chem. Commun., 2018, 54, 2244–2247 RSC; (b) C. Hänisch, D. Fenske, F. Weigend and R. Ahlrichs, Chem.–Eur. J., 1997, 3, 1494–1498 CrossRef; (c) C. Hänisch, D. Fenske, F. Weigend and R. Ahlrichs, Chem.–Eur. J., 1997, 3, 1494–1498 CrossRef; (d) G. Friedrich, O. J. Scherer and G. Wolmershäuser, Z. Anorg. Allg. Chem., 1996, 622, 1478–1486 CrossRef CAS; (e) L. Qiao, D. Chen, J. Zhu, A. Muñoz-Castro and Z.-M. Sun, Chem. Commun., 2021, 57, 3656–3659 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2079755–2079760. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02498a |
| This journal is © The Royal Society of Chemistry 2021 |