
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic parallel kinetic resolution of α-ferrocenyl cation initiated by chiral Brønsted acid catalyst†
Yasunori
Toda‡
 a,
Toshinobu
Korenaga
b,
Ren
Obayashi
a,
Jun
Kikuchi
a and
Masahiro
Terada
*a
a,
Toshinobu
Korenaga
b,
Ren
Obayashi
a,
Jun
Kikuchi
a and
Masahiro
Terada
*a
aDepartment of Chemistry, Graduate School of Science, Tohoku University, Aoba-ku, Sendai 980-8578, Japan. E-mail: mterada@tohoku.ac.jp; ytoda@shinshu-u.ac.jp; Web: http://www.orgreact.sakura.ne.jp/en-index.html]
bDepartment of Chemistry and Biological Sciences, Faculty of Science and Engineering, Iwate University, Morioka 020-8551, Japan
First published on 29th June 2021
Abstract
The dynamic parallel kinetic resolution (DPKR) of an α-ferrocenyl cation intermediate under the influence of a chiral conjugate base of a chiral phosphoric acid catalyst has been demonstrated in an SN1 type substitution reaction of a racemic ferrocenyl derivative with a nitrogen nucleophile. The present method provides efficient access to a ferrocenylethylamine derivative in a highly enantioselective manner, which is potentially useful as a key precursor of chiral ligands for metal catalysis. The mechanism of the present intriguing resolution system was elucidated by control experiments using the enantio-pure precursor of relevant α-ferrocenyl cation intermediates and the hydroamination of vinylferrocene. Further theoretical studies enabled the elucidation of the origin of the stereochemical outcome as well as the efficient DPKR. The present DPKR, which opens a new frontier for kinetic resolution, involves the racemization process through the formation of vinylferrocene and the chemo-divergent parallel kinetic resolution of the enantiomeric α-ferrocenyl cations generated by the protonation/deprotonation sequence of vinylferrocene.
Introduction
The kinetic resolution (KR) of racemic mixtures is one of the most fundamental and powerful methods for the preparation of enantiomerically enriched compounds.1 In general, KR occurs because the two enantiomers of a racemic substrate react at different rates. To date, electronically neutral substrates, namely, non-charged organic molecules, have been employed in KR reactions.1d,2 On the other hand, cationic molecules, despite being attractive chemical species, have rarely been used as KR reactants3,4 and thus the development of an efficient method for the resolution of racemic cations under catalytic conditions is expected to open a new frontier for KR. Cationic molecules are attractive because they are common reactive species and often invoked as key intermediates in typical Brønsted acid catalysis. In particular, during the past decades, chiral phosphoric acids (CPAs) have emerged as effective Brønsted acid catalysts for enantioselective electrophilic activation. In fact, CPAs have enabled a wide variety of enantioselective transformations5,6 in which the chiral conjugate base of a CPA catalyst functions as a powerful stereocontrolling element.7 Hence, it is naturally expected that the catalytic KR of a racemic cationic species using the chiral conjugate base of CPA would expand the scope of KR.8 The method would not only establish a novel class of fundamental strategies for asymmetric synthesis, but also empower chiral Brønsted acid catalysis as the master of cation control.The challenge of governing the reactivity of racemic cationic molecules as active intermediates using a chiral conjugate base has inspired the design of a fascinating resolution system. A CPA-catalysed SN1 type substitution reaction of α-ferrocenyl alcohol derivatives is a viable and attractive approach to accomplish the KR of a racemic cationic molecule. In principle, the substitution at the α-position of ferrocenyl alcohol derivatives should proceed through a stepwise pathway (Fig. 1).9 Elimination occurs as the initial step, generating an α-ferrocenyl cation,10 and subsequent addition of a nucleophile to the cation affords a substitution product. More importantly, the chirality at the α-position is completely preserved in a retentive manner during the course of the reaction because the central chirality at the α-position of ferrocenyl derivatives is cleanly transformed into the “planar” chirality of the cation via neighbouring group participation of the iron centre. It should be pointed out that the chiral cation does not undergo racemization in this particular type of substitution reaction. Taking into consideration the unique features of this cationic intermediate, we envisioned that cation A generated from racemic ferrocenyl derivative 1 using CPA catalyst 2 would be resolved in the reaction with nitrogen nucleophile 3 (Fig. 2).11,12 In the present characteristic KR, the competition between the substitution and elimination reactions would take place in parallel during the resolution. We thus assumed that both the substitution product, ferrocenylethylamine derivative 4, and the elimination product, vinylferrocene (5), would be ideally formed in a comparable fashion and hence the chemo-divergent parallel kinetic resolution (PKR)13 of α-ferrocenyl cation A would be feasible. Because only a limited number of PKRs have been reported, it is of particular interest whether the use of racemic cation A enables the present unique PKR under the influence of the chiral conjugate base of CPA 2. In addition, vinylferrocene (5) can be readily converted into racemic methyl ether 1 through the addition of methanol to the vinyl moiety of 5 in the presence of a strong Brønsted acid,§ and regenerated 1 is subjected to the same reaction sequence. Therefore, a formal dynamic kinetic resolution (DKR)14 would be established by the present method.
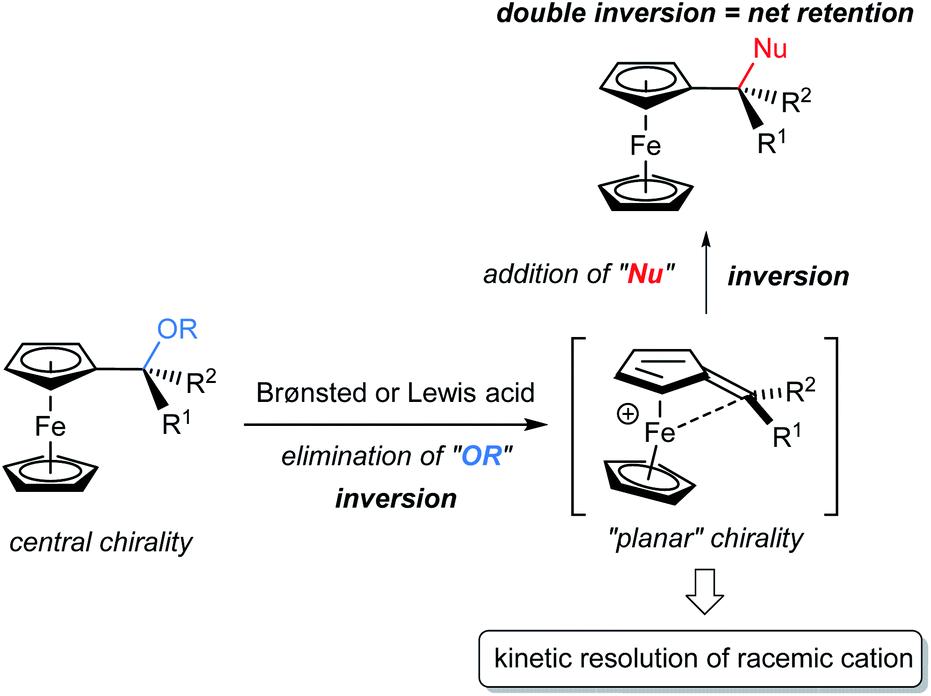 | ||
| Fig. 1 Stereoretentive substitution at α-position of ferrocenyl derivatives and application of cationic intermediate to KR. | ||
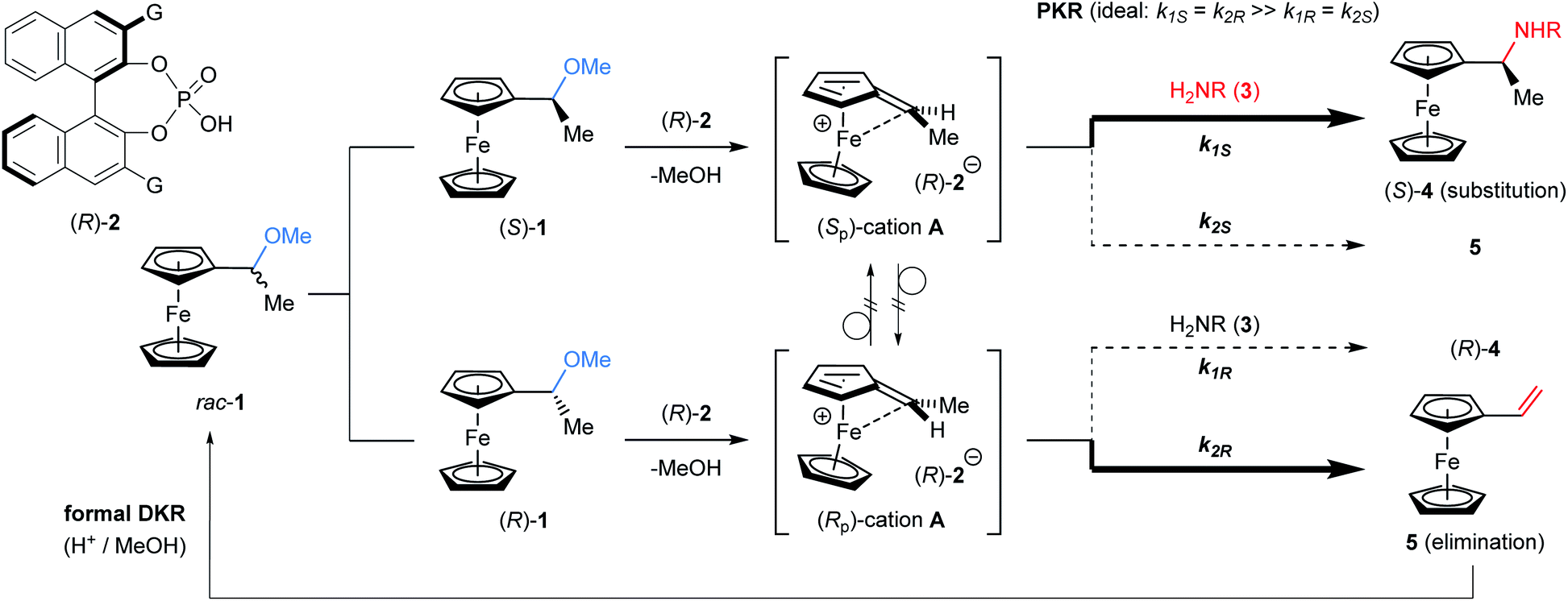 | ||
| Fig. 2 Proposal: parallel kinetic resolution of α-ferrocenyl cation catalysed by CPA. | ||
Keeping the above postulate in mind, we performed the SN1 type substitution reaction of racemic ferrocenyl derivative 1 with nitrogen nucleophile 3 catalysed by CPA 2 (G = 9-anthryl). Herein we disclose the formation of highly enantio-enriched ferrocenylethylamine derivative 4, which has been potentially useful as a key precursor of chiral ligands for metal catalysis,9,15 by virtue of the distinctive KR of racemic α-ferrocenyl cation A. In the present resolution system, it was confirmed that DKR, namely, the involvement of a racemization process between enantiomeric cations A, also occurs simultaneously during PKR of these cations. Mechanistic studies revealed that the formation of vinylferrocene (5) and its protonation/deprotonation sequence are the key to accomplishing the present intriguing resolution system, termed “dynamic PKR (DPKR)”.
Results and discussion
The initial experiment was performed using racemic 1, 5 mol% of CPA (R)-2 (G = 9-anthryl), 0.5 equivalent of 4-toluenesulfonamide (3a), and molecular sieve (MS) 5A in toluene at room temperature for 6 h (Table 1, entry 1). The reaction cleanly afforded substitution product 4 with moderate enantioselectivity and elimination product 5. Then, screening for catalysts, solvents, and temperatures was conducted; however, none of the conditions led to an improvement in the initial result (see ESI† for details). Importantly, investigation of amine nucleophiles 3 revealed that the N-protecting group strongly affected both reactivity and enantioselectivity (Table 1, entries 1–7).11a Whereas relatively nucleophilic 4-anisidine (3b) and other nitrogen nucleophiles, such as phosphinamide 3c and carbamate 3d, exhibited poor reactivity (Table 1, entries 2–4), less nucleophilic 3f and 3g bearing electron-withdrawing 4- and 2-nitrobenzenesulfonyl groups, respectively, were crucial for achieving high enantioselectivity (Table 1, entries 6 and 7). In particular, the reaction of 2-nitrobenzenesulfonamide (nosylamide: 3g) afforded substitution product (S)-4g in 95% ee (Table 1, entry 7), and the best chemo- and stereocontrol was achieved when 2.0 equivalents of nosylamide (3g) was employed (Table 1, entry 8).|
|
||||
|---|---|---|---|---|
| Entry | 3: R | Time (h) | Yieldb (%) 4/5 | eec (%) |
| a Unless otherwise noted, all reactions were performed using 0.20 mmol of 1, 5 mol% of catalyst 2, and 0.5 equivalent of 3 in toluene (0.2 M) at room temperature. b Determined by crude 1H NMR analysis (in C6D6) using 1,3-benzodioxole as the internal standard. Isolated yields are shown in parentheses. c Determined by chiral stationary phase HPLC analysis. d 2.0 equivalents of 3g was used. The absolute configuration of 4g was determined by derivatization into a known compound. See ESI for details. | ||||
| 1 | 3a: Ts | 6 | 50/50 | 40 |
| 2 | 3b: 4-MeOC6H4 | 24 | <5 | — |
| 3 | 3c: Ph2(O)P | 24 | <5 | — |
| 4 | 3d: Cbz | 24 | 34/26 | <1 |
| 5 | 3e: 4-MeOC6H4SO2 | 6 | 50/50 | 28 |
| 6 | 3f: 4-NO2C6H4SO2 | 2 | 44/52 | 82 |
| 7 | 3g: 2-NO2C6H4SO2 (Ns) | 2 | 40/60 | 95 |
| 8d | 3g | 2 | 47(47)/51(43) | 95 |
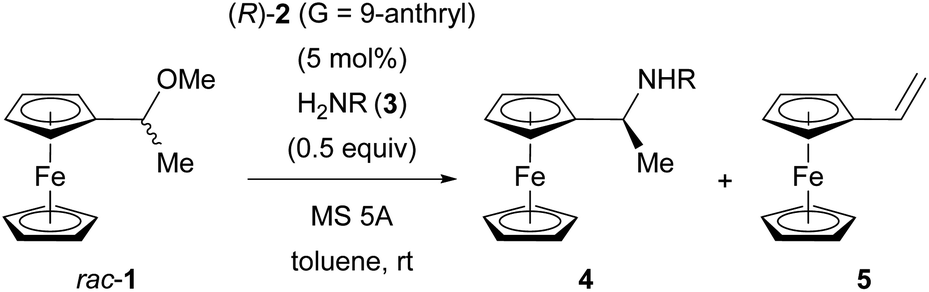
The present reaction proceeds in a stepwise manner. First, the C–O bond cleavage by CPA (R)-2 generates α-ferrocenyl cation A, and then the C–N bond formation (amination) by nosylamide (3g) yields substitution product (S)-4g or the deprotonation affords 5. As shown in Table 1, the amine nucleophile had the greatest impact on the enantioselectivity with respect to both the substituent and the number of equivalents, and it seems that the second step would be the key to achieving efficient KR. To confirm the proposed mechanism, a control experiment was performed in the reaction without using nucleophile 3g and the reaction was quenched prior to complete conversion (Fig. 3a). As expected, the reaction in the absence of 3g resulted in the recovery of nearly racemic 1, clearly suggesting that there is only a slight difference in the reaction rate between enantiomers 1 in the C–O bond cleavage step. Again, it is worth noting that the influence of the nucleophile on the enantioselectivity is a further indication that the resolution occurs in the second step, in which competitive amination and deprotonation reactions take place.
 | ||
| Fig. 3 Mechanistic studies using (R)-2 (G = 9-anthryl): (a) the kinetic resolution of racemic 1 in the absence of 3g. (b) The substitution reaction of enantio-pure (S)-1 with 3g. (c) The substitution reaction of enantio-pure (R)-1 with 3g. (d) The hydroamination of 5 with 3g. | ||
Next, the mechanism was further investigated using enantio-pure starting material 1. Based on the efficiency of the observed KR, it was assumed that (Sp)-cation A generated from (S)-1 and chiral conjugate base (R)-2− would undergo C–N bond formation (amination) with 3g to afford (S)-4g smoothly (C–N matched), whereas (R)-1 would selectively lead to the formation of vinylferrocene (5) via (Rp)-cation A (C–N mismatched). As expected, (S)-1 (99% ee) gave (S)-4g (98% ee) with nearly complete retention of chirality. However, it is worth mentioning that the formation of a considerable amount of vinylferrocene (5) was also observed (Fig. 3b). In contrast, in the reaction of the mismatched combination, namely, enantio-pure (R)-1 and CPA (R)-2, elimination product 5 was formed as the major product (Fig. 3c). More interestingly, a small amount of (S)-4g was obtained with inversion at the stereogenic center (89% ee), despite the fact that the nucleophilic substitution of ferrocenyl alcohol derivatives generally proceeds in a retentive manner.
The formation of anomalous inversion product (S)-4g from (R)-1 is presumably rationalized by the enantioselective hydroamination of 5 catalysed by CPA 2 after elimination of methanol from (R)-1, as shown in Fig. 4: (i) (Rp)-cation A generated from (R)-1 is resolved by chiral conjugate base (R)-2−, affording 5 as the major product along with a small amount of retentive substitution product (R)-4g; (ii) elimination product 5 is further protonated at the double bond by CPA 2 to generate enantiomeric (Sp)- and (Rp)-cations A; (iii) because of the matched combination of (Sp)-cation A and (R)-2−, the nucleophilic attack of 3g on (Sp)-cation A gives rise to (S)-4g with inversion at the stereogenic centre from initial (R)-1, although the deprotonation also takes place, affording 5 to some extent, as shown in Fig. 3b; and (iv) (Rp)-cation A repeats the same resolution process as proposed above.
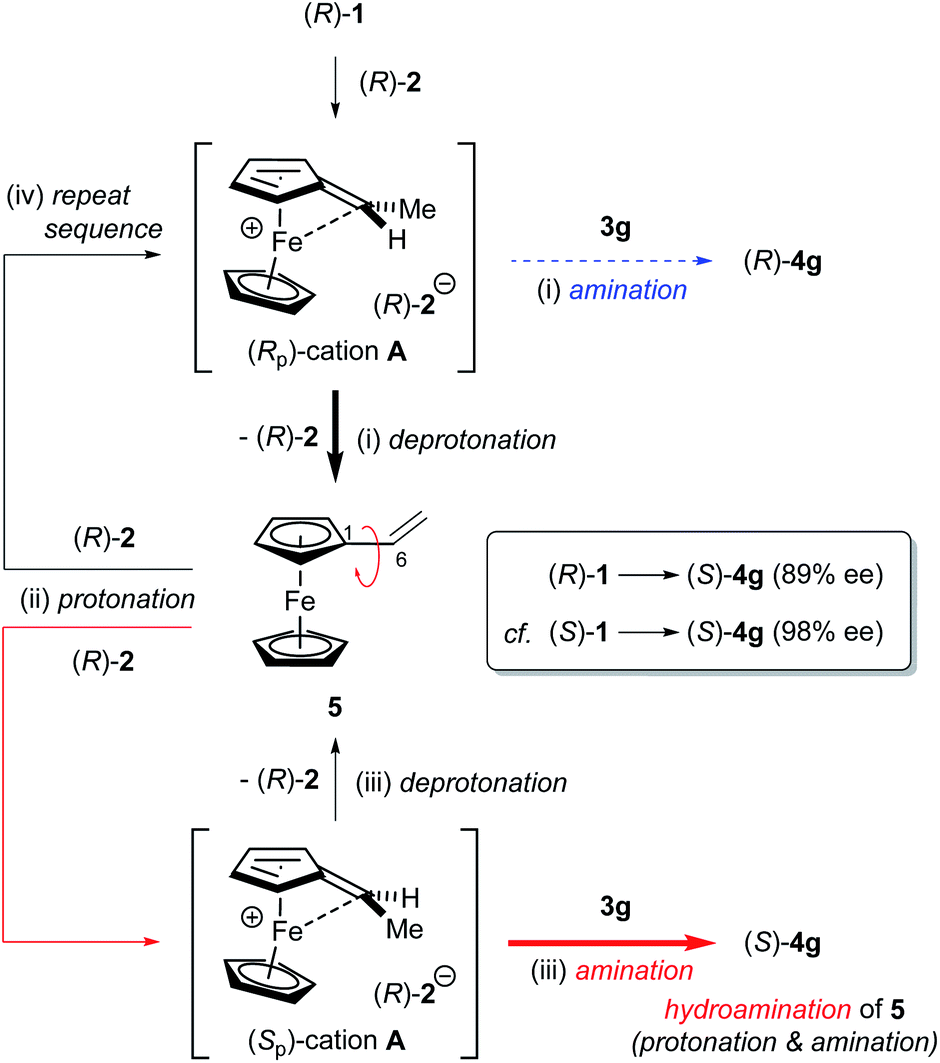 | ||
| Fig. 4 Proposed mechanism for the formation of anomalous inversion product (S)-4g from (R)-1 and the racemization process between (Sp)- and (Rp)-cations A through vinylferrocene (5). | ||
To confirm whether the enantioselective hydroamination of 5 by CPA 2 is involved in the present reaction, 5 was treated with nosylamide (3g) under the optimized reaction conditions (Fig. 3d). As expected, hydroamination product (S)-4g was obtained with high enantioselectivity (95% ee) even though the reaction was considerably sluggish and thus resulted in a diminished yield of (S)-4g. The enantiomeric excess (ee) of 4g [89% ee (S), Fig. 3c] obtained from (R)-1 was lower than that [95% ee (S), Fig. 3d] in the hydroamination of 5 because a small amount of product (R)-4g was furnished via the retentive substitution reaction of (R)-1. The ee observed in Fig. 3c is the sum of the stereochemical outcomes obtained for this retentive substitution and the hydroamination of 5. Hence, these results are consistent with the proposed mechanism.
As depicted in Fig. 4, the overall scheme for the enantioselective hydroamination of vinylferrocene (5) with 3g under the influence of CPA catalyst (R)-2 is also well described. It is pointed out that during the protonation/deprotonation sequence of 5, the free rotation around the C1–C6 single bond of 5 enables one enantiomer of A to isomerize into the other. Thus, the racemization between (Sp)- and (Rp)-cations A is allowed through the formation of vinylferrocene (5), although the direct racemization of enantiomeric cations A is absolutely prevented by the ferrocenyl neighboring effect. More importantly, the substitution of racemic 1 and the hydroamination of 5 exhibited the same enantioselectivity (95% ee) and hence it is considered that both the hydroamination and the substitution reaction proceed through common cation A.
In order to acquire insights into the origin of the stereochemical outcome and elucidate the present resolution system, we theoretically pursued the transition states in the C–N bond formation (amination) step, namely, the resolution step, of enantiomeric cations A with sulfonamide nucleophile 3g under the influence of chiral conjugate base (R)-2−. Geometrical optimization of transition states TSs and TSr leading to a pair of enantiomers (S)- and (R)-4g, respectively, was thoroughly conducted by DFT calculations.16–19¶ As shown in Fig. 5, in optimized transition structures TSs and TSr, chiral conjugate base (R)-2− interacts not only with the N–H proton of sulfonamide nucleophile 3g but also with a couple of protons of the vinylferrocene unit through hydrogen bonds. Thus, TSs and TSr feature multi-coordinating hydrogen bonds, which are an essential interaction mode observed widely in CPA-catalysed reactions5–7 and are beneficial for controlling the stereochemical outcome.20,21 Among the interactions observed, it is noteworthy that the hydrogen bond is formed between the vinyl proton of cation A and the oxygen atom of chiral conjugate base (R)-2− in both transition states TSs and TSr.
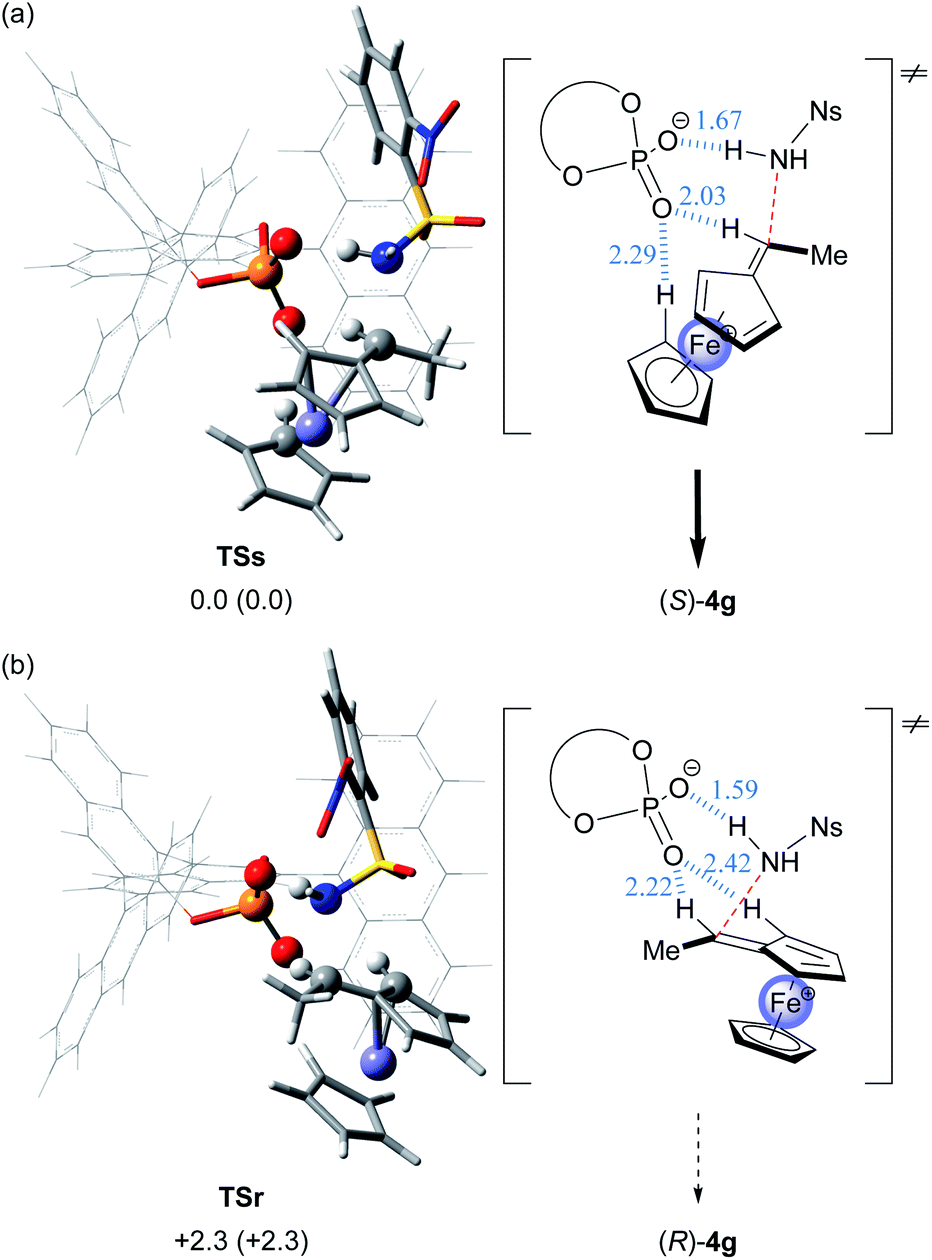 | ||
| Fig. 5 3D structures and schematic representation models of the most energetically favorable transition states for the C–N bond formation step, TSs and TSr, are shown. The 3D structures of each fragment are represented as follows: ferrocene and nosylamide fragments and phosphoric acid subunit: “tube” model; iron and atoms involved in the hydrogen bonding interaction: “ball & bond type” model; and binaphthyl backbone and substituent (G): “wire” model. Relative free energies (kcal mol−1) obtained by single-point energy calculations are shown for the optimized transition states at the M06-2X/6-311+G** level18 in the solution phase according to the SCRF method based on CPCM19 (toluene). Relative free energies (kcal mol−1) of the optimized structures at the B3LYP/6-31G* level17 in the gas phase are shown in parentheses. Hydrogen bond lengths are indicated in blue (angstroms): (a) (Sp)-cation A/3g/(R)-2− (G = 9-anthryl) for TSs. (b) (Rp)-cation A/3g/(R)-2− (G = 9-anthryl) for TSr. | ||
The relative energy difference between TSs and TSr is sufficient for good stereochemical outcome in favor of TSs (ΔΔG≠ = 2.3 kcal mol−1) and consistent with the experimental observation, namely, the formation of (S)-4g as the major product. However, in the present characteristic KR, the C–N bond formation (amination) and the deprotonation compete with each other in both reactions using enantiomeric substrates 1, as shown in the control experiments (Fig. 3b and c). In the matched combination of (Sp)-cation A/(R)-2−, these reactions are comparable and thus the transition state of the deprotonation step and TSs are energetically similar. In contrast, the mismatched combination of (Rp)-cation A/(R)-2− markedly facilitates the deprotonation and thus TSr is energetically unfavourable relative to the transition state of the deprotonation step. From the above considerations, it was assumed that in the deprotonation of enantiomeric cations A by chiral conjugate base (R)-2−, the energy difference between (Sp)- and (Rp)-cations A would not be significant. In fact, DFT calculations of the transition states revealed that the deprotonation of (Sp)-cation A by chiral conjugate base (R)-2− is energetically comparable to that of (Rp)-cation A (ΔΔG≠ = 0.3 kcal mol−1).|| More importantly, the backward reaction, namely, the protonation of vinylferrocene (5) by CPA (R)-2, also proceeds through the same transition states because of the equilibrium between cation A and 5. Hence, the protonation of 5 by CPA (R)-2 affords a nearly racemic mixture of cation A.
As shown in Fig. 3d, the control experiment revealed that the protonation of vinylferrocene (5) by CPA (R)-2 occurred albeit in low efficiency. Furthermore, in the protonation of 5 by CPA (R)-2, the formation of a nearly racemic mixture of cation A is predicted by DFT studies. However, the efficient PKR of racemic cation A has been established in the substitution reaction of racemic 1 with 3g as well as the hydroamination of 5 with 3g, both of which proceed through the same cation A. More importantly, the racemization between (Sp)- and (Rp)-cations A is allowed through the formation of vinylferrocene (5). Integrating these features enabled us to establish the innovative system termed DPKR because the racemization process takes place prior to the resolution step. In order to achieve the intended DPKR of enantiomeric cations A, we further explored the reaction conditions to accelerate the protonation of 5 by CPA (R)-2 and also the formation of product 4g efficiently. As shown in Fig. 6a, the use of chlorobenzene, instead of toluene, as the solvent facilitated the hydroamination of 5 with 3g under the influence of CPA (R)-2 presumably because of the stabilization of cations A generated in the slightly polar solvent, chlorobenzene.** The modification of the conditions resulted in the complete consumption of 5, affording (S)-4g in high yield without any detrimental effect on the enantioselectivity.
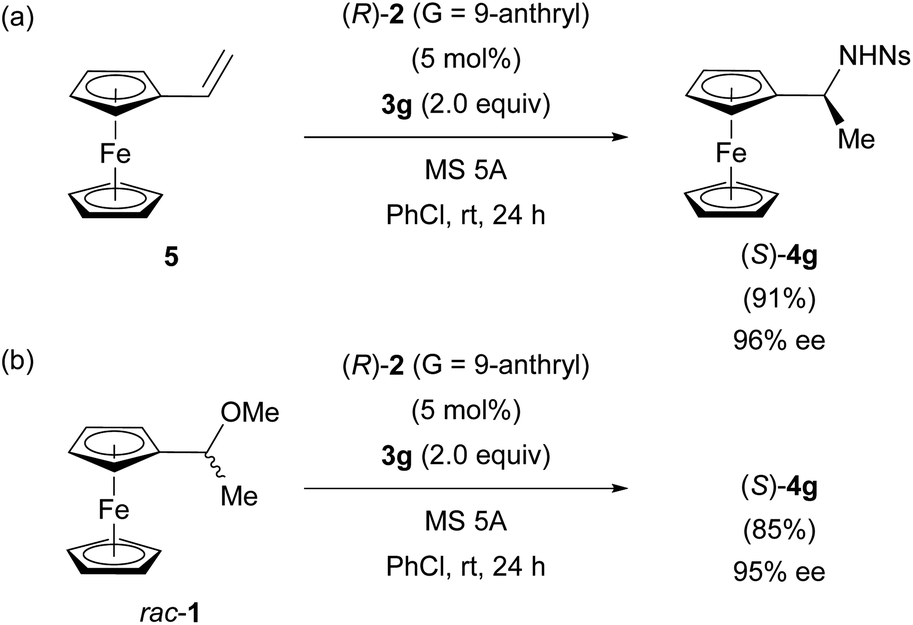 | ||
| Fig. 6 DPKR of enantiomeric cations A using (R)-2 (G = 9-anthryl): (a) the hydroamination of 5 with 3g. (b) The substitution reaction of racemic 1 with 3g achieving enantio-convergent process. | ||
The amended method is also applicable to the substitution reaction of racemic 1 with 3g, affording (S)-4g in comparable yield to the hydroamination while maintaining the high enantioselectivity (Fig. 6bvs.6a). The established DPKR realized the enantio-convergent process in which both enantiomers of racemic 1 were transformed into single enantiomer (S)-4g in a nearly quantitative manner.22†† In addition, the nosyl (Ns) group attached to the nitrogen atom of substitution product 4g can be readily removed to afford the primary amine derivative (see ESI† for details), which has been widely used as an important precursor of chiral ligands for metal catalysis.9,15 It should be emphasized again that the formation of vinylferrocene (5) is the key to allowing the racemization of enantiomeric cations A generated from racemic 1 or the protonation of 5. Otherwise, it is impossible to achieve the present enantio-convergent process.
Conclusions
We have demonstrated the dynamic parallel kinetic resolution (DPKR) of an α-ferrocenyl cation under the influence of a chiral conjugate base of a phosphoric acid catalyst. The mechanism of the present intriguing resolution system was elucidated by control experiments using the enantio-pure precursor of the relevant α-ferrocenyl cation intermediates and the hydroamination of vinylferrocene. Further theoretical studies enabled us to understand the origin of the stereochemical outcome as well as to establish an efficient DPKR. The present resolution system was accomplished by virtue of the dynamic racemization process of enantiomeric α-ferrocenyl cations through vinylferrocene and the chemo-divergent PKR of these cations. The present method enables the formation of a ferrocenylethylamine derivative, which is a key precursor of chiral ligands for metal catalysis, in a nearly quantitative manner with high enantioselectivity. The established DPKR features the integrated aspects of conventional KR and related variants. Further development of an efficient and distinctive kinetic resolution system using chiral phosphoric acid and its derivatives is in progress.Data availability
The exploratory investigation, experimental procedures, computational data, and characterization data are available.Author contributions
Y. T.: conceptualization, data curation, formal analysis, investigation (experimental studies), and writing – original draft. T. K.: data curation, formal analysis, and investigation (theoretical studies). R. O.: data curation, formal analysis, and investigation (experimental studies). J. K.: data curation, formal analysis, and investigation (experimental studies). M. T.: conceptualization, project administration, writing – review & editing, supervision, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Advanced Molecular Transformations by Organocatalysts” (No. 23105002 & 26105704) and “Hybrid Catalysis for Enabling Molecular Synthesis on Demand” (No. JP17H06447) from MEXT, Japan. We also thank the Japan Society for the Promotion of Sciences for the JSPS Research Fellowship for Young Scientists (Y. T.).Notes and references
- (a) H. B. Kagan and J. C. Fiaud, in Topics in Stereochemistry, ed. E. L. Eliel, Wiley & Sons, New York, 1988, vol. 18, pp. 249–330 Search PubMed; (b) A. H. Hoveyda and M. T. Didiuk, Curr. Org. Chem., 1998, 2, 489–526 CAS; (c) J. M. Kieth, J. F. Larrow and E. N. Jacobsen, Adv. Synth. Catal., 2001, 1, 5–26 CrossRef; (d) E. Vedejs and M. Jure, Angew. Chem., Int. Ed., 2005, 44, 3974–4001 CrossRef CAS PubMed; (e) H. Pellissier, in Separation of Enantiomers: Synthetic Methods, ed. M. H. Todd, Wiley & Sons, New York, 2014, pp. 75–89 Search PubMed.
- Dynamic thermodynamic resolution has been reported by Beak et al., see: (a) P. Beak, D. R. Anderson, M. D. Curtis, J. M. Laumer, D. J. Pippel and G. A. Weisenburger, Acc. Chem. Res., 2000, 33, 715–727 CrossRef CAS PubMed; (b) W. K. Lee, Y. S. Park and P. Beak, Acc. Chem. Res., 2009, 42, 224–234 CrossRef CAS PubMed.
- Some catalytic asymmetric reactions may proceed via the KR pathway for cationic intermediates under equilibrium conditions between the cations and the achiral reactants. In most of those cases, it is considered that enantio-enriched cationic intermediates are formed from the achiral reactants under the influence of chiral catalysts. See: (a) S. E. Denmark, W. E. Kuester and M. T. Burk, Angew. Chem., Int. Ed., 2012, 51, 10938–10953 CrossRef CAS; (b) K. Murai and H. Fujioka, Heterocycles, 2013, 87, 763–805 CrossRef CAS.
- Jacobsen et al. reported the dynamic kinetic resolution of seleniranium ions, see: H. Zhang, S. Lin and E. N. Jacobsen, J. Am. Chem. Soc., 2014, 136, 16485–16488 CrossRef CAS PubMed.
- For seminal studies, see: (a) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed; (b) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed.
- For selected reviews of chiral Brønsted acid catalysis, see: (a) H. Yamamoto and N. Payette, in Hydrogen Bonding in Organic Synthesis, ed. P. M. Pihko, Wiley-VCH, Weinheim, 2009, pp. 73–140 Search PubMed; (b) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed; (c) M. Terada, Synthesis, 2010, 1929–1982 CrossRef CAS; (d) D. Kampen, C. M. Reisinger and B. List, Top. Curr. Chem., 2010, 291, 395–456 CrossRef CAS; (e) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- For selected reviews of chiral anions in asymmetric catalysis, see: (a) R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603–614 CrossRef CAS PubMed; (b) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518–533 CrossRef CAS PubMed; (c) K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534–561 CrossRef CAS PubMed.
- For the enantio-convergent Nicholas reaction catalysed by chiral phosphoric acids, see: (a) M. Terada, Y. Ota, F. Li, Y. Toda and A. Kondoh, J. Am. Chem. Soc., 2016, 138, 11038–11043 CrossRef CAS PubMed; (b) Y. Ota, A. Kondoh and M. Terada, Angew. Chem., Int. Ed., 2018, 57, 13917–13921 CrossRef CAS PubMed.
- (a) T. Hayashi, in Ferrocenes, ed. A. Togni and T. Hayashi, VCH, Weinheim, Germany, 1995, pp. 105–142 Search PubMed; (b) A. Togni, in Metallocenes, ed. A. Togni and R. L. Halterman, Wiley-VCH, Weinheim, Germany, 1998, vol. 2, pp. 685–721 CrossRef. See also: (c) A. Togni, C. Breutel, A. Schnyder, F. Spindler, H. Landert and A. Taijani, J. Am. Chem. Soc., 1994, 116, 4062–4066 CrossRef CAS; (d) P. Vicennati and P. G. Cozzi, Eur. J. Org. Chem., 2007, 2248–2253 CrossRef CAS.
- (a) A. A. Koridze, P. V. Petrovskii, S. P. Gubin, V. I. Sokolov and A. I. Mokhov, J. Organomet. Chem., 1977, 136, 65–71 CrossRef CAS; (b) C. Bleiholder, F. Rominger and R. Gleiter, Organometallics, 2009, 28, 1014–1017 CrossRef CAS.
- For the enantioselective substitution reaction with nitrogen nucleophile using chiral phosphoric acids, see: (a) M. Rueping, U. Uria, M.-Y. Lin and I. Atodiresei, J. Am. Chem. Soc., 2011, 133, 3732–3735 CrossRef CAS PubMed; (b) P.-S. Wang, X.-L. Zhou and L.-Z. Gong, Org. Lett., 2014, 16, 976–979 CrossRef CAS PubMed; (c) M. Zhuang and H. Du, Org. Biomol. Chem., 2014, 12, 4590–4593 RSC; (d) Y. Kuroda, S. Harada, A. Oonishi, Y. Yamaoka, K. Yamada and K. Takasu, Angew. Chem., Int. Ed., 2015, 54, 8263–8266 CrossRef CAS PubMed; (e) J. Zhou and H. Xie, Org. Biomol. Chem., 2018, 16, 380–383 RSC; (f) M. Shimizu, J. Kikuchi, A. Kondoh and M. Terada, Chem. Sci., 2018, 9, 5747–5757 RSC.
- T. Izumi and S. Aratani, J. Chem. Technol. Biotechnol., 1995, 63, 25–32 CrossRef CAS.
- (a) J. Eames, Angew. Chem., Int. Ed., 2002, 39, 885–888 CrossRef; (b) J. R. Dehli and V. Gotor, Chem. Soc. Rev., 2002, 31, 365–370 RSC.
- For selected reviews of (dynamic) kinetic resolution, see: (a) K. S. Petersen, Asian J. Org. Chem., 2016, 5, 308–320 CrossRef CAS PubMed; (b) R. Gurubrahamam, Y.-S. Cheng, W.-Y. Huang and K. Chen, ChemCatChem, 2016, 8, 86–96 CrossRef CAS; (c) V. Bhat, E. R. Welin, X. Guo and B. M. Stoltz, Chem. Rev., 2017, 117, 4528–4561 CrossRef CAS PubMed.
- (a) R. G. Arrayás, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2006, 45, 7674–7715 CrossRef PubMed; (b) D. Schaarschmidt and H. Lang, Organometallics, 2013, 32, 5668–5704 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed, see ESI† for the full list of the authors..
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed; (b) C. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- For M06-2X, see: Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- For CPCM, see: V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- For a review of computational studies of chiral Brønsted acid catalysis, see: (a) R. Maji, S. C. Mallojjala and S. E. Wheeler, Chem. Soc. Rev., 2018, 47, 1142–1158 RSC ; For recent representative examples of computational studies of chiral phosphoric acid catalysis, see:; (b) M. N. Grayson and J. M. Goodman, J. Am. Chem. Soc., 2013, 135, 6142–6148 CrossRef CAS; (c) K. Mori, Y. Ichikawa, M. Kobayashi, Y. Shibata, M. Yamanaka and T. Akiyama, J. Am. Chem. Soc., 2013, 135, 3964–3970 CrossRef CAS PubMed; (d) I. Čorić, J. H. Kim, T. Vlaar, M. Patil, W. Thiel and B. List, Angew. Chem., Int. Ed., 2013, 52, 3490–3493 CrossRef PubMed; (e) K. Kanomata, Y. Toda, Y. Shibata, M. Yamanaka, S. Tuzuki, I. D. Gridnev and M. Terada, Chem. Sci., 2014, 5, 3515–3523 RSC; (f) M. Terada, T. Komuro, Y. Toda and T. Korenaga, J. Am. Chem. Soc., 2014, 136, 7044–7057 CrossRef CAS PubMed; (g) Y. Y. Khomutnyk, A. J. Argüelles, G. A. Winschel, Z. Sun, P. M. Zimmerman and P. Nagorny, J. Am. Chem. Soc., 2016, 138, 444–456 CrossRef CAS PubMed; (h) F. Li, T. Korenaga, T. Nakanishi, J. Kikuchi and M. Terada, J. Am. Chem. Soc., 2018, 140, 2629–2642 CrossRef CAS PubMed; (i) J. Zhang, P. Yu, S.-Y. Li, H. Sun, S.-H. Xiang, J. Wang, K. N. Houk and B. Tan, Science, 2018, 361, 8707–8710 CrossRef PubMed; (j) J. Kikuchi, H. Aramaki, H. Okamoto and M. Terada, Chem. Sci., 2019, 10, 1426–1433 RSC; (k) Y. Kwon, J. Li, J. P. Reid, J. M. Crawford, R. Jacob, M. S. Sigman, F. D. Toste and S. J. Miller, J. Am. Chem. Soc., 2019, 141, 6698–6705 CrossRef CAS PubMed; (l) K. Kanomata, Y. Nagasawa, M. Yamanaka, Y. Shibata, F. Egawa, J. Kikuchi and M. Terada, Chem.–Eur. J., 2020, 26, 3367–3372 CrossRef PubMed.
- J. Lapić, A. Višnjevac, M. Cetina, S. Djaković, V. Vrček and V. Rapić, J. Mol. Struct., 2012, 1019, 7–15 CrossRef.
- For a review of enantio-convergent substitution reactions using organocatalysts, see: J. Kikuchi and M. Terada, Chem.–Eur. J. DOI:10.1002/chem.202100439.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data for all new compounds. See DOI: 10.1039/d1sc02122b |
| ‡ Present address: Department of Materials Chemistry, Faculty of Engineering, Shinshu University, Nagano 380-8553, Japan. |
| § The acid-catalysed methanol addition to vinylferrocene (5) successfully regenerated methyl ether 1 in over 90% yield. See ESI† for details. |
| ¶ All calculations were performed with the Gaussian 09 package.16 Geometrical optimization of transition states TSs and TSr was conducted at the B3LYP/6-31G* level17 and characterized using frequency calculations, and the free energies were computed for the gas phase. Single-point energy calculations for the optimized transition states (at the B3LYP/6-31G* level) were also evaluated at the M06-2X/6-311+G** level18 in the solution phase according to the SCRF method based on CPCM (ε = 2.3741 for toluene).19 The free energies in toluene were calculated from the sum of the single-point energies in toluene and the value of thermal correction to Gibbs free energy in the gas phase. |
| || All calculations were performed with the Gaussian 09 package.16 The transition states of the deprotonation step of enantiomeric cations A by chiral conjugate base (R)-2− were optimized at the B3LYP/6-31G* level17 in the gas phase and characterized using frequency calculations. Single-point energy calculations for the optimized transition states were conducted at the M06-2X/6-311+G** level18 in the solution phase according to the SCRF method based on CPCM (ε = 2.3741 for toluene).19 The free energies in toluene were calculated from the sum of the single-point energies in toluene and the value of thermal correction to Gibbs free energy in the gas phase. |
| ** The reaction in toluene under the same reaction conditions (at room temperature for 24 h) afforded (S)-4g in 24% yield with 94% ee. |
| †† In order to enhance the utility of the developed DPKR, we attempted the reaction of ferrocenyl derivative methyl ether having ethyl substituent, instead of methyl substituent of 1. Initially, the optimized reaction conditions were applied to this derivative. However, demethoxylation product, namely, 1-propenylferrocene, was formed as the sole product and no desired substitution product was obtained at all, despite the complete consumption of the starting methyl ether. Similarly, the use of more nucleophilic TsNH2 (3a) rather than NsNH2 (3g) was also unsuccessful, affording 1-propenylferrocene quantitatively. These results indicate that protonation to 1-propenylferrocene became markedly sluggish using parent CPA (R)-2. Therefore CPAs having strong acidity, such as a triflylamide derivative and a bisphosphoric acid, were further investigated. As expected, these strong acids gave rise to the corresponding substitution product in moderate yield, albeit low enantioselectivity. The present intriguing DPKR is established by the well-balanced system between the deprotonation of cation A and the selective introduction of a nucleophile to enantiomeric cations A. Hence, to achieve the efficient DPKR with high enantioselectivity, optimization of CPAs and reaction conditions would be strictly required for each substrate. See ESI† for details. |
| This journal is © The Royal Society of Chemistry 2021 |