
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA high-spin diradical dianion and its bridged chemically switchable single-molecule magnet†
Haiyan
Cui‡
 ab,
Zhao-Bo
Hu‡
a,
Chao
Chen‡
a,
Huapeng
Ruan
a,
Yong
Fang
a,
Li
Zhang
d,
Yue
Zhao
a,
Gengwen
Tan
*c,
You
Song
*a and
Xinping
Wang
*a
ab,
Zhao-Bo
Hu‡
a,
Chao
Chen‡
a,
Huapeng
Ruan
a,
Yong
Fang
a,
Li
Zhang
d,
Yue
Zhao
a,
Gengwen
Tan
*c,
You
Song
*a and
Xinping
Wang
*a
aState Key Laboratory of Coordination Chemistry, Jiangsu Key Laboratory of Advanced Organic Materials, School of Chemistry and Chemical Engineering, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210023, China. E-mail: xpwang@nju.edu.cn; yousong@nju.edu.cn
bJiangsu Key Laboratory of Pesticide Science, College of Sciences, Nanjing Agricultural University, Nanjing 210095, China
cCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China. E-mail: gwtan@suda.edu.cn
dCenter of Materials Science and Engineering, Guangxi University of Science and Technology, Liuzhou 545006, China
First published on 23rd June 2021
Abstract
Triplet diradicals have attracted tremendous attention due to their promising application in organic spintronics, organic magnets and spin filters. However, very few examples of triplet diradicals with singlet–triplet energy gaps (ΔEST) over 0.59 kcal mol−1 (298 K) have been reported to date. In this work, we first proved that the dianion of 2,7-di-tert-butyl-pyrene-4,5,9,10-tetraone (2,7-tBu2-PTO) was a triplet ground state diradical in the magnesium complex 1 with a singlet–triplet energy gap ΔEST = 0.94 kcal mol−1 (473 K). This is a rare example of stable diradicals with singlet–triplet energy gaps exceeding the thermal energy at room temperature (298 K). Moreover, the iron analog 2 containing the 2,7-tBu2-PTO diradical dianion was isolated, which was the first single-molecule magnet bridged by a diradical dianion. When 2 was doubly reduced to the dianion salt 2K2, single-molecule magnetism was switched off, highlighting the importance of diradicals in single-molecule magnetism.
Introduction
There is growing interest in radical chemistry because of the potential applications of radicals in various research fields such as organic chemistry, inorganic chemistry and materials science.1 Diradicals are species containing two unpaired electrons that play pivotal roles in understanding the nature of chemical bonds and are of great interest in organic electronic device applications.2 For example, Kubo and coworkers found that bisphenalenyl-based singlet diradicaloids exhibited high ambipolar mobilities in organic field-effect transistors3 and strong two-photon absorption properties.4 Attributed to the small HOMO–LUMO gaps, several singlet diradicaloids show strong absorption in the near-infrared (NIR) region and can be applied to NIR-organic photodetectors.5 Triplet diradicals are particularly attractive regarding their promising usage in organic magnets, organic spintronics,6 spin filters,7 memory devices,8etc. The well-known triplet diradical m-xylylene possesses a large singlet–triplet energy gap (ΔEST) of ca. 9.6 kcal mol−1,9 but it is only observable in solution at room temperature for hundreds of nanoseconds.10 Rajca group reported a triplet aza-m-xylylene diradical with ΔEST around 10 kcal mol−1, which was persistent at ambient temperature in solution on the time scale of minutes.11 To date, the number of stable triplet diradicals with ΔEST over 0.59 kcal mol−1 (298 K), i.e., surpassing the thermal energy at room temperature, remains limited due to the intrinsic high reactivity.2f,12In contrast, the application of diradicals in synthesizing single-molecule magnets (SMMs) is much less explored, although monoradicals have been widely utilized in designing SMMs.13 In the studied diradical-containing SMMs, the diradicals are mainly composed of neutral bis(imino nitroxide) moieties.14 However, neutral diradicals often suffer from weak coordination to metal ions. In recent years, a few stable triplet ground state diradicals with ΔEST over 298 K have been realized by electron delocalization and steric protection strategies.15 Thus, it is highly desirable to apply these high-spin diradicals to design SMMs. Shultz and coworkers obtained a series of bis(semiquinone) diradical dianion metal complexes with large ΔEST over 298 K, but none of them are SMMs.16
In this contribution, we report a readily accessible 2,7-di-tert-butyl-pyrene-4,5,9,10-tetraone (2,7-tBu2-PTO) diradical dianion in the magnesium and iron complexes LMgII(2,7-tBu2-PTO)MgIIL (L = CH(MeCNAr)2, Ar = 2,6-iPr2C6H3) 1 and LFeII(2,7-tBu2-PTO)FeIIL 2, respectively. Complex 1 features a triplet ground state with ΔEST = 0.94 kcal mol−1 (473 K). Complex 2 represents the first SMM connected by a strong ferromagnetically coupled diradical dianion. Moreover, 1 and 2 can be further doubly reduced to dianion salts 1K2 and 2K2, where the 2,7-tBu2-PTO units are in the diamagnetic tetraanion state, and the single-molecule magnetism of 2K2 is switched off.
Results and discussion
Syntheses and two-electron reduction of the 2,7-tBu2-PTO diradical dianion ligated magnesium and iron complexes
Pyrene-4,5,9,10-tetraones have been widely used as electronic materials due to their intrinsic strong π-electron acceptor ability.17 They can accept as many as four electrons. However, the molecular and electronic structures of the reduced species, especially the doubly reduced species, have never been fully elucidated.18 The cyclic voltammograms reveal that 2,7-tBu2-PTO can undergo four reversible one-electron reduction events with reduction potentials at −0.431 V, −0.710 V, −0.884 V and −1.556 V versus the Ag/Ag+ electrode (Fig. S1†), which indicates that the reduced species may be isolable.The β-diketiminato-ligated magnesium dimer reported by Jones and coworkers,19 which has been proven to be an elegant reducing agent in synthetic chemistry,20 was utilized to reduce 2,7-tBu2-PTO. The reaction of 2,7-tBu2-PTO and one molar equivalent of LMgI–MgIL in toluene at room temperature resulted in deep green crystals of 1 after workup (Fig. 1). It has been spectroscopically and structurally characterized. Intrigued by the facile access of 1 and our previous phosphorus radical anion-coordinated FeII complex exhibiting single-molecule magnetic properties,21 we were interested in applying the 2,7-tBu2-PTO diradical dianion to construct SMMs. Then, the reaction of 2,7-tBu2-PTO and two molar equivalents of LFeI(toluene)22 was performed in toluene at room temperature, from which reddish brown crystals of 2 were isolated (Fig. 1). Complexes 1 and 2 are highly sensitive to oxygen and moisture but can be stored for months in an N2-filled glovebox at ambient temperature.
 | ||
| Fig. 1 Syntheses of complexes 1 and 2. | ||
The cyclic voltammetric investigations (Fig. S2 and S3†) show that compounds 1 and 2 exhibit two reversible reduction waves (Ered1/2 = −1.811 V and −1.548 V for 1; Ered1/2 = −0.194 V and −0.974 V for 2vs. Ag/Ag+), indicating that 1 and 2 can be doubly reduced. In addition, a reversible oxidation process was observed at E1/2 = 0.077 V for 2, suggesting that it can also be oxidized, which might be attributed to the oxidation of the reduced form of 2,7-tBu2-PTO. Consequently, reactions of 1 and 2 with two molar equivalents of elemental potassium in THF at room temperature led to the formation of dianion salts 1K2 and 2K2, respectively, which were isolated in moderate yields (Fig. 2). The solution NMR spectroscopic studies of 1K2 at room temperature show well-resolved 1H and 13C signals in the diamagnetic window (Fig. S11 and S12,† respectively). The 1H NMR spectrum exhibits a singlet resonance at δ 8.42 ppm for the hydrogen atoms of the pyrene moiety. The proton signal for the γ-H of the β-diketiminato ligand (δ = 5.10 ppm) in 1K2 is slightly downfield shifted compared to that of the β-diketiminato ligated magnesium dimer (δ = 4.81 ppm).
 | ||
| Fig. 2 Two-electron reduction of 1 and 2 affording 1K2 and 2K2, respectively. | ||
Crystal structures
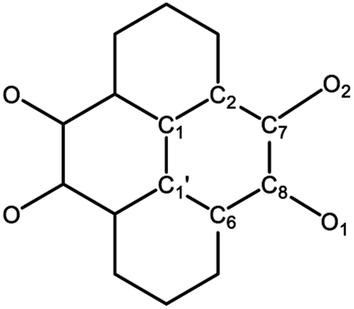
Crystals of 1 were obtained by cooling the toluene solution at −20 °C (Fig. 3A).23 It crystallizes in the monoclinic space group P21/c and features a centrosymmetric geometry. The Mg atoms feature a tetrahedral geometry. The Mg–O distances (1.9803(19) and 1.9714(19) Å) are comparable to those of the magnesium oxalate compound LMg(μ-C2O4)MgL (1.9855(15) and 1.9858(14) Å).24 The C–O bonds are elongated, while the C7–C8 bond is shortened in comparison to those in 2,7-tBu2-PTO (Table 1)25 in accordance with the reduction of carbonyl compounds. The C7–C8 bond length (1.443(3) Å) is between those of the C–C single bond (1.54 Å) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond (1.34 Å), which shows the semiquinone character of the C2O2 moieties. Moreover, due to the steric bulkiness of the L ligands, no intermolecular interaction is observed, which is normally present in pyrenes.26
C double bond (1.34 Å), which shows the semiquinone character of the C2O2 moieties. Moreover, due to the steric bulkiness of the L ligands, no intermolecular interaction is observed, which is normally present in pyrenes.26
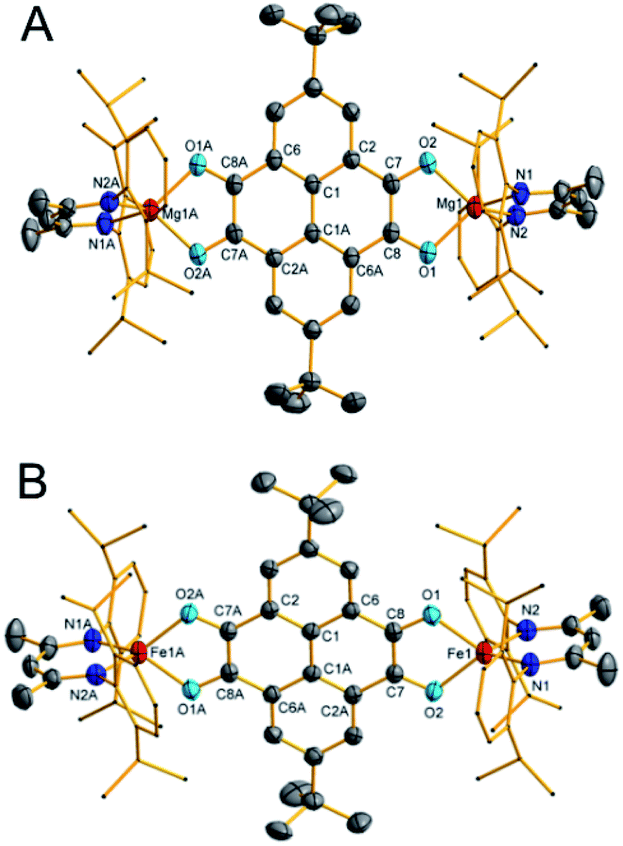 | ||
| Fig. 3 Thermal ellipsoid drawing of the molecular structures of 1 (A) and 2 (B) at 50% probability. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): 1: Mg1–O1 1.9803(19), Mg1–O2 1.9714(19), Mg1–N1 2.007(2), Mg1–N2 2.011(2), O1–C8 1.292(3), O2–C7 1.289(3), C1A–C6A 1.414(3), C1A–C2 1.405(3), C2–C7 1.441(3), C6A–C8 1.438(3), C7–C8 1.443(3), O2–Mg1–O1 83.88(7), O2–Mg1–N1 117.37(9), O1–Mg1–N1 127.47(9), O2–Mg1–N2 118.65(9), O1–Mg1–N2 116.80(9), and N1–Mg1–N2 95.13(9). 2: Fe1–N1 1.963(2), Fe1–N2 1.968(2), Fe1–O2 1.992(2), Fe1–O1 2.0014(19), O1–C8 1.291(3), O2–C7 1.295(3), C1A–C2A 1.408(4), C1–C1A 1.449(5), C1–C6 1.400(4), C2A–C7 1.445(4), C7–C8 1.422(4), C6–C8 1.443(4), N1–Fe1–N2 95.08(10), N1–Fe1–O2 128.34(9), N2–Fe1–O2 118.58(9), N1–Fe1–O1 117.52(9), N2–Fe1–O1 118.75(9), and O2–Fe1–O1 80.97(8). Symmetry transformations were used to generate equivalent atoms labeled with ‘A’: −x + 1, y + 1, and z + 1. | ||
|
|
|||||
|---|---|---|---|---|---|
| 2,7-tBu2-PTO25 | 1 | 2 | 1K2 | 2K2 | |
| C7–O2 | 1.221(7) | 1.289(3) | 1.295(3) | 1.362(3) | 1.344(3) |
| C8–O1 | 1.209(7) | 1.292(3) | 1.291(3) | 1.343(3) | 1.347(2) |
| C7–C8 | 1.545(8) | 1.443(3) | 1.422(4) | 1.382(3) | 1.390(3) |
| C6–C8 | 1.497(8) | 1.438(3) | 1.443(4) | 1.441(3) | 1.429(3) |
| C1′–C6 | 1.416(8) | 1.414(3) | 1.400(4) | 1.419(3) | 1.423(3) |
| C1–C1′ | 1.474(11) | 1.437(4) | 1.449(5) | 1.411(4) | 1.415(4) |
| C1–C2 | 1.401(7) | 1.405(3) | 1.408(4) | 1.425(3) | 1.420(3) |
| C2–C7 | 1.472(8) | 1.441(3) | 1.445(4) | 1.434(3) | 1.435(3) |
The molecular configuration of 2 is akin to that of 1 (Fig. 3B). The iron centers feature a tetrahedral geometry. The Fe–N bond lengths (1.963(2) and 1.968(2) Å) are comparable to those in the diazafluorenylidene-substituted phosphaalkene radical anion coordinated iron complex (1.980(2) and 1.968(2) Å).21 The bond lengths in the 2,7-tBu2-PTO moiety of 2 are comparable to those in 1 (Table 1), which suggest that 2,7-tBu2-PTO is also most likely in the diradical dianion state. Therefore, two-electron transfer to 2,7-tBu2-PTO occurs in the reaction, and the iron atoms are in the oxidation state of two. Additionally, the zero-field 57Fe Mössbauer spectrum recorded at 80 K afforded an isomer shift value δ = 0.86 mm s−1 and a quadrupole splitting value ΔEQ = 1.87 mm s−1 (Fig. S4†), which confirms the presence of high-spin tetrahedral FeII ions in 2.21,27
Crystals of 1K2 and 2K2 suitable for X-ray diffraction analysis were grown from THF solutions at −20 °C (Fig. 4). They crystallize in the monoclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . In comparison to precursors 1 and 2, the M–N bonds (2.044(2) and 2.045(2) Å for 1K2; 1.997(2) and 1.9938(19) Å for 2K2) are slightly elongated, while the M–O bonds (1.9300(18) and 1.9506(18) Å for 1K2; 1.9808(15) and 1.9811(16) Å for 2K2) are shortened. The most pronounced change is the further increased C–O bond lengths (Table 1), which are close to those in the pyrocatecholate aluminum compound (1.369(5) and 1.380(4) Å).28 Meanwhile, C7–C8 bonds (1.382(3) Å for 1K2; 1.390(3) Å for 2K2) are shortened, compared with those of 1 and 2. So 2,7-tBu2-PTO is in the tetraanion form. This result is consistent with the diamagnetic nature of 1K2 demonstrated by NMR spectroscopy. Moreover, the isomer shift value δ = 0.86 mm s−1 and quadrupole splitting value ΔEQ = 1.57 mm s−1 obtained from the zero-field 57Fe Mössbauer spectrum at 80 K of 2K2 (Fig. S5†) are close to those of 2, which prove the retention of the high-spin +2 oxidation state of the iron centers.
. In comparison to precursors 1 and 2, the M–N bonds (2.044(2) and 2.045(2) Å for 1K2; 1.997(2) and 1.9938(19) Å for 2K2) are slightly elongated, while the M–O bonds (1.9300(18) and 1.9506(18) Å for 1K2; 1.9808(15) and 1.9811(16) Å for 2K2) are shortened. The most pronounced change is the further increased C–O bond lengths (Table 1), which are close to those in the pyrocatecholate aluminum compound (1.369(5) and 1.380(4) Å).28 Meanwhile, C7–C8 bonds (1.382(3) Å for 1K2; 1.390(3) Å for 2K2) are shortened, compared with those of 1 and 2. So 2,7-tBu2-PTO is in the tetraanion form. This result is consistent with the diamagnetic nature of 1K2 demonstrated by NMR spectroscopy. Moreover, the isomer shift value δ = 0.86 mm s−1 and quadrupole splitting value ΔEQ = 1.57 mm s−1 obtained from the zero-field 57Fe Mössbauer spectrum at 80 K of 2K2 (Fig. S5†) are close to those of 2, which prove the retention of the high-spin +2 oxidation state of the iron centers.
 | ||
| Fig. 4 Thermal ellipsoid drawing of the molecular structures of 1K2 (A) and 2K2 (B) at 50% probability. Hydrogen atoms are omitted for clarity. Symmetry transformations were used to generate equivalent atoms labeled with ‘A’: −x + 1, y + 1, and z + 1. Selected bond lengths (Å) and angles (°): 1: Mg1–O1 1.9300(18), Mg1–O2 1.9506(18), Mg1–N1 2.044(2), Mg1–N2 2.045(2), O1–C8 1.343(3), O2–C7 1.362(3), C7–C8 1.382(3), O1–Mg1–O2 88.12(7), O1–Mg1–N1 124.02(8), O2–Mg1–N1 111.77(8), O1–Mg1–N2 119.64(9), O2–Mg1–N2 123.41(8), and N1–Mg1–N2 92.79(8). 2: Fe1–O2 1.9808(15), Fe1–O1 1.9811(16), Fe1–N2 1.997(2), Fe1–N1 1.9938(19), O1–C8 1.347(2), O2–C7 1.344(3), C7–C8 1.390(3), O2–Fe1–O1 83.55(6), O2–Fe1–N2 122.09(7), O1–Fe1–N2 124.11(8), O2–Fe1–N1 115.24(7), O1–Fe1–N1 120.34(7), and N2–Fe1–N1 94.10(8). | ||
Calculated electron spin density distribution and singlet–triplet gap of 1
The theoretical calculations of 1 were performed. Geometry optimizations were performed at the (U)B3LYP/6-31G(d) level, and the stationary points were checked by frequency calculations.29 The results from theoretical calculations are interpreted to suggest that 1 has a triplet ground state, and the calculated singlet–triplet gap is 0.91 kcal mol−1. Moreover, the spin density distribution shows that it is mainly delocalized over the two C2O2 moieties with contributions from the two central benzene rings (Fig. 5). | ||
| Fig. 5 Spin density distribution of 1 in the triplet state calculated at the UB3LYP/6-31G(d) level (isovalue = 0.002 a.u.). | ||
Magnetic characterization
Electro-paramagnetic resonance (EPR) spectroscopy and superconducting quantum interference device (SQUID) measurements were performed to clarify the electronic structures of newly formed complexes. The EPR spectrum of 1 in the frozen toluene solution at 90 K (Fig. 6A) reveals a clear half-field signal of the forbidden transition (Δms = ±2), which indicates the presence of a triplet-spin state species. The signals attributed to the Δms = ±1 transition are finely resolved, and parameters gx = gy = 2.0028, gz = 2.0042, D = 139 G and E = 32 G are extracted from the spectral simulation with the SimFonia software package. From the zero-field splitting parameter D, the distance between the spin centers is estimated to be 5.85 Å,2f which is close to the mean distance (6.05 Å) of C7 to C8A and O2 to O1A in the solid-state structure, which suggests that the spin density is delocalized over the C2O2 moieties. In contrast, iron complexes 2 and 2K2 are EPR-silent at room temperature and 90 K. | ||
| Fig. 6 (A) Experimental (black line) and simulated (red line) EPR spectra of 1 in toluene solution at 90 K. The central peak is attributed to the S = 1/2 monoradical impurity. (B) χMT–T plots of 1 with the fitting results (red line). | ||
SQUID measurements were performed on the powder sample of 1 (Fig. 6B). The χMT value at 300 K is 0.83 cm3 mol−1 K, which is larger than the theoretical value of 0.75 cm3 mol−1 K (S = 1/2, g = 2.0) for 2 free radicals. With decreasing temperature, the χMT values slowly increase to a maximum of 1.04 cm3 mol−1 K at 8 K. With the further decrease in temperature, the χMT values slightly decline to 1.0 cm3 mol−1 K at 1.8 K. This behavior obviously indicates a strong ferromagnetic coupling between two S = 1/2 centers in 1, and the spins are completely parallel below 8 K. Moreover, the isothermal magnetization of 1.96 NμB at 70 kOe and 1.8 K is close to the theoretical value of 2 NμB (S = 1, g = 2) (Fig. S6†), which provides evidence of ferromagnetic coupling. The temperature-dependent magnetizations were fitted using the PHI program based on equation Ĥ = −2JŜ1Ŝ2 + gμBŜH (S1 = S2 = 1/2). The best fitting results are g = 2.04(1), J = 165.1(5) cm−1, zj = −0.0053(1) cm−1 and TIP = 4.17 × 10−4 cm3 mol−1. Hence, complex 1 is a triplet ground state diradical with a singlet–triplet energy gap of 0.94 kcal mol−1 (473 K), and there is negligible intermolecular interaction. The experimental results are consistent with the theoretical calculations. Complex 1 is a rare example of stable diradicals with singlet–triplet energy gaps larger than 0.6 kcal mol−1 (300 K).
SQUID measurements on the powder samples of 2 and 2K2 were also performed to gain more insights into their electronic structures and magnetic properties. The χMT value of 5.26 cm3 mol−1 K at 300 K for 2 (Fig. 7) is far less than 6.75 cm3 mol−1 K for two high-spin FeII ions (S = 2, g = 2.0) and two organic radicals (S = 1, g = 2.0). This phenomenon may be due to a strong antiferromagnetic coupling between the metal ions and the diradical ions, which results in dominant magnetic properties even above room temperature. With decreasing temperature, the χMT values first gradually increase from 300 to 30 K, subsequently show a pronounced decrease, and finally reach the ultimate value of 5.48 cm3 mol−1 K at 1.8 K. This phenomenon should be attributed to the competition between the intramolecular ferromagnetic and antiferromagnetic coupling, which forms a ferromagnetic experimental curve. The temperature and field dependent magnetizations were fitted to quantify the anisotropy parameters based on eqn (1) using the PHI program:
 | (1) |
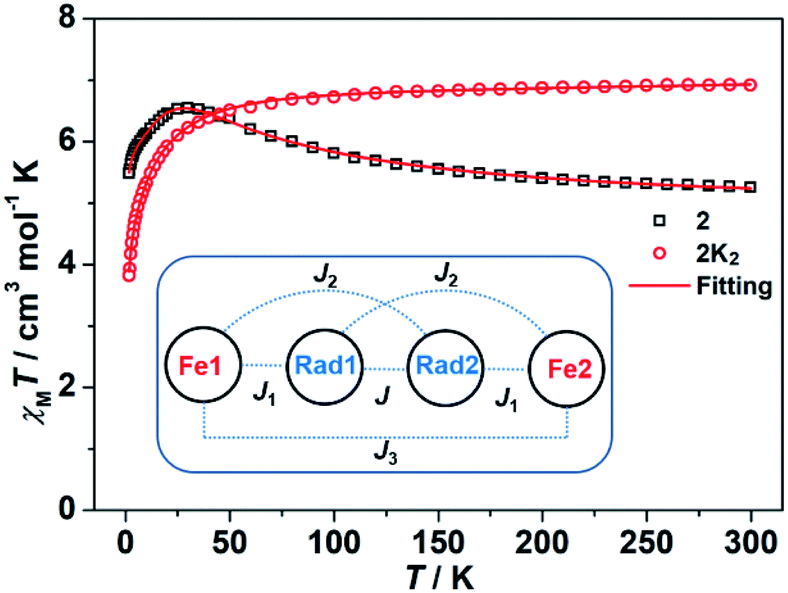 | ||
| Fig. 7 χ M T–T plots of 2 and 2K2 with the fitting results (red line) using the PHI program. The inset shows the magnetic coupling situation in 2. | ||
The χMT value of 2K2 at 300 K is 6.92 cm3 mol−1 K, and the value first slowly decreases from room temperature to 60 K and subsequently rapidly decreases to 3.82 cm3 mol−1 K at 1.8 K (Fig. 7). These results suggest that the two high-spin FeII ions are weakly antiferromagnetically coupled in 2K2. Fitting the χMT–T plots to eqn (2) using the PHI program31 yields the parameters of D = −6.45(10) cm−1, g = 2.12(2), J3 = −0.028(1) cm−1 and TIP = 7.22 × 10−4 cm3 mol−1. The weak coupling between the FeII ions is due to the closed-shell character of the 2,7-tBu2-PTO tetraanion, as observed in 1K2. The large difference in the D value in complexes 2 and 2K2 can be attributed to the change in the local ligand field after the two-electron reduction.
 | (2) |
Alternating current (ac) susceptibility measurements were performed to evaluate the magnetization dynamics of 2 and 2K2. Obvious out-of-phase signals (χM′′) were observed for 2 under a zero field, whereas no peak was observed below 1000 Hz (Fig. S9†), which suggests the existence of quantum tunneling of magnetization (QTM). In contrast, the susceptibility measurements of 2K2 showed that no χM′′ signals were observed with or without external fields, which suggests switching off the single-molecule magnetism after the two-electron reduction of 2.
To suppress the QTM effect, a 1.0 kOe field was applied to study the slow relaxation behavior of 2. The frequency-dependent in-of-phase signals (χM′, Fig. S10†) and χM′′ (Fig. 8A) are observed in the temperature range of 1.8–5 K. The Cole–Cole plots of χM′′ versus χM′ were fitted using the CC-FIT program and a modified Debye function (Fig. 8B). The extracted α values are listed in Table S2 in the ESI† and are less than 0.1, which indicates a narrow distribution of relaxation times. The relaxation time τ0 and effective barrier energy were afforded by fitting the Arrhenius-like diagrams (Fig. 8C). The entire temperature dataset was fitted using the equation τ−1 = AT + CT−n + τ0−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ueff/kBT), where A is the coefficient of the direct process, C is the coefficient of the Raman process, Ueff is the energy barrier for magnetization reversal, and kB is the Boltzmann constant. The best fitting parameters are A = 29.68 K−1 s−1, C = 4.66 K−3.03 s−1, n = 3.03, τ0 = 7.33 × 10−11 s and Ueff = 71.1 K. The fitting results indicate that the QTM effect has been suppressed well by the applied magnetic field.
exp(−Ueff/kBT), where A is the coefficient of the direct process, C is the coefficient of the Raman process, Ueff is the energy barrier for magnetization reversal, and kB is the Boltzmann constant. The best fitting parameters are A = 29.68 K−1 s−1, C = 4.66 K−3.03 s−1, n = 3.03, τ0 = 7.33 × 10−11 s and Ueff = 71.1 K. The fitting results indicate that the QTM effect has been suppressed well by the applied magnetic field.
 | ||
| Fig. 8 (A) Frequency dependence of the out-of-phase (χM′′) ac-susceptibilities for 2 at different temperatures under a 1.0 kOe field. (B) Cole–Cole curves under a 1.0 kOe field for 2. (C) Plots of ln(τ/s) versus T−1 under a 1.0 kOe field for 2. | ||
Conclusions
We have first demonstrated that the dianion of 2,7-tBu2-PTO is a triplet ground state diradical in the coordination sphere of magnesium. This is a rare example of stable diradicals with singlet–triplet energy gaps larger than 0.59 kcal mol−1 (298 K), which surpasses the thermal energy at room temperature. The diradical dianion was applied to synthesize iron complex 2, which serves as the first SMM bridged by a diradical dianion. Complexes 1 and 2 can be reduced to dianion salts 1K2 and 2K2, respectively, where the 2,7-tBu2-PTO units are in diamagnetic tetraanion form. This work provides more insights into the molecular and electronic structures of the reduced species of PTO and demonstrates the potential of diradical dianions in constructing interesting magnetic materials. Syntheses and magnetic studies of other complexes bearing the reduced species of PTO are ongoing in our laboratory.Data availability
Crystallographic data for 1, 2, 1K2 and 2K2 have been deposited in the Cambridge Crystallographic Data Center under CCDC No. 1998479–1998482.Author contributions
X. W. conceived the project. H. C. conducted the synthesis and physical characterizations. Z. H. and Y. S. performed the magnetic analysis. C. C. performed the SQUID measurements. H. R. and Y. Z. performed the crystallographic measurements and data refinements. Y. F. performed the EPR measurements. L. Z. performed the DFT calculations. G. T. wrote the manuscript. H. C. wrote the experimental part. G. T., Y. S. and X. W. revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Key R&D Program of China (2016YFA0300404, 2017YFA0303203 and 2018YFA0306004), National Natural Science Foundation of China (21402094, 21525102, 21690062 and 21973038), China Postdoctoral Science Foundation (2017M611774) and the Joint Fund for Regional Innovation and Development (U20A2073) for financial support. L. Z. thanks the Guangxi Department of Science and Technology (AD19245107) and Guangxi University of Science and Technology (03190227 and 03190228) for financial support. The calculations were performed at the High-Performance Computing Center of Nanjing University. We thank Ms Jie Xiao and Prof. Liang Deng of Shanghai Institute of Organic Chemistry for measuring the Mössbauer spectra.Notes and references
- (a) P. P. Power, Chem. Rev., 2003, 103, 789–809 CrossRef CAS; (b) R. G. Hicks, in Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds, John Wiley & Sons, Ltd, 2010 CrossRef; (c) M. Mas-Torrent, N. Crivillers, C. Rovira and J. Veciana, Chem. Rev., 2012, 112, 2506–2527 CrossRef CAS PubMed; (d) I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC; (e) T. Chivers and J. Konu, in Comprehensive Inorganic Chemistry II, ed. K. Poeppelmeier, Elsevier, Amsterdam, 2nd edn, 2013, pp. 349–373 Search PubMed; (f) G. Tan and X. Wang, Chin. J. Chem., 2018, 36, 573–586 CrossRef CAS.
- (a) L. Salem and C. Rowland, Angew. Chem., Int. Ed., 1972, 11, 92–111 CrossRef CAS; (b) W. T. Borden, Diradicals, Wiley-Interscience, New York, 1982 Search PubMed; (c) A. Rajca, Chem. Rev., 1994, 94, 871–893 CrossRef CAS; (d) F. Breher, Coord. Chem. Rev., 2007, 251, 1007–1043 CrossRef CAS; (e) Z. Sun, Q. Ye, C. Chi and J. Wu, Chem. Soc. Rev., 2012, 41, 7857–7889 RSC; (f) M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed; (g) N. M. Gallagher, A. Olankitwanit and A. Rajca, J. Org. Chem., 2015, 80, 1291–1298 CrossRef CAS; (h) Z. Zeng, X. Shi, C. Chi, J. T. Lopez Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC; (i) G. Tan and X. Wang, Acc. Chem. Res., 2017, 50, 1997–2006 CrossRef CAS PubMed.
- (a) T. Kubo, A. Shimizu, M. Sakamoto, M. Uruichi, K. Yakushi, M. Nakano, D. Shiomi, K. Sato, T. Takui, Y. Morita and K. Nakasuji, Angew. Chem., Int. Ed., 2005, 44, 6564–6568 CrossRef CAS PubMed; (b) H. Koike, M. Chikamatsu, R. Azumi, J. y. Tsutsumi, K. Ogawa, W. Yamane, T. Nishiuchi, T. Kubo, T. Hasegawa and K. Kanai, Adv. Funct. Mater., 2016, 26, 277–283 CrossRef CAS.
- K. Kamada, K. Ohta, T. Kubo, A. Shimizu, Y. Morita, K. Nakasuji, R. Kishi, S. Ohta, S.-i. Furukawa, H. Takahashi and M. Nakano, Angew. Chem., Int. Ed., 2007, 46, 3544–3546 CrossRef CAS PubMed.
- (a) A. Konishi, Y. Hirao, M. Nakano, A. Shimizu, E. Botek, B. Champagne, D. Shiomi, K. Sato, T. Takui, K. Matsumoto, H. Kurata and T. Kubo, J. Am. Chem. Soc., 2010, 132, 11021–11023 CrossRef CAS PubMed; (b) Z. Zeng, S. Lee, J. L. Zafra, M. Ishida, X. Zhu, Z. Sun, Y. Ni, R. D. Webster, R.-W. Li, J. T. López Navarrete, C. Chi, J. Ding, J. Casado, D. Kim and J. Wu, Angew. Chem., Int. Ed., 2013, 52, 8561–8565 CrossRef CAS PubMed; (c) Y. Zheng, M.-s. Miao, G. Dantelle, N. D. Eisenmenger, G. Wu, I. Yavuz, M. L. Chabinyc, K. N. Houk and F. Wudl, Adv. Mater., 2015, 27, 1718–1723 CrossRef CAS PubMed; (d) Y. Ni, S. Lee, M. Son, N. Aratani, M. Ishida, A. Samanta, H. Yamada, Y.-T. Chang, H. Furuta, D. Kim and J. Wu, Angew. Chem., Int. Ed., 2016, 55, 2815–2819 CrossRef CAS PubMed.
- (a) A. R. Rocha, V. M. Garcia-suarez, S. W. Bailey, C. J. Lambert, J. Ferrer and S. Sanvito, Nat. Mater., 2005, 4, 335–339 CrossRef CAS PubMed; (b) S. Sanvito, Chem. Soc. Rev., 2011, 40, 3336–3355 RSC.
- (a) C. Herrmann, G. C. Solomon and M. A. Ratner, J. Am. Chem. Soc., 2010, 132, 3682–3684 CrossRef CAS PubMed; (b) S. Shil, D. Bhattacharya, A. Misra and D. J. Klein, Phys. Chem. Chem. Phys., 2015, 17, 23378–23383 RSC.
- (a) Y. Yonekuta, K. Susuki, K. Oyaizu and K. Honda, J. Am. Chem. Soc., 2007, 129, 14128–14129 CrossRef CAS PubMed; (b) J. Lehmann, A. Gaita-Ariño, E. Coronado and D. Loss, J. Mater. Chem., 2009, 19, 1672–1677 RSC; (c) K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 2339–2344 CrossRef CAS; (d) R. Gaudenzi, J. de Bruijckere, D. Reta, I. d. P. R. Moreira, C. Rovira, J. Veciana, H. S. J. van der Zant and E. Burzurí, ACS Nano, 2017, 11, 5879–5883 CrossRef CAS PubMed.
- (a) B. B. Wright and M. S. Platz, J. Am. Chem. Soc., 1983, 105, 628–630 CrossRef CAS; (b) P. G. Wenthold, J. B. Kim and W. C. Lineberger, J. Am. Chem. Soc., 1997, 119, 1354–1359 CrossRef CAS; (c) P. Neuhaus, D. Grote and W. Sander, J. Am. Chem. Soc., 2008, 130, 2993–3000 CrossRef CAS PubMed.
- K. W. Haider, E. Migirdicyan, M. S. Platz, N. Soundararajan and A. Despres, J. Am. Chem. Soc., 1990, 112, 733–738 CrossRef CAS.
- A. Rajca, A. Olankitwanit and S. Rajca, J. Am. Chem. Soc., 2011, 133, 4750–4753 CrossRef CAS PubMed.
- T. Stuyver, B. Chen, T. Zeng, P. Geerlings, F. De Proft and R. Hoffmann, Chem. Rev., 2019, 119, 11291–11351 CrossRef CAS PubMed.
- (a) S. Demir, I.-R. Jeon, J. R. Long and T. D. Harris, Coord. Chem. Rev., 2015, 289–290, 149–176 CrossRef CAS; (b) L. Wang, J. Li, L. Zhang, Y. Fang, C. Chen, Y. Zhao, Y. Song, L. Deng, G. Tan, X. Wang and P. P. Power, J. Am. Chem. Soc., 2017, 139, 17723–17726 CrossRef CAS PubMed; (c) X. Meng, W. Shi and P. Cheng, Coord. Chem. Rev., 2019, 378, 134–150 CrossRef CAS.
- (a) S. G. Reis, M. Briganti, D. O. T. A. Martins, H. Akpinar, S. Calancea, G. P. Guedes, S. Soriano, M. Andruh, R. A. A. Cassaro, P. M. Lahti, F. Totti and M. G. F. Vaz, Dalton Trans., 2016, 45, 2936–2944 RSC; (b) H. Li, Z. Sun, J. Sun, L. Xi, J. Guo, G. Sun, J. Xie, Y. Ma and L. Li, J. Mater. Chem. C, 2018, 6, 2060–2068 RSC; (c) K. Wang, J. Sun, L. Xi, J. Lu, P. Jing and L. Li, Dalton Trans., 2019, 48, 14383–14389 RSC.
- (a) A. Rassat and H. U. Sieveking, Angew. Chem., Int. Ed., 1972, 11, 303–304 CrossRef CAS; (b) J. Veciana, C. Rovira, M. I. Crespo, O. Armet, V. M. Domingo and F. Palacio, J. Am. Chem. Soc., 1991, 113, 2552–2561 CrossRef CAS; (c) A. Rajca and S. Utamapanya, J. Org. Chem., 1992, 57, 1760–1767 CrossRef CAS; (d) K. Inoue and H. Iwamura, Angew. Chem., Int. Ed., 1995, 34, 927–928 CrossRef CAS; (e) D. A. Shultz, S. H. Bodnar and J. W. Kampf, Chem. Commun., 2001, 93–94 RSC; (f) E. Fukuzaki and H. Nishide, J. Am. Chem. Soc., 2006, 128, 996–1001 CrossRef CAS; (g) A. Rajca, M. Takahashi, M. Pink, G. Spagnol and S. Rajca, J. Am. Chem. Soc., 2007, 129, 10159–10170 CrossRef CAS PubMed; (h) A. Rajca, K. Shiraishi and S. Rajca, Chem. Commun., 2009, 4372–4374 RSC; (i) P. J. Boratyński, M. Pink, S. Rajca and A. Rajca, Angew. Chem., Int. Ed., 2010, 49, 5459–5462 CrossRef PubMed; (j) S. Suzuki, T. Furui, M. Kuratsu, M. Kozaki, D. Shiomi, K. Sato, T. Takui and K. Okada, J. Am. Chem. Soc., 2010, 132, 15908–15910 CrossRef CAS PubMed; (k) W. Wang, L. Wang, S. Chen, W. Yang, Z. Zhang and X. Wang, Sci. China: Chem., 2018, 61, 300–305 CrossRef CAS; (l) N. Gallagher, H. Zhang, T. Junghoefer, E. Giangrisostomi, R. Ovsyannikov, M. Pink, S. Rajca, M. B. Casu and A. Rajca, J. Am. Chem. Soc., 2019, 141, 4764–4774 CrossRef CAS.
- (a) D. A. Shultz, S. H. Bodnar, H. Lee, J. W. Kampf, C. D. Incarvito and A. L. Rheingold, J. Am. Chem. Soc., 2002, 124, 10054–10061 CrossRef CAS; (b) M. L. Kirk and D. A. Shultz, Coord. Chem. Rev., 2013, 257, 218–233 CrossRef CAS.
- (a) T. Nokami, T. Matsuo, Y. Inatomi, N. Hojo, T. Tsukagoshi, H. Yoshizawa, A. Shimizu, H. Kuramoto, K. Komae, H. Tsuyama and J.-i. Yoshida, J. Am. Chem. Soc., 2012, 134, 19694–19700 CrossRef CAS; (b) Y. Liang, Y. Jing, S. Gheytani, K.-Y. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841–848 CrossRef CAS PubMed; (c) Y. Liang and Y. Yao, Joule, 2018, 2, 1690–1706 CrossRef CAS.
- K. M. Kadish, W. E, R. Zhan, T. Khoury, L. J. Govenlock, J. K. Prashar, P. J. Sintic, K. Ohkubo, S. Fukuzumi and M. J. Crossley, J. Am. Chem. Soc., 2007, 129, 6576–6588 CrossRef CAS PubMed.
- J. Hicks, M. Juckel, A. Paparo, D. Dange and C. Jones, Organometallics, 2018, 37, 4810–4813 CrossRef CAS.
- (a) S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed; (b) C. Jones, Nat. Rev. Chem., 2017, 1, 0059 CrossRef CAS.
- G. Tan, J. Li, L. Zhang, C. Chen, Y. Zhao, X. Wang, Y. Song, Y.-Q. Zhang and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 12741–12745 CrossRef CAS PubMed.
- F. Spitzer, C. Graßl, G. Balázs, E. M. Zolnhofer, K. Meyer and M. Scheer, Angew. Chem., Int. Ed., 2016, 55, 4340–4344 CrossRef CAS.
- (a) G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed; (b) CCDC 1998479–1998482 contain the supplementary crystallographic data for this paper.†.
- R. Lalrempuia, A. Stasch and C. Jones, Chem. Sci., 2013, 4, 4383–4388 RSC.
- Z. Wang, V. Enkelmann, F. Negri and K. Müllen, Angew. Chem., Int. Ed., 2004, 43, 1972–1975 CrossRef CAS PubMed.
- S.-i. Kawano, M. Baumgarten, D. Chercka, V. Enkelmann and K. Müllen, Chem. Commun., 2013, 49, 5058–5060 RSC.
- (a) R. E. Cowley, J. Elhaïk, N. A. Eckert, W. W. Brennessel, E. Bill and P. L. Holland, J. Am. Chem. Soc., 2008, 130, 6074–6075 CrossRef CAS; (b) S. Meyer, C. M. Orben, S. Demeshko, S. Dechert and F. Meyer, Organometallics, 2011, 30, 6692–6702 CrossRef CAS.
- P. Hao, Z. Yang, X. Ma, X. Wang, Z. Liu, H. W. Roesky, K. Sun, J. Li and M. Zhong, Dalton Trans., 2012, 41, 13520–13524 RSC.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, M. B. F. Ogliaro, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Kieth, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, in Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- I.-R. Jeon, J. G. Park, D. J. Xiao and T. D. Harris, J. Am. Chem. Soc., 2013, 135, 16845–16848 CrossRef CAS PubMed.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, all characterization data and theoretical calculation details. CCDC 1998479–1998482. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01932e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |