
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The exchange of the fast substrate water in the S2 state of photosystem II is limited by diffusion of bulk water through channels – implications for the water oxidation mechanism†
Casper
de Lichtenberg
 ab,
Christopher J.
Kim
c,
Petko
Chernev
b,
Richard J.
Debus
*c and
Johannes
Messinger
*ab
ab,
Christopher J.
Kim
c,
Petko
Chernev
b,
Richard J.
Debus
*c and
Johannes
Messinger
*ab
aDepartment of Chemistry, Umeå University, Linnaeus väg 6 (KBC huset), SE-901 87, Umeå, Sweden. E-mail: johannes.messinger@kemi.uu.se
bMolecular Biomimetics, Department of Chemistry – Ångström Laboratory, Uppsala University, POB 523, SE-75120 Uppsala, Sweden. E-mail: johannes.messinger@kemi.uu.se
cDepartment of Biochemistry, University of California, Riverside, California 92521, USA. E-mail: richard.debus@ucr.edu
First published on 1st September 2021
Abstract
The molecular oxygen we breathe is produced from water-derived oxygen species bound to the Mn4CaO5 cluster in photosystem II (PSII). Present research points to the central oxo-bridge O5 as the ‘slow exchanging substrate water (Ws)’, while, in the S2 state, the terminal water ligands W2 and W3 are both discussed as the ‘fast exchanging substrate water (Wf)’. A critical point for the assignment of Wf is whether or not its exchange with bulk water is limited by barriers in the channels leading to the Mn4CaO5 cluster. In this study, we measured the rates of H216O/H218O substrate water exchange in the S2 and S3 states of PSII core complexes from wild-type (WT) Synechocystis sp. PCC 6803, and from two mutants, D1-D61A and D1-E189Q, that are expected to alter water access via the Cl1/O4 channels and the O1 channel, respectively. We found that the exchange rates of Wf and Ws were unaffected by the E189Q mutation (O1 channel), but strongly perturbed by the D61A mutation (Cl1/O4 channel). It is concluded that all channels have restrictions limiting the isotopic equilibration of the inner water pool near the Mn4CaO5 cluster, and that D61 participates in one such barrier. In the D61A mutant this barrier is lowered so that Wf exchange occurs more rapidly. This finding removes the main argument against Ca-bound W3 as fast substrate water in the S2 state, namely the indifference of the rate of Wf exchange towards Ca/Sr substitution.
Introduction
Photosynthesis performed by plants, algae and cyanobacteria is critical for life on Earth as it releases molecular oxygen into the atmosphere and stores solar energy as biomass. Utilizing sunlight, the protein complex photosystem II (PSII) generates and stabilizes charge pairs that are employed for the extraction of 4 electrons and 4 protons from 2 water molecules, and to reduce plastoquinone to plastohydroquinone.1,2 The solar-to-chemical energy conversion efficiency of PSII has been estimated to reach values of up to 15%.3The water oxidation reaction is catalyzed by a metal–oxygen cluster comprising the metals manganese and calcium in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry as well as five oxo bridges (O1–O5).4–6 During the reaction cycle, the Mn4CaO5 cluster is stepwise oxidized by light-induced charge separations in the chlorophyll containing reaction center of PSII. Thereby, it attains four discrete reaction intermediates (S0–S3) and one highly reactive transient (S4).7–10 The S1 state is dark-stable, and the S2 → S3 transition involves the association of a new water molecule (WN1), yielding a Mn4CaO6 cluster as the last stable intermediate before O2 formation.11–17 The next light-induced charge separation triggers the S3 → S4 → S0 transition, which not only involves the O–O bond formation, but also O2 release and the concomitant filling of the open coordination site by one of the terminal water ligands (W3 or W2) as well as the binding of a new water molecule (WN2).9,12,18,19 All S state transitions, with the exception of S1 → S2, are coupled to proton release into the bulk, keeping the total charge of the cluster at 0 or +1, respectively.20 Proton release is facilitated by an intricate H-bonding network that is pivotal to the function of PSII and its earth-abundant water oxidation catalyst.17,21–24
:1 stoichiometry as well as five oxo bridges (O1–O5).4–6 During the reaction cycle, the Mn4CaO5 cluster is stepwise oxidized by light-induced charge separations in the chlorophyll containing reaction center of PSII. Thereby, it attains four discrete reaction intermediates (S0–S3) and one highly reactive transient (S4).7–10 The S1 state is dark-stable, and the S2 → S3 transition involves the association of a new water molecule (WN1), yielding a Mn4CaO6 cluster as the last stable intermediate before O2 formation.11–17 The next light-induced charge separation triggers the S3 → S4 → S0 transition, which not only involves the O–O bond formation, but also O2 release and the concomitant filling of the open coordination site by one of the terminal water ligands (W3 or W2) as well as the binding of a new water molecule (WN2).9,12,18,19 All S state transitions, with the exception of S1 → S2, are coupled to proton release into the bulk, keeping the total charge of the cluster at 0 or +1, respectively.20 Proton release is facilitated by an intricate H-bonding network that is pivotal to the function of PSII and its earth-abundant water oxidation catalyst.17,21–24
The Mn4CaO5 cluster is frequently described as having a ‘chair’-like structure, with the base formed by a Mn3CaO4 hetero-cubane and the back by the fourth Mn ion (Mn4) that is connected to the base via the oxygen bridges O5 and O4 (Fig. 1).6 As there is no bond between O5 and Mn1, the structure is referred to as ‘open cubane’.5,15 Importantly, this structure binds four water molecules, two at Mn4 (W1, W2) and two at Ca (W3, W4), while all other coordination sites, except one at Mn1, are filled by five oxo-bridges, six bridging carboxylates and one histidine ligand.6,15 In the S0 state, the four Mn ions have the oxidation states Mn4(III,IV,III,III) (oxidation states given in the order Mn1 through Mn4), and up to S3 all transitions involve a Mn(III) → Mn(IV) oxidation (for review see ref. 25, 26), although for the S3 state also a small equilibrium concentration of a peroxidic intermediate has been proposed to exist.1,21,27,28 By contrast, the S4 state likely involves oxygen radical formation.12 Alternatively, electronic compositions of Mn(IV,IV,IV,V), Mn(III,III,IV,VII), or superoxo intermediates have been proposed for S4 (for review see ref. 29, 30 and ESI Fig. S1†).
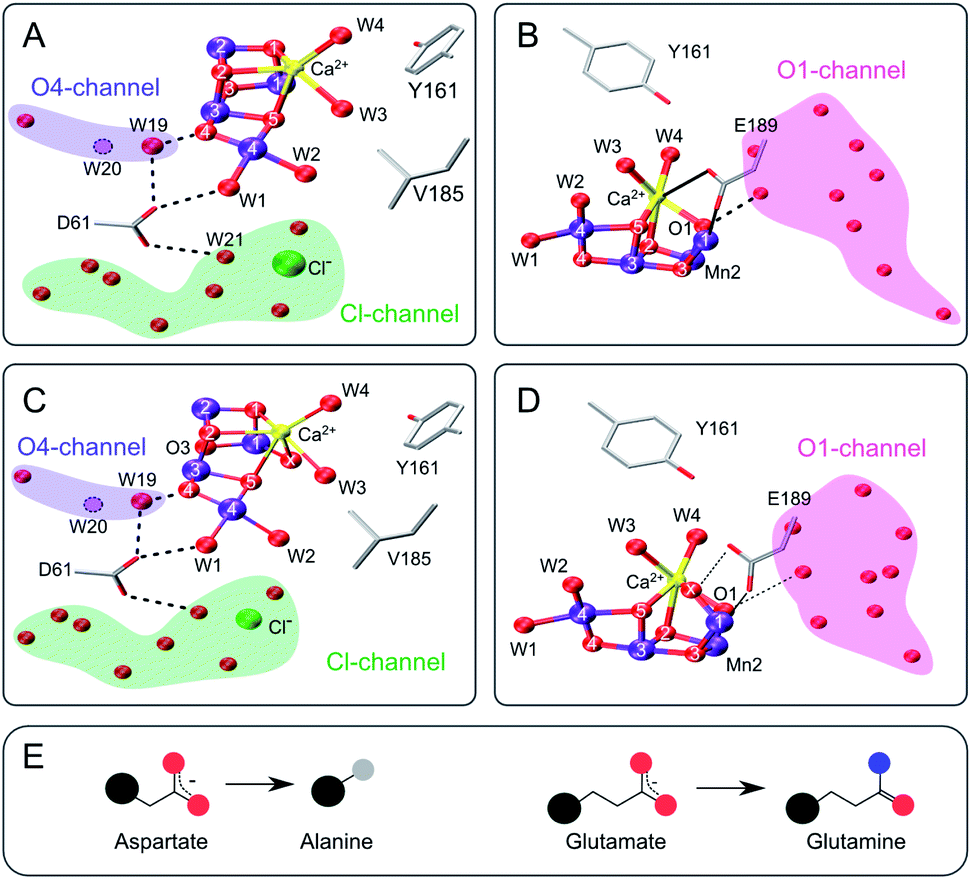 | ||
| Fig. 1 Structure of the Mn4CaO5 cluster with selected ligands and water molecules in the S2 (panels A and B) and S3 (C and D) states of photosystem II (PDB: 6DHF & 6DHO). Note that the cluster has a sixth oxygen bridge labelled X in the S3 state. Panels A & C highlight the position of D61 and panels B & D that of E189 in relation to the Mn4CaO5/6-cluster and the O4 (blue), Cl1 (green) and O1 (pink) water/proton channels. Potential hydrogen bonds are shown as dashed lines, while the coordination of E189 to Ca and Mn is indicated with solid lines. The position of W20, which is not resolved in the S2- and S3-state structures, is indicated by a dashed circle. E: Cartoon of the D to A mutation (left) and the E to Q mutation (right). Color code: large black sphere – peptide backbone; red - oxygen; blue - nitrogen; purple – manganese; yellow – calcium; green – chloride; grey - methyl group. The molecular representations were generated with VMD.102 | ||
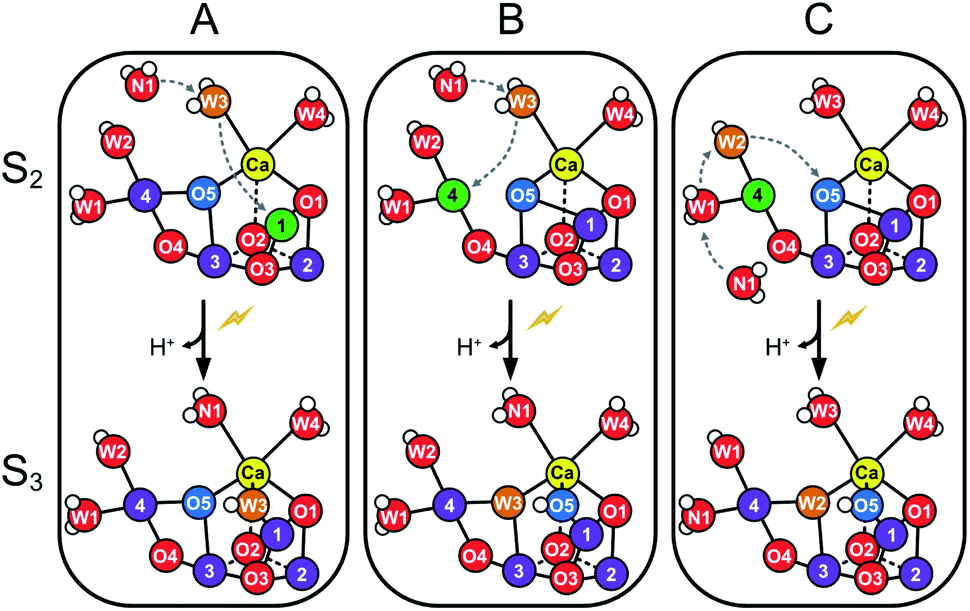
The structure of the Mn4CaO5 cluster is flexible. In the S2 → S3 transition it takes up one additional water molecule (WN1) and a new hydroxo/oxo bridge (Ox/O6; here after Ox) is formed between Ca and Mn1.14,15,17,21,31 The precise mechanism for this is under debate and the three discussed options are depicted in Scheme 1.32–37 In addition to this water uptake (denoted by a W superscript; Scheme 2), in each S state the cluster can attain at least two different conformations.18,38–41,56 This is best documented for the S2 state, where the two conformations give rise to the low spin (LS) S2g = 2 multiline and the broad high-spin (HS) g = 4–6 EPR signals, respectively. The well-characterized open cube (S2A) structure gives rise to the S2LS signal, while for the S2HS state several structures have been proposed: the closed cube S2B,34,41 the open cube water bound S2AW42-46 and the protonation isomer S2API47,48 (Scheme 2). Among these, the S2B and the S2API structures provide the best computational explanation for the g = 4 EPR signal, while the S2AW state, which has a structure akin to the S3AW state, is favored on the basis of substrate water exchange experiments, and because it provides a straightforward explanation for the low transition temperature of S2HS to S3AW.44–46,49 For all S states, the SA structures dominate under most conditions, except for the S3 state, where S3AW is most stable.19,29
 | ||
| Scheme 1 Suggested routes for insertion of WN1 and formation of the Ox hydroxo bridge during the S2 → S3 transition. Panels A and B show two proposed pathways for W3 insertion. Pathway A starts from the more stable, open cube (S2A) conformation of the Mn4CaO5-cluster. W3 is inserted into the Ox site between Ca and Mn1, while WN1 replaces W3.32,33 B: The Mn4CaO5-cluster attains first the S2B conformation before W3 binds to Mn4. W3 then flips into the O5 binding site, while O5 moves into the Ox position and WN1 replenishes the original W3 coordination site at Ca.32,33 C: The pivot or carousel mechanism requires also that the cluster attains first the less stable S2B conformation. Binding of WN1 to the five-coordinate Mn4(III) induces a cascade of water/oxygen relocations allowing W1 to replace W2, W2 to flip into the O5 position, and O5 to occupy the Ox site.36,37 | ||
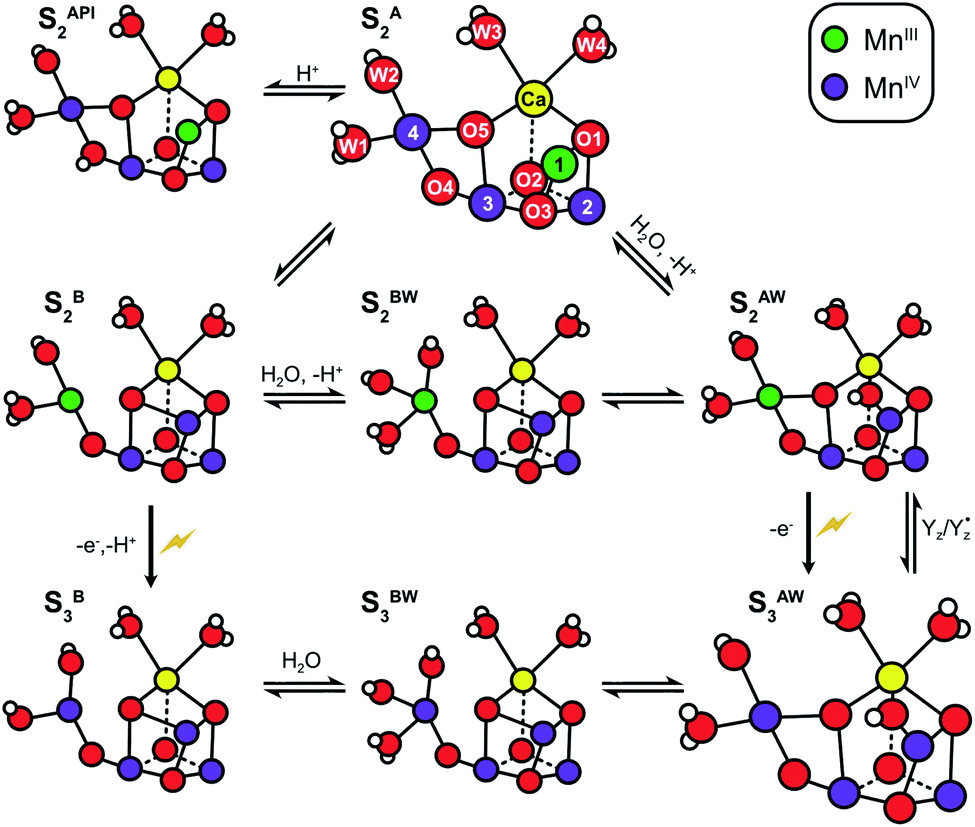 | ||
| Scheme 2 Structural flexibility of the Mn4CaO5/6 cluster in photosystem II. S2A and S3AW are the most stable structures of the S2 and S3 states and have been observed by crystallography at room temperature.14,15,17 S2B, S2BW, S2AW and S2API are computational structures that were proposed to give rise to the S2HS EPR signal, and or have been suggested as intermediates during O5 exchange against bulk water.41,46,48,55 S3B and S3BW have been supported by EPR spectra obtained when S2 samples were advanced to S3 under conditions that may block water insertion, and have been suggested as intermediates during the S2 → S3 transition.36,92 They are also involved in substrate water exchange.44,46,55 For example, in the S2A state O5 (Ws) exchanges via the S2AW, S2BW and S2B states, while its exchange in the S3AW state requires the equilibrium of S3AW YZ with S2AW YZ˙ or alternatively the transition into the S3BW and S3B states. Color code: MnIV purple, MnIII green, Ca yellow, O red, H white. The flash indicates a light-induced charge separation in PSII. | ||
Identification of the two substrate water binding sites in the four discrete intermediates of the reaction cycle would provide a solid basis for decoding the mechanism of biological water oxidation. While there are several ways to identify water molecules bound to or near the Mn4CaO5/6 cluster, only the determination of the isotopic composition of the O2 produced after a rapid enrichment of the sample with H218O by membrane inlet mass spectrometry (MIMS) allows obtaining a unique experimental signature for the two substrates: their exchange rates with bulk water.18,50,51
Using this approach, it was shown that the two substrates are bound differently in the S2 and S3 states.18,52 The faster exchanging substrate water is referred to as Wf, while the slower one is denoted as Ws. For the S0 and S1 states, only the exchange rates of Ws were determined. However, since no water binding events are known for the S0 → S1 and S1 → S2 transitions, both substrates must be bound already also in these early S states.46 Connecting the water exchange data with emerging structural and spectroscopic information led to the proposal that Ws is the central μ3-oxo bridge, today known as O5.9 This was subsequently supported by theoretical and spectroscopic12,53,54 as well as further MIMS studies.18 Exchange of O5 has been shown to be a multistep process in which O5 is brought into a terminal position on Mn4 where it is fully protonated. In this process, the Mn4CaO5/6 cluster attains several of the alternative conformations shown in Scheme 2.44,46,55 For example, O5 (Ws) would exchange in the S2A state by first forming S2AW through the uptake of one water, and then changing conformation to a S2BW state, which via equilibrium with the S2B state allows exchange of O5 with bulk water.44,46,55
By contrast, the assignment of Wf is controversial. FTIR and snapshot crystallographic studies as well as a number of DFT calculations suggest that Wf is bound as W3 to Ca in the S2 state, but then forms a bridge between Ca and Mn upon S3 state formation via insertion pathways A or B (Scheme 1).14–17,21,33
On the other hand, present MIMS experiments favor the terminal water ligand W2 as Wf, because the exchange of Wf is firstly several orders of magnitude slower than would be expected for a terminal water ligand on Ca,9,18,57 and secondly independent of Ca/Sr-substitution in both the S246 and S318,58,59 states. In addition, Wf exchange becomes observable first in the S2 state, and then slows upon S3 and S3YZ˙ state formation, making a diffusion limitation that could obscure the Ca/Sr dependence seemingly unlikely. By contrast, these two observations can be well explained with W2 as Wf by the known oxidation of Mn4 during the S1 → S2 transition and the need to involve electron back donation of YZ for Wf exchange in the S3 state.9,18,59 Absence of a diffusion limitation is apparently further supported by molecular dynamics (MD) calculations that predict water access in the 50 ns to 100 μs time range,60,61i.e. orders of magnitude faster than Wf exchange (50–100 ms).44,46,57
Three channels have been identified that lead to the Mn4CaO5 cluster: the O1 or ‘large’ channel, the O4 or ‘narrow’ channel, and the Cl1 or ‘broad’ channel (Fig. 1). While the O1 and Cl1 channels both split into two branches (A, B),15,17 all three channels have been variously proposed to be involved in either proton, dioxygen and/or water transport during various S state transitions, for review see.17,21,60,62,63 Recent room temperature and cryogenic X-ray crystallography studies favor that water access to the catalytic site occurs via the O1 channel as it shows the largest variation in water positions between studies and S states.15,17,64 By contrast, previous theoretical studies suggested that water is delivered through the O4 channel to the Mn4 site and is inserted during the S2 → S3 transition via the pivot/carousel mechanism (Scheme 1C).36,37 Recent mass spectrometric studies analyzing the oxidative damage to the D1, D2 and CP47 proteins caused by the formation of reactive oxygen species (ROS) at the Mn4CaO5/6 cluster under illumination support both the B branch of the Cl1 channel and the O1 channel as water access pathways.63,65,66
To probe if the fast water exchange (Wf) in the S2 state is limited by diffusion through channels or by the chemical exchange process, we study here the effects of the D1-D61A and D1-E189Q mutations on the rates of substrate water exchange with bulk water in the S2 and S3 states.
The D61 residue is located close to Mn4 at the apex between the potential O4 and Cl1 substrate channels (Fig. 1). D61 hydrogen bonds W1 and some further waters in its surroundings. If this aspartate (D) residue is mutated to either asparagine (N) or alanine (A), O2 production decreases by ∼75–80%, and the S1 → S2 and S2 → S3 transitions are decelerated by factors of 2–3.67 Meanwhile, O2 release in the S3 → S0 transition is retarded 20–30 fold.67–69 These functional effects were attributed to poor proton abstraction from the mutants, identifying this residue as an important proton relay.68,70,71 It may be speculated that if W2 were a substrate, its exchange would be greatly affected by the D61A mutation. The S3 state exchange rates were previously measured for the D61N mutant, showing 6-fold and 3-fold slower exchange rates for Wf and Ws, respectively.72
E189 is located at the end of the O1 channel. In the S1 and S2 states, E189 is a ligand of Mn1, and it also weakly ligates Ca. Recently it was shown, by time-resolved X-ray crystallography, that during the S2 → S3 transition E189 detaches from Ca before Ox is inserted, and afterwards hydrogen bonds Ox (Fig. 1B and D).15,17,21 Consequently, this glutamate residue (E189) may be important for the insertion of Ox during the S2 → S3 transition, the exchange of Ox by bulk water in the S3 state, and O–O bond formation. Only a handful mutations of E189 yield active PSII centers, namely isoleucine (I), lysine (K), leucine (L), glutamine (Q) and arginine (R).73 E189Q is a conservative mutant, as it is of similar size and retains the ability to act as bidentate ligand (Fig. 1E). While the S2LS signal is not perturbed by the mutation, the oxygen evolution activity is decreased by ∼30%,73 indicating that some transition in the catalytic cycle does not function optimally. For the S3 state, an up to 2-fold faster substrate water exchange was reported previously.74
Experimental procedures
Preparation of photosystem II core complexes
Synechocystis sp. PCC 6803 strains, with a 6xHis-tag fused to the CP47 gene, expressing the psbA2-gene (WT, D1-D61A or D1-E189Q) were propagated in BG11 medium supplemented with glucose in glass carboys and grown as previously described.70 Thylakoid membranes and core complexes were prepared as described previously.70 The PSII core complexes were suspended in 1.2 M betaine, 10% (v/v) glycerol, 50 mM MES-NaOH (pH 6.0), 20 mM CaCl2, 5 mM MgCl2, 50 mM histidine, 1 mM EDTA, and 0.03% (w/v) n-dodecyl β-D-maltoside, and were concentrated to ∼1 mg of Chl mL−1. The samples were then divided into 100 μL aliquots and flash-frozen in liquid N2. Finally, samples were stored at −80 °C.Time-resolved membrane-inlet mass spectrometry
Substrate–water exchange rates were measured at 10 °C employing an isotope ratio mass spectrometer (Finnigan Delta Plus XP) featuring 7 Faraday cups (m/z 32, 34, 36, 40, 44, 46 & 48) and a 165 μL rapid mixing reaction cell that was connected to the spectrometer through a stainless steel pipe that passed through a Dewar filled with liquid N2.51 After thawing, the PSII core complexes were washed (total dilution factor: 100–1000) in 50 mM MES-NaOH pH/pD 6.5, 1 M betaine, 15 mM CaCl2, 15 mM MgCl2 using an Amicon Ultra-0.5 centrifugal filter unit and finally concentrated to 0.15–0.2 mg Chl per mL. After a saturating preflash (5 μs FWHM), the sample was dark-adapted for 1 hour at room temperature. Prior to loading in dim green light, 0.3 mM (final concentration) 2,6-dichloro-1,4-benzoquinone was added.A modified gas-tight syringe (Hamilton CR-700-50) with an air pressure driven, computer triggered piston, previously loaded under N2 atmosphere with ∼22 μL 97% H218O, was employed for rapid (∼6 ms) isotope enrichment to a final level of ∼12%.57
Residual O2 in the H218O was estimated and removed from the data as described previously.44 The measurement sequences for all samples and S states are shown in ESI Fig. S2.† The substrate exchange rates (kf1, kf2, ks1 and ks2) for the fast and slow substrate waters were determined by a simultaneous fit of the m/z 34 and the m/z 36 data (for details see ESI Text 1 and Table S1†).
Results
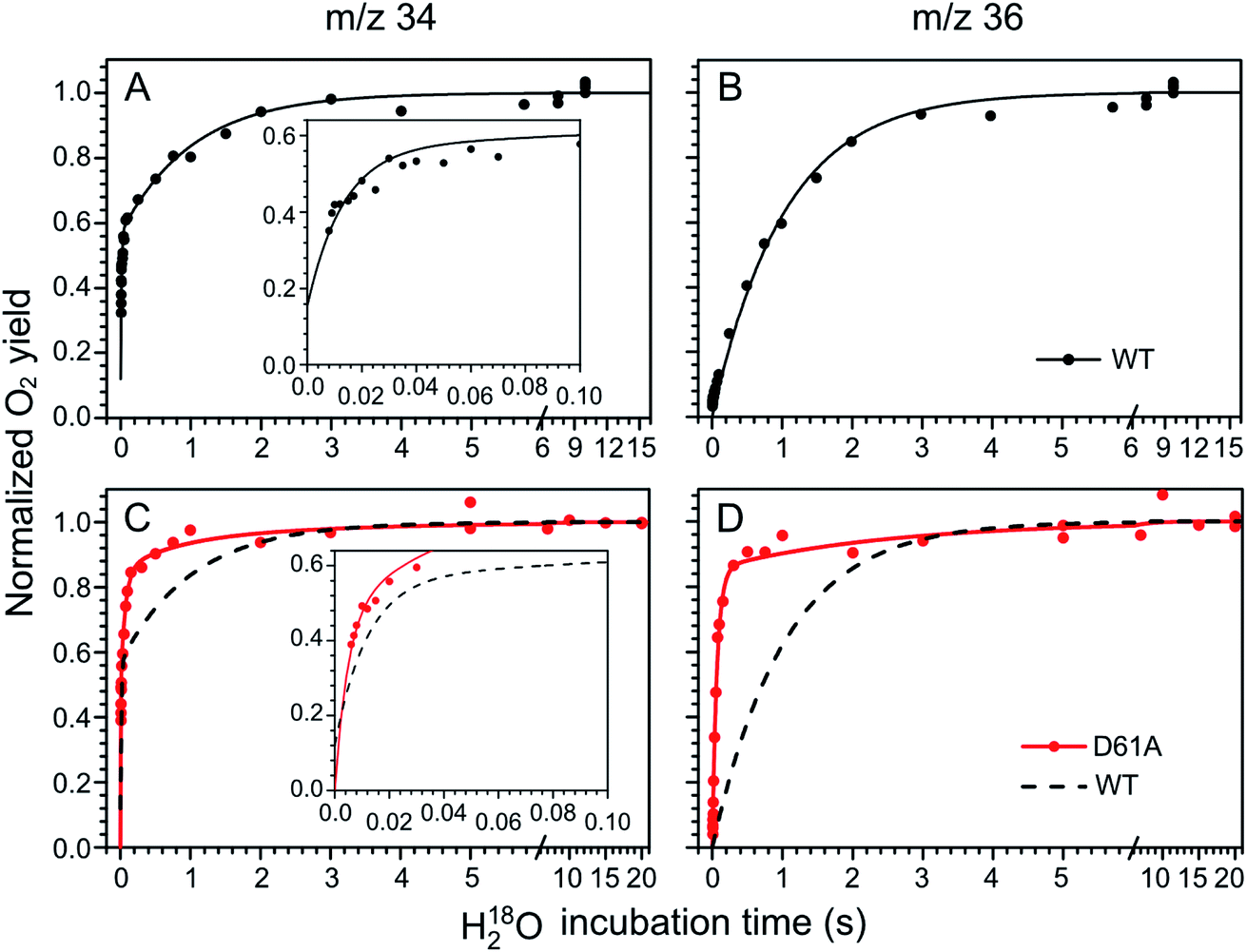
The substrate water exchange rates of WT-, D61A- and E189Q-PSII core complexes from Synechocystis sp. PCC 6803 were studied in the S2 and S3 states of the oxygen-evolving complex at 10 °C, pH 6.5. For WT-PSII, the canonical biphasic exponential rise with a fast and slow phase18 was observed for the 16,18O2 signal from the m/z 34 cup in the S2 and S3 states (symbols in Fig. 2A and 3A). The biphasic rise shows that the two substrate waters are bound differently to the Mn4CaO5 cluster in these S states. Accordingly, they are referred to as the fast, Wf, and slow, Ws, exchanging substrate waters. The corresponding rates, kf and ks, obtained from the kinetic fits (solid lines) are given in Table 1. For the 18,18O2 signal (m/z 36), which requires that both substrate waters exchange against H218O added to the bulk water, a mono-exponential rise with the rate ks was detected (Fig. 2B and 3B). This is expected, as this process is limited by the slower exchange process. The monophasic rise of the m/z 36 signal confirms that the two kinetic phases in the m/z 34 signal do not arise from sample heterogeneity.51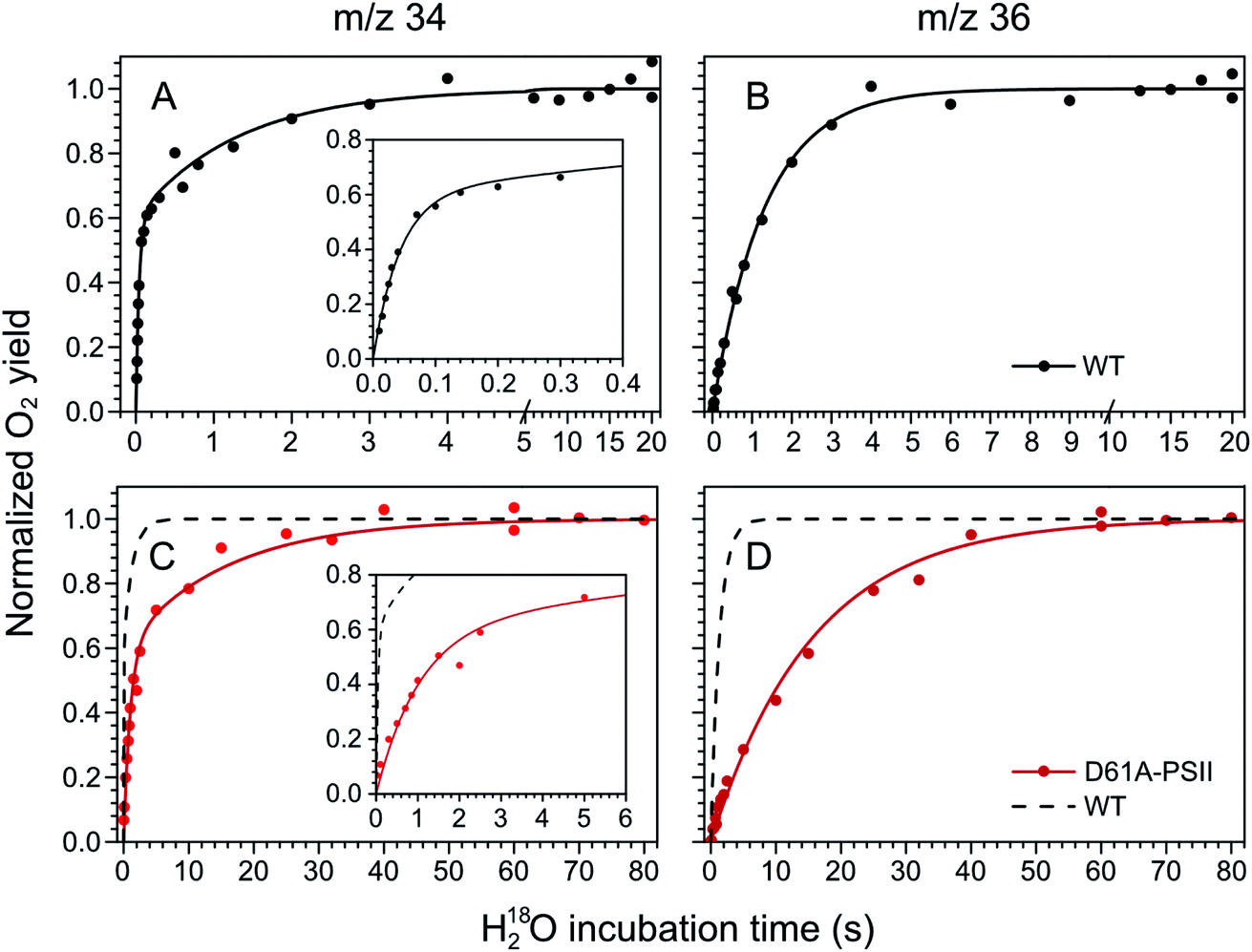 | ||
| Fig. 2 Substrate water exchange measurements in the S3 state of WT- (black) and D61A- (red) PSII core complexes of Synechocystis sp. PCC6803. The normalized oxygen yield of a flash given after different incubation times with H218O in the S3 state are plotted. A and C show the results for single labelled oxygen (m/z 34), while panels B and D those for double labelled oxygen (m/z 36). Dots represent individual measurements, while solid lines the results of kinetic fits (Table 1). The fits of the WT-PSII substrate exchange are shown as a dashed line next to the D61A-PSII data for visual comparison. The inserts show an enlarged view of the fast exchange phase in the m/z 34 data. Observe differences in the time scales. The data were recorded at 10 °C, pH 6.5. | ||
 | ||
| Fig. 3 Substrate water exchange measurements in the S2 state of WT- (black) and D61A- (red) PSII core complexes of Synechocystis sp. PCC6803. The normalized oxygen yield of a double flash given after different incubation times with H218O in the S2 state are plotted. A and C show the results for single labelled oxygen (m/z 34), while panels B and D those for double labelled oxygen (m/z 36). Dots represent individual measurements, while solid lines the results of kinetic fits (Table 1). The fits of the WT-PSII substrate exchange are shown as a dashed line next to the D61A-PSII data for visual comparison. The inserts show an enlarged view of the fast exchange phase in the m/z 34 data. Observe differences in the time scales. The data were recorded at 10 °C, pH 6.5. | ||
| WT-PSII | D61A-PSII | E189Q-PSII | |||||||
|---|---|---|---|---|---|---|---|---|---|
| k f | k s | k f1 | k s1 | k f2 | k s2 | k f | k s | ||
| S3 | Fraction, % | 100 | 100 | 0 | 100 | ||||
| Rate, s−1 | 23.4 ± 1.4 | 0.76 ± 0.03 | 0.97 ± 0.07 | 0.064 ± 0.003 | — | — | 24.6 ± 1.8 | 0.76 ± 0.04 | |
| Mutant/WT | — | — | 0.041 ± 0.004 | 0.084 ± 0.005 | — | — | 1.07 ± 0.08 | 1.00 ± 0.07 | |
| S2 | Fraction, % | 100 | 85 | 15 | 100 | ||||
| Rate, s−1 | 84 ± 5 | 0.97 ± 0.03 | >300 | 15 ± 1 | 1.4 ± 0.9 | 0.4 ± 0.1 | 66 ± 5 | 0.94 ± 0.04 | |
| Mutant/WT | — | — | >3.5 | 15 ± 1 | 0.017 ± 0.011 | 0.4 ± 0.1 | 0.79 ± 0.08 | 0.97 ± 0.05 | |
In the S3 state, mutation of the D1–D61 residue to alanine led to a 24- and 12-fold slowing of Wf and Ws exchange (Fig. 2C and D, Table 1). This slowing is one of the largest effects of a mutation or biochemical change on substrate exchange kinetics observed thus far.18 For example, this change is 4-fold larger than the previously reported 6- and 3-fold decelerations for the D61N mutant.72 Notably, the monophasic rise of the m/z 36 signal was preserved (Fig. 2D).
In the S2 state, the same mutation had the opposite effect, i.e. a strong acceleration of the exchange was found for both substrates (Fig. 3C and D): 15-fold for Ws and more than 3.5-fold for Wf, of which the rate could no longer be resolved with our present mixing system (Table 1).
However, detailed analysis showed that the exchange of both Wf and Ws were biphasic, and that in the smaller fraction, about 15%, the exchange of Wf and Ws occurred with rates that were slower than those of WT-PSII (Table 1). Thus, the m/z 34 data were fit with 4 kinetic phases instead of 2. This showed that in the S2 state of D61A-PSII two stable populations of the Mn4CaO5 cluster with possibly different substrates, exchange pathways or water accessibility must exist.
To probe the effects of H-bonding and of O–H bond breaking/formation on the exchange of substrate water in the S2 state of WT- and D61A-PSII, we performed the same experiments also in D2O (Fig. S3 and Table S2†). In general, the exchange rates of Wf and Ws were slower in D2O. Wf showed a corrected H/D isotope effect of ≤1.3. By contrast, Ws displayed an H/D isotope effect of 1.5 (WT) to 1.9 (D61A, larger fraction) and 2.8 (D61A, smaller fraction). In D61A-PSII, the smaller phase of Wf and Ws exchange increased from 15% (H2O) to 24% (D2O) (Table S2†).
Water exchange in the S2 and S3 states of the D1-E189Q mutant occurred with nearly identical rates as in WT-PSII. Only the exchange of Wf was retarded by ∼20% in the S2 state of the E189Q samples (Table 1; Fig. S4†). We note that a ∼2-fold acceleration was previously observed in the S3 state exchange rates of E189Q-PSII thylakoid membranes.74
Discussion
In this study, we observed that the mutation of D61 to alanine had a strong effect on the exchange of both substrate waters in the S2 and S3 states, while the mutation of E189 to glutamine had essentially no influence on either Wf or Ws exchange. As D61 is close to W2, while E189 is near W3 and Ox, these results appear, at first glance, to favor W2 over W3 as fast exchanging substrate Wf. However, because we previously showed that the substrate water exchange rates in PSII are strongly affected by conformational equilibria of the Mn4CaO5/6 cluster, and because the mutations are also located at the end points of water channels and may thereby affect the diffusion of water to the catalytic site, a more detailed analysis is required.For example, our recent studies have shown that the exchange rate of Ws in the S2 state depends on the equilibria between the S2A, S2AW, S2BW and S2B states of the Mn4CaO5 cluster (Scheme 2).44,46,55 This allows O5 to reach a terminal position on a MnIII ion (Mn4) and to be exchanged with bulk water. For Wf the situation is less clear as previous data allow for two options: either the Wf exchange rate also depends on conformational equilibria, or its exchange is limited by diffusion of bulk water through the channels leading to the Mn4CaO5 cluster. Knowing which exchange mechanism applies may help identifying Wf and thus for experimentally elucidating the mechanism of water oxidation.
If conformational changes determine the exchange kinetics, then the Mn4-ligated W2 must be Wf because these equilibria only affect the exchange of W2 and not that of the Ca-ligated W3. The absolute rate for Wf exchange, which is orders of magnitude slower than previously reported for water ligands of Ca ions and too fast for a water ligand of a Mn(IV) ion, can in this case be explained via the equilibrium between the S2A and S2B states, because in the S2B state Mn4 has the oxidation state Mn(III) that allows for rapid water exchange (Mn(III) is exchange-labile; Mn(IV) is exchange inert – for discussion see ref. 9,18 and 57). Binding to Mn would also explain the insensitivity of the Wf exchange rate to Ca/Sr substitution.
If diffusion of water through channels determines the exchange kinetics, then the Ca-ligated W3 would remain an option for Wf, because this limitation would explain that Wf exchange is comparatively slow for a Ca-bound water ligand and that its exchange is unaffected by Ca/Sr substitution. In this case, it would be impossible to distinguish W2 or W3 as the fast exchanging substrate in wild-type PSII in the S2 state on the basis of substrate water exchange rates, unless some treatment shifted the equilibrium between S2A and S2B strongly towards S2A, as this would keep W2 bound to an exchange-inert Mn(IV) ion, leading to a very slow exchange of W2.
In the following, we will first analyze if the faster water exchange in D61A-PSII is due to a shift of conformational equilibria, or if the truncation of this amino acid from aspartate to alanine increases water accessibility to the catalytic site. Subsequently, we will elucidate the consequences of this result for (i) understanding the exchange rates in the other S states and (ii) the assignment of Wf. Finally, we will discuss the remaining options for the mechanism of water oxidation.
Wf exchange in the S2 state
In the S2 state, Wf exchanges significantly faster than in WT-PSII in the majority of D61A-PSII centers (85%; Table 1). If a shift in conformational equilibria accounts for this observation, the Mn4-bound W2 would be the most likely assignment for Wf, as outlined above. In this case, the D61A mutation would induce a change in the conformational equilibria of the Mn4CaO5 cluster towards the S2B state (or another S2HS state), because this allows W2 to exchange much more readily compared to WT-PSII.46 Therefore, a faster exchange of W2 in D61A-PSII would imply that the activation barrier for reaching the S2B state would be lower and/or the relative stability of the S2B state would be increased in the mutant. However, previous experimental data show that a stabilization of the HS S2B state can be excluded, as only the LS S2 multiline signal was observed in the D61A-PSII samples and its signal intensity was comparable to that of WT-PSII (see ESE-EPR spectra in ref. 75). This is supported by theoretical calculations that find the equilibrium between the S2A and S2B state unchanged or even slightly shifted in favor of the S2A state.71 These calculations also indicate that in D61A-PSII one proton is lost from the W1/W2/Mn4 site of the cluster.71 Such a proton loss would slow the W2 exchange. In conclusion, the direct chemical changes that can be expected to occur would either leave the water exchange the same or likely even slow the exchange of W2, the opposite to what is observed experimentally for Wf exchange. This analysis shows that a shift of the conformational equilibrium between S2A and S2B cannot explain the present data.On this basis, we conclude that the exchange of Wf by isotopically labelled bulk water must be slowed by a steric constraint in all the channels that supply substrate to the Mn4CaO5 cluster in WT-PSII.57 The D61A mutation then appears to remove one of these diffusion barriers so that Wf exchange can occur at the experimentally observed faster rate. Indeed, barriers for water transport were described previously for all channels, and D1-D61 was identified as forming a barrier for water access together with D2-K317 and Cl1.61 We propose that shortening D1-D61 via the D61A mutation creates a void that is filled by one or two water molecules, which promotes faster water diffusion to the Mn4CaO5 cluster. This idea is in line with a recent theoretical study that shows water redistributions and faster movements of water molecules in the D61A mutant.76
Model for Wf exchange via the Cl1 channel in the S2 state
Our data strongly indicate that D61 forms a steric barrier for water access to the catalytic site that contributes to limiting the rate of Wf exchange in the S2 state. However, comparison of the measured water exchange rates to water transport rates estimated from MD simulations appears to contradict this conclusion: in WT-PSII, the rate for Wf exchange is about 80 s−1 (at 283 K), while barriers of 10–14 kcal mol−1 calculated for all channels for moving a water molecule from the bulk to the Mn4CaO5 cluster would predict exchange rates up to a 1000-fold faster than our observation (see ESI TEXT 3†).61 However, the two processes are not directly comparable. MD simulations of water movements always employ a force to achieve concerted or directed water movement along a certain trajectory. This force can be provided for example by inserting extra water molecules near the Mn4CaO5 cluster, or by pulling water molecules through the channels at a constant velocity.61 By contrast, isotopic equilibration involves random swapping of neighboring water molecules driven by thermal energy. It thus requires many swapping events to reach isotopic equilibrium between an inner water pool and bulk water.As D61 is located at a branching point of the O4 channel and the Cl1 channel, the faster water access may occur through either or both of these channels. The O4 pathway (channel 2 in ref. 61) has been proposed to facilitate substrate water entry36,77–80 because binding sites for the substrate analogues ammonia75,81–83 and methanol78,84–86 are located in the vicinity of Mn4, O4, and D1-D61. Also, the D1 residue at position 87, which is near the origin of the O4 pathway, is Ala in spinach and Asn in cyanobacteria, a fact that appears to correlate with the finding that methanol has a much larger effect on EPR signals of the Mn4CaO5 cluster of plants than cyanobacteria.77,78 However, other reports find that the O4 channel is rather narrow and possibly unsuitable for water transport and instead favor the Cl1 channel (or O1 channel) as main water access pathway.60,63,84,87
To test the validity of our conclusion we examined the expected substrate water exchange rates through the shorter (25 Å) arm of the Cl1 channel (‘channel 1’ in ref. 61). This channel is reported to have two barriers: the first is formed by the D1-E65/D1-R334/D2-E312 triad and has a barrier of 11.5 kcal mol−1, while the second is formed by D1-D61, D2-K317 and Cl1 and has a barrier of 7 kcal mol−1 in the inward direction, and about 11 kcal mol−1 in the outward direction (Scheme 3). Using these parameters, we constructed a model that included two significant barriers, while other waters can exchange essentially freely. Eight water molecules, including W3 (but not W1, W2 and W4), formed the inner pool. To further simulate the water channel characteristics observed in crystal structures,6,15,17,61 four water molecules were placed between the two barriers, and five crystal waters are in rapid exchange with bulk water (Scheme 3; ESI Text 3†). We achieved excellent agreement with our experimental data by assuming that the inner barrier, formed by D1-D61, D2-E317 and Cl1, has an energy of 12.8 kcal mol−1, and the barrier closer to the bulk formed by D1-E65, D1-P66, D1-V67 and D2-E312 has a height of 11.5 kcal mol−1 (Fig. S5 and Table S3†). The inner barrier is slightly higher than determined for the outward direction by MD simulations, but this value is presumably within the accuracy of the MD method. It is also possible that the barrier for swapping two water molecules is actually higher (or the frequency factor lower; see SI Text S3) than for pulling water molecules through a channel,61 as this process requires two water molecules to pass each other in a bottleneck. This simulation thus shows that our proposal of an access limitation of the fast water exchange in the S2 state is realistic.
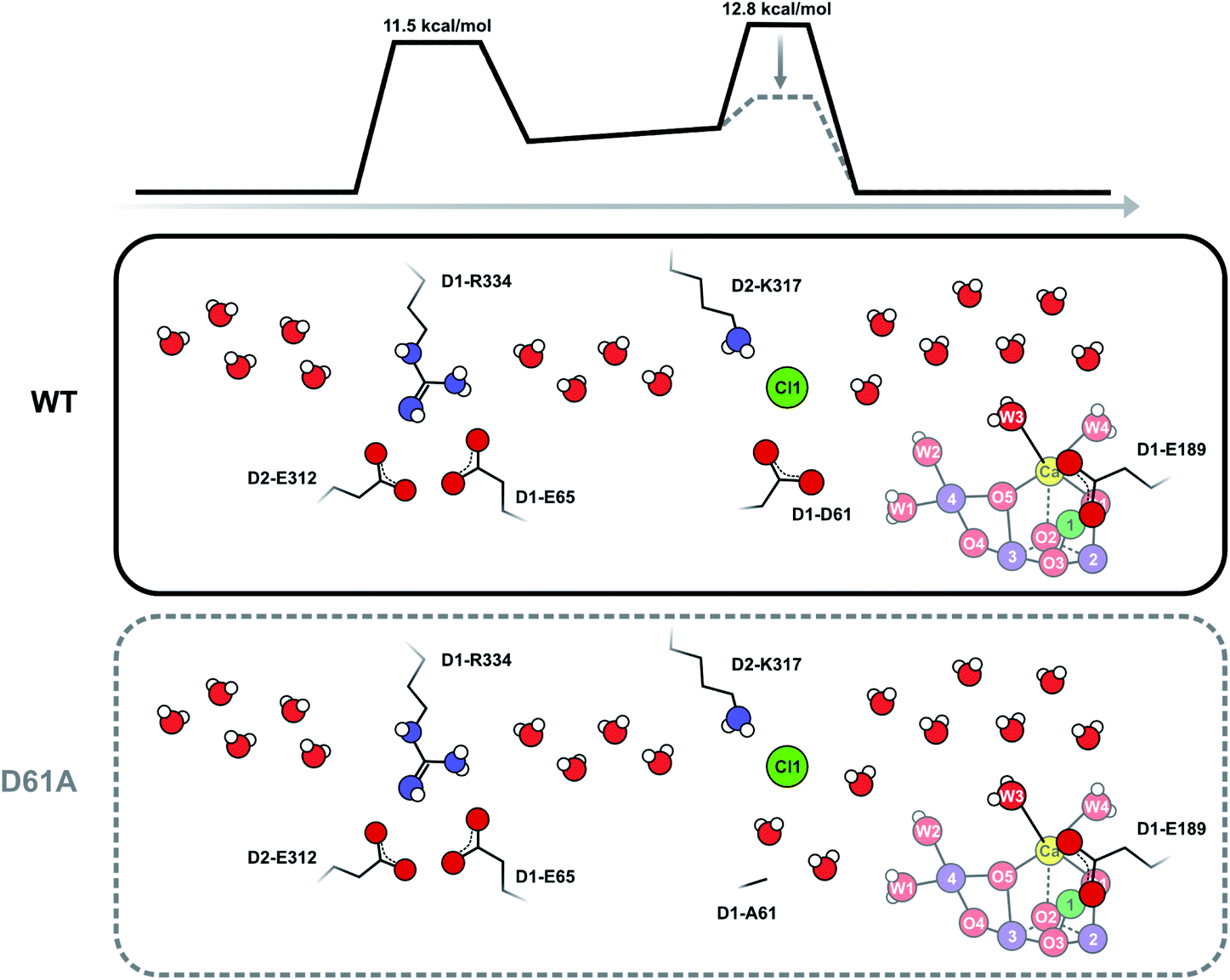 | ||
| Scheme 3 Schematic view of the Cl1 channel that is based on crystal structure information15,17,21 and MD simulations.61 The energy barriers have been slightly adjusted from the previous estimates in accordance with the present results. It is proposed that D1-D61 together with D2-K317 and Cl1 participate in forming the inner barrier that determines the rate of water exchange in WT-PSII. Shortening of D1-D61 to alanine reduces the second barrier by creating new water binding sites. In this case, the outer barrier, formed by the D1-E65, D2-E312 and D2-R334 residues, becomes rate limiting. As this outer barrier has a lower height, water exchange becomes faster in D61A-PSII. Barrier heights are estimated from the measured exchange rates as described in ESI Text 3.† These estimates assume that the frequency factor of the Eyring equation is 1.0; we note that it is possible that in reality a lower frequency factor should be used for the exchange processes, which could lower the barrier height. | ||
Wf exchange in the S0, S1 and S3 states of the majority of D61A-PSII centers
In the S3 state, Wf exchange is slower than in the S2 state and thus no longer controlled by water access. This implies that Wf is now more tightly bound, in line with the suggested movement of Wf into the Ox or O5 positions (Scheme 1). Because in the S3 state all Mn ions are in oxidation state Mn(IV), the rate of the fast water exchange is limited instead by the redox equilibrium between the S3AWYZ and S2AWYZ˙ states.46,55 The exchange of Wf in the S3 state most likely occurs by a reversal of the insertion pathway (Scheme 1).The exchange of Wf becomes observable for the first time in the S2 state, which might be taken as indication of a faster exchange of Wf in the S0 and S1 states. This would be inconsistent with an S state independent water access barrier. However, simulations show that the required dark-times of 10 ms between the subsequent flashes employed for producing O2 (Fig. S2†) are long enough to scramble basically all isotopic information regarding the exchange kinetics of Wf in S0 and S1 (see44 and ESI Text 2†). Thus, the unresolved Wf exchange in the S0 and S1 states is consistent with an S state independent water access barrier; that is, with a diffusion limited exchange in the S0, S1 and S2 states, and thus with W3 or W2 as Wf in these states.
Ws exchange in the S2 state of the majority of D61A-PSII centers
While improved substrate access provides a satisfying rationale for the unresolved and therefore more than 3-fold faster Wf exchange in D61A-PSII, it does not explain the 15-fold faster exchange of Ws in the dominant fraction of D61A-PSII centers. We recently observed a similar acceleration in WT-PSII at pH 8.6 and in Sr-PSII core complexes at pH 8.3.46 In this earlier study, the accelerated exchange correlated well with a stabilization of the S2HS state, indicating that at normal pH the conversion from the S2LS configuration into the S2HS configuration is limiting the rate of Ws exchange. We assigned the alkaline-induced S2HS state to the S2AW state, as this state allows an easy transition into the S2BW state (Scheme 2) in which O5 exchange can occur rapidly.44,46,55 As discussed above, the situation is different in the D61A mutant because the available data clearly exclude the stabilization of a S2HS form.71,75 However, since water exchange in the S2BW state is presumably very fast, and the S2AW to S2BW transition also has a comparatively low barrier,42,55 a similar acceleration of Ws exchange can be achieved by lowering the barrier for the rate limiting transformation of S2A into the S2AW state.As shown in Scheme 1, water insertion during the S2 → S3 transition requires the deprotonation of W3. The same is true for the formation of S2AW from S2A, which likely occurs in a similar fashion to mechanism A in Scheme 1. In the S2 state of WT-PSII, this proton needs to be transported away from the positively charged catalytic site into the bulk phase. In D61A-PSII, W1/W2 have collectively lost one proton,71 and should thus be able to transiently act as a nearby base that accepts the W3 proton during S2AW and S2BW formation. We propose that this lowers the energy barrier for S2AW formation enough to allow the observed 15-fold increase in Ws exchange rate. That the breakage of an OH bond is rate determining for O5 exchange in the S2 state is supported by the H/D isotope effect of 1.9 ± 0.2 determined for Ws exchange in the mutant (Fig. S3; Table S2†).
S2 state water exchange in the minority of D61A-PSII centers
We found that in about 15% of the D61A centers the exchange rates for Wf and Ws were similar to each other and to Ws exchange in WT-PSII (Table 1). This means that Wf exchange in this minority fraction was 10-fold slower than Ws exchange in the majority fraction, 60-fold slower than Wf exchange in WT-PSII, and more than 200-fold slower than Wf exchange in the majority fraction. By contrast, Ws exchange was slowed only 2–3 fold compared to WT-PSII, but nearly 40-fold relative to the majority fraction.We see two options to explain the slow and comparatively similar rates of exchange of Wf and Ws in this fraction of the D61A-PSII. Firstly (Option 1), in these centers the D61A mutation induces a secondary structural change that restricts the water access at a different point of the channel even more than in WT-PSII. For example, if the Cl1 channel would be the dominant substrate entry pathway, such a secondary structural change might occur at the D1-E65/D1-R334/D2-E312 triad, which was suggested previously to be another bottleneck for water transport through the Cl1 channel.61 As this triad provides a rather narrow path for water, a small change in protein conformation or dynamics may be enough to further restrict water passage. As the D1-D61A mutation is only 4 amino acids away from D1-E65, such an allosteric effect cannot be excluded. Secondly (Option 2), both W3 and W2 serve as Wf, but in different populations of D61A PSII centers, with one serving as Wf in the majority fraction and the other serving as Wf in the minority fraction. This idea is motivated by the similar rates of exchange found for Wf and Ws in the minority fraction of D61A-PSII, which suggest that their exchange may be limited by the same critical steps. This would be the case for W2 and O5, as substrate exchange via the S2A, S2AW and S2BW route places both at terminal positions of Mn4(III) in the S2BW state. This option would indicate a substantially increased barrier for the S2A to S2B conversion in the D61A-PSII (from 6–10 kcal mol−1 in WT41 to >16 kcal mol−1).
Ws exchange in the S3 state
In contrast to the S2 state, no heterogeneity is observed in the Ws exchange in the S3 state. Thus, if it is correct that Wf is different for the two fractions in the S2 state (Option 2, above), then rearrangements of the cluster must happen during the transition from S2 → S3 that bring the substrates into the same binding sites in the S3 state. This would indeed be possible, if in one fraction of PSII centers W3 and O5 are the substrates in the S2 state and W3 is inserted into the Ox position in the S2 → S3 transition (Scheme 1A), while in the other fraction W2 and O5 are the substrates that reach the same two binding sites via a pivot insertion (Scheme 1C).Exchange of Ws (O5) in the S3 state thus occurs most likely via the S2AWYZ˙ state, which further transforms into the S2BWYZ˙ state where O5 is bound in a terminal position at Mn4 and can be replaced via the S2BYZ˙ intermediate (Scheme 2).44,46,55 The much slower exchange of Ws in the S3 state of D61A-PSII as compared to WT-PSII indicates that in the D61A mutant the back donation of an electron from YZ to the Mn4CaO6 cluster is less efficient than in WT-PSII.55 One alternative for O5 exchange in the S3 state would be its exchange via the S3BW and S3B states (Scheme 1).44,46 In this case, the slowed Ws exchange implies a destabilization of one or both of these states as compared to the S3AW state.
Absence of effects of E189Q mutation
The analyses of recent XFEL studies favor water delivery via the O1 channel, and some of the authors suggest a gating of water access by the observed movement of E189 during the S2 → S3 transition.14,15,17,21 As the Wf exchange rates in the S2 and S3 states are nearly identical between WT-PSII and E189Q-PSII, the present data do not support a role of E189 as gate keeper for water access to the Mn4CaO5/6 cluster, at least not during water exchange in the studied semi-stable S states. This is in line with the MD calculations by Vassiliev, which suggest that D1-E329, D1-D342, CP43-V410 and CP43-T412 form the main bottleneck for water transport through the O1 channel system,61 and thus any subtle effects of E189 would be masked.Relation of water exchange and water binding
While our data show that in D61A-PSII water exchange occurs through the O4 and/or one or both branches of the Cl1 channel, they do not reveal which of the channels, including the O1 channel, has the lowest barrier in WT-PSII. Furthermore, it is important to note that water binding during the S2 → S3 transition is a fundamentally different and much faster (100–400 μs)15,17 process than water exchange in the S2 and S3 states (10–500 ms). During water binding, a nearby water attaches to an open binding site of the cluster and thereby initiates a bucket brigade of refilling vacant sites, while reaching the isotopic equilibrium with bulk water requires full equilibration of all exchangeable water molecules in the channels and around the catalytic site. Thus, our present data do not identify through which of the three water channels the substrate water is delivered in WT-PSII.Is the control of water access functionally important?
It has previously been hypothesized that regulation of substrate accessibility is crucial to minimize side reactions that would lead to the production of reactive oxygen species at the Mn4CaO5 cluster.88,89 This hypothesis assumed that in intact PSII complexes only substrate water can interact in a specific way with the Mn4CaO5 cluster. Recent crystal structures have shown that the Mn4CaO5 cluster is surrounded by several additional water molecules. Nevertheless, the present data and the previous calculations by Vassiliev61 show that water access is not completely free. This somewhat regulated access likely evolved to stabilize the Mn4CaO5 cluster, and to allow for the formation of a highly specific hydrogen bonding network, which is crucial for removing protons from substrate water during the water oxidation reactions. By contrast, the access of water is fast when compared to the maximal turnover frequency of PSII, which is limited by the acceptor side reactions of PSII to about 50 O2 s−1 (20 ms),90 while water is delivered through the channels with a time constant in the order of 100 μs.61 Interestingly, the time constant for water delivery is in the same order as that for water binding during the S2 → S3 transition. It might thus be speculated that the restriction of water access is a compromise between excluding other redox active molecules and ions from the Mn4CaO5 cluster, while allowing fast enough water access to promote efficient S state turnover. This idea is supported by the finding that partial dehydration of PSII increases the misses specifically of the S state transitions that involve binding of water molecules.91 Similarly, addition of the water analog methanol increases the miss parameter and allows the observation of a water deprived S3 state.92,93Implications for the mechanism of water oxidation
The significance of the present results is that they remove the strongest arguments against the assignment of Wf to W3 in the S2 state, namely (i) the indifference of the Wf exchange rate to Ca/Sr substitution and (ii) the significant mismatch with reported exchange rates for water ligated to Ca.46,94The present data are fully consistent with O5 as slowly exchanging substrate water Ws, and W2 or W3 as fast exchanging substrate water Wf. A further distinction between W2 and W3 as Wf is not possible on the basis of substrate water exchange data alone because the rate limitation provided by the barriers in the channels obscures small perturbations such as Ca/Sr substitution that could otherwise be used to distinguish the binding sites. However, other recent experimental data favor W3 over W2 as substrate water. FTIR experiments by the groups of Noguchi and Debus have provided evidence for the involvement of W3 in water binding during the S2 → S3 transition.11,16,95 Similarly, femtosecond X-ray crystallography measurements have revealed that the largest changes in water positions during this transition occur in the O1 channel that leads to the Ca site and found no evidence for the predicted closed cube S2B-like intermediate that would be required if W2 were the fast substrate (Scheme 1).15,17 By contrast, the support for W2 is mostly based on substrate analogs like methanol or ammonia,75,77–83 which we regard as more indirect. On this basis, we propose that O5 and W3 are the two substrate water molecules under normal circumstances, but that W2 may serve as the fast exchanging substrate under some circumstances, such as in a minority of D61A PSII centers. The resulting experimentally supported ‘molecular S state cycle’ is summarized in Scheme 4.
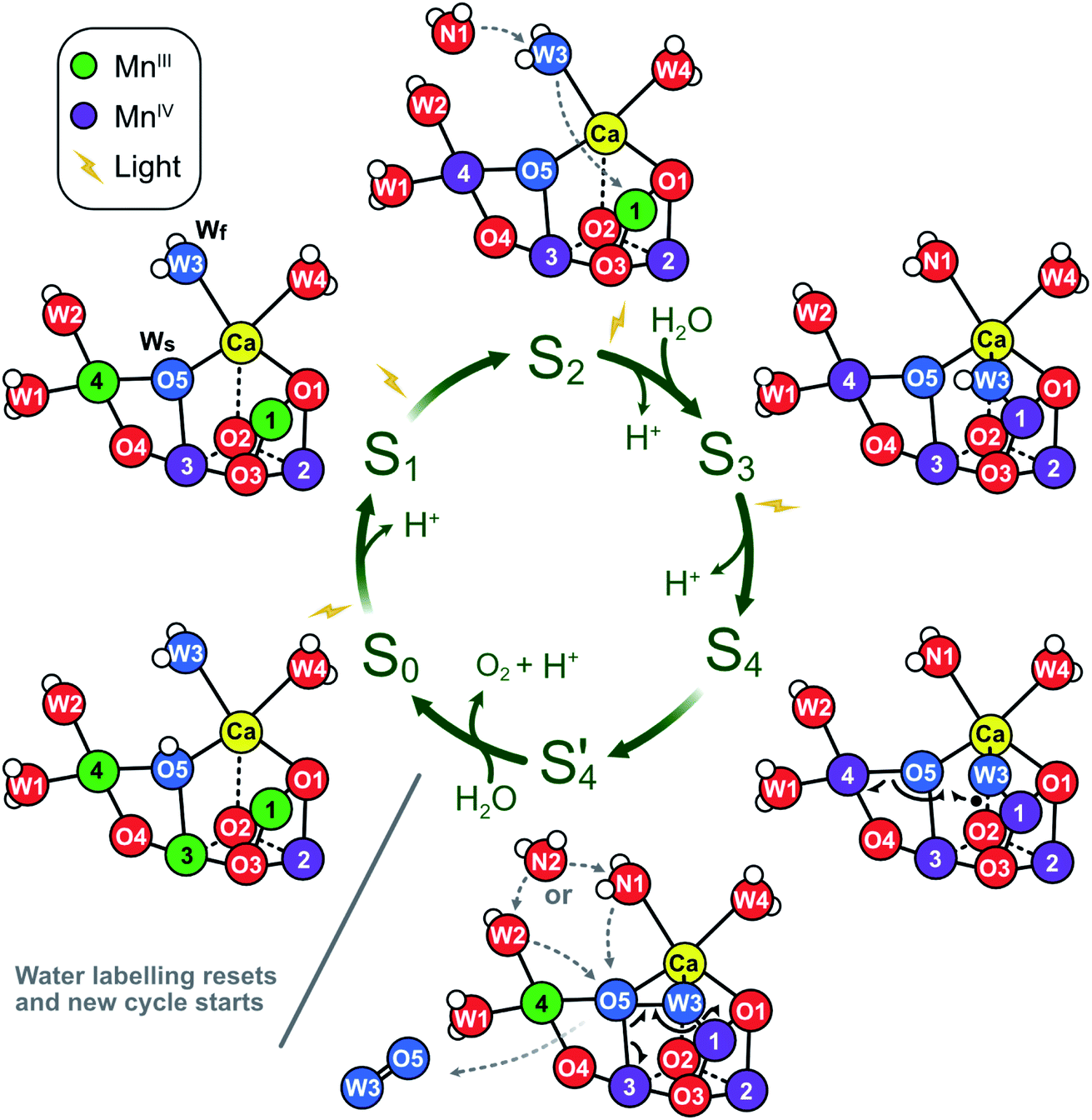 | ||
| Scheme 4 Proposed molecular Kok cycle illustrating the binding of the two substrate waters Wf and Ws in the various S states. The center shows the traditional S state scheme indicating water binding as well as proton and dioxygen release, while the outer circle depicts schematically the corresponding dominant structures of the Mn4CaO5/6 complex based on X-ray crystallography6,15,17,21 as well as calculated structural models of key intermediates during O–O bond formation.12 In dark-adapted PSII, the reaction cycle starts with the S1 state that has two MnIII and two MnIV ions and in which all bridges are deprotonated.10 During the S1 → S2 transition, Mn4 is oxidized. While the S2A state is in equilibrium with other conformations (see Scheme 2), it is proposed that W3 is inserted directly into the Ox binding site between Ca and Mn1, concomitant with Mn1 oxidation and the binding of a new water, WN1, to the W3 site (dashed grey arrows; for details, see Scheme 1A).17 In S3, the dominant state is S3AW. Upon further oxidation, the S3AWYZ˙ state is formed, which under proton release converts into the S3AWYZ˙’ state (lag phase; not shown).103 This may be coupled to unknown rearrangements within the H-bonding network of the OEC. Only thereafter, the Mn4CaO6 cluster can be oxidized to S4. Instead of Mn oxidation, S4 state formation involves the oxidation of the fast substrate water, indicated by a black dot on W3 (in the Ox position).12 By rearranging the electrons of the chemical bonds (black half-arrows), the S4 state rapidly converts into the S4’ state, which contains a complexed peroxide. The further conversion of S4’ into S0 + O2 requires the binding of one water and the release of a proton. We suggest that a pre-bound water ligand (W2 or W3) fills the empty O5 binding site,9 and that this ligand is concomitantly replaced by a new water (WN2; dashed grey arrows). In the S0 state, the O5 bridge is protonated, in line with the faster exchange of Ws and spectroscopic data.4,104,105 Oxygen atoms are labeled red, and the two substrate ‘waters’ are shown in blue. Hydrogen atoms are shown as small white spheres (protonation states based on S2 state assignment in ref. 106). | ||
Presently no experimental data are available that allow to determine the actual O–O bond formation mechanism during the S3 → S4 → S0 transition, but the present data are fully consistent with the best worked out theoretical mechanism for O–O bond formation, which involves oxo-oxyl radical coupling between oxygens in the O5 and Ox binding sites via a low-energy path paved by favorable spin paring.12,53
However, the recently revived idea that the formation of a peroxidic intermediate (<5–10%) in the S3 state is required for further oxidation to the S4 state cannot be excluded on the basis of our present data (Fig. S1E and F†),1,21,27,28 because the same substrates and main state conformations are involved, and such a small equilibrium population of a peroxidic intermediate would easily escape detection by, for example, femtosecond X-ray crystallography. Nevertheless, a very recent theoretical study considers a peroxidic intermediate in the S3 state as unlikely.96 By contrast, our substrate water exchange data are inconsistent with nucleophilic attack mechanisms between W3 and W2,51,97–99 and geminal coupling between W2 and O5 at Mn4 (ref. 30, 100) (for details see ESI Text 4 and Fig. S1†).
Conclusions
In this study, we demonstrate that the fast water exchange in the S0, S1 and S2 states is rate limited by specific diffusion barriers in all the channels connecting bulk water with the Mn4CaO5 cluster in PSII, and that the D61A mutation reduces one of these barriers so that Wf exchange is accelerated. This finding removes previous arguments that appeared to exclude W3 as the fast exchanging substrate water. Combining our present results with recent FTIR and XFEL data supporting the insertion of W3 into the Ox position during the S2 → S3 transition,11,15–17,95,101 now make W3 the prime candidate for Wf. As our previous experiments identified O5 as the slow substrate water,9,18,44,46,54 this study clarifies the fate of the substrate waters during the S state cycle, and thereby limits the possible mechanisms for O–O bond formation to a few that all involve coupling between O5 and W3, while they are bound in the O5 and Ox positions of the S3AW or S4AW states (Scheme 4).Data availability
All relevant data is presented in the paper and ESI.† Raw data is available upon request by email to JM.Author contributions
CDL, RJD and JM conceived and designed the research; CDL, CJK and PC performed the research; CDL and JM analyzed the data; CDL, RJD and JM wrote the paper with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by ÅForsk (to CDL), Vetenskapsrådet (2016-05183 and 2020-03809; to JM) and the US Department of Energy (DE-SC0005291; to RJD). The authors thank Anh P. Nguyen for growing the Synechocystis cells and helping with the purification of the PSII core complexes.References
- G. Renger, Biochim. Biophys. Acta, 2012, 1817, 1164–1176 CrossRef CAS PubMed.
- W. Junge, Q. Rev. Biophys., 2019, 52(e1), 1–17 CrossRef PubMed.
- H. Dau and I. Zaharieva, Acc. Chem. Res., 2009, 42, 1861–1870 CrossRef CAS PubMed.
- J. H. Robblee, J. Messinger, R. M. Cinco, K. L. McFarlane, C. Fernandez, S. A. Pizarro, K. Sauer and V. K. Yachandra, J. Am. Chem. Soc., 2002, 124, 7459–7471 CrossRef CAS PubMed.
- J. Yano, J. Kern, K. Sauer, M. J. Latimer, Y. Pushkar, J. Biesiadka, B. Loll, W. Saenger, J. Messinger, A. Zouni and V. K. Yachandra, Science, 2006, 314, 821–825 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–61 CrossRef CAS PubMed.
- B. Kok, B. Forbush and M. McGloin, Photochem. Photobiol., 1970, 11, 457–476 CrossRef CAS PubMed.
- J. Messinger and G. Renger, in Primary Processes of Photosynthesis – Part 2: Basic Principles and Apparatus, ed. G. Renger, The Royal Society of Chemistry, Cambridge, UK, 2008, pp. 291–349 Search PubMed.
- J. Messinger, Phys. Chem. Chem. Phys., 2004, 6, 4764–4771 RSC.
- N. Cox, D. A. Pantazis and W. Lubitz, Annu. Rev. Biochem., 2020, 89, 795–820 CrossRef CAS PubMed.
- H. Suzuki, M. Sugiura and T. Noguchi, Biochemistry, 2008, 47, 11024–11030 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Acc. Chem. Res., 2009, 42, 1871–1880 CrossRef CAS PubMed.
- N. Cox, M. Retegan, F. Neese, D. A. Pantazis, A. Boussac and W. Lubitz, Science, 2014, 345, 804–808 CrossRef CAS PubMed.
- M. Suga, F. Akita, M. Sugahara, M. Kubo, Y. Nakajima, T. Nakane, K. Yamashita, Y. Umena, M. Nakabayashi, T. Yamane, T. Nakano, M. Suzuki, T. Masuda, S. Inoue, T. Kimura, T. Nomura, S. Yonekura, L. J. Yu, T. Sakamoto, T. Motomura, J. H. Chen, Y. Kato, T. Noguchi, K. Tono, Y. Joti, T. Kameshima, T. Hatsui, E. Nango, R. Tanaka, H. Naitow, Y. Matsuura, A. Yamashita, M. Yamamoto, O. Nureki, M. Yabashi, T. Ishikawa, S. Iwata and J. R. Shen, Nature, 2017, 543, 131–135 CrossRef CAS PubMed.
- J. Kern, R. Chatterjee, I. D. Young, F. D. Fuller, L. Lassalle, M. Ibrahim, S. Gul, T. Fransson, A. S. Brewster, R. Alonso-Mori, R. Hussein, M. Zhang, L. Douthit, C. de Lichtenberg, M. H. Cheah, D. Shevela, J. Wersig, I. Seuffert, D. Sokaras, E. Pastor, C. Weninger, T. Kroll, R. G. Sierra, P. Aller, A. Butryn, A. M. Orville, M. N. Liang, A. Batyuk, J. E. Koglin, S. Carbajo, S. Boutet, N. W. Moriarty, J. M. Holton, H. Dobbek, P. D. Adams, U. Bergmann, N. K. Sauter, A. Zouni, J. Messinger, J. Yano and V. K. Yachandra, Nature, 2018, 563, 421–425 CrossRef CAS PubMed.
- C. J. Kim and R. J. Debus, Biochemistry, 2019, 58, 3185–3192 CrossRef CAS PubMed.
- M. Ibrahim, T. Fransson, R. Chatterjee, M. H. Cheah, R. Hussein, L. Lassalle, K. D. Sutherlin, I. D. Young, F. D. Fuller, S. Gul, I. S. Kim, P. S. Simon, C. de Lichtenberg, P. Chernev, I. Bogacz, C. C. Pham, A. M. Orville, N. Saichek, T. Northen, A. Batyuk, S. Carbajo, R. Alonso-Mori, K. Tono, S. Owada, A. Bhowmick, R. Bolotovsky, D. Mendez, N. W. Moriarty, J. M. Holton, H. Dobbek, A. S. Brewster, P. D. Adams, N. K. Sauter, U. Bergmann, A. Zouni, J. Messinger, J. Kern, V. K. Yachandra and J. Yano, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12624–12635 CrossRef CAS PubMed.
- N. Cox and J. Messinger, Biochim. Biophys. Acta, 2013, 1827, 1020–1030 CrossRef CAS PubMed.
- Y. Guo, H. Li, L. L. He, D. X. Zhao, L. D. Gong and Z. Z. Yang, Biochim. Biophys. Acta, 2017, 1858, 833–846 CrossRef CAS PubMed.
- H. Dau and M. Haumann, Coord. Chem. Rev., 2008, 252, 273–295 CrossRef CAS.
- M. Suga, F. Akita, K. Yamashita, Y. Nakajima, G. Ueno, H. J. Li, T. Yamane, K. Hirata, Y. Umena, S. Yonekura, L. J. Yu, H. Murakami, T. Nomura, T. Kimura, M. Kubo, S. Baba, T. Kumasaka, K. Tono, M. Yabashi, H. Isobe, K. Yamaguchi, M. Yamamoto, H. Ago and J. R. Shen, Science, 2019, 366, 334–338 CrossRef CAS PubMed.
- A. N. Bondar and H. Dau, Biochim. Biophys. Acta, 2012, 1817, 1177–1190 CrossRef CAS PubMed.
- H. Bao, P. L. Dilbeck and R. L. Burnap, Photosynth. Res., 2013, 116, 215–229 CrossRef CAS PubMed.
- M. Amin, L. Vogt, W. Szejgis, S. Vassiliev, G. W. Brudvig, D. Bruce and M. R. Gunner, J. Phys. Chem. B, 2015, 119(24), 7366–7377 CrossRef CAS PubMed.
- V. Krewald, M. Retegan, N. Cox, J. Messinger, W. Lubitz, S. DeBeer, F. Neese and D. A. Pantazis, Chem. Sci., 2015, 6, 1676–1695 RSC.
- M. H. Cheah, M. Zhang, D. Shevela, F. Mamedov, A. Zouni and J. Messinger, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 141–145 CrossRef CAS PubMed.
- T. A. Corry and P. J. O'Malley, J. Phys. Chem. Lett., 2020, 11, 4221–4225 CrossRef CAS PubMed.
- H. Isobe, M. Shoji, T. Suzuki, J. R. Shen and K. Yamaguchi, J. Photochem. Photobiol., A, 2021, 405 Search PubMed.
- D. A. Pantazis, ACS Catal., 2018, 8, 9477–9507 CrossRef CAS.
- B. B. Zhang and L. C. Sun, Dalton Trans., 2018, 47, 14381–14387 RSC.
- M. Ibrahim, N. W. Moriarty, J. Kern, J. M. Holton, A. S. Brewster, A. Bhowmick, U. Bergmann, A. Zouni, J. Messinger, V. K. Yachandra, J. Yano, H. Dobbek, N. K. Sauter and P. D. Adams, Proc. Natl. Acad. Sci. U. S. A., 2021, 118 Search PubMed.
- M. Shoji, H. Isobe and K. Yamaguchi, Chem. Phys. Lett., 2015, 636, 172–179 CrossRef CAS.
- I. Ugur, A. W. Rutherford and V. R. I. Kaila, Biochim. Biophys. Acta, 2016, 1857, 740–748 CrossRef CAS PubMed.
- D. Bovi, D. Narzi and L. Guidoni, Angew. Chem., Int. Ed., 2013, 52, 11744–11749 CrossRef CAS PubMed.
- M. Amin, D. Kaur, K. R. Yang, J. M. Wang, Z. Mohamed, G. W. Brudvig, M. R. Gunner and V. Batista, Phys. Chem. Chem. Phys., 2019, 21, 20840–20848 RSC.
- M. Retegan, V. Krewald, F. Mamedov, F. Neese, W. Lubitz, N. Cox and D. A. Pantazis, Chem. Sci., 2016, 7, 72–84 RSC.
- M. Askerka, D. J. Vinyard, G. W. Brudvig and V. S. Batista, Biochemistry, 2015, 54, 5783–5786 CrossRef CAS PubMed.
- A. Boussac, A. W. Rutherford and M. Sugiura, Biochim. Biophys. Acta, 2015, 1847, 576–586 CrossRef CAS PubMed.
- A. Boussac, I. Ugur, A. Marion, M. Sugiura, V. R. I. Kaila and A. W. Rutherford, Biochim. Biophys. Acta, 2018, 1859, 342–356 CrossRef CAS PubMed.
- M. Shoji, H. Isobe, T. Nakajima, Y. Shigeta, M. Suga, F. Akita, J. R. Shen and K. Yamaguchi, Faraday Discuss., 2017, 198, 83–106 RSC.
- D. A. Pantazis, W. Ames, N. Cox, W. Lubitz and F. Neese, Angew. Chem., Int. Ed., 2012, 51, 9935–9940 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Phys. Chem. Chem. Phys., 2018, 20, 22926–22931 RSC.
- Y. Pushkar, A. K. Ravari, S. C. Jensen and M. Palenik, J. Phys. Chem. Lett., 2019, 10, 5284–5291 CrossRef CAS PubMed.
- C. de Lichtenberg, A. P. Avramov, M. Zhang, F. Mamedov, R. L. Burnap and J. Messinger, Biochim. Biophys. Acta, 2021, 1862, 148319 CrossRef CAS PubMed.
- S. Taguchi, T. Noguchi and H. Mino, J. Phys. Chem. B, 2020, 124, 5531–5537 CrossRef CAS PubMed.
- C. de Lichtenberg and J. Messinger, Phys. Chem. Chem. Phys., 2020, 22, 12894–12908 RSC.
- T. A. Corry and P. J. O'Malley, J. Phys. Chem. Lett., 2019, 10, 5226–5230 CrossRef CAS PubMed.
- T. A. Corry and P. J. O'Malley, J. Am. Chem. Soc., 2020, 142, 10240–10243 CrossRef CAS PubMed.
- A. Boussac, Biochim. Biophys. Acta, 2019, 1860, 508–518 CrossRef CAS PubMed.
- R. Radmer and O. Ollinger, FEBS Lett., 1980, 110, 57–61 CrossRef CAS.
- J. Messinger, M. Badger and T. Wydrzynski, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 3209–3213 CrossRef CAS PubMed.
- W. Hillier and T. Wydrzynski, Coord. Chem. Rev., 2008, 252, 306–317 CrossRef CAS.
- P. E. M. Siegbahn, Chem.–Eur. J., 2006, 12, 9217–9227 CrossRef CAS PubMed.
- L. Rapatskiy, N. Cox, A. Savitsky, W. M. Ames, J. Sander, M. M. Nowaczyk, M. Rögner, A. Boussac, F. Neese, J. Messinger and W. Lubitz, J. Am. Chem. Soc., 2012, 134, 16619–16634 CrossRef CAS PubMed.
- P. E. M. Siegbahn, J. Am. Chem. Soc., 2013, 135, 9442–9449 CrossRef CAS PubMed.
- M. Drosou, G. Zahariou and D. A. Pantazis, Angew. Chem., Int. Ed., 2021, 60, 13493–13499 CrossRef CAS PubMed.
- H. Nilsson, T. Krupnik, J. Kargul and J. Messinger, Biochim. Biophys. Acta, 2014, 1837, 1257–1262 CrossRef CAS PubMed.
- G. Hendry and T. Wydrzynski, Biochemistry, 2003, 42, 6209–6217 CrossRef CAS PubMed.
- H. Nilsson, F. Rappaport, A. Boussac and J. Messinger, Nat. Commun., 2014, 5, 4305 CrossRef CAS PubMed.
- N. Sakashita, H. C. Watanabe, T. Ikeda, K. Saito and H. Ishikita, Biochemistry, 2017, 56, 3049–3057 CrossRef CAS PubMed.
- S. Vassiliev, T. Zaraiskaya and D. Bruce, Biochim. Biophys. Acta, 2012, 1817, 1671–1678 CrossRef CAS PubMed.
- K. Linke and F. M. Ho, Biochim. Biophys. Acta, 2014, 1837, 14–32 CrossRef CAS PubMed.
- D. A. Weisz, M. L. Gross and H. B. Pakrasi, Sci. Adv., 2017, 3, eaao3013 CrossRef PubMed.
- H. J. Li, Y. Nakajima, T. Nomura, M. Sugahara, S. Yonekura, S. K. Chan, T. Nakane, T. Yamane, Y. Umena, M. Suzuki, T. Masuda, T. Motomura, H. Naitow, Y. Matsuura, T. Kimura, K. Tono, S. Owada, Y. Joti, R. Tanaka, E. Nango, F. Akita, M. Kubo, S. Iwata, J. R. Shen and M. Suga, Iucrj, 2021, 8, 431–443 CrossRef CAS PubMed.
- R. Kale, A. E. Hebert, L. K. Frankel, L. Sallans, T. M. Bricker and P. Pospisil, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2988–2993 CrossRef CAS PubMed.
- L. K. Frankel, L. Sallans, P. A. Limbach and T. M. Bricker, Biochemistry, 2012, 51, 6371–6377 CrossRef CAS PubMed.
- M. Hundelt, A.-M. A. Hays, R. J. Debus and W. Junge, Biochemistry, 1998, 37, 14450–14456 CrossRef CAS PubMed.
- P. L. Dilbeck, H. J. Hwang, I. Zaharieva, L. Gerencser, H. Dau and R. L. Burnap, Biochemistry, 2012, 51, 1079–1091 CrossRef CAS PubMed.
- H. Bao and R. L. Burnap, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E6139–E6147 CrossRef CAS PubMed.
- R. J. Debus, Biochemistry, 2014, 53, 2941–2955 CrossRef CAS PubMed.
- D. Kaur, W. Szejgis, J. J. Mao, M. Amin, K. M. Reiss, M. Askerka, X. H. Cai, U. Khaniya, Y. Y. Zhang, G. W. Brudvig, V. S. Batista and M. R. Gunner, Photosynth. Res., 2019, 141, 331–341 CrossRef CAS PubMed.
- S. Singh, R. J. Debus, T. Wydrzynski and W. Hillier, Philos. Trans. R. Soc. London, Ser. B, 2008, 363, 1229–1234 CrossRef CAS PubMed.
- R. J. Debus, K. A. Campbell, D. P. Pham, A. M. A. Hays and R. D. Britt, Biochemistry, 2000, 39, 6275–6287 CrossRef CAS PubMed.
- W. Hillier, I. McConnell, S. Singh, R. Debus, A. Boussac and T. Wydrzynski, in Photosynthesis. Energy from the Sun: 14th International Congress on Photosynthesis, eds. J. F. Allen, E. Gantt, J. H. Golbeck and B. Osmond, Springer, 2008, pp. 427–430 Search PubMed.
- P. H. Oyala, T. A. Stich, R. J. Debus and R. D. Britt, J. Am. Chem. Soc., 2015, 137, 8829–8837 CrossRef CAS PubMed.
- K. Ogata, M. Hatakeyama, Y. Sakamoto and S. Nakamura, J. Phys. Chem. B, 2019, 123, 6444–6452 CrossRef CAS PubMed.
- M. Retegan and D. A. Pantazis, J. Am. Chem. Soc., 2017, 139, 14340–14343 CrossRef CAS PubMed.
- M. Retegan and D. A. Pantazis, Chem. Sci., 2016, 7, 6463–6476 RSC.
- M. Askerka, G. W. Brudvig and V. S. Batista, Acc. Chem. Res., 2017, 50, 41–48 CrossRef CAS PubMed.
- M. Askerka, J. M. Wang, D. J. Vinyard, G. W. Brudvig and V. S. Batista, Biochemistry, 2016, 55, 981–984 CrossRef CAS PubMed.
- J. Schraut and M. Kaupp, Chem.–Eur. J., 2014, 20, 7300–7308 CrossRef CAS PubMed.
- M. Pérez Navarro, W. M. Ames, H. Nilsson, T. Lohmiller, D. A. Pantazis, L. Rapatskiy, M. M. Nowaczyk, F. Neese, A. Boussac, J. Messinger, W. Lubitz and N. Cox, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15561–15566 CrossRef PubMed.
- M. Mandal, M. Askerka, G. Banerjee, M. Amin, G. W. Brudvig, V. S. Batista and M. R. Gunner, Dalton Trans., 2017, 46, 16089–16095 RSC.
- H. Yata and T. Noguchi, Biochemistry, 2018, 57, 4803–4815 CrossRef CAS PubMed.
- H. Nagashima and H. Mino, J. Phys. Chem. Lett., 2017, 8, 621–625 CrossRef CAS PubMed.
- P. H. Oyala, T. A. Stich, J. A. Stull, F. T. Yu, V. L. Pecoraro and R. D. Britt, Biochemistry, 2014, 53, 7914–7928 CrossRef CAS PubMed.
- Y. Shimada, T. Kitajima-Ihara, R. Nagao and T. Noguchi, J. Phys. Chem. B, 2020, 124, 1470–1480 CrossRef CAS PubMed.
- T. Wydrzynski, W. Hillier and J. Messinger, Physiol. Plant., 1996, 96, 342–350 CrossRef CAS.
- A. Arato, N. Bondarava and A. Krieger-Liszkay, Biochim. Biophys. Acta, 2004, 1608, 171–180 CrossRef CAS PubMed.
- W. J. Lee and J. Whitmarsh, Plant Physiol., 1989, 89, 932–940 CrossRef CAS PubMed.
- T. Noguchi, Philos. Trans. R. Soc. London, Ser. B, 2008, 363, 1189–1194 CrossRef CAS PubMed.
- M. Chrysina, E. Heyno, Y. Kutin, M. Reus, H. Nilsson, M. M. Nowaczyk, S. DeBeer, F. Neese, J. Messinger, W. Lubitz and N. Cox, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16841–16846 CrossRef CAS PubMed.
- B. Nöring, D. Shevela, G. Renger and J. Messinger, Photosynth. Res., 2008, 98, 251–260 CrossRef PubMed.
- L. Helm and A. E. Merbach, Chem. Rev., 2005, 105, 1923–1959 CrossRef CAS PubMed.
- C. J. Kim and R. J. Debus, Biochemistry, 2017, 56, 2558–2570 CrossRef CAS PubMed.
- M. Drosou and D. A. Pantazis, Chem.–Eur. J., 2021 DOI:10.1002/chem.202101567.
- V. L. Pecoraro, M. J. Baldwin, M. T. Caudle, W.-Y. Hsieh and N. A. Law, Pure Appl. Chem., 1998, 70, 925–929 CAS.
- J. P. McEvoy and G. W. Brudvig, Phys. Chem. Chem. Phys., 2004, 6, 4754–4763 RSC.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
- M. Kusunoki, Biochim. Biophys. Acta, 2007, 1767, 484–492 CrossRef CAS PubMed.
- Y. Okamoto, Y. Shimada, R. Nagao and T. Noguchi, J. Phys. Chem. B, 2021, 125(25), 6864–6873 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38 CrossRef CAS.
- M. Haumann, P. Liebisch, C. Müller, M. Barra, M. Grabolle and H. Dau, Science, 2005, 310, 1019–1021 CrossRef CAS PubMed.
- L. V. Kulik, B. Epel, W. Lubitz and J. Messinger, J. Am. Chem. Soc., 2007, 129, 13421–13435 CrossRef CAS PubMed.
- T. Lohmiller, V. Krewald, A. Sedoud, A. W. Rutherford, F. Neese, W. Lubitz, D. A. Pantazis and N. Cox, J. Am. Chem. Soc., 2017, 139, 14412–14424 CrossRef CAS PubMed.
- W. Ames, D. A. Pantazis, V. Krewald, N. Cox, J. Messinger, W. Lubitz and F. Neese, J. Am. Chem. Soc., 2011, 133, 19743–19757 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Data analysis and simulation procedures (ESI Text 1-3; Fig. S2 and S5; Tables S1 and S3), H/D dependence (Fig. S3 and Table S2), alternative mechanisms (ESI Text 4 and Fig. S1). See DOI: 10.1039/d1sc02265b |
| This journal is © The Royal Society of Chemistry 2021 |