
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAbsolute configuration retention of a configurationally labile ligand during dynamic processes of thiolate protected gold clusters†
Yanan
Wang
 a,
Esko
Makkonen
b,
Xi
Chen
*b and
Thomas
Bürgi
*a
a,
Esko
Makkonen
b,
Xi
Chen
*b and
Thomas
Bürgi
*a
aDepartment of Physical Chemistry, University of Geneva, 30 Quai Ernest-Ansermet, 1211, Geneva 4, Switzerland. E-mail: Thomas.Buergi@unige.ch
bDepartment of Applied Physics, Aalto University, Otakaari 1, FI-02150, Espoo, Finland. E-mail: xi.6.chen@aalto.fi
First published on 4th June 2021
Abstract
Monolayer protected metal clusters are dynamic nanoscale objects. For example, the chiral Au38(2-PET)24 cluster (2-PET: 2-phenylethylthiolate) racemizes at moderate temperature. In addition, ligands and metal atoms can easily exchange between clusters. Such processes are important for applications of monolayer protected metal clusters; however, the mechanistic study of such processes turns out to be challenging. Here we use a configurationally labile, axially chiral ligand, biphenyl-2,2′-dithiol (R/S-BiDi), as a probe to study dynamic cluster processes. It is shown that the ligand exchange of free R/S-BiDi on a chiral Au38(2-PET)24 cluster is diastereospecific. Using chiral chromatography, isolated single diastereomers of the type anticlockwise/clockwise-Au38(2-PET)22(R/S-BiDi)1 could be isolated. Upon heating, the cluster framework racemizes, while the R/S-BiDi ligand does not. These findings demonstrate that during cluster racemization and/or ligand exchange between clusters, the R/S-BiDi ligand is sufficiently confined, thus preventing its racemization, and exclude the possibility that the ligand desorbs from the cluster surface.
Introduction
Precise gold nanoclusters, as a kind of material with a well-defined composition and structure, have attracted widespread interest over the past few decades.1–3 So far, examples such as Au25(SR)18, Au38(SR)24, Au40(SR)24, Au102(SR)44 and Au140(SR)60 (SR stands for thiolate) have already become excellent candidates as catalysts,4,5 as optically active materials,6,7 for chemical sensing,8,9 and for biological applications.10,11 Preparation of Aun(SR)m nanoclusters is normally done through direct synthesis using neutral and anionic thiolate ligands.12 However, due to the low solubility and steric and electrostatic effects, some ligands cannot be used in direct synthesis, which severely limits the diversity of surface properties of the accessible clusters.13,14Ligand exchange reactions (LERs), which introduce new ligands to the parent nanoclusters,15–20 are widely applied for adding functionalities or chemical properties to clusters as a versatile post-modification method.14,21 This methodology drastically increases the possibility to attain the required surface properties without changing the metal core structure.14 Up to now, these dynamic exchange processes were mostly investigated with Au25(SR)18 and Au38(SR)24 clusters,22,23 which benefit from simple synthesis and stability. The Au25(SR)18 cluster has an icosahedral Au13 core surrounded by six dimeric SR-Au-SR-Au-SR staple units.18,20,24–27 Au38(SR)24 has an elongated structure with three monomeric SR-Au-SR staples at the equator of the cluster and three dimeric staples at each pole.28 The SR groups in one dimeric staple can be classified into terminal SR groups, which are linked to the gold core, and a central SR group.29 Owing to the geometry of these clusters, the SR groups in staples possess different chemical environments, and thus there exist preferential exchange sites during LERs.17,20,22,30
Experimental and computational studies for Au25(SR)18 and Au38(SR)24 clusters indicate that LERs start preferentially at the terminal SR groups,20,29,30 which are directly bound to the metal core, via an associative SN2-like mechanism.17 Negishi and co-workers also found that Pd doping of Au25(SR)18 drastically increases the rate of ligand exchange reaction.25 Dass and co-workers systematically studied LERs with aliphatic dithiol ligands HS-(CH2)n-SH with various chain length n.31 They documented that C3 and C4 prefer interstaple coupling, and C5 and C6 are good candidates for intrastaple binding, while C2 is too short for the bidentate binding. In addition to aliphatic ligands, aromatic dithiols have also been used for LERs, such as 1,1′-binaphthalene-2,2′-dithiol (BINAS), which is a rigid chiral ligand.18,32 Experimental and theoretical studies of Au25 and Au38 clusters18,32–35 show that the bidentate ligand connects preferentially to two neighboring staples thus forming a bridge between the latter.32 Furthermore, the LER between the R-BINAS and intrinsically chiral Au38 is diastereoselective which has been revealed by in situ HPL chromatography.28
Apart from the ligand exchange reaction via free thiols, it has been shown that ligands can exchange between clusters in the absence of free thiols. In 2013, intercluster LERs between Au25(SC10H21)18 and Au25(SC12H25)18 were observed and the authors assumed that this exchange resulted from the detachment of ligand or gold–ligand species from the cluster.25 Later, Salassa and co-workers carried out ligand exchange between Au25(SBut)18 and Au25(2-PET)18 (2-PET: 2-phenylethylthiolate, a monothiol) at room temperature. Their results indicated that the exchange proceeds via a cluster collision mechanism without release of free thiol or a thiol–gold complex from the cluster.13
Recently, we confirmed that also bidentate ligands exchange between clusters by observing the mass peak belonging to Au25(2-PET)16(R-BINAS)1 after mixing Au38(2-PET)24-2x(R-BINAS)x and Au25(2-PET)18 clusters at 70 °C for 24 hours.36 This study also showed that higher temperatures are needed for the intercluster ligand exchange with dithiols compared to monothiols. In addition, it has been shown that the intercluster BINAS exchange between chiral Au38(SR)24 clusters is diastereoselective. This, together with fast racemization of Au38(2-PET)24, led to an amplification of the enantiomeric excess of the cluster.36 Au38 clusters can racemize in solution.37 Experimental activation energies show that this process proceeds without complete Au–S bond breaking.37 Recent calculations indicate that this racemization involves a concerted rotation of three gold atoms of the core, which drag along the staples.38 After addition of BINAS dithiol to the ligand shell the racemization is still observed experimentally, although at considerably higher temperatures.34
There are still many open questions concerning intercluster ligand exchange reaction and racemization, especially when dithiol ligands are involved. Here we use a new bidentate ligand, biphenyl-2,2′-dithiol (in the following R/S-BiDi for short), as a molecular probe in order to shed some more light on these processes. Compared with BINAS, which has a rigid structure, R/S-BiDi is more flexible owing to the small steric hindrance of the two benzene rings and therefore R/S-BiDi racemizes rapidly in solution, i.e. BiDi is configurationally labile, while BINAS is not. It can therefore be used as a probe in order to test whether, during the dynamic cluster processes (exchange and racemization), the ligands are confined or not. In the latter case, we may expect a racemization of the R/S-BiDi ligand during these processes.
Results and discussion
As one prominent example of an intrinsically chiral cluster, Au38(2-PET)24 has been used for many studies. The cluster is easily prepared and stable. Following a previous report,1 rac-Au38(2-PET)24 was synthesized and purified. The purity of the cluster was demonstrated by UV-vis spectra (Fig. S1A†) and MALDI-TOF mass spectra (Fig. S1B†). R/S-BiDi was prepared analogously to BINAS starting from the biphenyl-2,2′-diol.39 Compared with BINAS, biphenyl-2,2′-dithiol (R/S-BiDi) is more flexible due to the lower steric hindrance as shown in Fig. 1A. We optimized R/S-BiDi structures with different dihedral angles of the carbon rings using Density Functional Theory (DFT) with the PBE + TS functional and found that R/S-BiDi has two global minimum structures with dihedral angles of 91.45° and 268.36°, corresponding to the two enantiomers (Fig. 1B). Note that the dihedral angles may change after linking to the cluster surface. In order to estimate the barrier for interconversion between the two enantiomers (racemization), nudged elastic band (NEB) calculations were performed (Fig. 1B).40 The racemization barrier for R/S-BiDi is about 0.69 eV (16 kcal mol−1), which is less than the barrier (23 kcal mol−1) of other biphenyl derivatives to undergo torsional isomerization.41,42 The corresponding rate constant k can be estimated from the Arrhenius formula k = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ea/kBT). If we use typical molecular vibrational frequency 1013 to 1014 s−1 as A, the constant k of R/S-BiDi is around 26 s−1 to 256 s−1 at T = 300 K. In conclusion, R/S-BiDi racemizes fast at room temperature and the enantiomers are therefore not separable. As expected R/S-BiDi does not show CD signals (Fig. S2†) and the absorption spectrum of R/S-BiDi is characterized by a peak at 280 nm with a shoulder at 295 nm (Fig. 1C).
exp(−Ea/kBT). If we use typical molecular vibrational frequency 1013 to 1014 s−1 as A, the constant k of R/S-BiDi is around 26 s−1 to 256 s−1 at T = 300 K. In conclusion, R/S-BiDi racemizes fast at room temperature and the enantiomers are therefore not separable. As expected R/S-BiDi does not show CD signals (Fig. S2†) and the absorption spectrum of R/S-BiDi is characterized by a peak at 280 nm with a shoulder at 295 nm (Fig. 1C).
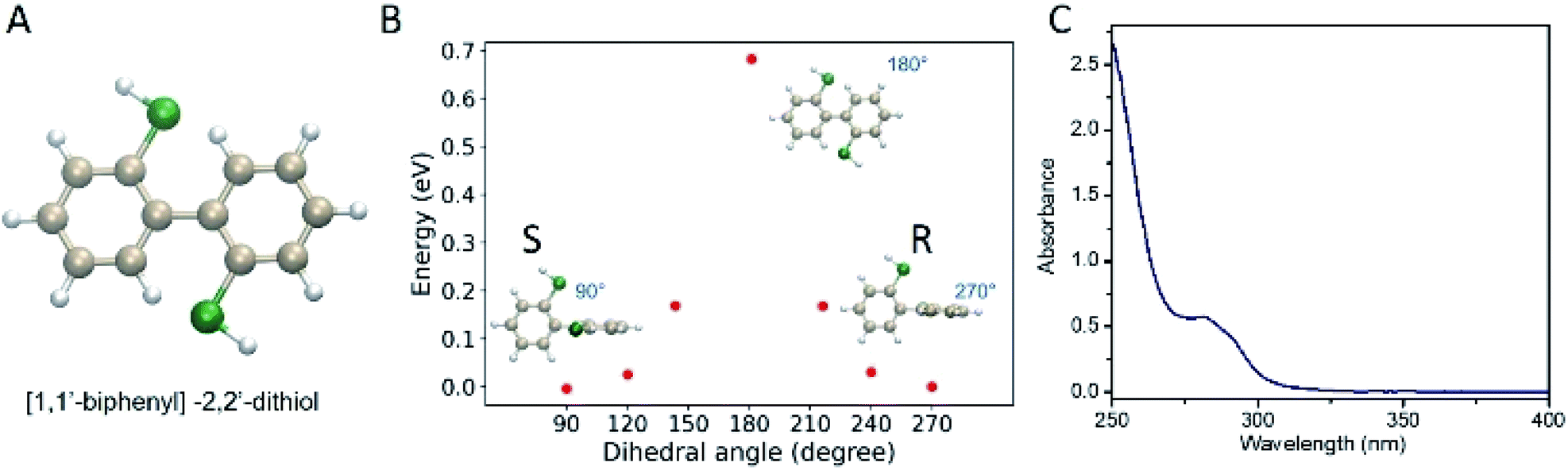 | ||
| Fig. 1 Characterization of [1,1′-biphenyl]-2,2′-dithiol. (A) Structure of [1,1′-biphenyl]-2,2′-dithiol. Color code: tan: C atoms, white: H atoms and green: S atoms. (B) Calculated energies for BiDi configuration at different dihedral angles. (C) UV-vis spectrum of the BiDi ligand in dichloromethane. | ||
R/S-BiDi was introduced to the Au38(2-PET)24 clusters by ligand exchange reactions, and the evolution of the process was followed by chiral HPL chromatography. As a reference, rac-Au38(2-PET)24 showed two well-defined peaks with retention times of 38 min (anti-clockwise, A) and 78 min (clockwise, C) in chiral HPLC, corresponding to the two enantiomers of the cluster (Fig. S3†).43 Under the same conditions (see Methods) the free ligand R/S-BiDi eluted at a retention time of 10 min (Fig. S3†). For the ligand exchange reactions, rac-Au38(2-PET)24 was mixed with free R/S-BiDi at a molar ratio of 1:50. The sample was kept at room temperature and analyzed by HPLC every 200 minutes. The time-resolved chromatograms (Fig. S4†) show that after 200 min, new peaks arise at 48 min, 83 min and 120 min, while the two peaks from the parent cluster decreased with increasing reaction time. We anticipated that the new peaks originate from clusters with one ligand exchange, Au38(2-PET)22(R/S-BiDi)1, which is confirmed by the MALDI-TOF mass spectrum of a sample taken at 18 h (Fig. S5†). In view of this composition after ligand exchange, each enantiomer of Au38(2-PET)24 may be substituted with one of the two enantiomers of R/S-BiDi, resulting in four stereoisomers as shown in Fig. 2. Here we assume that R/S-BiDi prefers the same adsorption site as BINAS (bridging two dimeric staples, see Fig. 2), which seems reasonable given the structural similarity of the two molecules (see Fig. S6†). Note that once adsorbed, the R/S-BiDi molecule cannot easily racemize anymore. Due to peak overlap only three out of the four possible Au38(2-PET)22(R/S-BiDi)1 stereoisomers are clearly observed in the chromatogram (Fig. S4†), which hinders further analysis.
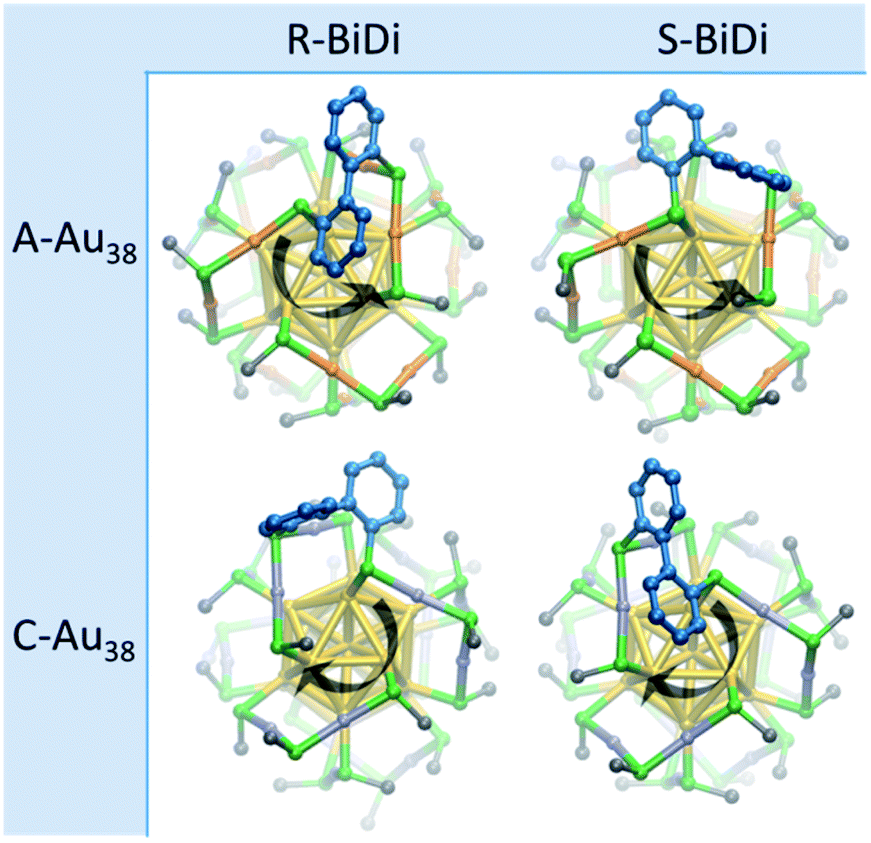 | ||
| Fig. 2 Structures of Au38(SR)24 with one BiDi adsorbed. Different atoms are depicted in gray (C atom), gold (Au atom), and green (S atom) colors. The color of the gold atoms in staples of clockwise (anti-clockwise) clusters, named as C-Au38 (A-Au38), is violet (orange) and the color of C atoms in R/S-BiDi is cornflower blue. H-atoms are emitted for clarity. The four structures above are named as A-Au38-R-BiDi (left-top), A-Au38-S-BiDi (right-top), C-Au38-R-BiDi (left-bottom), and C-Au38-S-BiDi (right-bottom). | ||
In order to partially avoid peak overlap and to help with the assignment of the peaks, ligand exchange reactions were carried out with enantiopure Au38 clusters. The two enantiomers of Au38(2-PET)24 were separated following the reported procedure,43 and the purity of the enantiomers was confirmed by chiral HPLC (Fig. S3†). A-Au38(2-PET)24 (Fig. 3) and C-Au38(2-PET)24 (Fig. S7†) clusters, respectively, were mixed with free R/S-BiDi at a molar ratio of 1:50 at room temperature and HPL chromatograms were recorded every 4 hours. Fig. 3A shows that two new peaks arise simultaneously at retention times of 48 min (A-PI) and 83 min (A-PII) after only 4 hours. The two peaks arise simultaneously, as expected for parallel reactions, indicating that they belong to the two diastereomers of A-Au38(2-PET)22(R/S-BiDi)1. With increasing reaction time, the intensity of A-Au38(2-PET)24 decreases and those of A-PI and A-PII increase progressively. Interestingly, the intensities of the A-PI and A-PII peaks are not equal, A-PII being predominant. The peaks were integrated (Fig. S8†) and a plot of the ratio of peak areas A-PII/A-PI as a function of time is shown in Fig. 3B. At short reaction times (before 20 h) the ratio between A-PII and A-PI was nearly stable and increased afterwards, which may be due to further subsequent exchange reactions (second exchange, etc). The above experiment shows that the ligand exchange is diastereoselective in a similar fashion as was reported for the rigid chiral dithiol BINAS. In that case R-BINAS preferably reacted with A-Au38(2-PET)24.28 Here the initial A-PII/A-PI peak ratio is 2.15 (Fig. 3), which corresponds to the ratio of the rate constants at 4 h (parallel reaction). This translates into a diastereomeric excess of 36%. For the BINAS case the diastereomeric excess was 60%.28 The higher value for BINAS seems reasonable given the larger size of BINAS compared to R/S-BiDi, which enhances the influence of steric factors.
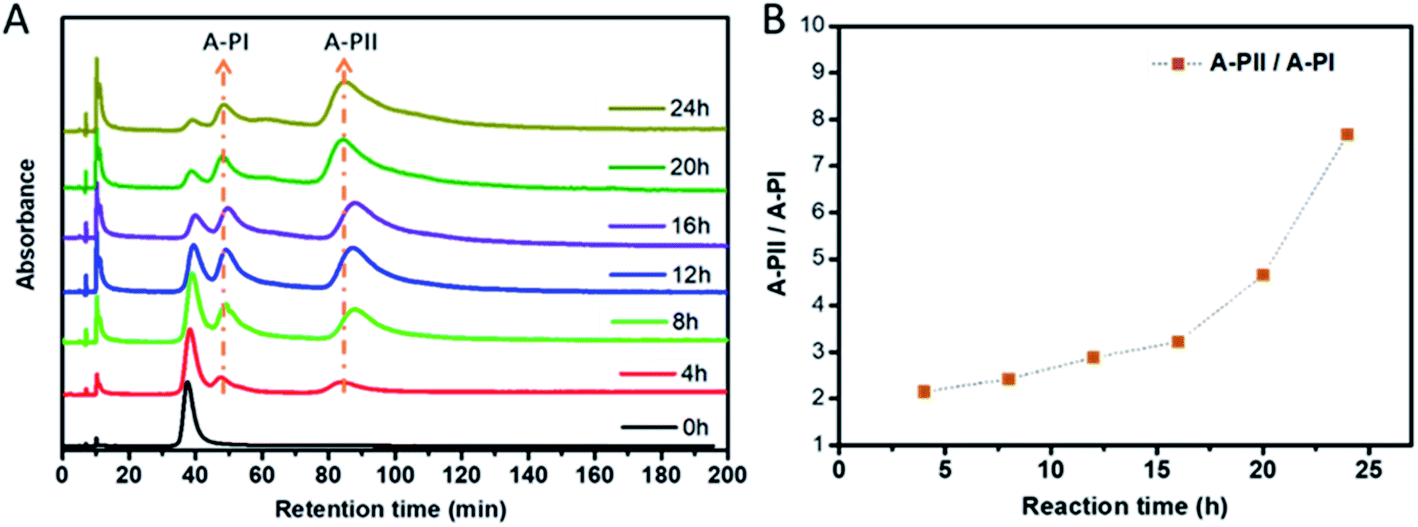 | ||
| Fig. 3 Characterization of A-Au38(2-PET)24 ligand exchange with BiDi. (A) Evolution of the chiral HPL chromatogram during the ligand exchange reaction between A-Au38(2-PET)24 and R/S-BiDi. The sample was at room temperature and the chromatogram was recorded every 4 hours. Two main peaks are marked as A-PI (48 min) and A-PII (83 min). (B) Plot of the ratio of peak areas A-PII/A-PI as a function of time. | ||
The reaction between C-Au38(2-PET)24 and R/S-BiDi shows similar behavior as shown in Fig. S7.† A new peak at a retention time of 115 min (C-PII) is evident. Another peak (C-PI) appears as a shoulder peak around 80 min partly overlapping with the parent cluster. From the evolution of the peaks with time it becomes clear that C-PII is the predominant peak. Further peaks appear as these reactions progress at 60 min and 110 min (Fig. 3) from A-Au38(2-PET)24, and at 95 min and 125 min (Fig. S7†) from C-Au38(2-PET)24. The peaks are proposed to belong to exchange species with two BiDi molecules in their ligand shell.
The diastereoselective ligand exchange process can also be evidenced by circular dichroism (CD), as showed in Fig. S9.† CD spectra of A-Au38(2-PET)24 during ligand exchange with R/S-BiDi have been recorded over time. The normalized spectra (at 375 nm, −1) clearly showed changes with reaction time. In order to investigate the evolution of optical activity, the concentration-independent anisotropy factors g = θ[mdeg]/(32980 × A) were calculated (Fig. S10†). With longer reaction time, the anisotropy factor of the sample increased at 360 nm and 490 nm; in addition, a new peak appeared at around 570 nm. As we stated before, the free R/S-BiDi molecule is not optically active (Fig. S2†), so the changes observed in the CD spectra result from the ligand exchange of R/S-BiDi on the cluster. The change in the optical activity is due to the fact that the two formed diastereomers A-Au38(2-PET)22(R-BiDi)1 and A-Au38(2-PET)22(S-BiDi)1 have different CD spectra and in addition they are not equally abundant. Furthermore, instead of A-Au38(2-PET)24, achiral Au25(2-PET)18 clusters were used for the ligand exchange reaction with R/S-BiDi (Fig. S11†). After the ligand exchange reaction the clusters were passed through a SEC column and the composition of different fractions was revealed with MALDI-TOF (Fig. S11A†), which indicated that fraction 1 and fraction 2 mainly contain Au25 clusters with one R/S-BiDi ligand in their ligand shell. As expected the corresponding CD spectra show no signal (Fig. S11B†).
In order to assign the predominant diastereomer, A-Au38(2-PET)22(S-BiDi)1 or A-Au38(2-PET)22(R-BiDi)1 (C-Au38(2-PET)22(R-BiDi)1 or C-Au38(2-PET)22(S-BiDi)1), Density Functional Theory (DFT) was used to calculate the corresponding energy of the four clusters as illustrated in Fig. 2. Here, –SCH3 was used as the ligand to reduce computational cost. Table 1 gives the energies of the different stereoisomers, relative to the most stable one, C-Au38(SCH3)22(S-BiDi)1. From the relative energy of those four stereoisomers, it can be inferred that A-Au38(2-PET)22(R-BiDi)1 is more stable than A-Au38(2-PET)22(S-BiDi)1 and C-Au38(2-PET)22(S-BiDi)1 is more stable than C-Au38(2-PET)22(R-BiDi)1. We note that the calculated energy of the enantiomeric structures is not identical, which is due to the slightly different orientation of the –CH3 groups in the corresponding structures after DFT optimization. It is important to note that the experiment shows that the rate constants for the formation of the two possible cluster diastereomers are different while the DFT calculations refer to the relative stability of the diastereomers.
| Enantiomer | Cluster | Potential energy |
|---|---|---|
| A-Au38 | A-Au38-R-BiDi | 0.032 eV |
| A-Au38-S-BiDi | 0.128 eV | |
| C-Au38 | C-Au38-R-BiDi | 0.100 eV |
| C-Au38-S-BiDi | 0.0 eV |
To support this assignment, CD spectroscopy was used. We collected the predominant cluster species in HPLC experiments (clusters from A-PII (Fig. 3) and C-PII (Fig. S7†)) and recorded the corresponding CD spectra separately (Fig. 4A). CD spectra were then calculated using a model Au38(SCH3)24 cluster instead of Au38(2-PET)24 used in the experiments. This approximation was justified by the good agreement between the experimental CD spectra of enantiopure Au38(2-PET)24 (Fig. S12A†) and the calculated CD spectra of Au38(SCH3)24 clusters (Fig. S12B†). As shown in Fig. 4A, upon exchange with R/S-BiDi the band at 350 nm (band f in the figures) became more dominant with respect to the 400 nm band, such that the latter became a shoulder peak.
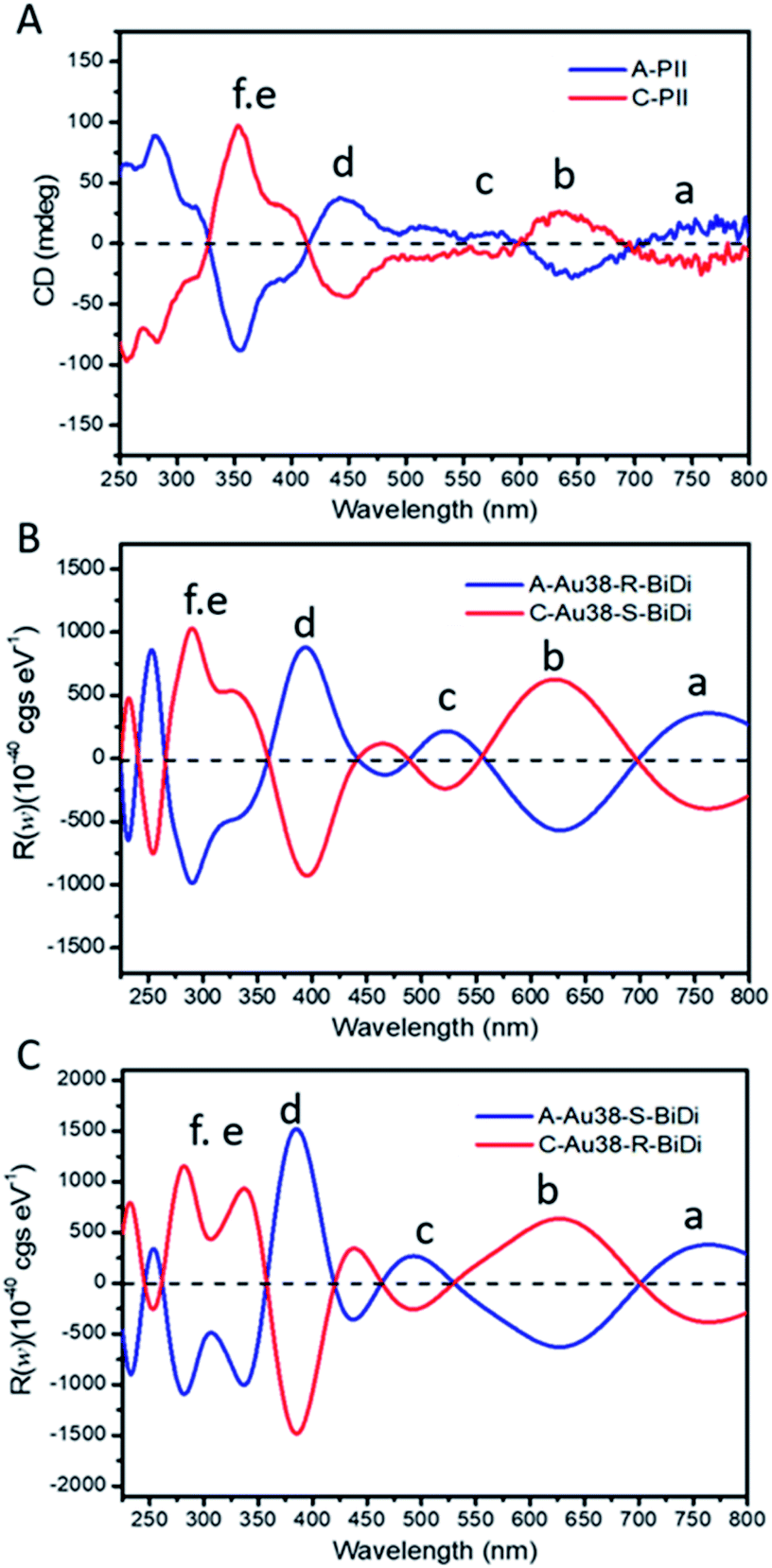 | ||
| Fig. 4 Experimental and calculated CD spectra of Au38 species. (A) Experimental samples were collected from A-PII and C-PII, (B) calculated CD spectra of A-Au38(SCH3)22(R-BiDi)1 and C-Au38(SCH3)22(S-BiDi)1, and (C) calculated CD spectra of A-Au38(SCH3)22(S-BiDi)1 and C-Au38(SCH3)22(R-BiDi)1. Calculations were carried out via the real-time propagation time-dependent DFT method using the GPAW code.44 For the calculations 2-PET ligands were substituted by –SCH3. | ||
The salient chiroptical signal at 280 nm corresponds to absorption of free R/S-BiDi (Fig. 1B). CD spectra calculated for the more stable species A-Au38(SCH3)22(R-BiDi)1 and C-Au38(SCH3)22(S-BiDi)1 are shown in Fig. 4B. These spectra match very well with the experimental CD spectra of the dominant Au38(2-PET)22(R/S-BiDi)1 species in the ligand exchange experiment apart from a shift of maximum 50 nm. The calculated CD spectra of the minor species (according to the calculated energies) A-Au38(SCH3)22(S-BiDi)1 and C-Au38(SCH3)22(R-BiDi)1 are shown in Fig. 4C. The match with the experimental CD spectra in Fig. 4A is less good compared to the CD spectra of the more stable species, particularly in the region between 300 nm and 400 nm (bands d–f). Note that we were not able to record the CD spectra of the minor species (peaks A-PI and C-PI) due to the very low amount of sample and the peak overlaps in the HPLC separation.
In order to highlight the influence of the chiral ligand on the chiroptical properties, we subtracted the CD spectra of the pure Au38 cluster from those of the corresponding Au38(SR)22(R/S-BiDi)1 species both for the experimental and for the calculated spectra (Fig. S12A† and 4A for experimental spectra and S12B† and 4B for the calculated spectra). These difference spectra show still good agreement between experiments (Fig. S12C†) and calculation (Fig. S12D†). Based on the analysis above we assign the major peaks in the HPLC experiments, A-PII and C-PII, respectively, to A-Au38(2-PET)22(R-BiDi)1 and C-Au38(2-PET)22(S-BiDi)1, respectively. As a consequence, the minor peaks in the HPLC experiments A-PI and C-PI, respectively, correspond to A-Au38(2-PET)22(S-BiDi)1 and C-Au38(2-PET)22(R-BiDi)1, respectively. Therefore, R-BiDi preferentially exchanges with anti-clockwise A-Au38 and S-BiDi prefers the clockwise C-Au38 cluster. This is consistent with the diastereoselective ligand exchange between Au38 and BINAS, where the same preferences (R-BINAS prefers A-Au38) were found.28
With these assignments at hand we can now focus on dynamic processes of the ligand exchanged species Au38(2-PET)22(R/S-BiDi)1, namely racemization of the cluster framework and intercluster ligand exchange, with a focus on the configuration of R/S-BiDi during these processes. Intercluster ligand exchange of the dithiol BINAS takes place at 70 °C but not at room temperature as has been shown recently.36 Note that this is in contrast to monothiols like 1-butanethiol and 2-PET which easily exchange at room temperature.13 Here we studied the behavior of R/S-BiDi in intercluster ligand exchange at room temperature and at 70 °C. For this purpose, Au25(2-PET)18 and Au38(2-PET)24-2x(R/S-BiDi)x were mixed (average x = 1.95), then the sample was kept at 70 °C for 24 h followed by recording MALDI-TOF (Fig. S13†). A reference sample was kept at room temperature. The conclusion that intercluster R/S-BiDi ligand exchange needs elevated temperatures was clearly evidenced by the observation of Au25(2-PET)16(R/S-BiDi)1 at 70 °C and its absence at room temperature. However, we should mention that even at 70 °C the ligand exchange is not very fast considering that on average 0.42 R/S-BiDi ligands were observed on the Au25 clusters after 24 h while the initial Au38 sample contained on average 1.95 R/S-BiDi ligands.
In order to study the dynamic processes of Au38(2-PET)22(R/S-BiDi)1 (ligand exchange, racemization), we collected C-Au38(2-PET)22(S-BiDi)1 in several HPLC runs (peak C-PII, Fig. S7†). As illustrated in Fig. 5 this species can be isolated and is stable at room temperature as re-injection into the HPLC system shows only one major peak besides a trace amount of C-Au38(2-PET)24 (small peak at 80 min likely due to incomplete initial separation). The sample was then heated to 70 °C and HPL chromatograms were measured over time. After 18 hours, a new peak with a retention time of around 50 min was observed corresponding to peak A-PI (A-Au38(2-PET)22(S-BiDi)1), and the intensity of the new peak increased with heating time. However, there was no visible peak at 83 min (A-PII) which would indicate A-Au38(2-PET)22(R-BiDi)1. These experiments clearly show that the system evolves as some of the clusters invert the handedness of the Au-S framework. However, during these processes the configuration of the R/S-BiDi ligand is retained, despite the fact that free R/S-BiDi easily racemizes.
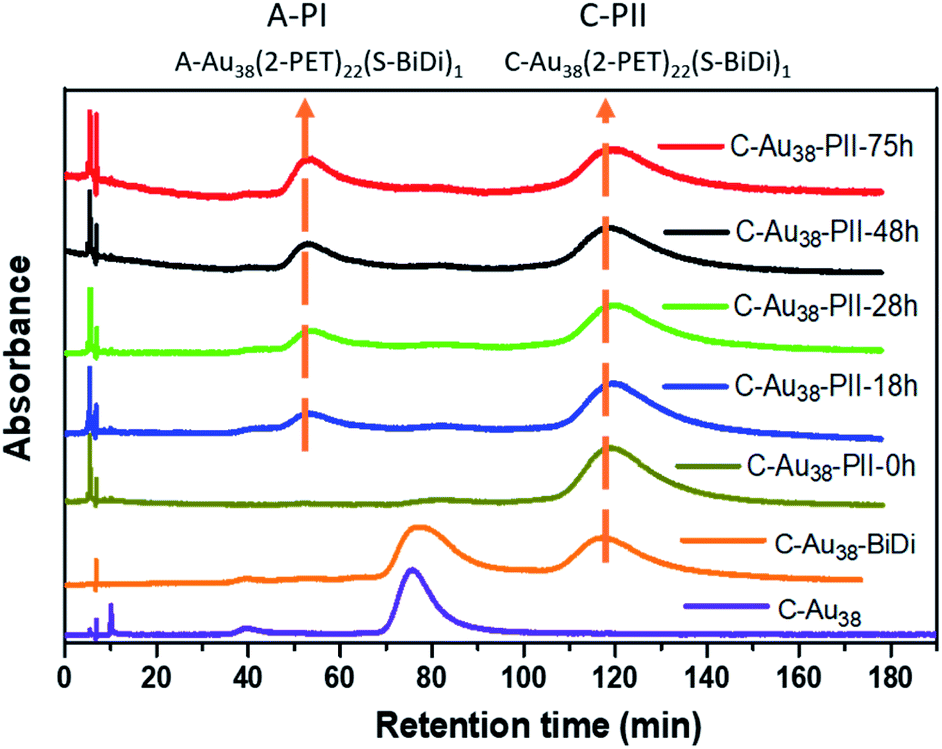 | ||
| Fig. 5 Evolution of chiral HPL chromatograms with time when heating C-Au38(2-PET)22(S-BiDi)1 (C-PII) to 70 °C. As a reference the chromatograms of C-Au38(2-PET)24 (bottom) and after ligand exchange with BiDi (second from bottom) are shown. The latter sample was used for the collection of C-Au38(2-PET)22(S-BiDi)1. | ||
We conclude that R/S-BiDi does not desorb from the cluster, which would cause the appearance of the other enantiomer of R/S-BiDi. In contrast, the conservation of the configuration of R/S-BiDi indicates that during the dynamic cluster processes the R/S-BiDi ligand is sufficiently confined to prevent inversion. An analogous experiment was performed with A-Au38(2-PET)22(R-BiDi)1 (Fig. S14†). After ligand exchange with A-Au38(2PET)24 and R/S-BiDi, A-PII was collected, the sample was heated to 70 °C, and the reaction was followed by HPL chromatography. Although in this case C-PI and A-PII peaks overlap, it is clear that with time peak C-PI appears, belonging to C-Au38(2-PET)22(R-BiDi)1 and no C-PII is visible at 120 min, which would indicate C-Au38(2-PET)22(S-BiDi)1. Hence, also in this experiment the absolute configuration of R/S-BiDi is retained.
The evolution of species as observed by HPLC (Fig. 5 and S14†) may involve several processes, notably the inversion of the Au-S framework of Au38(2-PET)22(R/S-BiDi)1 and the exchange of R/S-BiDi between clusters. The contribution of each process to the overall observation is difficult to judge. To explain the observations, we can propose two mechanisms as shown in Scheme 1 and both could actually contribute. Mechanism I is simply the inversion of the Au-S framework of the Au38(2-PET)22(R/S-BiDi)1 clusters (epimerization), for example C-Au38(2-PET)22(S-BiDi)1 is directly transformed into A-Au38(2-PET)22(S-BiDi)1. In mechanism II, the overall result is the same but it is a two-step mechanism involving ligand exchange. We note that in the experiments we see traces of A-Au38(2-PET)24 and C-Au38(2-PET)24 clusters, probably stemming from the HPLC separation/collection process. These clusters racemize very fast at 70 °C.37 This could be the main mechanism for the inversion of the Au-S framework. In addition, ligand exchange between clusters could take place: C-Au38(2-PET)22(S-BiDi)1 reacts with A-Au38(2-PET)24 to form A-Au38(2-PET)22(S-BiDi)1 and C-Au38(2-PET)24 (the latter can racemize easily).
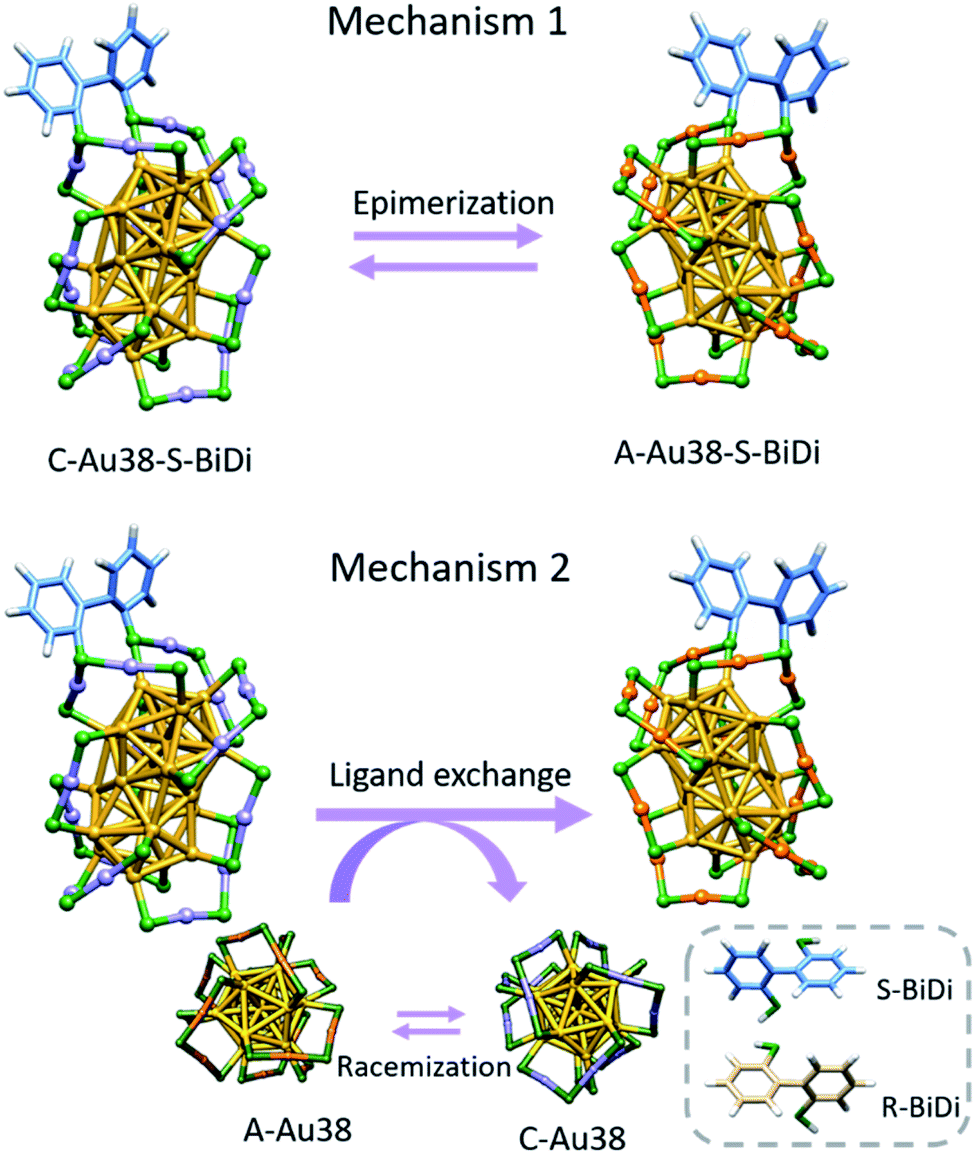 | ||
| Scheme 1 The evolution of the Au-S framework via simple inversion (M1) and a two-step mechanism (M2). For clockwise (counter-clockwise) clusters the color of the gold atom in staples is violet (orange). | ||
From the experimental data shown above it is clear that during the step involving the epimerization of Au38(2-PET)22(S-BiDi)1 and/or intercluster ligand exchange of R/S-BiDi, the absolute configuration of the ligand is retained. This excludes mechanisms where R/S-BiDi desorbs from the cluster (which would lead to its racemization, in contrast to observation), and furthermore indicates that during these processes the ligand is confined, which prevents its racemization. We note that this finding supports a mechanism where ligand exchange takes place during a collision between clusters within a dimeric species as suggested before.13,45 This ligand offers a powerful strategy to study molecular level details of dynamic processes involving thiolate-protected metal clusters.
Conclusion
In summary, ligand exchange with chiral dithiol (R/S-BiDi) has been shown to be diastereoselective. Calculations show that R-BiDi (thermodynamically) prefers A-Au38 clusters and S-BiDi is more stable on C-Au38 clusters. The same preference was found experimentally for the kinetics of the ligand exchange (R-BiDi reacts faster with A-Au38 clusters). More importantly, it was observed that the configurationally labile BiDi ligand retains its configuration during dynamic cluster processes i.e. cluster epimerization and/or intercluster ligand exchange. Racemization of the configurationally labile BiDi molecule is hindered after adsorption on the cluster surface. The absence of racemization of the ligand during epimerization of the cluster and/or ligand exchange shows that the ligand is confined during these processes and does not desorb into solution.Data availability
The data shown in the figures can be downloaded from https://doi.org/10.5281/zenodo.4959547.Author contributions
Thomas Bürgi designed the project. Yanan Wang carried out the experiments. Esko Makkonen and Xi Chen performed the DFT calculations. Xi Chen and Thomas Bürgi supervised the project. All authors discussed the results and contributed to writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TB acknowledges the generous support of the Swiss National Science Foundation (grants 200020_172511 and 200020_192232) and the University of Geneva. YW is thankful for a China Scholarship Council fellowship (201706450070). XC and EM are grateful for the financial support from the Academy of Finland, Projects 308647 and 314298, and for the computational resources provided by the CSC – IT Center for Science (Finland). We thank Raymond Azoulay for the synthesis of the ligand.Notes and references
- H. Qian, M. Zhu, Z. Wu and R. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 Search PubMed.
- T. Tsukuda, Bull. Chem. Soc. Jpn., 2012, 85, 151–168 Search PubMed.
- C. M. Aikens, Acc. Chem. Res., 2018, 51, 3065–3073 Search PubMed.
- G. Li and R. Jin, Acc. Chem. Res., 2013, 46, 1749–1758 Search PubMed.
- T. Kawawaki and Y. Negishi, Nanomaterials, 2020, 10, 238 Search PubMed.
- M. Zhou, T. Higaki, Y. Li, C. Zeng, Q. Li, M. Y. Sfeir and R. Jin, J. Am. Chem. Soc., 2019, 141, 19754–19764 Search PubMed.
- A. Sels, R. Azoulay, W. J. Buma, M. A. J. Koenis, V. P. Nicu and T. Bürgi, J. Phys. Chem. C, 2019, 123, 22586–22594 Search PubMed.
- Z. Wu, M. Wang, J. Yang, X. Zheng, W. Cai, G. Meng, H. Qian, H. Wang and R. Jin, Small, 2012, 8, 2028–2035 Search PubMed.
- T. Shu, J. Wang, L. Su and X. Zhang, Anal. Chem., 2016, 88, 11193–11198 Search PubMed.
- S. K. Katla, J. Zhang, E. Castro, R. A. Bernal and X. Li, ACS Appl. Mater. Interfaces, 2018, 10, 75–82 Search PubMed.
- D. Yang, G. Yang, S. Gai, F. He, G. An, Y. Dai, R. Lv and P. Yang, Nanoscale, 2015, 7, 19568–19578 Search PubMed.
- Z. Huang, Y. Ishida, K. Narita and T. Yonezawa, J. Phys. Chem. C, 2018, 122, 18142–18150 Search PubMed.
- G. Salassa, A. Sels, F. Mancin and T. Bürgi, ACS Nano, 2017, 11, 12609–12614 Search PubMed.
- E. S. Shibu, M. A. H. Muhammed, T. Tsukuda and T. Pradeep, J. Phys. Chem. C, 2008, 112, 12168–12176 Search PubMed.
- Y. Wang, B. Nieto-Ortega and T. Bürgi, Chem. Commun., 2019, 55, 14914–14917 Search PubMed.
- M. Kluenker, M. Mondeshki, M. N. Tahir and W. Tremel, Langmuir, 2018, 34, 1700–1710 Search PubMed.
- C. L. Heinecke, T. W. Ni, S. Malola, V. Mäkinen, O. A. Wong, H. Häkkinen and C. J. Ackerson, J. Am. Chem. Soc., 2012, 134, 13316–13322 Search PubMed.
- S. Knoppe and T. Bürgi, Phys. Chem. Chem. Phys., 2013, 15, 15816–15820 Search PubMed.
- T. W. Ni, M. A. Tofanelli, B. D. Phillips and C. J. Ackerson, Inorg. Chem., 2014, 53, 6500–6502 Search PubMed.
- Y. Niihori, Y. Kikuchi, A. Kato, M. Matsuzaki and Y. Negishi, ACS Nano, 2015, 9, 9347–9356 Search PubMed.
- V. Rojas-Cervellera, L. Raich, J. Akola and C. Rovira, Nanoscale, 2017, 9, 3121–3127 Search PubMed.
- L. Beqa, D. Deschamps, S. Perrio, A.-C. Gaumont, S. Knoppe and T. Bürgi, J. Phys. Chem. C, 2013, 117, 21619–21625 Search PubMed.
- X. Kang, H. Chong and M. Zhu, Nanoscale, 2018, 10, 10758–10834 Search PubMed.
- C. A. Fields-Zinna, J. F. Parker and R. W. Murray, J. Am. Chem. Soc., 2010, 132, 17193–17198 Search PubMed.
- Y. Niihori, W. Kurashige, M. Matsuzaki and Y. Negishi, Nanoscale, 2013, 5, 508–512 Search PubMed.
- P. Pengo, C. Bazzo, M. Boccalon and L. Pasquato, Chem. Commun., 2015, 51, 3204–3207 Search PubMed.
- S. Hossain, W. Kurashige, S. Wakayama, B. Kumar, L. V. Nair, Y. Niihori and Y. Negishi, J. Phys. Chem. C, 2016, 120, 25861–25869 Search PubMed.
- S. Knoppe, R. Azoulay, A. Dass and T. Bürgi, J. Am. Chem. Soc., 2012, 134, 20302–20305 Search PubMed.
- A. Fernando and C. M. Aikens, J. Phys. Chem. C, 2015, 119, 20179–20187 Search PubMed.
- A. Fernando and C. M. Aikens, J. Phys. Chem. C, 2016, 120, 14948–14961 Search PubMed.
- V. R. Jupally, R. Kota, E. V. Dornshuld, D. L. Mattern, G. S. Tschumper, D.-e. Jiang and A. Dass, J. Am. Chem. Soc., 2011, 133, 20258–20266 Search PubMed.
- B. Molina, A. Sánchez-Castillo, S. Knoppe, I. L. Garzón, T. Bürgi and A. Tlahuice-Flores, Nanoscale, 2013, 5, 10956–10962 Search PubMed.
- S. Knoppe, A. C. Dharmaratne, E. Schreiner, A. Dass and T. Bürgi, J. Am. Chem. Soc., 2010, 132, 16783–16789 Search PubMed.
- S. Knoppe, S. Michalet and T. Bürgi, J. Phys. Chem. C, 2013, 117, 15354–15361 Search PubMed.
- S. Si, C. Gautier, J. Boudon, R. Taras, S. Gladiali and T. Bürgi, J. Phys. Chem. C, 2009, 113, 12966–12969 Search PubMed.
- Y. Wang, B. Nieto-Ortega and T. Bürgi, Nat. Commun., 2020, 11, 4562 Search PubMed.
- S. Knoppe, I. Dolamic and T. Bürgi, J. Am. Chem. Soc., 2012, 134, 13114–13120 Search PubMed.
- S. Malola and H. Hakkinen, J. Am. Chem. Soc., 2019, 141, 6006–6012 Search PubMed.
- D. Fabbri, G. Delogu and O. De Lucchi, J. Org. Chem., 1993, 58, 1748–1750 Search PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 Search PubMed.
- H. Kessler, Angew. Chem., Int. Ed. Engl., 1970, 9, 219–235 Search PubMed.
- E. Masson, Org. Biomol. Chem., 2013, 11, 2859–2871 Search PubMed.
- I. Dolamic, S. Knoppe, A. Dass and T. Bürgi, Nat. Commun., 2012, 3, 798 Search PubMed.
- E. Makkonen, T. P. Rossi, A. H. Larsen, O. Lopez-Acevedo, P. Rinke, M. Kuisma and X. Chen, J. Chem. Phys., 2021, 154, 114102 Search PubMed.
- K. R. Krishnadas, A. Baksi, A. Ghosh, G. Natarajan, A. Som and T. Pradeep, Acc. Chem. Res., 2017, 50, 1988–1996 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01702k |
| This journal is © The Royal Society of Chemistry 2021 |