
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiverse protein manipulations with genetically encoded glutamic acid benzyl ester†
Xiaochen Yanga,
Hui Miaoa,
Ruotong Xiaoa,
Luyao Wangb,
Yan Zhaob,
Qifan Wu a,
Yanli Jia,
Juanjuan Dub,
Hongqiang Qinc and
Weimin Xuan*a
a,
Yanli Jia,
Juanjuan Dub,
Hongqiang Qinc and
Weimin Xuan*a
aState Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: wxuan@nankai.edu.cn
bSchool of Pharmaceutical Sciences, Tsinghua University, 30 Shuangqing Rd., Beijing, China
cCAS Key Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences (CAS), Dalian, 116023, China
First published on 17th June 2021
Abstract
Site-specific modification of proteins has significantly advanced the use of proteins in biological research and therapeutics development. Among various strategies aimed at this end, genetic code expansion (GCE) allows structurally and functionally distinct non-canonical amino acids (ncAAs) to be incorporated into specific sites of a protein. Herein, we genetically encode an esterified glutamic acid analogue (BnE) into proteins, and demonstrate that BnE can be applied in different types of site-specific protein modifications, including N-terminal pyroglutamation, caging Glu in the active site of a toxic protein, and endowing proteins with metal chelator hydroxamic acid and versatile reactive handle acyl hydrazide. Importantly, novel epigenetic mark Gln methylation is generated on histones via the derived acyl hydrazide handle. This work provides useful and unique tools to modify proteins at specific Glu or Gln residues, and complements the toolbox of GCE.
Introduction
There is an increasing demand for precisely decorated proteins in academic research and the pharmaceutical industry, so methods that enable the site-specific introduction of various functional entities into proteins are under extensive research and development.1 Typical strategies include the fusion of protein/peptide tags with catalytic activities (e.g. SNAP tags, HaloTags and SpyTags)2 or chemical reactivities (e.g. tetracysteine, aldehyde, CBT tags and π-clamps),3 enzyme-catalyzed protein modifications (e.g. sortase, lipoic acid ligase, transglutaminase and butelase 1),4 the use of elaborately designed chemical reagents to react with rare residues (e.g. Cys, Met and N-terminal residues),5 ligand-directed protein labelling,6 and protein synthesis via expressed protein ligation,7 native chemical ligation,8 or most recently KAHA ligation,9etc. These technologies have significantly advanced our capabilities in functionalizing proteins with photophysical probes2b,4a,4c and bioorthogonal handles,10 preparing proteins with post-translational modifications,11 as well as producing high-quality biologics.12Genetic code expansion (GCE) is a biotechnology whereby a non-canonical amino acid (ncAA) could be inserted into proteins through suppressing an artificially introduced stop codon or quadruplet codon with an orthogonal aminoacyl-tRNA synthetase (aaRS)/tRNA pair.13 After two decades of research and development,14 GCE is becoming one of the most powerful strategies in protein engineering, and provides unique tools to many research fields, such as bioorthogonal chemistry,15 artificial enzyme design,16 biocontainment development,17 vaccine development,18 protein/peptide-based drug discovery,19 and so on.
We are interested in further expanding the GCE toolbox to address important and yet unmet needs in protein engineering. When carefully examining the repertoire of genetically encoded ncAAs,20 it is not difficult to find out that ncAA designed as Glu or Gln derivative is very rare, thus rendering the manipulation of proteins at specific Glu or Gln residues challenging or even unlikely. Glu/Gln residues are often essential for proper protein folding, maturation, and activity, and there are increasing reports of post-translational modifications occurring on them.21 Additionally, ester, as one of the most basic functional groups, remains largely unexplored in the history of GCE, likely due to the concern of ncAA integrity in intracellular environments. With these considerations in mind, herein we would like to genetically encode a glutamic acid analogue with an esterified side-chain to enable certain protein manipulations at Glu and Gln residues. The ester side-chain is more amenable to nucleophilic attack as compared to the natural counterpart amide and carboxylate side-chains, and therefore holds great potential to realize several types of largely unexplored protein manipulations, including (1) protein N-terminal pyroglutamation by placing ncAA at the N-terminus; (2) a chemical cage of Glu; (3) site-specifically conferring hydroxamic acid (HA) on proteins; (4) generating an acyl hydrazide handle for diverse protein modifications (Scheme 1).
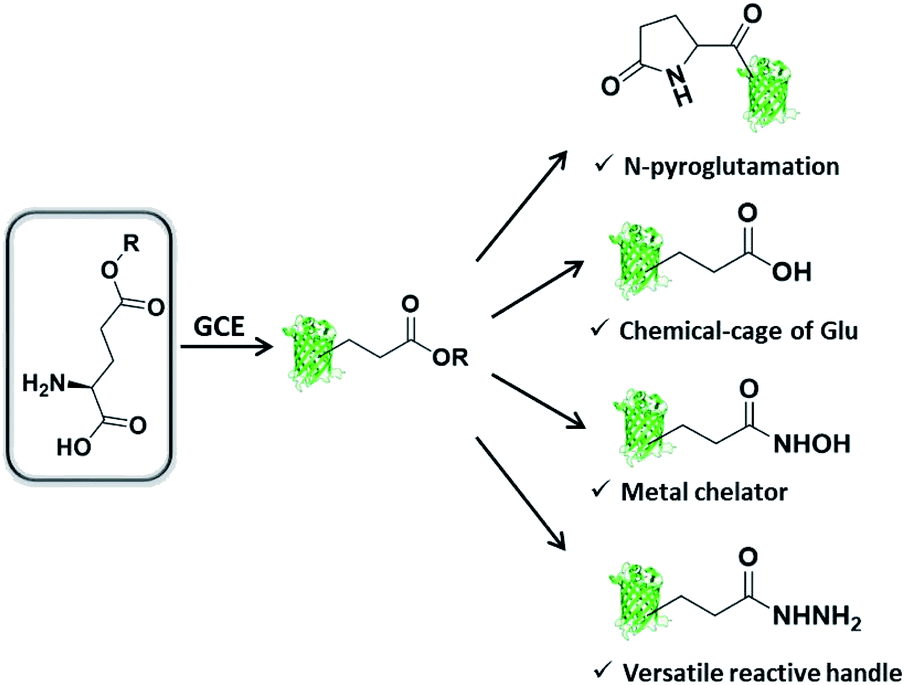 | ||
| Scheme 1 Genetic incorporation of side-chain esterified glutamic acid analogue enables multiple types of protein engineering. | ||
Results and discussion
Genetic incorporation of L-glutamic acid γ-benzyl ester (BnE)
Firstly, we chose the commercially available L-glutamic acid γ-benzyl ester as the target of investigation (BnE, Fig. 1A), as the unnatural benzyl ester is less likely to experience enzymatic hydrolysis in living cells, and empirically the moderate-sized hydrophobic side-chain is favourable for genetic incorporation. | ||
| Fig. 1 Genetic incorporation of BnE. (A) The structure of BnE. (B) SDS-PAGE analysis of sfGFP (Y151TAG) expressed in E. coli DH10B in the presence or absence of 5 mM BnE. (C) Mass spectrum of purified sfGFP-Y151BnE. The inset shows the mass spectrum before deconvolution. | ||
To encode BnE, a pyrrolysyl-tRNA synthetase/tRNA pair (PylRS/PylT) was employed based on its adaptability to recognize a large number of ncAAs and well-demonstrated orthogonality in different organisms. In detail, a Methanosarcina barkeri PylRS library with residues A267, Y271, N311, C313 and Y349 randomized to NNK (N = A, T, G or C; K = T or G),22 was subject to multiple rounds of positive and negative selection in E. coli DH10B. Gratifyingly, colonies with BnE dependent growth were obtained after the last round of positive selection, and DNA sequencing revealed three unique variants (BnERS1-3, Table S1†). A GFP-based assay revealed that all variants could enhance fluorescence intensity in the presence of BnE (Fig. S1†). BnERS1 harboring N311S, C313A and Y349F showed the highest amber suppression efficiency, and was used in the following studies (thereafter denoted as BnERS). To confirm the selective incorporation of BnE, a C-terminal His-tagged super-folder GFP variant with an amber mutation at a permissive site (sfGFP-Y151TAG) was expressed in E. coli DH10B in LB media supplemented with 5 mM BnE, and purified through a Ni-NTA column in 56 mg L−1 isolated yield. This efficiency is comparable to N-Boc-L-lysine in our study (Fig. S2†). The purified sfGFP-Y151BnE was analyzed by SDS-PAGE gel (Fig. 1B) and electrospray ionization quadrupole time of flight (ESI-QTOF) mass spectrometry (Fig. 1C). The observed mass peak (27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 653 Da) is consistent with the expected mass, thus confirming BnE incorporation. Of note, BnE at Y151 is located on the external surface of sfGFP, and no ester bond hydrolysis was observed, suggesting sufficient stability in an intracellular environment and during the protein purification processes.
653 Da) is consistent with the expected mass, thus confirming BnE incorporation. Of note, BnE at Y151 is located on the external surface of sfGFP, and no ester bond hydrolysis was observed, suggesting sufficient stability in an intracellular environment and during the protein purification processes.
We also attempted to encode BnE in mammalian cells. An EGFP variant (EGFP-Y39TAG) along with BnERS/PylT was transiently expressed in HEK293T cells, and obvious green fluorescence was observable only in the presence of BnE, indicating the BnE-dependent full-length expression of EGFP. The result was further validated by western blot analysis (Fig. S3†).
BnE enabling the recombinant expression of proteins with N-terminal pyroglutamation
N-terminal pyroglutamate (pGlu) frequently occurs in proteins and peptides, such as ribonuclease,23 amyloid-β24 and antibodies,25 and as reported pGlu formation can reduce the susceptibility of proteins to aminopeptidase digestion, and affect protein's structural integrity.26 pGlu is derived from N-terminal glutaminyl and glutamyl precursors, usually catalyzed by glutaminyl cyclase, and in certain cases through a very sluggish intramolecular cyclization.25bWe reasoned that BnE at the N-terminus of a protein should accelerate the spontaneous intramolecular cyclization to form pGlu. The initial sfGFP construct (sfGFP-S2BnE) revealed failed removal of the starting Met in E. coli DH10B (Fig. S4†), and then a BnE-exposing strategy was designed. Specifically, the protein of interest (POI) with N-terminal BnE was fused to the C-terminus of a small ubiquitin-like modifier protein (SUMO), which can be removed by SUMO-specific protease (Ulp1), thus exposing BnE (Fig. 2A). Experimentally, SUMO-sfGFP fusion containing BnE right after the SUMO tag was expressed in E. coli DH10B, and purified through the C-terminal His-Tag in 10 mg L−1 isolated yield. This protein was treated with recombinantly expressed Ulp1 in pH 8 buffer at 30 °C for 2.5 h, and analyzed by SDS-PAGE and ESI-QTOF mass spectrometry (Fig. 2B–D). The results show that the SUMO tag was completely removed, and mass peaks corresponding to BnE (27729 Da) and pGlu (27621 Da) respectively at the N-terminus of sfGFP were observed (Fig. 2D). In contrast, SUMO-sfGFP-S2E was expressed and treated with Ulp1, and no pGlu formation was observed in mass spectrometry (Fig. S5†). The result indicates that BnE at the N-terminus of a protein undergoes an obviously accelerated cyclization reaction.
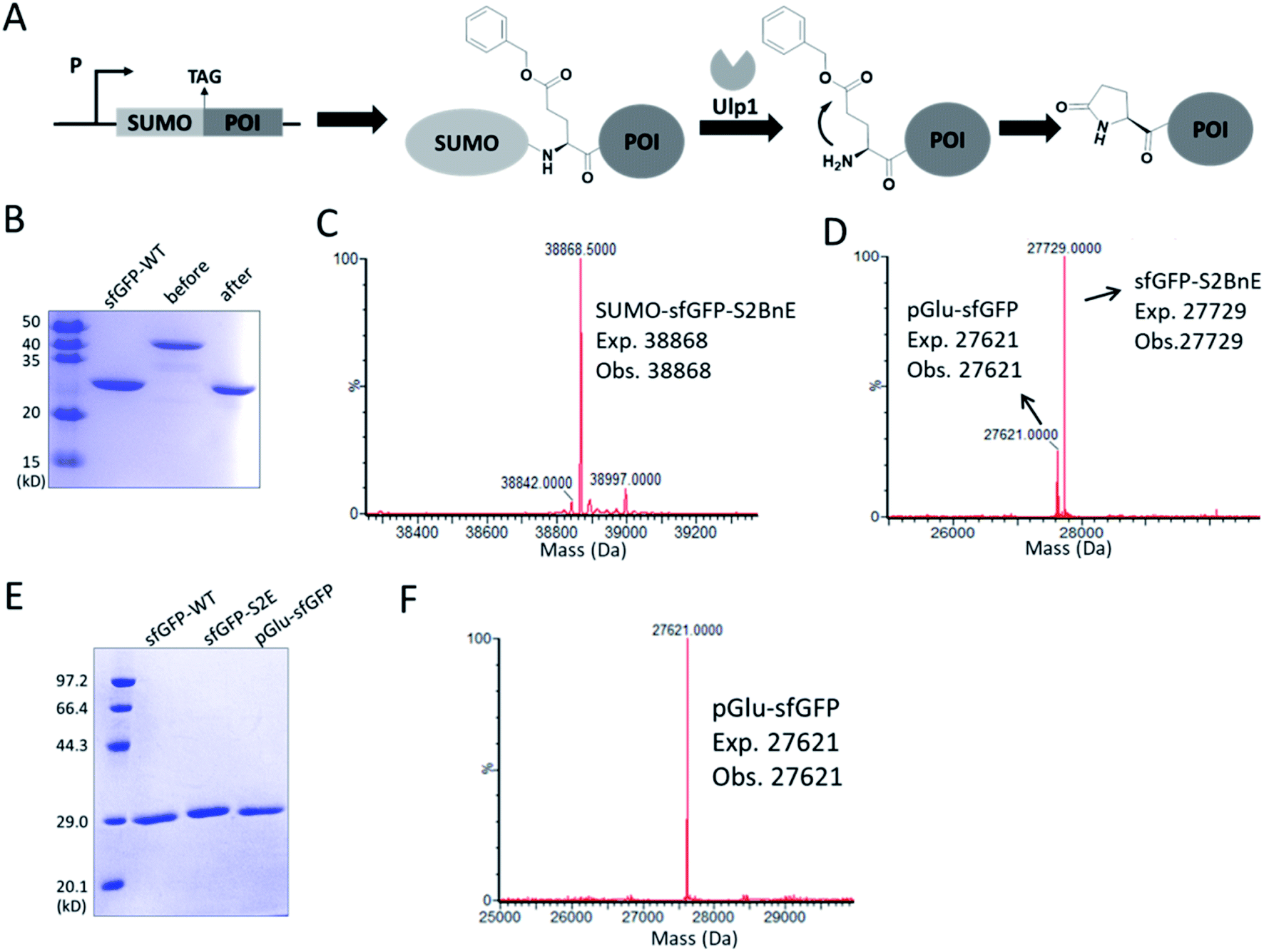 | ||
| Fig. 2 Generating pGlu at the N-terminus of proteins. (A) The strategy used to generate pGlu. POI: protein of interest. (B) SDS-PAGE analysis of SUMO-sfGFP-S2BnE before (lane 2) and after (lane 3) Ulp1 treatment. The mass spectra of purified SUMO-sfGFP-S2BnE before (C) and after (D) Ulp1 treatment. (E) SDS-PAGE analysis of purified pGlu-sfGFP (lane 3) during co-expression of Ulp1 and sfGFP-S2E (lane 2). (F) Mass spectrum of purified pGlu-sfGFP during co-expression of Ulp1. Wild-type sfGFP was used as a control in SDS-PAGE analysis. | ||
To complete the pGlu formation, Ulp1 was co-expressed with the SUMO-BnE-POI fusion, as in situ generated N-terminal BnE would allow instant pGlu formation. Specifically, Ulp1 was co-expressed along with BnERS/PylT and SUMO-sfGFP-S2BnE in a standard expression process, and the affinity purification by the C-terminal His-Tag afforded 5 mg L−1 of the recombinant protein. SDS-PAGE revealed a complete removal of the SUMO tag, and mass spectrometry indicated complete pGlu formation (Fig. 2E and F). As a control, co-expression of SUMO-sfGFP-S2Q with Ulp1 hardly produced pGlu-sfGFP based on mass spectrometry analysis of the purified protein (Fig. S6†). To demonstrate the generality of this method, thioredoxin and 14-3-3γ with an N-terminal pGlu were facilely prepared and confirmed by SDS-PAGE and mass spectrometry (Fig. S7†). The heavy chain of Herceptin Fab has an N-terminal Glu residue, and by the method here, a pGlu was quantitatively generated at this position (Fig. S8†).
Recently, Ball et al. demonstrated that pGlu-His dipeptide at the N-terminus of a protein could serve as an efficient reactive handle for Chan–Lam coupling.27 By the method developed in this work, the pGlu-His tag was installed at the N-terminus of sfGFP, and successfully reacted with an arylboronic acid reagent; without the adjacent His, no labelling product was observed (Fig. S9†). In short, this work provides an alternative method to equipping proteins with N-terminal pGlu,28 possibly promoting the relevant biological research and applications.
BnE can serve as a chemical cage of Glu
The ester bond is susceptible to alkaline hydrolysis (Fig. 3A). Indeed, after incubation in a pH 11 Tris buffer solution for 7 h, sfGFP-Y151BnE was completely converted to sfGFP-Y151E based on SDS-PAGE and ESI-QTOF analysis (Fig. 3B and C). Thus, BnE could be regarded as a chemical cage of Glu.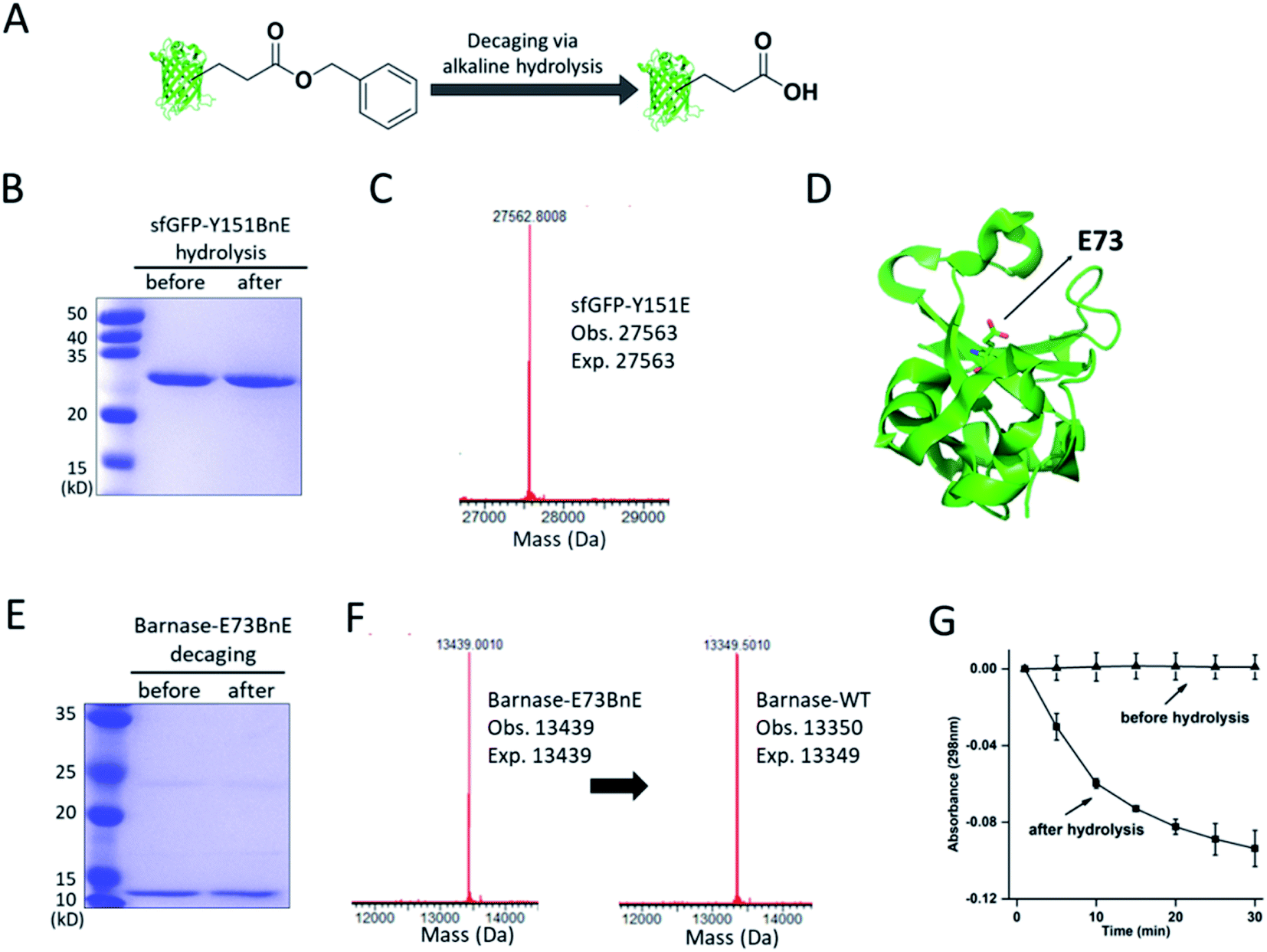 | ||
| Fig. 3 BnE is chemically caged Glu. (A) A general scheme of BnE alkaline hydrolysis on proteins. (B) SDS-PAGE analysis of sfGFP-Y151BnE before and after alkaline hydrolysis. (C) Mass spectrum of sfGFP-Y151BnE after alkaline hydrolysis. The mass change (loss of 90 Da) suggests the conversion of BnE to Glu. (D) The crystal structure of barnase (PDB: 1BRS), E73 was shown in stick. (E) SDS-PAGE analysis of barnase-E73BnE before and after alkaline hydrolysis. (F) Mass spectrum of barnase-E73BnE before and after alkaline hydrolysis. (G) A yeast RNA-based assay confirmed the formation of active barnase, and barnase-E73BnE is inactive. Error bars represent s.d. of three independent biological replicates. Site-specifically generating acyl hydrazide and hydroxamic acid on proteins with BnE. | ||
Barnase is a secreted ribonuclease from Bacillus amyloliquefaciens, and its recombinant expression in a bacterial host requires the presence of cytoplasmic inhibitor barstar, with which a complex can be formed (KD = ∼10−14 M).29 To obtain an active barnase protein, additional steps including denaturation and refolding are thus inevitable to get rid of barstar, and to some extent impede the application of barnase.30 There is an essential Glu (E73, Fig. 3D) at the active site of barnase, and we wondered if E73 can be caged with BnE to temporarily abate its toxicity to the expression host. To prove this concept, barnase-E73BnE under the control of a T5 promoter was expressed in E. coli DH10B in the presence of 5 mM BnE at 37 °C. The expression after induction hardly affected E. coli growth (Fig. S10†), and the affinity purification through the C-terminal His-Tag afforded barnase-E73BnE at a yield of 2.5 mg L−1. Then the purified protein was incubated in pH 11 Tris buffer at 37 °C for 7 h, and analyzed by SDS-PAGE (Fig. 3E) and ESI-QTOF. Mass spectra indicated a complete conversion of barnase-E73BnE to wild type barnase (Fig. 3F). Furthermore, a yeast RNA-based assay confirmed the enzymatic activity of generated barnase (Fig. 3G), and the calculated enzymatic activity (4 × 106 units per mg) is consistent with the reported value of wild type barnase.30 Herein, the benzyl cage can be quantitatively removed under relatively mild conditions, indicating potential for future applications.
Site-specifically generating acyl hydrazide and hydroxamic acid on proteins with BnE
Esters can react with hydrazine and hydroxylamine to form acyl hydrazide and HA respectively. HA has a high chelating power to many metal ions, and serves as reactive warheads in a number of bioactive compounds, such as natural iron chelator siderophores and inhibitors of Zn2+ dependent enzymes (e.g. matrix metalloproteinases and HDACs).31 Also, acyl hydrazide has widespread applications in bioorthogonal conjugation reactions32 and chemical protein synthesis.33Considering the attractive features, we attempted to equip proteins with acyl hydrazide and HA through derivatizing the encoded BnE (Fig. 4A). To prove this method, the purified sfGFP-Y151BnE was incubated with an excessive amount of hydrazine or hydroxylamine (4%, v/v) in pH 7.4 PBS solutions at 37 °C for 1 h, and then analyzed by SDS-PAGE (Fig. 4B) and ESI-QTOF. The observed mass peaks for both reactions are in good agreement with the expected values (Fig. 4C). As a control, no reaction occurred when wild type sfGFP was treated with hydrazine or hydroxylamine. This confirmed that BnE on a protein could be efficiently converted into acyl hydrazide or HA.
 | ||
| Fig. 4 BnE allowing the site-specific installation of acyl hydrazide and hydroxamic acid on proteins. (A) A general scheme for the reaction between POI with encoded BnE and hydrazine or hydroxylamine. (B) SDS-PAGE analysis of sfGFP-Y151BnE after reaction with hydrazine or hydroxylamine. (C) ESI-QTOF analysis of sfGFP-Y151BnE after reaction with hydrazine or hydroxylamine. | ||
Constructing HA-based artificial metal-binding centers on proteins
Metal-chelating ability is of great interest in protein engineering, and herein the site-specifically generated HA side-chains on a protein would be a unique tool in metalloprotein design. Natural siderophores usually employ three HA moieties to realize the extremely high binding affinities for Fe3+, thus making us wonder if multiple HA moieties could be constructed on a single protein. Experimentally, three closely spaced residues on sfGFP including K45, D210 and N212, were chosen to construct a tridentate metal-binding center (Fig. 5A). As a control, single and double HA installations were also included (sfGFP-N212, and sfGFP-D210-N212). To enhance amber suppression, sfGFP variants with two and three BnE were expressed in a RF1 knockout strain (C321.deltaA), and purified at a yield of 47 and 20 mg L−1 respectively. sfGFP-N212BnE was purified in E. coli BL21(DE3) at a yield of 53 mg L−1. All sfGFP variants were treated with hydroxylamine, and then subject to SDS-PAGE and ESI-QTOF analysis (Fig. 5B, C and S11†). Mass spectrometry revealed nearly quantitative conversion of BnE to HA in all cases. Metal cations can affect the fluorescence intensity of GFP,34 so the fluorescence intensity of these sfGFP variants was investigated in the presence of Cu2+ and Fe3+ respectively. Relative to the wild type, single or double HA installation moderately increased the sensitivity of sfGFP to Cu2+, while triple HA installation displayed much enhanced sensitivity, implying a synergistic effect of triple HA moieties (Fig. 5D). A similar phenomenon was also observed for Fe3+ (Fig. S12†). This experiment indicated the feasibility of constructing HA-based artificial metal binding centers on proteins by encoding BnE.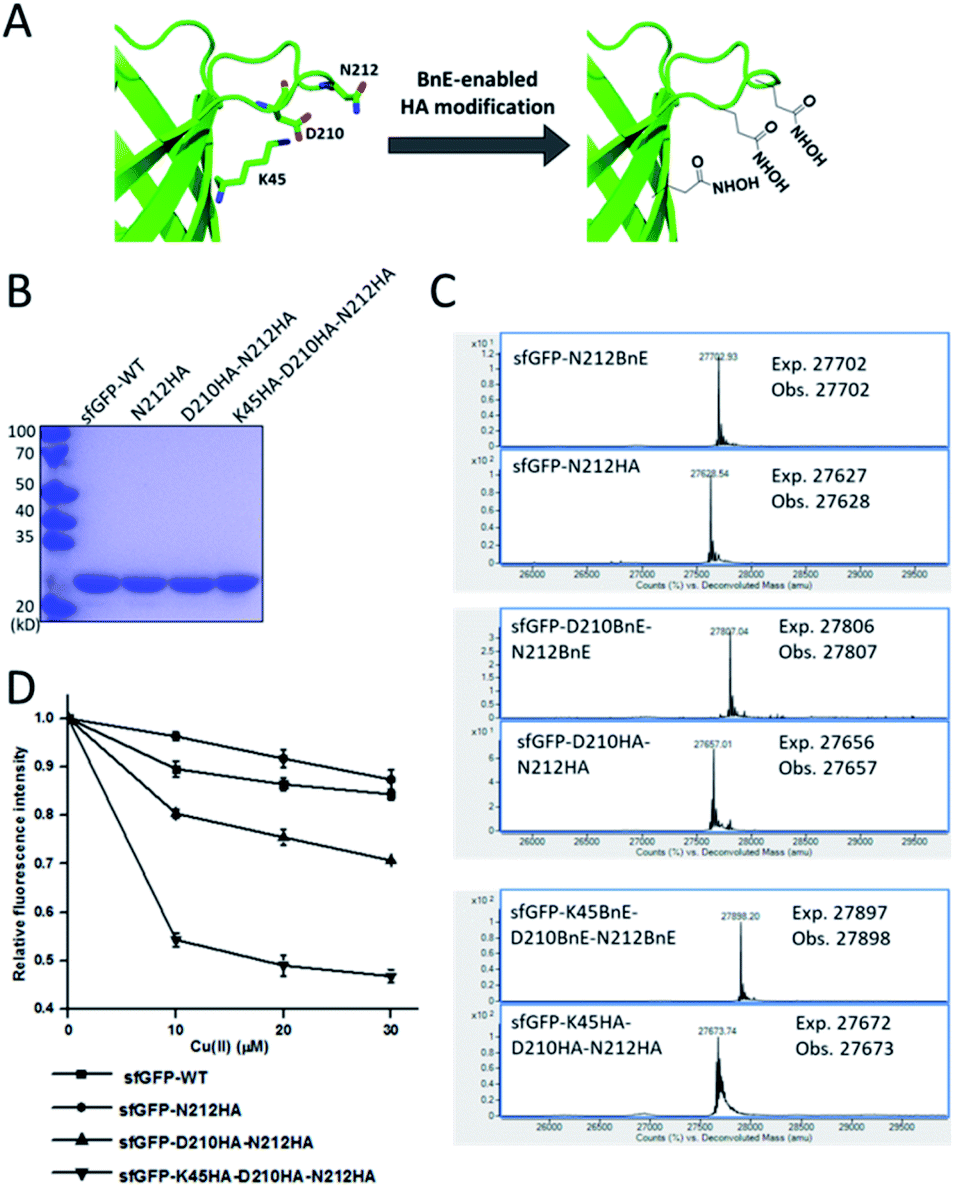 | ||
| Fig. 5 BnE-derived hydroxamic acid confers protein with metal-binding ability. (A) A scheme of generating tridentate metal-binding sites on sfGFP (PDB: 2B3P). (B) SDS-PAGE analysis of sfGFP variants with one, two and three HA groups. (C) ESI-QTOF analysis of sfGFP variants with one, two and three BnE groups before and after being treated with hydroxylamine. (D) The fluorescence change of sfGFP variants after incubation with different concentrations of Cu2+. Error bars represent s.d. of three independent biological replicates. | ||
BnE-derived acyl hydrazide on proteins is a versatile reactive handle
Lastly, we would like to explore the potential of BnE-derived acyl hydrazide in protein engineering. Liu et al. developed a chemoselective reaction between N-terminal Cys and C-terminal hydrazide of two different peptides for chemical protein synthesis, and demonstrated its application in many cases.8b,33a This inspired us to know whether the BnE-derived acyl hydrazide can be used in site-specific protein labelling (Fig. 6A). Specifically, sfGFP harboring an acyl hydrazide at Y151 was treated with 1 mM NaNO2 in 20 mM pH 3 phosphate buffer for 20 min, and then incubated with 2 mM Fl-cys at pH 7 for 4 h. The labelling product was subject to SDS-PAGE analysis, and a bright band was observed by fluorescence (Fig. 6B). Furthermore, ESI-QTOF analysis confirmed the formation of the desired product; the observed mass (27996 Da) is in good agreement with the expected mass (27995 Da), and only a small amount of hydrolyzed product was observed (Fig. 6C). This experiment verified the usefulness of BnE-derived acyl hydrazide in site-specific protein labelling.
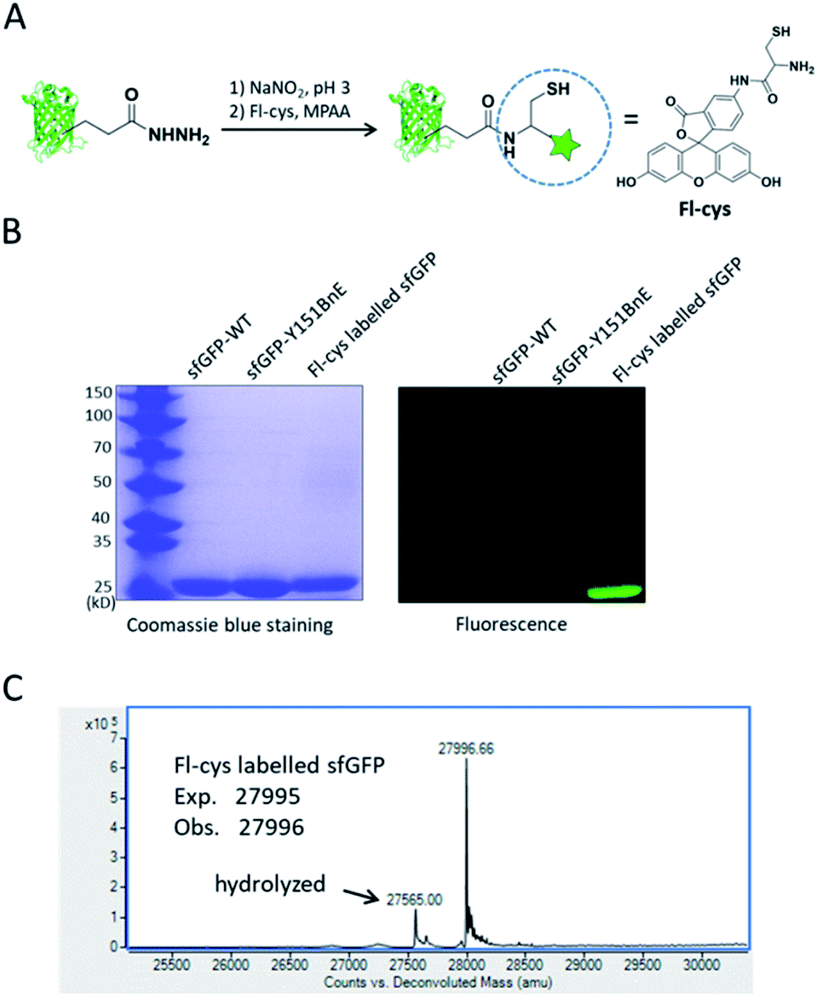 | ||
| Fig. 6 Site-specific protein labelling with BnE-derived acy hydrazide. (A) A scheme for the reaction between BnE-derived acyl hydrazide and Fl-cys. (B) SDS-PAGE analysis of the labelling product, and the gel was visualized by both coomassie blue staining and fluorescence. (C) ESI-QTOF analysis of the labelling product. The arrow indicates the hydrolyzed product (sfGFP-Y151E). | ||
A recent study revealed that Gln methylation on histones is a novel epigenetic mark and RNA-polymerase-I-dedicated modification.21b Ambitiously, we attempted to use BnE-derived acyl hydrazide to prepare histone variants with site-specific Gln methylation. To start with, a H3 variant with BnE at Q56 (H3-Q56BnE) was expressed in E. coli, and verified by mass spectrometry (Fig. 7B). Initial efforts in adjusting Liu's method by using methylamine instead of a Cys derivative did not afford the desired product, possibly due to the short half-life of acyl azide in aqueous solutions. Recently, Dawson et al. reported the use of Knorr pyrazole as the thioester surrogate in native chemical ligation.33b Derived from acyl hydrazide, the acyl pyrazole exhibits weaker electrophilicity, and hopefully would react with methylamine in aqueous solutions (Fig. 7A). BnE at H3-Q56 was converted into the corresponding acyl hydrazide, then reacted with acetyl acetone (acac) in a pH 3 guanidine solution at 37 °C for 1.5 h. ESI-QTOF analysis revealed an efficient formation of the acyl pyrazole intermediate (Fig. 7B). Methylamine was added into the acyl pyrazole solution with subsequently pH adjusted to 9, and then the mixture was incubated at 37 °C for 5 h. The final product was analyzed by SDS-PAGE (Fig. 7C) and ESI-QTOF. The mass spectra confirmed the formation of H3-Q56Qme, and only a small amount of hydrolyzed product was observed (Fig. 7B). To prove the generality of this method, Gln methylation was also generated at Q5 and Q94 on H3 with similar efficiency (Fig. S13 and S14†). Finally, enzymatic digestion and collision-induced fragmentation were applied to H3-Q56Qme and H3-Q94Qme, and confirmed the site-specific Gln methylation (Fig. 7D, and Fig. S14†). Notably, this work provided a useful method to construct amide bonds on protein side-chains, and may offer solutions to the generation of other PTMs on proteins, such as N-glycosylation.
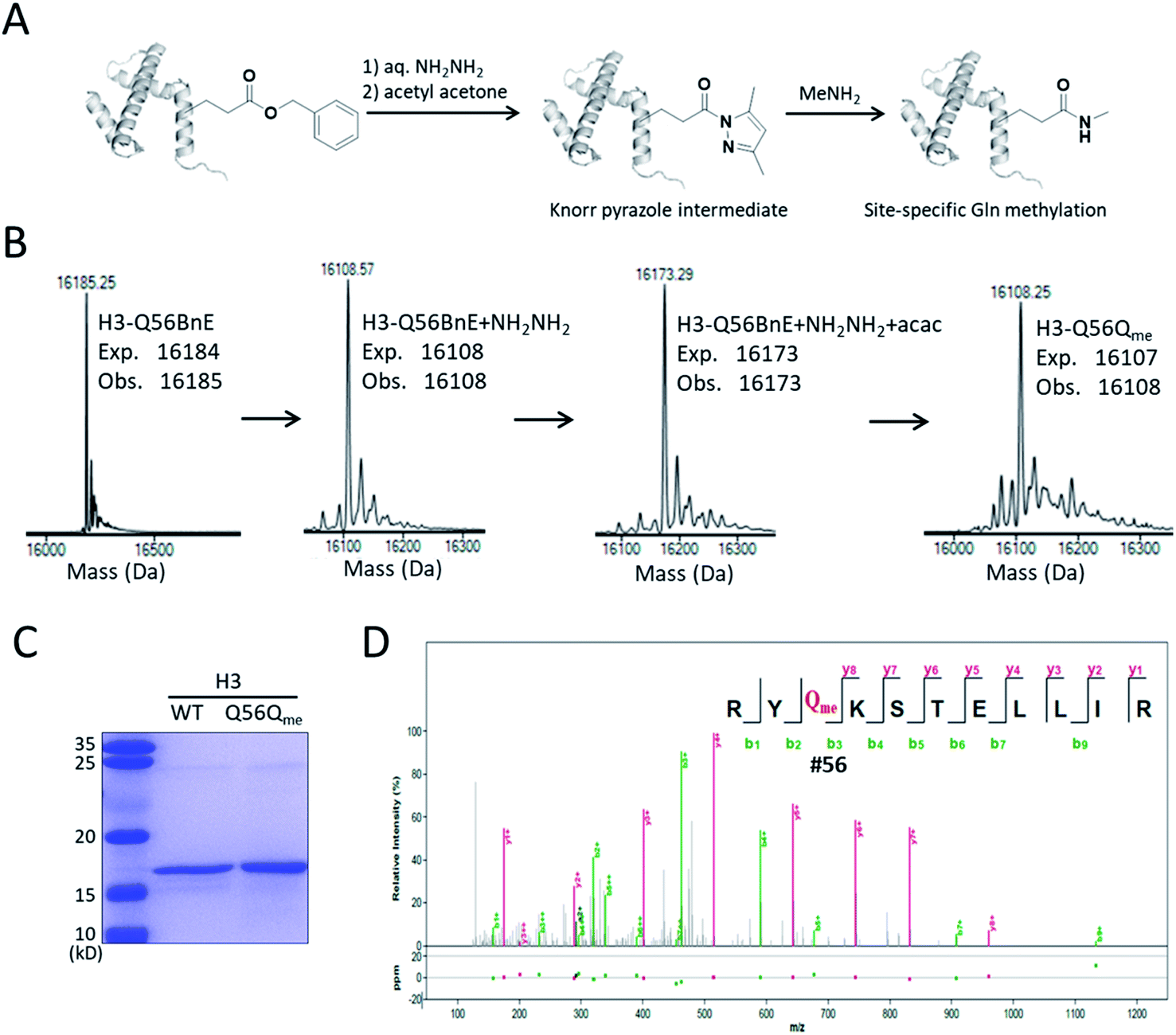 | ||
| Fig. 7 Site-specifically generating methylated Gln on H3. (A) A general procedure for generating methylated Gln on histone. (B) ESI-QTOF analysis of H3-Q56 variants in the process of preparing H3-Q56Qme. (C) SDS-PAGE analysis of H3 WT and H3-Q56Qme. (D) The MS/MS analysis of the obtained H3-Q56Qme. | ||
Conclusions
In summary, a side-chain esterified glutamic acid analogue (BnE) has been genetically encoded into proteins in bacteria and mammalian cells. With this tool, we demonstrated the generation of N-terminal pGlu on recombinant proteins expressed in E. coli, and proved that BnE can serve as a chemical cage of Glu, offering a method for the expression of cytotoxic barnase. Furthermore, we showed that BnE on proteins can be facilely converted into HA, a useful metal chelator, and into acyl hydrazide, a handle that allows versatile protein modifications, including histone Gln methylation. We believe this work provides unique tools to manipulate proteins, and would be useful in a number of fields.Author contributions
University of Nankai: Xiaochen Yang performed most of the protein expression, modification and characterization. Hui Miao performed the ncAA selection experiment. Ruotong Xiao and Yanli Ji performed the molecular cloning experiments and the fluorescence imaging experiments. Qifan Wu developed the Knoor pyrazole strategy to prepare Histones with site-specific Gln methylation. Weimin Xuan conceived of the project and wrote the manuscript. Tsinghua University: Luyao Wang and Yan Zhao discussed the project and participated in the barnase experiments. Juanjuan Du supervised Luyao Wang and Yanzhao, and partially conceived of the project. Dalian Institute of Chemical Physics, Chinese Academy of Sciences: Hongqiang Qin carried out the MS/MS analysis of the histone variants.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Prof. Peter G. Schultz for the plasmid gift. This work was supported by grants from the National Natural Science Foundation of China (21807061), the Natural Science Foundation of Tianjin (18JCYBJC41600), the Youth Innovation Promotion Association of CAS (2018212) and the start-up funding from Nankai University. This work is dedicated to the 100th anniversary of Chemistry at Nankai University.Notes and references
- (a) N. Krall, F. P. da Cruz, O. Boutureira and G. J. L. Bernardes, Nat. Chem., 2016, 8, 102–112 CrossRef PubMed; (b) P. G. Isenegger and B. G. Davis, J. Am. Chem. Soc., 2019, 141, 8005–8013 CAS; (c) K. C. Nicolaou and S. Rigol, Angew. Chem., Int. Ed., 2019, 58, 11206–11241 CrossRef CAS; (d) M. S. Kariolis, S. Kapur and J. R. Cochran, Curr. Opin. Biotechnol., 2013, 24, 1072–1077 CrossRef CAS.
- (a) G. V. Los, L. P. Encell, M. G. McDougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohane, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS PubMed; (b) A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS PubMed; (c) S. C. Reddington and M. Howarth, Curr. Opin. Chem. Biol., 2015, 29, 94–99 CrossRef CAS PubMed.
- (a) C. Zhang, M. Welborn, T. Y. Zhu, N. J. Yang, M. S. Santos, T. Van Voorhis and B. L. Pentelute, Nat. Chem., 2016, 8, 120–128 CrossRef CAS; (b) C. P. Ramil, P. An, Z. Yu and Q. Lin, J. Am. Chem. Soc., 2016, 138, 5499–5502 CrossRef CAS PubMed; (c) I. S. Carrico, B. L. Carlson and C. R. Bertozzi, Nat. Chem. Biol., 2007, 3, 321–322 CrossRef CAS PubMed.
- (a) C. Uttamapinant, K. A. White, H. Baruah, S. Thompson, M. Fernandez-Suarez, S. Puthenveetil and A. Y. Ting, Proc. Natl. Acad. Sci., 2010, 107, 10914–10919 CrossRef CAS PubMed; (b) T. I. Chio, B. R. Demestichas, B. M. Brems, S. L. Bane and L. N. Tumey, Angew. Chem., Int. Ed., 2020, 59, 13814–13820 CrossRef CAS PubMed; (c) X. B. Bi, J. Yin, G. K. T. Nguyen, C. Rao, N. B. A. Halim, X. Hemu, J. P. Tam and C. F. Liu, Angew. Chem., Int. Ed., 2017, 56, 7822–7825 CrossRef CAS PubMed; (d) V. Haridas, S. Sadanandan and N. U. Dheepthi, ChemBioChem, 2014, 15, 1857–1867 CrossRef CAS PubMed.
- (a) C. B. Rosen and M. B. Francis, Nat. Chem. Biol., 2017, 13, 697–705 CrossRef CAS; (b) S. B. Gunnoo and A. Madder, ChemBioChem, 2016, 17, 529–553 CrossRef CAS; (c) S. Lin, X. Yang, S. Jia, A. M. Weeks, M. Hornsby, P. S. Lee, R. V. Nichiporuk, A. T. Iavarone, J. A. Wells, F. D. Toste and C. J. Chang, Science, 2017, 355, 597–602 CrossRef CAS; (d) O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS.
- K. Matsuo, Y. Nishikawa, M. Masuda and I. Hamachi, Angew. Chem., Int. Ed., 2018, 57, 659–662 CrossRef CAS PubMed.
- N. H. Shah and T. W. Muir, Chem. Sci., 2014, 5, 446–461 RSC.
- (a) V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoet, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS; (b) J.-S. Zheng, S. Tang, Y.-C. Huang and L. Liu, Acc. Chem. Res., 2013, 46, 2475–2484 CrossRef CAS PubMed.
- (a) C. E. Murar, M. Ninomiya, S. Shimura, U. Karakus, O. Boyman and J. W. Bode, Angew. Chem., Int. Ed., 2020, 59, 8425–8429 CrossRef CAS PubMed; (b) S. Baldauf, D. Schauenburg and J. W. Bode, Angew. Chem., Int. Ed., 2019, 58, 12599–12603 CrossRef CAS PubMed.
- Y. Tian and Q. Lin, ACS Chem. Biol., 2019, 14, 2489–2496 CrossRef CAS.
- Y. David and T. W. Muir, J. Am. Chem. Soc., 2017, 139, 9090–9096 CrossRef CAS PubMed.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaia, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS.
- (a) D. D. Young and P. G. Schultz, ACS Chem. Biol., 2018, 13, 854–870 CrossRef CAS PubMed; (b) J. W. Chin, Nature, 2017, 550, 53–60 CrossRef CAS.
- L. Wang, A. Brock, B. Herberich and P. G. Schultz, Science, 2001, 292, 498–500 CrossRef CAS PubMed.
- (a) K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS; (b) J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129–137 CrossRef CAS PubMed.
- I. Drienovska and G. Roelfes, Nat. Catal., 2020, 3, 193–202 CrossRef CAS.
- A. Ravikumar and C. C. Liu, ChemBioChem, 2015, 16, 1149–1151 CrossRef CAS PubMed.
- J. A. Fok and C. Mayer, ChemBioChem, 2020, 21, 3291–3300 CrossRef CAS PubMed.
- (a) Q. K. Li, Q. Chen, P. C. Klauser, M. Y. Li, F. Zheng, N. X. Wang, X. Y. Li, Q. B. Zhang, X. M. Fu, Q. Wang, Y. Xu and L. Wang, Cell, 2020, 182, 85–97 CrossRef CAS; (b) X. S. S. Wang, P. H. C. Chen, J. T. Hampton, J. M. Tharp, C. A. Reed, S. K. Das, D. S. Wang, H. S. Hayatshahi, Y. Shen, J. Liu and W. R. Liu, Angew. Chem., Int. Ed., 2019, 58, 15904–15909 CrossRef CAS PubMed; (c) M. C. Kang, Y. C. Lu, S. G. Chen and F. Tian, Curr. Opin. Chem. Biol., 2018, 46, 123–129 CrossRef CAS PubMed.
- (a) J. C. Anderson, N. Wu, S. W. Santoro, V. Lakshman, D. S. King and P. G. Schultz, Proc. Natl. Acad. Sci., 2004, 101, 7566–7571 CrossRef CAS; (b) H. Xiao and P. G. Schultz, Cold Spring Harbor Perspect. Biol., 2016, 8, a023945 CrossRef PubMed; (c) A. Dumas, L. Lercher, C. D. Spicer and B. G. Davis, Chem. Sci., 2015, 6, 50–69 RSC.
- (a) B. Pan, N. Kamo, M. Shimogawa, Y. Huang, A. Kashina, E. Rhoades and E. J. Petersson, J. Am. Chem. Soc., 2020, 142, 21786–21798 CrossRef CAS; (b) P. Tessarz, H. Santos-Rosa, S. Robson, K. B. Sylvestersen, C. J. Nelson, M. L. Nielsen and T. Kouzarides, Nature, 2014, 505, 564–568 CrossRef CAS; (c) L. A. Farrelly, R. E. Thompson, S. Zhao, A. E. Lepack, Y. Lyu, N. V. Bhanu, B. Zhang, Y.-H. E. Loh, A. Ramakrishnan, K. C. Vadodaria, K. J. Heard, G. Erikson, T. Nakadai, R. M. Bastle, B. J. Lukasak, H. Zebroski, III, N. Alenina, M. Bader, O. Berton, R. G. Roeder, H. Molina, F. H. Gage, L. Shen, B. A. Garcia, H. Li, T. W. Muir and I. Maze, Nature, 2019, 567, 535–539 CrossRef CAS PubMed.
- W. Xuan, D. Collins, M. Koh, S. Shao, A. Yao, H. Xiao, P. Garner and P. G. Schultz, ACS Chem. Biol., 2018, 13, 578–581 CrossRef CAS PubMed.
- Y. D. Liao, S. C. Wang, Y. J. Leu, C. F. Wang, S. T. Chang, Y. T. Hong, Y. R. Pan and C. P. Chen, Nucleic Acids Res., 2003, 31, 5247–5255 CrossRef CAS PubMed.
- D. Schlenzig, H. Cynis, M. Hartlage-Ruebsamen, U. Zeitschel, K. Menge, A. Fothe, D. Ramsbeck, C. Spahn, M. Wermann, S. Rossner, M. Buchholz, S. Schilling and H.-U. Demuth, J. Alzheimer's Dis., 2018, 66, 359–375 CAS.
- (a) A. Hinterholzer, V. Stanojlovic, C. Cabrele and M. Schubert, Anal. Chem., 2019, 91, 14299–14305 CrossRef CAS PubMed; (b) D. Chelius, K. Jing, A. Lueras, D. S. Rehder, T. M. Dillon, A. Vizel, R. S. Rajan, T. S. Li, M. J. Treuheit and P. V. Bondarenko, Anal. Chem., 2006, 78, 2370–2376 CrossRef CAS PubMed.
- K. F. Huang, Y. L. Liu, W. J. Cheng, T. P. Ko and A. H. J. Wang, Proc. Natl. Acad. Sci., 2005, 102, 13117–13122 CrossRef CAS PubMed.
- K. Hanaya, M. K. Miller and Z. T. Ball, Org. Lett., 2019, 21, 2445–2448 CrossRef CAS PubMed.
- Y.-P. Shih, C.-C. Chou, Y.-L. Chen, K.-F. Huang and A. H. J. Wang, PLoS One, 2014, 9, e94812 CrossRef PubMed.
- S. M. Deyev, R. Waibel, E. N. Lebedenko, A. P. Schubiger and A. Pluckthun, Nat. Biotechnol., 2003, 21, 1486–1492 CrossRef CAS PubMed.
- E. Edelweiss, T. G. Balandin, J. L. Ivanova, G. V. Lutsenko, O. G. Leonova, V. I. Popenko, A. M. Sapozhnikov and S. M. Deyev, PLoS One, 2008, 3, e2434 CrossRef PubMed.
- (a) D. Al Shaer, O. Al Musaimi, B. G. de la Torre and F. Albericio, Eur. J. Med. Chem., 2020, 208 Search PubMed; (b) C. J. Marmion, D. Griffith and K. B. Nolan, Eur. J. Inorg. Chem., 2004, 3003–3016 CrossRef CAS.
- D. K. Kolmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef CAS PubMed.
- (a) G.-M. Fang, Y.-M. Li, F. Shen, Y.-C. Huang, J.-B. Li, Y. Lin, H.-K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 7645–7649 CrossRef CAS PubMed; (b) D. T. Flood, J. C. J. Hintzen, M. J. Bird, P. A. Cistrone, J. S. Chen and P. E. Dawson, Angew. Chem., Int. Ed., 2018, 57, 11634–11639 CrossRef CAS PubMed.
- N. Ayyadurai, N. S. Prabhu, K. Deepankumar, S.-G. Lee, H.-H. Jeong, C.-S. Lee and H. Yun, Angew. Chem., Int. Ed., 2011, 50, 6534–6537 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01882e |
| This journal is © The Royal Society of Chemistry 2021 |