
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNA-templated control of chirality and efficient energy transport in supramolecular DNA architectures with aggregation-induced emission†
Hülya
Ucar
and
Hans-Achim
Wagenknecht
 *
*
Institute of Organic Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, 76131 Karlsruhe, German. E-mail: Wagenknecht@kit.edu
First published on 21st June 2021
Abstract
Two conjugates of tetraphenylethylene with D-2′-deoxyuridine (1D) and L-2′-deoxyuridine (1L) were synthesized to construct new supramolecular DNA-architectures by self-assembly. The non-templated assemblies of 1D and 1L show strong aggregation-induced emission and their chirality is exclusively controlled by the configuration of their sugar part. In contrast, the chirality of the DNA-templated assemblies is governed by the configuration of the DNA, and there is no configuration-selective binding of 1D to D-A20 and 1L to L-A20. The quantum yield of the assembly of 1D along the single-stranded DNA A20 is 0.40; approximately every second available binding site on the DNA template is occupied by 1D. The strong aggregation-induced emission of these DNA architectures can be efficiently quenched and the excitation energy can be transported to Atto dyes at the 5′-terminus. A multistep energy transport “hopping” precedes the final energy transfer to the terminal acceptor. The building block 1D promotes this energy transport as stepping stones. This was elucidated by reference DNA double strands in which 1D was covalently incorporated at two distinct sites in the sequences, one near the Atto dye, and one farther away. This new type of completely self-assembled supramolecular DNA architecture is hierarchically ordered and the DNA template controls not only the binding but also the energy transport properties. The high intensity of the aggregation-induced emission and the excellent energy transport properties make these DNA-based materials promising candidates for optoelectronic applications.
Introduction
Conventional chromophores suffer from aggregation-caused quenching (ACQ), a phenomenon describing the quenched emission at high concentrations. For technical applications, particularly OLEDs, chromophores with strong fluorescence in the aggregated or condensed phase are crucial. The term “aggregation-induced emission” (AIE) was introduced by Tang’s group in 2001 describing the emissive behavior of 1,2,3,4,5-pentaphenylsilole in ethanol.1 Since then, the number of organic chromophores that show AIE is constantly increasing;2,3 in particular, tetraphenylethylene (Tpe) has been identified as a lead structure with AIE.4 Similar to the aforementioned silol, the propeller-shaped and non-planar Tpe is weakly emissive in dilute solution, but becomes highly emissive in the aggregated or solid state. It is not a distinct type of stacking interaction that increases the emission but rather the restriction of intramolecular rotations blocking the non-radiative decay pathways from the photoexcited state.5The supramolecular organization of chromophores gives access to nanostructured materials with unique optical properties.6 The bottom-up approach is a powerful concept to achieve this goal,7 and DNA is a unique template with sequence specificity through the recognition by hydrogen bonding to arrange chromophores in a precise way,8 and this DNA-like structure also persists in the solid state for optoelectronic applications.9 The most precise, covalent incorporation of chromophores into DNA is limited to 5–10 units in a row, until the yields drop.10,11 The non-covalent self-assembly of nucleosides along single-stranded DNA is an important alternative, for instance with naphthalenes12 and porphyrins13,14 through binding to T20 or T40. We recently described the sequence-selective assembly of pyrenes,15–17 perylenes18 and nile red16,17 as modified 2′-deoxynucleosides along single-stranded DNA templates and mainly ACQ was observed. However, AIE would increase the value of DNA-based materials for optoelectronics. This makes Tpe a promising candidate for DNA-based architectures with AIE.19 The fluorescence intensity increase of typical DNA staining agents, such as SYBR Green, is a phenomenon related to AIE since rigidification of the chromophore has an impact on the fluorescence in both cases.19 However, it should be noted here that, molecules, particularly derivatives of Tpe, showing AIE in assemblies are promising candidates for new materials. Achiral Tpe derivatives promote sensing of DNA by their emission20–22 and circularly polarized luminescence.23,24 The covalent conjugation of Tpe with DNA25 helps in the visualization of cellular RNA26 and gives DNA-grafted nanosheets,27 and the AIE can be controlled by DNA hybridization.28 To construct new supramolecular DNA-architectures with strong AIE by self-assembly, we present two chiral conjugates of Tpe, 1D and 1L (Fig. 1). The configuration of their 2′-deoxyribofuranosides differs to probe the chirality of both the non-templated assembly and the assembly along D- and L-DNA templates by means of optical spectroscopy. These supramolecular architectures are hierarchically ordered and the DNA template controls the binding selectivity, the chirality and the light harvesting through efficient energy transport to the Atto dye attached to the 5′-terminus.
 | ||
| Fig. 1 Tetraphenylethylene-modified 2′-deoxyuridines 1D and 1L. Illustration of the non-templated assembly and DNA-templated assembly of 1D and 1L. | ||
Results and discussion
Non-templated supramolecular assemblies
Both conjugates 1D and 1L were synthesized by Sonogashira-coupling as described in the ESI (Schemes S1, S2–S7).† Firstly, the formation of assemblies with 1D and 1L, respectively, was investigated in aqueous solution without any DNA template. Stock solutions of 1D and 1L in THF or DMSO (4.3 mM) were prepared to ensure full solubility of the modified 2′-deoxyuridines. The assembly of 1D and 1L was induced by diluting samples (8.8 μL) of the stock solutions with H2O (total volume of 1 mL, 37.5 μM in H2O with 0.9% THF or DMSO). The first samples of 1D and 1L were simply stored for 1 h at room temperature and characterized by optical spectroscopy (Fig. 2). A second set of samples was treated under the typical conditions of DNA annealing, including incubation at 90 °C for 5 min and slow cooling to room temperature (approximately 1 °C min−1). Interestingly, these different preparations yield different types of water-soluble assemblies. Representatively, we discuss the optical properties of the samples that were prepared with THF as cosolvent; the corresponding results with DMSO as cosolvent are similar (see Fig. S22†). The fluorescence of the assemblies of 1D and 1L prepared at room temperature show maxima at 478 nm, whereas the annealed assemblies show maxima at 442 nm and reduced intensities. The first type of assembly is dominated by Tpe interactions, whereas the second, the annealed type of assembly is, at least partially, controlled by stacking of the 5-ethynyl-2′-deoxyuridines. Accordingly, the electronic decoupling of Tpe from the 5-ethynyl 2′-deoxyuridine part by a rotational twist along the phenylene bridge causes a blue-shift of fluorescence. | ||
| Fig. 2 Fluorescence (left bottom), circular dichroism (CD, right top) and fluorescence-derived circular dichroism (FDCD, bottom right) spectra of the assemblies prepared with 1D and 1L in the absence of any DNA template (37.5 μM 1D/1L, λexc = 341 nm, H2O, 0.9% THF, for the UV/Vis absorption see Fig. S21,† for the experiments with 0.9% DMSO see Fig. S22†) at room temperature (r.t.) or by annealing (ann.) after heating to 90 °C for 5 min. | ||
Chirality is an essential feature of these supramolecular structures and therefore the CD spectra were recorded. Remarkably, the CD spectra of assemblies of 1D and 1L from both types of preparation show strong mirror bisignate signals in the absorption range between 305 nm and 450 nm of the Tpe chromophore. Interestingly, the D-configured conjugate 1D yields an assembly with left-handed helicity according to this bisignate signal,29 whereas 1L yields assemblies with right-handed chirality. The CD signals of the annealed assemblies of 1D and 1L look similar in the range between 305 nm and 400 nm and their crossing points are shifted from 350 nm to 370 nm. The absorbance range between 200 nm and 305 nm, which is the typical absorption range for the 5-ethynyl-uracil part of the conjugates, shows another bisignate signal. Here, the annealed assembly formed with 1D shows left-handed helicity whereas the annealed assembly formed with 1L shows right-handed chirality. Their crossing point is at 270 nm, which is near the absorbance maximum of 5-ethynyl-2′-deoxyuridine. This supports our aforementioned hypothesis that the annealed assemblies are controlled by the nucleoside stacking. The chirality order may be unexpected, but, in fact, it is not. For instance, the D-configured nile red-modified 2′-deoxyuridines yielded assemblies with left-handed chirality, too.30 The CD spectra show very clearly that the chirality of the assemblies of 1D and 1L is exclusively controlled by the configuration of the attached 2′-deoxyribofuranosides. The fluorescence detected circular dichroism (FDCD) combines conventional CD with the high sensitivity of fluorescence spectroscopy. It allows investigating the chiral behavior of fluorescent chromophores in a non-emissive chiral architecture, such as DNA.31 The detected FDCD signals are mirror bisignate signals and again show that the chirality of the assemblies of 1D and 1L is controlled by the different configurations of the attached chiral 2′-deoxyribofuranosides.
DNA-templated supramolecular architectures
The next level of hierarchically ordered supramolecular architectures is the structural control of the self-assembly of 1D and 1L in aqueous solution by DNA templates. Therefore, we followed our established protocol to prepare such DNA-based supramolecular architectures.32,33 We used 1D and 1L as building blocks and probed their assembly along D- and L-configured DNA templates with the sequences A20 and T20 by means of optical spectroscopy. Small volumes of 2′-deoxynucleoside stock solutions in DMSO were added to an aqueous solution of the DNA templates (1 mL, 1.25 μM). The concentrations of both stock solutions were prepared sufficiently high that not more than 0.9% DMSO as cosolvent was added to the final samples in water. This amount of cosolvent is typically tolerated by the DNA conformation. Both, 1D and 1L, were added in a 1.5-fold excess to promote the occupancy of the available binding sites along the templates. 250 mM NaCl is known to stabilize double-helical DNA and thus expected to also support the formation of the non-covalent supramolecular DNA architectures with 1D and 1L. Both conjugates are nearly insoluble in this high salt aqueous solution unless they are bound to single stranded DNA and therefore remain in the solution. The excess and unbound 1D or 1L was removed by short centrifugation (3 min @ 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g). The supernatant contained the DNA-templated assembly and was investigated by optical spectroscopy (Fig. 3). The absorbances in the range between 300 nm and 400 nm of the assemblies of 1D with D-A20 and 1L with L-A20 as templates are significantly higher than those with D-T20 and L-T20. This result reveals preferred binding of 1D and 1L to D-A20 and L-A20, respectively, which matches the rules of canonical base-pairing, because both modified 2′-deoxuridines are complementary to 2′-deoxyadenosines in the DNA template A20, but not complementary to T20. This selectivity is not as pronounced as for the Watson–Crick base pairing in unmodified double-stranded DNA because “mismatched” binding of 1D and 1L to the wrong templates D-T20 and L-T20 occurs to a certain extent. There is, however, no configuration-selective recognition by the DNA templates. The absorbances of the assemblies of 1D with D-A20 and L-A20 are equally high (Fig. 3), which is also the case for the assemblies of 1L with both D-A20 and L-A20 (Fig. S23†). The absorbances can be used to calculate the occupancy fraction f which is the number of occupied binding sites (by 1D or 1L) divided by the number of available binding sites on the template. For the assemblies of 1D and 1L along D-A20 and L-A20, f lies in the range of 0.55 ± 0.1; approximately 11 out of 20 available binding sites of the DNA templates are occupied. Obviously, the four phenyl groups of Tpe are sterically hindering and prevent a complete occupancy of all binding sites, as previously observed for planar chromophores, such as nile red.32 The fluorescence gives additional support for the selective binding of the chromophore-nucleosides. Since Tpe shows AIE and unbound chromophore conjugates were removed by centrifugation, the fluorescence can be clearly attributed to the DNA-templated assemblies of 1D and 1L. The assembly of 1D along D-A20 (and L-A20) shows the strongest fluorescence intensity with a maximum at 492 nm and a quantum yield of Φ = 0.40. The assemblies of 1L along L-A20 (and D-A20) show similarly strong fluorescence intensities (Fig. S23†), whereas the assemblies of 1L and 1D along the wrong templates L-T20 and D-T20 show significantly weaker fluorescence intensities. These results track well with the absorbance differences and the idea of selective binding of 1L and 1D to D-A20 and L-A20.
000g). The supernatant contained the DNA-templated assembly and was investigated by optical spectroscopy (Fig. 3). The absorbances in the range between 300 nm and 400 nm of the assemblies of 1D with D-A20 and 1L with L-A20 as templates are significantly higher than those with D-T20 and L-T20. This result reveals preferred binding of 1D and 1L to D-A20 and L-A20, respectively, which matches the rules of canonical base-pairing, because both modified 2′-deoxuridines are complementary to 2′-deoxyadenosines in the DNA template A20, but not complementary to T20. This selectivity is not as pronounced as for the Watson–Crick base pairing in unmodified double-stranded DNA because “mismatched” binding of 1D and 1L to the wrong templates D-T20 and L-T20 occurs to a certain extent. There is, however, no configuration-selective recognition by the DNA templates. The absorbances of the assemblies of 1D with D-A20 and L-A20 are equally high (Fig. 3), which is also the case for the assemblies of 1L with both D-A20 and L-A20 (Fig. S23†). The absorbances can be used to calculate the occupancy fraction f which is the number of occupied binding sites (by 1D or 1L) divided by the number of available binding sites on the template. For the assemblies of 1D and 1L along D-A20 and L-A20, f lies in the range of 0.55 ± 0.1; approximately 11 out of 20 available binding sites of the DNA templates are occupied. Obviously, the four phenyl groups of Tpe are sterically hindering and prevent a complete occupancy of all binding sites, as previously observed for planar chromophores, such as nile red.32 The fluorescence gives additional support for the selective binding of the chromophore-nucleosides. Since Tpe shows AIE and unbound chromophore conjugates were removed by centrifugation, the fluorescence can be clearly attributed to the DNA-templated assemblies of 1D and 1L. The assembly of 1D along D-A20 (and L-A20) shows the strongest fluorescence intensity with a maximum at 492 nm and a quantum yield of Φ = 0.40. The assemblies of 1L along L-A20 (and D-A20) show similarly strong fluorescence intensities (Fig. S23†), whereas the assemblies of 1L and 1D along the wrong templates L-T20 and D-T20 show significantly weaker fluorescence intensities. These results track well with the absorbance differences and the idea of selective binding of 1L and 1D to D-A20 and L-A20.
 | ||
| Fig. 3 UV/Vis absorbance (top right), fluorescence (bottom right) and circular dichroism (bottom left) spectra of the DNA-templated assemblies of 1D with D-A20, L-A20, D-T20 and L-T20 (1.25 μM DNA in water + 0.9% DMSO, 250 mM NaCl, λexc = 341 nm, and supernatant after centrifugation 3 min @ 16000g). The corresponding spectra of 1L are shown Fig. S23.† | ||
Although configuration selectivity was not observed for the binding of 1D and 1L to the DNA templates, the chirality of the formed helical assemblies was probed by CD spectroscopy. The CD spectra of the DNA-templated assemblies of 1D and 1L show generally weaker signals than those of the non-templated assemblies, and there are only signals of 2′-deoxynucleosides in the absorbance range between 200 nm and 300 nm (dU in the Tpe-conjugates and A or T in the templates). In contrast to the non-templated assemblies of 1D and 1L, as discussed above, the chirality in the templated assemblies is not controlled by the configuration of 2′-deoxyribofuranosides in 1D and 1L, but instead by the configuration of the DNA templates. The CD signal of all four DNA templates shows the slightly different helical preorganized conformations of the single strands (Fig. S24†) which are not changed by the binding of 1D (Fig. 3) or 1L (Fig. S23†). According to the characteristic bisignate signals with a zero crossing at approximately 280 nm, assembling 1D with D-A20 and D-T20 yields right-handed helicity, whereas with L-A20 and L-T20 it gives left-handed helicity in accordance with the expected chirality for such DNA-like helical structures. There is no measurable CD above 305 nm and thus no information on the ordered chirality of the Tpe chromophores along the DNA templates. Only approximately every second binding site of the DNA templates is occupied by 1D or 1L, preventing the chromophores from CD-active excitonic interactions. Based on the determined general helicity of the DNA-like architectures as determined by their CD signals between 200 nm and 300 nm, we assume that the Tpe chromophores are also helically arranged.
The next step towards DNA-based light harvesting systems is to transfer the excitation energy from the aggregation-emissive Tpe to an appropriate energy acceptor. For the preparation of such energy transfer systems, we focused solely on the components with D-configuration (1D and D-A20) due to the commercial availability of appropriate dye–DNA conjugates. But in principle, we assume that the following experiments would also work with the components with L-configuration. The DNA template D-A20 bears the Atto565 and the Atto633 dye conjugated to the 5′-termini that serve as energy acceptors in the supramolecular assemblies of 1D. The preparation was carried out following the previous protocol including centrifugation to remove unbound 1D by precipitation from the solution. The fluorescence of the Tpe chromophores in the assemblies along the A20–Atto565 and A20–Atto633 templates at 492 nm is strongly reduced compared to that in the assembly of 1D along D-A20. As a result of energy transfer, the strong fluorescence signals of the Atto dyes appear at 598 nm and 660 nm, respectively. When the donor 1D, as part of the assembled DNA architectures, is excited at 389 nm, the quantum yields in the fluorescence range of the donor (400 nm–550 nm, 1D with D-A20, without acceptor: ΦD = 0.400; 1D with A20–Atto565: ΦDA = 0.056; and 1D with A20–Atto633: ΦDA = 0.079) and the quantum yields in the fluorescence range of the acceptor (550–675 nm, A20–Atto565: ΦA = 0.575 and A20–Atto633: ΦA = 0.366) were determined. This allows the calculation of the energy transfer efficiencies according to  which gives remarkable values of E = 86% for the DNA architecture with D-A20–Atto565 and E = 80% for that with D-A20–Atto633. The fluorescence lifetime of 1D along the template A20 at 492 nm is 3.78 ns when excited at 366 nm. It is shortened to 2.11 ns in the DNA architecture with A20–Atto565 and to 2.59 ns with A20–Atto633, which is caused by the energy transfer. The lifetimes of the energy acceptors are 2.78 ns at 598 nm (Atto565), and 3.41 ns at 660 nm (Atto633) in the DNA architectures with 1D when excited at the donor wavelength. We tried to measure the lifetimes of A20–Atto565 and A20–Atto633 without 1D at the donor excitation wavelength (366 nm) too, but the fluorescence intensities were too low for any lifetime determination. As an alternative, we measured the steady-state fluorescence of A20–Atto565 and A20–Atto633 in the absence of 1D, yielding only very small intensities when excited at the donor wavelength (341 nm, Fig. 4, top right), which rules out direct excitation of the acceptors and gives clear evidence for the efficient energy migration from 1D to the Atto dyes in these assembled DNA architectures.
which gives remarkable values of E = 86% for the DNA architecture with D-A20–Atto565 and E = 80% for that with D-A20–Atto633. The fluorescence lifetime of 1D along the template A20 at 492 nm is 3.78 ns when excited at 366 nm. It is shortened to 2.11 ns in the DNA architecture with A20–Atto565 and to 2.59 ns with A20–Atto633, which is caused by the energy transfer. The lifetimes of the energy acceptors are 2.78 ns at 598 nm (Atto565), and 3.41 ns at 660 nm (Atto633) in the DNA architectures with 1D when excited at the donor wavelength. We tried to measure the lifetimes of A20–Atto565 and A20–Atto633 without 1D at the donor excitation wavelength (366 nm) too, but the fluorescence intensities were too low for any lifetime determination. As an alternative, we measured the steady-state fluorescence of A20–Atto565 and A20–Atto633 in the absence of 1D, yielding only very small intensities when excited at the donor wavelength (341 nm, Fig. 4, top right), which rules out direct excitation of the acceptors and gives clear evidence for the efficient energy migration from 1D to the Atto dyes in these assembled DNA architectures.
 | ||
| Fig. 4 Formation of supramolecular light-harvesting DNA architectures with 1D and D-A20 templates that are modified at the 5′-terminus with the Atto565 or the Atto633 dye; fluorescence (top right), 1.25 μM DNA in water + 0.9% DMSO, 250 mM NaCl, and λexc = 341 nm), excitation spectra (bottom right), detected at an emission wavelength of λem = 598 or 660 nm) and images of cuvettes under the UV lamp (bottom left). For the UV/Vis absorption see Fig. S25.† | ||
When control experiments with the “wrong” templates T20 were performed, T20–Atto565 and T20–Atto633 show significantly less energy transfer since very few molecules of 1D are bound to these “wrong” DNA templates according to the low absorbance at 345 nm after removal of unbound 1D by centrifugation (Fig. S26†). As mentioned above, the assembly of 1D along D-A20 (without any attached Atto dye) has a quantum yield of ΦF = 0.40. Accordingly, the emission of the 1D assemblies along A20–Atto565 and A20–Atto633 as templates is quenched by 86% and 80%, respectively. The excitation spectra clearly show that the emission of the Atto dyes originates from the excited 1D units. If we assume an occupancy fraction of f = 0.55 for the 20 binding sites available on these templates, the emission of approximately 9 out of 11 DNA-bound molecules of 1D is quenched. Based on a regular stacking distance of 3.4 Å, the farthest 1D engaged in energy transfer would be approximately 58 Å away from the Atto dye. The efficient quenching over such long distances is likely not the result of one-step energy transfer processes from the individual Tpe that is assembled as a donor at different distances to the 5′-terminal Atto dye as the acceptor. In contrast, the efficient quenching indicates a step-wise energy transport between the Tpe molecules taking place before the final energy transfer to the Atto acceptor dyes occurs.
In order to probe the distance dependence of the energy transfer between 1D and the Atto dyes, we incorporated 1D as a modified nucleotide into synthetic single-stranded DNA. The optical properties of this covalent modification were compared with the non-covalent supramolecular assemblies (Fig. 5). The phosphoramidite as a DNA building block was synthesized by standard procedures and used for automated oligonucleotide synthesis on a solid phase as described in the ESI (Fig. S8–S12).† The synthetic oligouncleotides TPE1a, TPE1b, TPE2a and TPE2b were purified by semi-preparative HPLC and were checked by MALDI-TOF mass spectrometry. TPE1a bears a single 1D modification in the middle of the DNA sequence, and TPE1b, nearer to the 5′-terminus.
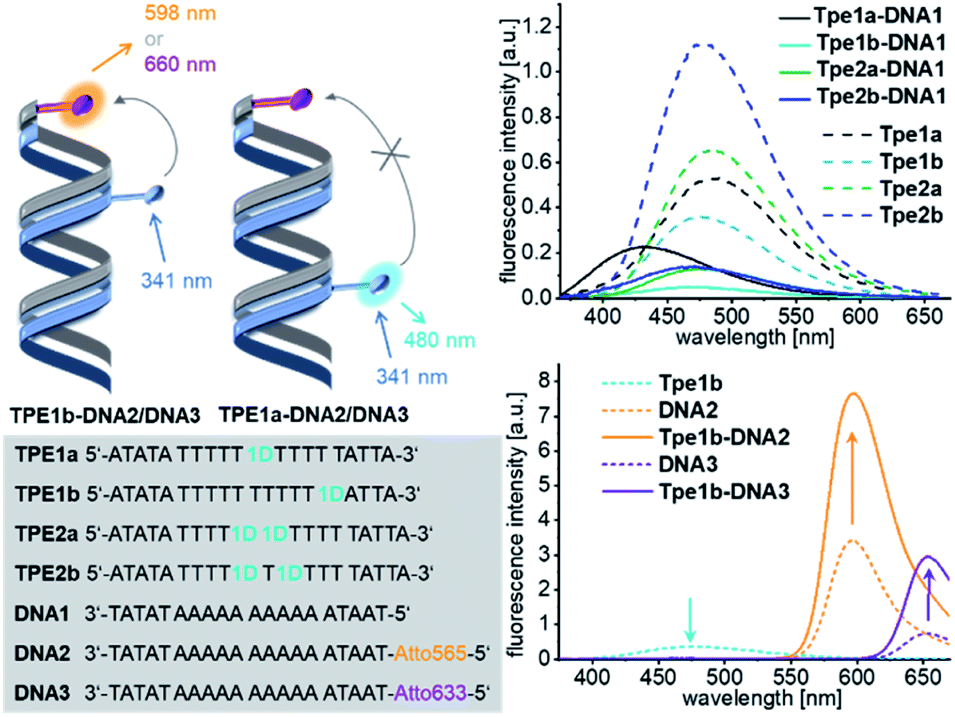 | ||
| Fig. 5 Sequences of DNA strands with covalent 1D modification TPE1a to TPE2b and complementary counterstrands DNA1 to DNA3 (bottom left), forming DNA hybrids, e.g.TPE1–DNA1; fluorescence of hybrids with unmodified counterstrand DNA1 (top right) and with Atto-counterstrands DNA2/DNA3 (bottom right), c(DNA) = c(cs) = 2.5 μM, 250 mM NaCl, 10 mM NaPi, and λexc = 341 nm. For UV/Vis absorption see Fig. S27.† | ||
The fluorescence of these single-stranded oligonucleotides show maxima at 489 nm. The fluorescence intensity is significantly reduced and the maximum is shifted to 430 nm in the double stranded hybrid TPE1a–DNA1, but not in TPE1b–DNA1, when the modified single strands were annealed with the complementary and unmodified counterstrand DNA1. This was similarly observed in the annealed assemblies of 1D (see above). We explain this observation again by stacking the 5-ethynyl-uracil part in a DNA-like double-helical assembly which induces a rotational twist to the phenylene group and decouples the Tpe chromophore from the ethynyl-nucleoside part. This stacking of the core part of the DNA architectures is induced by annealing with complementary counter strand DNA1 with strong effects on the fluorescence – less fluorescence intensity and blue-shift. The conformation of the Tpe chromphore is obviously influenced also by the base on the 3′-side of the 1D modification, since the fluorescence of the DNA hybrid TPE1b–DNA1 is only quenched, but not blue-shifted. TPE1a and TPE1b were subsequently annealed with DNA2 bearing the Atto565 dye or with DNA3 bearing the Atto633 dye as acceptor dyes. The TPE emission of the hybrids TPE1a–DNA2 and TPE1a–DNA3 is not quenched at all and there is no observable energy transfer. Obviously, the distance of 34 Å is too long for energy transfer. In comparison, the hybrids TPE1b–DNA2 and TPE1b–DNA3 show 48% and 46% quenching of the TPE emission and a detectable energy transfer to the Atto dyes over a distance of 17 Å. In comparison, in the non-covalent DNA architectures 1D20–D-A20–Atto565 and 1D20–D-A20–Atto, the pronounced fluorescence quenching involves an energy transfer distance of up to 58 Å, as discussed above. The typical sigmoidal Förster dependence of the energy transfer efficiency on the distance with a characteristic Förster radius cannot be applied to DNA architectures with chromophores that are attached with short and rigid linkers.34–36 Taken together, it implies that a multistep homo-energy transport between the assembled Tpe chromophores precedes the final energy transfer to the Atto dyes as acceptors in the supramolecular structures. Such homo-energy transport cannot be promoted by the unmodified A–T pairs, separating the Tpe from the Atto dyes in the hybrids TPE1a–DNA2 and TPE1a–DNA3. This result underscores the significant role of the DNA templates that control not only the direction of the energy transport but also significantly improve the energy transport properties by their building blocks within the supramolecular DNA architectures, such that the light is harvested much better. The strands TPE2a and TPE2b were modified twice with the nucleotide 1D. The hybridization with the counter strand DNA1 affects the fluorescence in the same manner like in case of TPE1b. TPE2a or TPE2b was annealed with DNA2 modified with the Atto565 dye or with DNA3 modified with the Atto633 dye. In both cases, there was no energy transfer observable between 1D and the Atto dyes in these double-stranded DNA hybrids.
Conclusions
The two new conjugates 1D and 1L with 2′-deoxyuridine as the recognition unit for 2′-deoxyadenosine in single-stranded DNA templates and with Tpe as a fluorescent chromophore can be applied to prepare a new type of supramolecular DNA architecture by self-assembly. Most importantly, all assemblies, including the non-templated and the DNA-templated ones, show significant AIE. The quantum yield of the assembly of 1D along A20 is 0.40; approximately 55% of the available binding sites on the DNA template are occupied by 1D. The two conjugates 1D and 1L differ by the configurations of 2′-deoxyribofuranosides to probe the chirality of both their non-templated assembly and their templated assembly along D- and L-configured DNA templates by means of optical spectroscopy. The chirality of the non-templated assemblies of 1D and 1L is exclusively controlled by the configuration of their sugar parts. In contrast, the chirality of the templated assemblies is governed by the configuration of the sugar part of the DNA templates, and there is no configuration-selective binding of 1D to D-A20 and 1L to L-A20. These supramolecular DNA architectures are hierarchically ordered and the DNA template controls not only the chirality, but also the binding selectivity and the energy transport properties. Moreover, the strong AIE of these DNA architectures with 1D and 1L can be efficiently quenched and the excitation energy can be transported to Atto dyes that were attached to the 5′-termini of the DNA templates. The building blocks of the self-assembled DNA architectures participate themselves in the energy transport along the DNA template; a multistep energy “hopping” transport precedes the final energy transfer to the terminal Atto dyes. This was elucidated by the reference DNA double strands in which we incorporated 1D covalently at two distinct sites in the sequences, one near the Atto dye (4 base pairs, ca. 17 Å), and one farther away (9 base pairs, 34 Å). Only the first DNA construct showed energy transfer properties. The high intensity of the AIE of this new type of supramolecular DNA architecture and the excellent energy transport properties efficiently harvest light and make these DNA-based materials promising candidates for optoelectronic applications.Author contributions
HU performed the experiments and wrote parts of the manuscript. HAW wrote the manuscript and supervised the research project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (grant Wa 1386/20-1) and KIT is gratefully acknowledged. We thank the Biedermann group (INT at KIT) for measurements of FDCD.Notes and references
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhuc and B. Z. Tang, Chem. Commun., 2001, 1740–1741, 10.1039/B105159H.
- J. Li, J. Wang, H. Li, N. Song, D. Wang and B. Z. Tang, Chem. Soc. Rev., 2020, 49, 1144–1172 RSC.
- D. Wang and B. Z. Tang, Acc. Chem. Res., 2019, 52, 2559–2570 CrossRef CAS PubMed.
- X. Gu, J. Yao, G. Zhang, Y. Yan, Y. Zhao and D. Zhang, Chem. –Asian J., 2013, 8, 2362–2369 CrossRef CAS PubMed.
- K. Kokado and K. Sada, Angew. Chem., Int. Ed., 2019, 58, 8632–8639 CrossRef CAS PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- T. F. A. D. Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef PubMed.
- A. Ruiz-Carretero, P. G. A. Janssen, A. Kaeser and A. P. H. J. Schenning, Chem. Commun., 2011, 47, 4340–4347 RSC.
- S. Müller, F. Manger, L. G. v. Reventlow, A. Colsmann and H.-A. Wagenknecht, Front. Chem., 2021, 9, 645006 CrossRef PubMed.
- D. Baumstark and H.-A. Wagenknecht, Chem.–Eur. J., 2008, 14, 6640–6645 CrossRef CAS PubMed.
- E. Mayer-Enthart and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2006, 45, 3372–3375 CrossRef CAS PubMed.
- M. Surin, P. G. A. Janssen, R. Lazzaroni, P. Leclère, E. W. Meijer and A. P. H. J. Schenning, Adv. Mater., 2009, 21, 1126–1130 CrossRef CAS.
- G. Sargsyan, B. M. Leonard, J. Kubelka and M. Balaz, Chem.–Eur. J., 2014, 20, 1878–1892 CrossRef CAS PubMed.
- G. Sargsyan, A. A. Schatz, J. Kubelka and M. Balaz, Chem. Commun., 2013, 49, 1020–1022 RSC.
- S. Sezi and H.-A. Wagenknecht, Chem. Commun., 2013, 49, 9257–9259 RSC.
- R. Hofsass, S. Sinn, F. Biedermann and H. A. Wagenknecht, Chemistry, 2018, 24, 16257–16261 CrossRef PubMed.
- P. Ensslen, Y. Fritz and H.-A. Wagenknecht, Org. Biomol. Chem., 2015, 13, 487–492 RSC.
- S. Muller, Y. Fritz and H. A. Wagenknecht, ChemistryOpen, 2020, 9, 389–392 CrossRef PubMed.
- F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 14192–14196 CrossRef PubMed.
- X. Lou, C. Wai, T. Leung, C. Dong, Y. Hong, S. Chen, E. Zhao, J. W. Y. Lam and B. Z. Tang, RSC Adv., 2014, 4, 33307–33311 RSC.
- D. D. La, S. V. Bhosale, L. A. Jones and S. V. Bhosale, ACS Appl. Mater. Interfaces, 2018, 10, 12189–12216 CrossRef CAS PubMed.
- S. Wang, D. Wei, Z. Zhu and C. Yang, Sens. Actuators, B, 2016, 235, 280–286 CrossRef CAS.
- Q. Jiang, X. Xu, P.-A. Yin, K. Ma, Y. Zhen, P. Duan, Q. Peng, W.-Q. Chen and B. Ding, J. Am. Chem. Soc., 2019, 141, 9490–9494 CrossRef CAS PubMed.
- Y.-X. Yuan, H.-C. Zhang, M. Hu, Q. Zhou, B.-X. Wu, F.-l. Wang, M.-h. Liu and Y.-S. Zheng, Org. Lett., 2020, 22, 1836–1840 CrossRef CAS PubMed.
- F. Xia, J. Wu, X. Wu, Q. Hu, J. Dai and X. Lou, Acc. Chem. Res., 2019, 52, 3064–3074 CrossRef CAS PubMed.
- X. Gao, X. Shu, Y. Song, J. Cao, M. Gao, F. Wang, Y. Wang, J. Z. Sun, J. Liu and B. Z. Tang, Chem. Commun., 2019, 55, 8321–8324 RSC.
- N. Krishnan, M. Golla, H. V. P. Thelu, S. K. Albert, S. Atchimnaidu, D. Perumal and R. Varghese, Nanoscale, 2018, 10, 17174–17181 RSC.
- S. Li, S. M. Langenegger and R. Häner, Chem. Commun., 2013, 49, 5835–5837 RSC.
- K. Swathi, C. Sissa, A. Painelli and K. G. Thomas, Chem. Commun., 2020, 56, 8281–8284 RSC.
- R. Varghese and H.-A. Wagenknecht, Chem.–Eur. J., 2010, 16, 9040–9046 CrossRef CAS PubMed.
- B. Ranjbar and P. Gill, Chem. Biol. Drug Des., 2009, 74, 101–120 CrossRef CAS PubMed.
- R. Hofsäβ, P. Ensslen and H.-A. Wagenknecht, Chem. Commun., 2019, 55, 1330–1333 RSC.
- S. Müller, Y. Fritz and H.-A. Wagenknecht, ChemistryOpen, 2020, 9, 389–392 CrossRef PubMed.
- K. Börjesson, S. Preus, A. H. El-Sagheer, T. Brown, B. Albinsson and L. M. Wilhelmsson, J. Am. Chem. Soc., 2009, 131, 4288–4293 CrossRef PubMed.
- T. Kato, H. Kashida, H. Kishida, H. Yada, H. Okamoto and H. Asanuma, J. Am. Chem. Soc., 2013, 135, 741–750 CrossRef CAS PubMed.
- S. Sindbert, S. Kalinin, H. Nguyen, A. Kienzler, L. Clima, W. Bannwarth, B. Appel, S. Müller and C. A. M. Seidel, J. Am. Chem. Soc., 2011, 133, 2463–2480 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02351a |
| This journal is © The Royal Society of Chemistry 2021 |