
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRigid, biconical hydrogen-bonded dimers that strongly encapsulate cationic guests in solution and the solid state†
Jordan N.
Smith
 ab,
Courtney
Ennis
a and
Nigel T.
Lucas
*ab
ab,
Courtney
Ennis
a and
Nigel T.
Lucas
*ab
aDepartment of Chemistry, University of Otago, Union Place, Dunedin, New Zealand. E-mail: nigel.lucas@otago.ac.nz
bMacDiarmid Institute for Advanced Materials and Nanotechnology, New Zealand
First published on 11th August 2021
Abstract
The octol of a new rigid, tetraarylene-bridged cavitand was investigated for self-assembly behaviour in solution. 1H and DOSY NMR spectroscopic experiments show that the cavitand readily dimerizes through an unusual seam of interdigitated hydrogen-bonds that is resistant to disruption by polar co-solvents. The well-defined cavity encapsulates small cationic guests, but not their neutral counterparts, restricting the conformation of sequestered tetraethylammonium in solution and the solid state.
Introduction
Confined spaces within self-assembled capsules are distinct from the bulk solvent, sequestering complementary guests1 or catalysing chemical reactions.2–4 The volume and morphology of the cavity depends upon the size and shape of the monomers: Rebek's highly-curved arcs homo-dimerize into the spherical tennis balls5 and softballs6 (Fig. 1a), whereas the shallow and flexible bowls resorcin[4]arenes and pyrogallol[4]arenes form hexamers7–14 or dimers15–22 (Fig. 1b) depending on the solvent and added guests. While hydrogen-bonded assemblies of other molecular bowls are known (trioximes23 tribenzo-triquinacenes,24 calix[4]arenes,25 cyclotriveratrylenes26 and cyclotricatechylenes27), the majority of reported capsules are built upon the readily available resorcin[4]arene scaffold, the intrinsic concave surface and an exoannular array of hydroxyl groups providing the elements for capsular self-assembly. Importantly, the solubility of the macrocycle in polar and non-polar solvents is modulated through the length of the alkyl “feet”.28 The upper-rim of resorcin[4]arenes is readily functionalized, with additional hydrogen-bond donors and acceptors increasing the cavity size and/or strengthening the interaction.29–33 For example, Rebek's diimide34 and de Mendoza's benzimidazolone35 cavitands interact through a seam of 16 bifurcated N–H⋯O bonds to generate large, cylindrical dimers around complementary guests (Fig. 1c).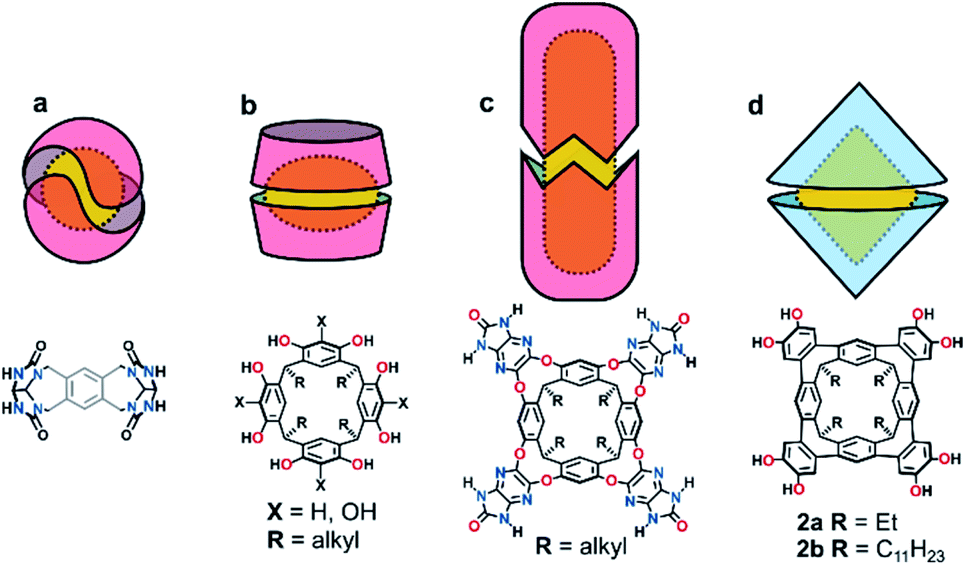 | ||
| Fig. 1 Cartoon representations of hydrogen-bonded homo-dimeric capsules and chemical diagrams of their monomer units; (a) Rebek's tennis and softballs, (b) resorcin[4]arene dimers, (c) Rebek and de Medoza's cylindrical capsules and (d) the biconical capsules presented in this work. | ||
We recently reported the synthesis of the novel resorcin[4]arene-derived octamethoxy tetraarylene-bridged cavitand 1a (Scheme 1), with an enforced, conical cavity that is not subject to the conformational fluxionality experienced by many resorcin[4]arene bowls.36 As part of further studies into this new class of cavitand, we were interested as to whether its octol, 2a, would self-assemble into multicomponent aggregates through the four catechol units at the upper-rim (Fig. 1d). The unusual rigidity of the cavitand, combined with the preorganized and inflexible array of exoannular hydroxyl groups, was predicted to give hydrogen-bonded constructs of distinct geometry and high stability. Herein we report the findings of this study, in which 2a and its undecyl-footed analogue 2b homo-dimerize through a remarkable circular seam of hydrogen-bonds to give a rigid, biconical capsule with a well-defined cavity (Fig. 1d).
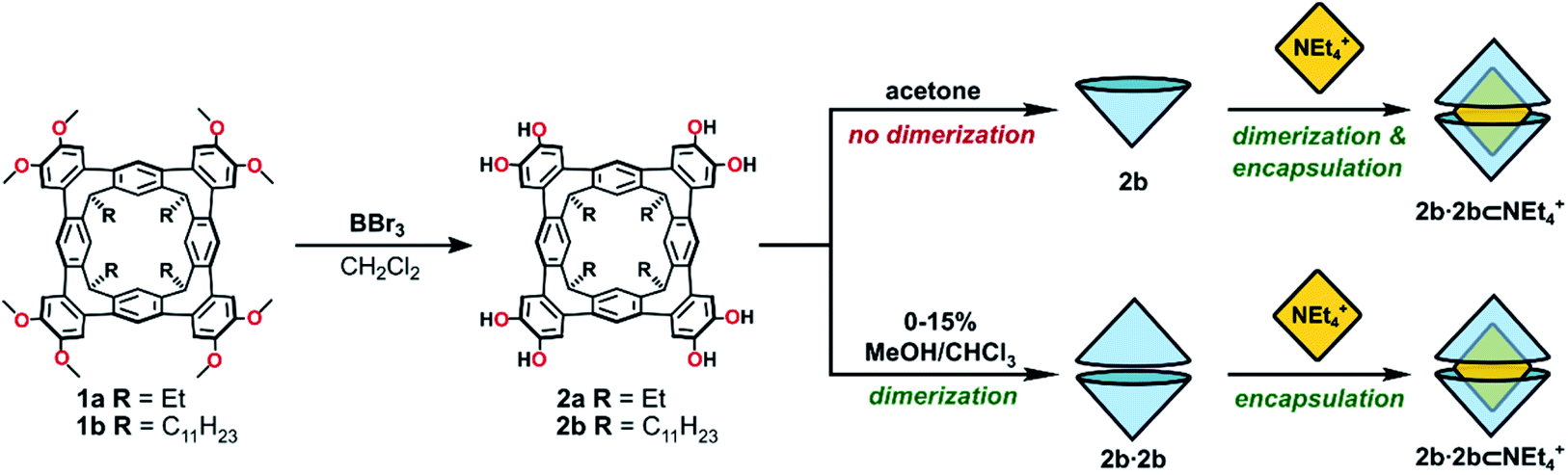 | ||
| Scheme 1 The synthesis of octol cavitands 2a and 2b, and a comparison of self-assembly behaviour of 2b in acetone or MeOH/CHCl3 mixtures. | ||
Results and discussion
Cleavage of the eight exoannular methyl ethers of 1a with BBr3 in CH2Cl2 was facile; however, the product 2a was insoluble in the non-polar solvents CHCl3 or CH2Cl2 (Scheme 1). To improve the solubility in non-polar solvents, we used our recent synthesis of C2v-symmetric resorcin[4]arene derivatives37 to prepare the undecyl analogue 1b in eight steps (see ESI†).Eightfold methyl ether cleavage afforded 2b in 93% yield, and the macrocycle was found to be soluble in CHCl3, CH2Cl2 and acetone. The 1H NMR spectrum of a freshly prepared solution of 2b in CDCl3 showed three cavitand species in slow exchange on the NMR timescale. Within ca. 60 minutes, equilibrium was established showing only a single species of C4v-symmetry, with no further change over days. The nature of the stable species formed from 2b was investigated by DOSY NMR spectroscopy. Self-assembled aggregates are predicted to diffuse more slowly than their monomers, as shown by the Stokes–Einstein equation in which the diffusion coefficient (D) is inversely proportional to the spherical hydrodynamic radius38 (see ESI†). Experiments in CDCl3 (Table 1) showed 2b diffuses more slowly than octamethoxy 1b—which cannot form multicomponent adducts through intermolecular hydrogen-bonding and is an appropriate surrogate for the monomer—but faster than the related resorcin[4]arene hexamers reported by Cohen (2.8 × 10−6 cm2 s−1; R = C11H23).11,38 The estimated diffusion volume of octol 2b is 1.7 times that of “monomer” 1b (see ESI†), consistent with homo-dimerization to 2b·2b driven by mutual hydrogen-bonding interactions at the cavitands' wide rims. An energy-optimised model of the dimer 2′·2′ supports this analysis (the prime symbol in 2′·2′ denotes calculated structures throughout this text), showing a unidirectional, cyclic seam of eight intermolecular and eight intramolecular hydrogen bonds (Fig. 2). In the 1H NMR spectrum of 2b (CDCl3, 298 K) a single OH resonance appears at ca. 6.89 ppm, its shift largely independent of 2b concentration across the range 1–14 mM (Δδ = 0.05 ppm; see ESI†). This lone OH signal is consistent with the rapid interconversion between the two equivalent arrangements of hydrogen-bonds on the NMR timescale (Fig. 2).
| Entry | Host | Added guest | Solvent | D (10−6 cm2 s−1) | Diffusing species |
|---|---|---|---|---|---|
| 1 | 1b | — | CDCl3 | 4.10 | 1b |
| 2 | 2b | — | CDCl3 | 3.45 | 2b·2b |
| 3 | 2b | NEt4Br | CDCl3 | 3.35 | 2b·2b⊂NEt4+ |
| 4 | 1b | — | (CD3)2CO | 6.95 | 1b |
| 5 | 2b | — | (CD3)2CO | 6.95 | 2b |
| 6 | 2b | NEt4Br | (CD3)2CO | 5.50 | 2b·2b⊂NEt4+ |
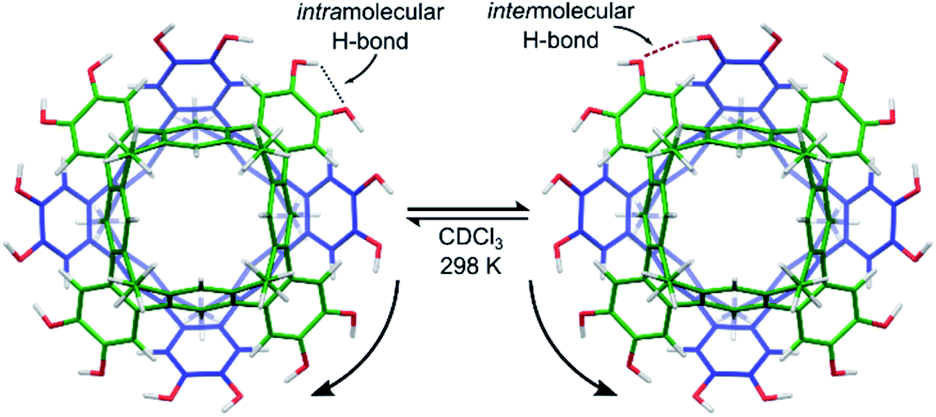 | ||
| Fig. 2 Energy-optimised model of the dimer complex 2′·2′ (B3LYP-D3/6-31G(d,p) with idealized S8-symmetry). The fast interconversion between two symmetry-equivalent hydrogen-bonding arrangements is shown. | ||
Further 1H NMR experiments in 20–50% v/v CHCl3/CDCl3 mixtures show solvent is encapsulated by the host, as evidenced by the appearance of CHCl3 resonance at 3.03 ppm (Δδ = −4.23 ppm; see ESI†). Similarly, in 50% v/v C6D6/CDCl3 two species are present in a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio at equilibrium, corresponding to the complexes 2b·2b⊂CDCl3 or 2b·2b⊂C6D6. The absence of a third species likely excludes the co-capture of two guest molecules.
:1 ratio at equilibrium, corresponding to the complexes 2b·2b⊂CDCl3 or 2b·2b⊂C6D6. The absence of a third species likely excludes the co-capture of two guest molecules.
To probe the stability of the dimer in protic, polar solvents, the diffusion coefficient of 2b·2b in CDCl3 with increasing proportions of CD3OD (0–800 equiv. per 2b) was measured by DOSY NMR spectroscopy (see ESI†).14D remained consistent with a dimer across the series (3.4–3.5 × 10−6 cm2 s−1), in mixtures of up to 15% CD3OD (v/v). This high stability of the complex contrasts with the resorcin[4]arene and pyrogallol[4]arene hexamers studied by Cohen,14 which show rapid increases in diffusion coefficient upon the addition of small amounts of MeOH. At the limit of solubility (1600 equiv. CD3OD), a second species appears in slow exchange with the dimer; however, its diffusion coefficient could not be differentiated. Unfortunately, the dimerization constant, Kdimer, for 2b in CDCl3 was not readily established, as concentration titrations of 2b monitored by UV-visible spectroscopy in CHCl3 or 5% MeOH/CHCl3 showed only a linear response in the Beer–Lambert region (see ESI†), and the weak emission of the cavitand prevented collection of reliable fluorescence spectroscopic titration data. Due to the unique hydrogen-bonding motif, we know of no suitable dimerization constants for direct comparison; however, on the basis of the 1H and DOSY NMR and UV-vis data presented herein, and the high stability of the complex in protic, polar solvents, Kdimer for 2b is likely >106 M−1 and may exceed >109 M−1. Similar values were reported for a dimeric capsule stabilised by eight hydrogen-bonds.39
To assess the guest-binding properties of the cavitand to non-solvent guests, 2b was combined with NEt4Br in 1:1, 2:1 and 4:1 ratios in CDCl3 at 298 K and the 1H NMR spectra examined for evidence of encapsulation (Fig. 3). The 1:1 mixture showed slow exchange between 2b·2b and 2b·2b⊂NEt4+ species, reaching quantitative encapsulation within 60 minutes (Fig. 3b). The free and bound NEt4+ species appear as two sets of signals in a 1:1 ratio (denoted with blue and yellow circles, respectively; Fig. 3b), as expected for the formation of 2b·2b⊂NEt4+. Alkyl signals for the bound cation are dramatically shifted upfield as far as −4.90 ppm (Δδ = −6.3 ppm) in response to the shielding influence of the host upon the internalized guest. The high-field ethyl signals of NEt4+ appear in two distinct environments as the conformation of the tightly bound guest is restricted within the host, behaviour supported by X-ray crystallography (vide infra). When 2b and NEt4Br were combined in a 2:1 ratio, only the host–guest adduct 2b·2b⊂NEt4+ is observed (Fig. 3c). DOSY NMR spectroscopy confirmed equal diffusion rates of the host and encapsulated guest at 3.35 × 10−6 cm2 s−1 (Table 1, entry 3), a rate similar to that of the free host 2b·2b. Increasing the host–guest ratio to 4:1 shows complete encapsulation of the guest, with an equimolar amount of residual free dimer host 2b·2b (Fig. 3d). These data support both the apparent exclusive formation of the homodimer 2b·2b and quantitative uptake of NEt4+ to give 2b·2b⊂NEt4+, with both species in slow exchange in CDCl3. The association constant, Ka, for the binding of NEt4+ by 2b·2b is 4.6 ± 0.3 × 105 M−1 (ΔG = −31.9 ± 1.6 kJ mol−1 in CHCl3, 293 K), as determined by UV-vis titrations40 of the host with the guest, and assuming a 1:1 host:guest binding model on the assumption Kdimer ≫ Ka (see ESI†).
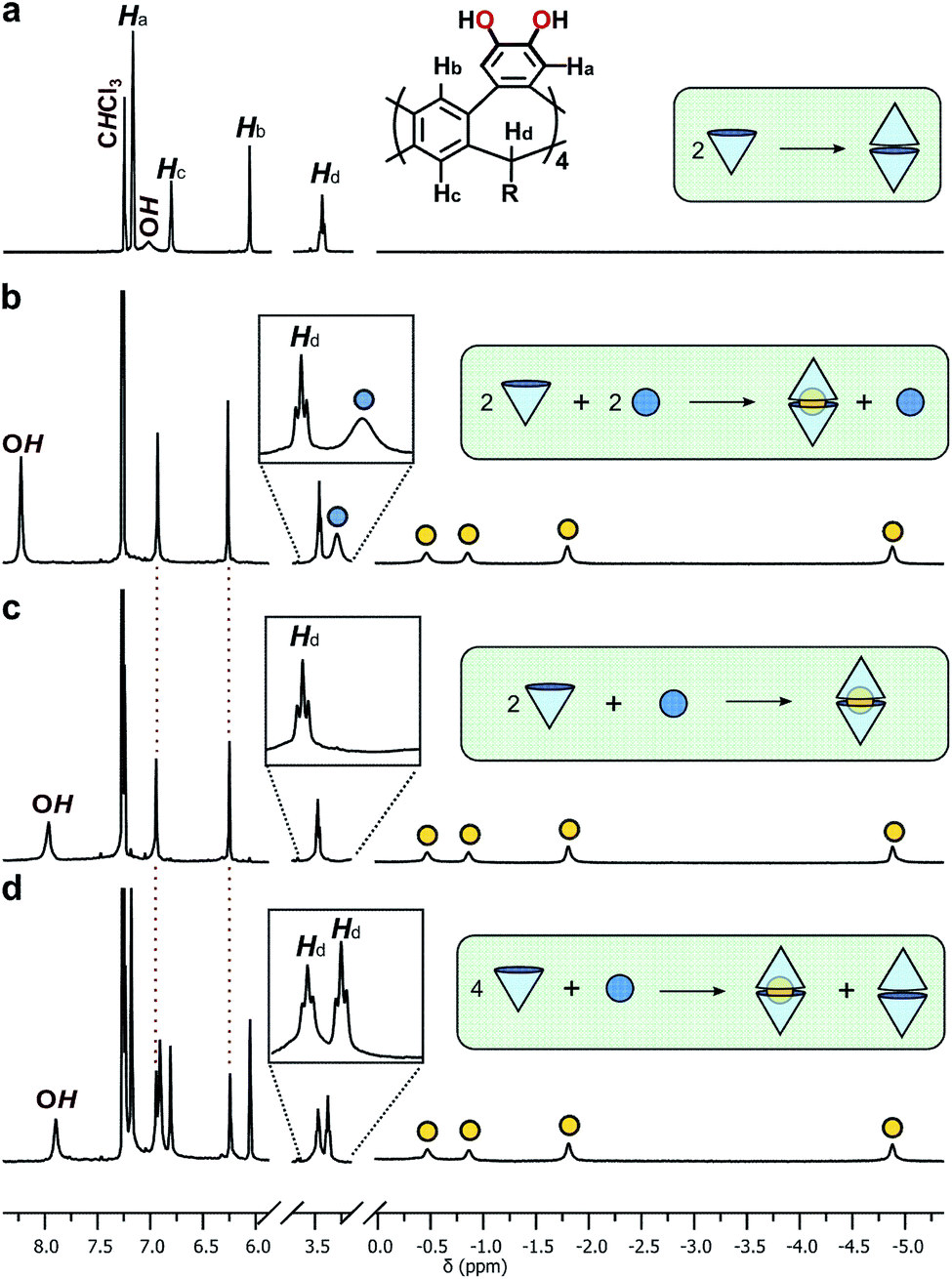 | ||
| Fig. 3 Truncated 1H NMR spectra (CDCl3, 298 K, 400 MHz) of (a) 2b, and 2b and NEt4Br in (b) 1:1, (c) 2:1 and (d) 4:1 ratios. Bound and free NEt4+ cations are represented by yellow and blue circles, respectively. Insets show expansions of the methine proton of the host (Hd) and the methylene protons of the unbound guest. | ||
The coordination of catechol units with halide anions is well known,41 and similar interactions have been shown to template the formation of resorcin[4]arene and pyrogallol[4]arene capsules.42 Homo-dimeric resorcin[4]arene-derived capsules formed through intermolecular hydrogen-bonding alone are rare; solvent molecules or the halide counterions of cationic guests are typically incorporated into the hydrogen-bonded network to template the dimerization processes, usually observed only in the solid state.22 Here, dimerization readily occurs in solution in the absence of templating molecules or anions, and moreover, is unaffected by their presence. Presumably, the low concentration of bromide counterions relative to the complex, combined with the competitive solvent, appear to limit interactions to the periphery of the dimer, causing only a moderate downfield shift in the OH proton resonance that increases with bromide concentration (Fig. 3). The limited role of the bromide ion in bonding at the seam is supported by crystallography (vide infra). While beyond the scope of the present study, it is noted that coordinating halide anions at higher concentrations may template different/larger multicomponent adducts and is an interesting avenue for further investigation.
In the bulk polar aprotic solvent d6-acetone, 2b failed to dimerize, as shown by the similar diffusion coefficients for 1b and 2b (Table 1, entries 4 and 5; Scheme 1; Fig. 4). The addition of excess NEt4Br to the monomer solution of 2b saw quantitative formation of the complex 2b·2b⊂NEt4+, as evidenced by the shift in OH proton resonance (Δδ = +1.29 ppm) consistent with increased hydrogen-bonding and the reduced diffusion coefficient (Table 1, entry 6). From these data it is clear interactions with the cationic guest play a role in the enthalpic-stabilisation of the dimer in bulk polar media.
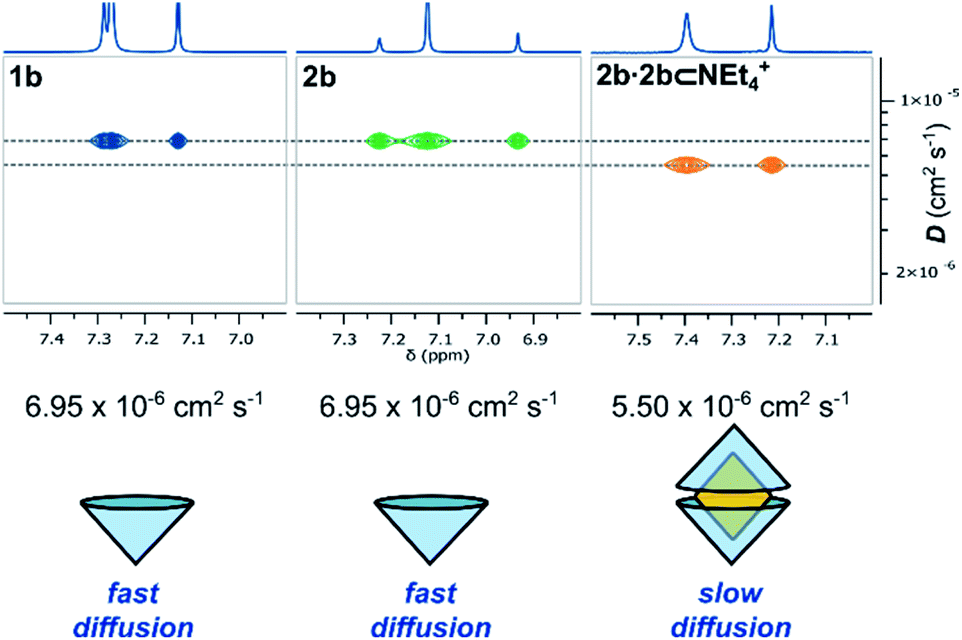 | ||
| Fig. 4 Truncated 1H and DOSY NMR spectra (d6-acetone, 298 K, 500 MHz) of 1b, 2b and 2b⊂NEt4Br and their diffusion coefficients. | ||
The ethyl-footed analogue 2a and NEt4+ (2:1) failed to form a soluble 2a·2a⊂NEt4+ complex in neat CDCl3; however, following the addition of 20% CD3OD, the suspension dissolved within 12 hours. 1H NMR analysis again showed complete formation of a 2:1 complex in this competitive media (see ESI†). The charged complexes 2a·2a⊂NEt4+ and 2b·2b⊂NEt4+ observed in solution were similarly detected in the gas phase by ESI-MS (see ESI†).
Pleasingly, X-ray quality crystals of the complex adduct could be grown, an analysis confirming that the solid-state behaviour mirrors that of the solution-phase experiments (Fig. 5).‡ The cavitands interact via a cyclic seam of hydrogen bonds to give a pseudo-octagonal bipyramidal capsule with a volume of ca. 1500 Å3 and dimensions of ca. 10.7 × 14.4 Å (excluding the alkyl feet, Fig. 5a and b). The internal cavity is 235 Å3 of which 65% is occupied by the guest (152 Å3), notably higher than the optimal 55% (ref. 43) (see ESI† for details of cavity and guest volume calculations).44–46 This tight fit, combined with the rigid biconical topology of the cavity (Fig. 5d and e) forces the ethyl groups of the guest into magnetically anisotropic environments—axial (along the long axis of the capsule) and equatorial (in the plane of the cavitand rims)—resulting in the breaking of the guest's symmetry observed by NMR. The methyl groups of the axially-orientated substituents are buried deeply within the cavitand and localised over four aromatic rings, consistent with the large upfield shifts observed in the 1H NMR spectrum. The phenolic protons of the host were well defined in the Fourier difference map and are positionally restrained only by their O⋯H distance (0.84 Å; Fig. 5c). The cavitands interact through seven intermolecular hydrogen-bonds (O⋯O distance 2.73–2.81 Å), six of which follow a head-to-tail arrangement around the capsule. Those groups not involved in cavitand–cavitand stabilization are free to form intramolecular hydrogen-bonds within the catechol subunit (i.e. O1⋯O2), or interact with MeOH solvent or the bromide counterion (Fig. 5c). In solution, these non-intermolecular hydrogen-bonds likely offer redundancy to the cavitand–cavitand interactions and may act to defend the dimer against interference by competitive media or bromide counterions.
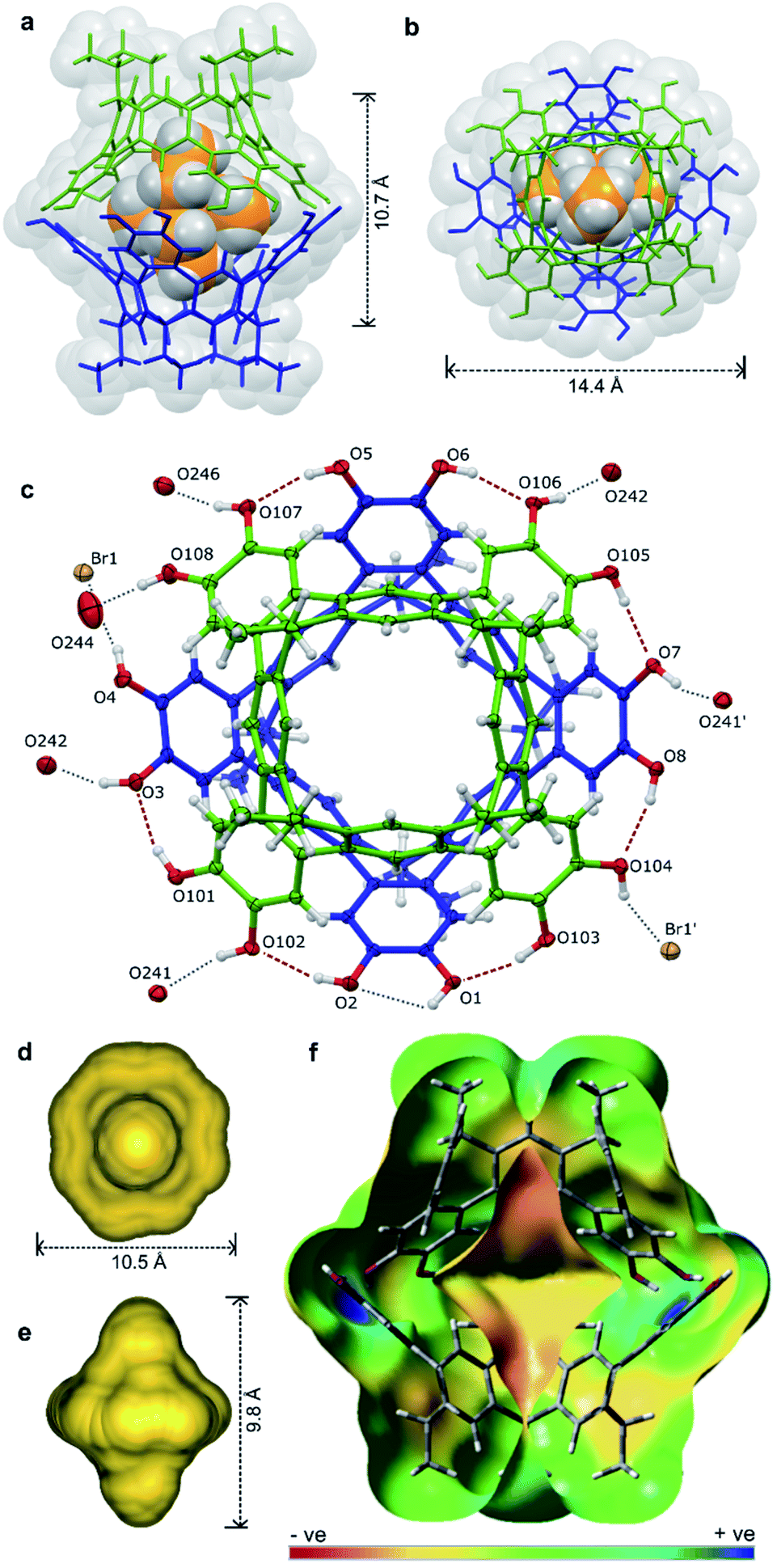 | ||
| Fig. 5 The X-ray structure of [2a·2a⊂NEt4]-Br·7.63MeOH·toluene·0.37CHCl3 with guest, solvent and disorder omitted for clarity where necessary: the host guest complex (a) from the side and (b) top, (c) an ORTEP diagram showing the arrangement of hydrogen-bond donors and acceptors with the NEt4+ guest omitted for clarity, the calculated cavity surface (d) shown from the top and (e) side, and (f) a calculated electronic surface potential map of 2′·2′, showing the electronegative surface of the cavity (see ESI† for details). | ||
The hydrogen-bonding motif is unprecedented for resorcin[4]arene-derived cavitands due to the coplanar arrangement of exoannuar hydroxyl groups, and may be thought of as an extension of the intercalated cyclotricatechylene (CTC) “clamshell” dimers reported by Abrahams.27 Here, the four catechol units of the interacting cavitands are rotationally offset by ca. 45° relative to the cavitand partner (compared to 60° for CTC dimers), the resulting greater cavity volume able to encapsulate larger guests with retention of the full hydrogen-bonding array.
An energy-optimised model of 2′·2′⊂CHCl3 (B3LYP-D3/6-31G(d,p)) shows a cavity volume of 175 Å3, 25% less than that measured experimentally with a bound NEt4+. As the monomer units are rigid, the volume difference arises through increased separation of the dimers (ca. +0.4 Å; measured between centroids calculated for O atoms of each cavitand) allowing the host to contract or expand at the hydrogen-bond equator to accommodate a range of guests with different volumes. Solution experiments support this analysis: the smaller guests NMe4+ (94 Å3) and choline+ (122 Å3) are quantitatively sequestered despite volumes ca. 20–40% less than that of NEt4+. Notably, the complexes were stable in the polar solvent mixtures required to dissolve the salts (5% CD3OD/CDCl3), each evidencing slow exchange with the free guest (see ESI†). The 1H NMR spectrum of 2b·2b⊂choline+ shows only a single host species of S8-symmetry, consistent with rapid tumbling of the guest on the NMR timescale at 298 K. The larger cation NPr4+ is not encapsulated, presumably due to the greater volume (227 Å3) significantly disrupting the hydrogen-bonding array between interacting cavitands.
Neutral guests of a size and geometry complementary to the host cavity including adamantane (147 Å3), SiEt4 (176 Å3) and terephthalonitrile (116 Å3) showed no interaction, nor did the electron-poor aromatic C6F6 (113 Å3). Calculated binding energies for the complexes 2′·2′⊂CHCl3 and 2′·2′⊂SiEt4 are indistinguishable within the range of error, whilst the cationic complexes 2′·2′⊂NEt4+ and 2′·2′⊂choline+ enjoy significant stabilisation (Table 2; B3LYP-D3/6-311G++(2d,2p), in vacuo, see ESI† for full details). Electronic surface potential maps show the curvature of the host imparts a substantial electronegative bias to the internal surface of the cavity (Fig. 5f; see ESI†); it is likely that a favourable enthalpic contribution from π-basic⋯cation interactions drives the exchange of CDCl3 for NEt4+, but not SiEt4. The calculations show a strong energetic preference for NEt4+ over choline+ (−33 kJ mol−1; Table 2), behaviour replicated in solution as demonstrated by competition experiments between NMe4+, NEt4+ and choline+. When 1 equivalent of each guest is combined together with the dimer host in 5% CD3OD/CDCl3, NMe4+ and NEt4+ are bound in a ca. 1:1.2 ratio, whereas the choline+ complex is not detected (see ESI†). While the preference NEt4+ ≥ NMe4+ ≫ choline+ is difficult to reconcile based on the volume of the guests alone, it seems likely that the OH group of choline+ interferes with the hydrogen-bonding motif between cavitands, destabilizing the complex. It should be noted that this geometry is not reflected in the calculated structure of the 2′·2′⊂choline+ complex—possibly due to bias in the starting geometry—likely resulting in an overestimation of the binding energy.
| Guest | Guest vol. (Å3) | E binding (kJ mol−1) |
|---|---|---|
| a Relative energy of guest molecule displacing an encapsulated CHCl3. | ||
| CHCl3 | 71 | 0.0 |
| NMe4+ | 94 | −144 |
| C6F6 | 113 | — |
| Terephthalonitrile | 116 | — |
| Choline+ | 122 | −138 |
| Adamantane | 147 | — |
| NEt4+ | 152 | −171 |
| SiEt4 | 176 | +2.1 |
| NPr4+ | 227 | — |
Conclusions
We have presented our observations into highly stable, self-assembled capsules stabilised by a cyclic seam of eight hydrogen bonds. The rigid cavity selectively sequesters and restricts the conformation of cationic guests, behaviour observed in both bulk solutions and the solid state, and analytically in the gas phase by ESI-MS. The hydrogen-bond array acts to articulate the interface between the rigid cavitands, allowing for some flexibility in the cavity volume and dimensions.Data availability
All supporting data is provided in the ESI.†Author contributions
N. T. L. conceived and supervised the project. J. N. S. carried out the experimental work and analyses, and C. E. performed the DFT calculations. J. N. S. drafted then refined the manuscript with input from C. E. and N. T. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by The MacDiarmid Institute for Advanced Materials and Nanotechnology, and the Marsden Fund Council, managed by the Royal Society Te Apārangi, New Zealand. J. N. S. thanks the University of Otago for the award of a PhD scholarship. C. E. thanks the National Computational Infrastructure (NCI: supported by the Australian Government) for high performance computing resources and services provided via project grant ay7.Notes and references
- L. J. Liu and J. Rebek, in Hydrogen Bonded Supramolecular Structures, ed. Z.-T. Li and L.-Z. Wu, Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, pp. 227–248 Search PubMed.
- J. Kang and J. Rebek, Nature, 1997, 385, 50–52 CrossRef CAS PubMed.
- Q. Zhang, L. Catti and K. Tiefenbacher, Acc. Chem. Res., 2018, 51, 2107–2114 CrossRef CAS PubMed.
- V. Angamuthu, M. Petroselli, F.-U. Rahman, Y. Yu and J. Rebek, Org. Biomol. Chem., 2019, 17, 5279–5282 RSC.
- R. Wyler, J. de Mendoza and J. Rebek Jr, Angew. Chem., Int. Ed. Engl., 1993, 32, 1699–1701 CrossRef.
- J. Kang and J. Rebek, Nature, 1996, 382, 239–241 CrossRef CAS.
- L. R. MacGillivray and J. L. Atwood, Nature, 1997, 389, 469 CrossRef CAS.
- T. Gerkensmeier, W. Iwanek, C. Agena, R. Fröhlich, S. Kotila, C. Näther and J. Mattay, Eur. J. Org. Chem., 1999, 1999, 2257–2262 CrossRef.
- A. Shivanyuk and J. Rebek, Chem. Commun., 2001, 2424–2425 RSC.
- A. Shivanyuk and J. Rebek, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 7662 CrossRef CAS PubMed.
- L. Avram and Y. Cohen, J. Am. Chem. Soc., 2002, 124, 15148–15149 CrossRef CAS.
- L. Avram and Y. Cohen, Org. Lett., 2002, 4, 4365–4368 CrossRef CAS.
- L. Avram and Y. Cohen, Org. Lett., 2003, 5, 1099–1102 CrossRef CAS PubMed.
- L. Avram and Y. Cohen, J. Am. Chem. Soc., 2004, 126, 11556–11563 CrossRef CAS PubMed.
- K. Murayama and K. Aoki, Chem. Commun., 1998, 607–608 RSC.
- K. N. Rose, L. J. Barbour, G. W. Orr and J. L. Atwood, Chem. Commun., 1998, 407–408 RSC.
- A. Shivanyuk, E. F. Paulus and V. Böhmer, Angew. Chem., Int. Ed., 1999, 38, 2906–2909 CrossRef CAS.
- A. Shivanyuk, K. Rissanen and E. Kolehmainen, Chem. Commun., 2000, 1107–1108 RSC.
- A. Shivanyuk, E. F. Paulus, K. Rissanen, E. Kolehmainen and V. Böhmer, Chem.–Eur. J., 2001, 7, 1944–1951 CrossRef CAS.
- M. Luostarinen, A. Åhman, M. Nissinen and K. Rissanen, Supramol. Chem., 2004, 16, 505–512 CrossRef CAS.
- H. Mansikkamäki, C. A. Schalley, M. Nissinen and K. Rissanen, New J. Chem., 2005, 29, 116–127 RSC.
- N. K. Beyeh and K. Rissanen, Isr. J. Chem., 2011, 51, 769–780 CrossRef CAS.
- A. Scarso, L. Pellizzaro, O. De Lucchi, A. Linden and F. Fabris, Angew. Chem., Int. Ed., 2007, 46, 4972–4975 CrossRef CAS.
- D. Beaudoin, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2016, 55, 15599–15603 CrossRef CAS.
- B. C. Hamann, K. D. Shimizu and J. Rebek, Angew. Chem., Int. Ed. Engl., 1996, 35, 1326–1329 CrossRef CAS.
- E. Huerta, G. A. Metselaar, A. Fragoso, E. Santos, C. Bo and J. de Mendoza, Angew. Chem., Int. Ed., 2007, 46, 202–205 CrossRef CAS PubMed.
- B. F. Abrahams, N. J. FitzGerald, T. A. Hudson, R. Robson and T. Waters, Angew. Chem., Int. Ed., 2009, 48, 3129–3132 CrossRef CAS PubMed.
- D. J. Cram, S. Karbach, Y. H. Kim, L. Baczynskyj, K. Marti, R. M. Sampson and G. W. Kalleymeyn, J. Am. Chem. Soc., 1988, 110, 2554–2560 CrossRef CAS.
- T. Gerkensmeier, J. Mattay and C. Näther, Chem.–Eur. J., 2001, 7, 465–474 CrossRef CAS.
- A. Shivanyuk and J. J. Rebek, Chem. Commun., 2001, 2374–2375 RSC.
- M. C. Letzel, B. Decker, A. B. Rozhenko, W. W. Schoeller and J. Mattay, J. Am. Chem. Soc., 2004, 126, 9669–9674 CrossRef CAS PubMed.
- Y. S. Park and K. Paek, Org. Lett., 2008, 10, 4867–4870 CrossRef CAS PubMed.
- C. B. Aakeröy, A. Rajbanshi and J. Desper, Chem. Commun., 2011, 47, 11411–11413 RSC.
- T. Heinz, D. M. Rudkevich and J. Rebek, Nature, 1998, 394, 764–766 CrossRef CAS.
- M. H. K. Ebbing, M.-J. Villa, J.-M. Valpuesta, P. Prados and J. de Mendoza, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4962–4966 CrossRef CAS.
- J. N. Smith and N. T. Lucas, Chem. Commun., 2018, 54, 4716–4719 RSC.
- J. N. Smith, T. K. Brind, S. B. Petrie, M. S. Grant and N. T. Lucas, J. Org. Chem., 2020, 85, 4574–4580 CrossRef CAS.
- L. Avram and Y. Cohen, Chem. Soc. Rev., 2015, 44, 586–602 RSC.
- T. Szabo, G. Hilmersson and J. Rebek, J. Am. Chem. Soc., 1998, 120, 6193–6194 CrossRef CAS.
- D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- K. J. Winstanley, A. M. Sayer and D. K. Smith, Org. Biomol. Chem., 2006, 4, 1760–1767 RSC.
- M. Chwastek, P. Cmoch and A. Szumna, Angew. Chem., Int. Ed., 2021, 60, 4540–4544 CrossRef PubMed.
- J. Rebek, Acc. Chem. Res., 1999, 32, 278–286 CrossRef CAS.
- A. Pedretti, L. Villa and G. Vistoli, J. Mol. Graphics Modell., 2002, 21, 47–49 CrossRef CAS.
- A. Pedretti, L. Villa and G. Vistoli, J. Comput.-Aided Mol. Des., 2004, 18, 167–173 CrossRef CAS PubMed.
- A. Jurcik, D. Bednar, J. Byska, S. M. Marques, K. Furmanova, L. Daniel, P. Kokkonen, J. Brezovsky, O. Strnad, J. Stourac, A. Pavelka, M. Manak, J. Damborsky and B. Kozlikova, Bioinformatics, 2018, 34, 3586–3588 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterization data of all new compounds; details of solution spectroscopic/crystallographic characterization of dimer and host–guest complexes; details of calculations and electrostatic potential maps. CCDC 2070238. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01802g |
‡ Crystal data for 2a·2a·C8H20N·Br·7.63(CH3OH)·0.37(CHCl3)·C7H8: C143H154.89BrCl1.11NO23.63, M = 2384.90, pale-yellow block, 0.45 × 0.29 × 0.21 mm3, triclinic, a = 16.8535(2) Å, b = 17.7481(2) Å, c = 26.0471(4) Å, α = 82.9010(10)°, β = 71.9630(10)°, γ = 64.5340(16)°, V = 6688.07(16) Å3, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (#2), Z = 2, μ(Mo-Kα) = 0.397 mm−1, 2θmax = 57.13°, 2θfull = 50.00° (99.7% complete), 128545 reflections measured, 29947 independent reflections (Rint = 0.0392). The final R1(F) = 0.0616 (I > 2σ(I)); 0.0824 (all data). The final wR2(F2) = 0.1654 (I > 2σ(I)); 0.1815 (all data). GoF = 1.035. (#2), Z = 2, μ(Mo-Kα) = 0.397 mm−1, 2θmax = 57.13°, 2θfull = 50.00° (99.7% complete), 128545 reflections measured, 29947 independent reflections (Rint = 0.0392). The final R1(F) = 0.0616 (I > 2σ(I)); 0.0824 (all data). The final wR2(F2) = 0.1654 (I > 2σ(I)); 0.1815 (all data). GoF = 1.035. |
| This journal is © The Royal Society of Chemistry 2021 |