
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantification and molecular imaging of fatty acid isomers from complex biological samples by mass spectrometry†
Hua
Zhang
 a,
Meng
Xu
b,
Xudong
Shi
c,
Yuan
Liu
a,
Zihui
Li
b,
Justin C.
Jagodinsky
d,
Min
Ma
a,
Nathan V.
Welham
c,
Zachary S.
Morris
d and
Lingjun
Li
*ab
a,
Meng
Xu
b,
Xudong
Shi
c,
Yuan
Liu
a,
Zihui
Li
b,
Justin C.
Jagodinsky
d,
Min
Ma
a,
Nathan V.
Welham
c,
Zachary S.
Morris
d and
Lingjun
Li
*ab
aSchool of Pharmacy, University of Wisconsin–Madison, Madison, WI 53705, USA. E-mail: lingjun.li@wisc.edu
bDepartment of Chemistry, University of Wisconsin–Madison, Madison, WI 53706, USA
cDivision of Otolaryngology, Department of Surgery, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, WI 53792, USA
dDepartment of Human Oncology, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, WI 53705, USA
First published on 4th May 2021
Abstract
Elucidating the isomeric structure of free fatty acids (FAs) in biological samples is essential to comprehend their biological functions in various physiological and pathological processes. Herein, we report a novel approach of using peracetic acid (PAA) induced epoxidation coupled with mass spectrometry (MS) for localization of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in unsaturated FAs, which enables both quantification and spatial visualization of FA isomers from biological samples. Abundant diagnostic fragment ions indicative of the CC positions were produced upon fragmentation of the FA epoxides derived from either in-solution or on-tissue PAA epoxidation of free FAs. The performance of the proposed approach was evaluated by analysis of FAs in human cell lines as well as mapping the FA isomers from cancer tissue samples with MALDI-TOF/TOF-MS. Merits of the newly developed method include high sensitivity, simplicity, high reaction efficiency, and capability of spatial characterization of FA isomers in tissue samples.
C bond in unsaturated FAs, which enables both quantification and spatial visualization of FA isomers from biological samples. Abundant diagnostic fragment ions indicative of the CC positions were produced upon fragmentation of the FA epoxides derived from either in-solution or on-tissue PAA epoxidation of free FAs. The performance of the proposed approach was evaluated by analysis of FAs in human cell lines as well as mapping the FA isomers from cancer tissue samples with MALDI-TOF/TOF-MS. Merits of the newly developed method include high sensitivity, simplicity, high reaction efficiency, and capability of spatial characterization of FA isomers in tissue samples.
Introduction
Structural diversity and varying expression levels of fatty acids (FAs) are closely related to their unique roles in cell metabolic pathways. Positional isomers of FA with different carbon–carbon double bond (CC) positions possess considerably different roles in various biological functions.1–3 Metabolic dysregulations of FA CC bond positional isomers may contribute to the pathogenesis of cancer,4 cardiovascular diseases,5 Alzheimer's and other neurodegenerative diseases.6 Therefore, precise elucidation of FA isomeric structures in biological samples is critical for a deeper understanding of their biological functions.
Mass spectrometry (MS) is emerging as one of the most informative techniques for the study of FAs due to its superior analytical power.7–9 Although significant advancements have occurred in lipidomics research using various MS techniques including shotgun MS10 and hyphenated chromatographic MS techniques (LC/GC-MS),7 challenges remain in elucidating the fine structures of FA CC bond positional isomers. Conventional low-energy collisional activation methods, such as collision-induced-dissociation (CID) tandem MS, are ineffective in recognizing the CC bond positions in FA isomers due to the high energy required to cleave the CC bond.11,12 To circumvent this limitation, extensive efforts have been devoted to achieve unambiguous identification of FA isomers from biological samples in recent years.13–17 For instance, alternative ion activation methods14,16,18–22 have been explored to determine lipid isomers owing to the great advancements in instrumentation. However, these methods usually require specific MS instrument modifications or specialized instrumentation. Moreover, cleaving the CC bond with high fragmentation energy is usually accompanied by the breakage of carbon–carbon single bonds, which increases the complexity of the spectra, and lowers the abundance of diagnostic ions.18,23
Another emerging strategy to locate the CC bond position is implementation of a CC derivatization procedure prior to CID MS/MS interrogation. CC bond chemical derivatization methods based on the Paternò–Büchi (PB) reactions,13,15,24,25 ozonolysis,26,27meta-chloroperoxybenzoic acid (m-CPBA)-epoxidation,28,29 and plasma/electrochemical induced epoxidation,30–33 have been utilized to induce structurally informative dissociation of FAs in CID MS/MS in recent years. Additionally, covalent adduct chemical ionization (CACI) GC MS/MS34 and gas-phase ion/ion reactions35 have been adopted for the characterization of isomeric FAs. Nevertheless, most of these approaches are performed under in-solution (bulk) conditions, therefore sacrificing FA spatial information. Visualization of FA isomers across tissue samples provides critical information to better understand their region-specific functions. It is noted that the Paternò–Büchi reaction,15,17,36 ozone-induced dissociation (OzID),16 ozonolysis37 and m-CPBA epoxidation29 have been adopted for MS-based mapping of lipid isomers on tissues, however, challenges still remain including low yields, side reactions, and interference from excess on-tissue derivatization reagent. Therefore, the development of novel methodologies for spatial mapping and quantification of FA isomers within biological samples is still in great demand.
Herein, we report a novel approach of using peracetic acid (PAA)-induced epoxidation in MS for localization of CC bond in unsaturated FAs, which enables both quantification and spatial visualization of FA isomers from biological samples without need for instrumental modifications and can be broadly transferrable to various MS platforms. The performance of the proposed approach was demonstrated by analysis of FA isomer standards, FAs in HPV16-E6E7 immortalized human pancreatic duct epithelial (HPDE/E6E7) cells and pancreatic cancer cells (PANC-1) using shotgun MS, as well as mapping of FA isomers from cancer tissues based on a matrix-assisted laser desorption/ionization time-of-flight/time-of-flight MS (MALDI-TOF/TOF-MS) platform. In these experiments, FA isomers were unambiguously and sensitively identified and quantified. Additionally, the heterogeneity of FA isomers was mapped on tumor tissue sections from a murine tumor model. Overall, our results indicate that the PAA epoxidation coupling with MS approach provides a versatile platform for elucidating the fine structures of FAs, which help to better understand their physiological functions.
Results and discussion
Localization C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C bond in FA by PAA epoxidation coupled with tandem MS
C bond in FA by PAA epoxidation coupled with tandem MS
The mechanism of pinpointing CC bond in FAs by PAA-initiated epoxidation and tandem MS is shown in Fig. 1. The unsaturated FAs were converted into FA epoxides after the treatment of PAA under either in-solution reaction or on-tissue epoxidation using the vapor of PAA (Fig. S1†). The epoxy-FAs were subsequently subjected to tandem MS fragmentation, under which the three-membered oxirane ring derived from the CC bond was readily cleaved yielding an aldehyde and an olefin fragment ion pair. Affirmative CC bond positional information was then deduced from the characteristic fragment ion pair.
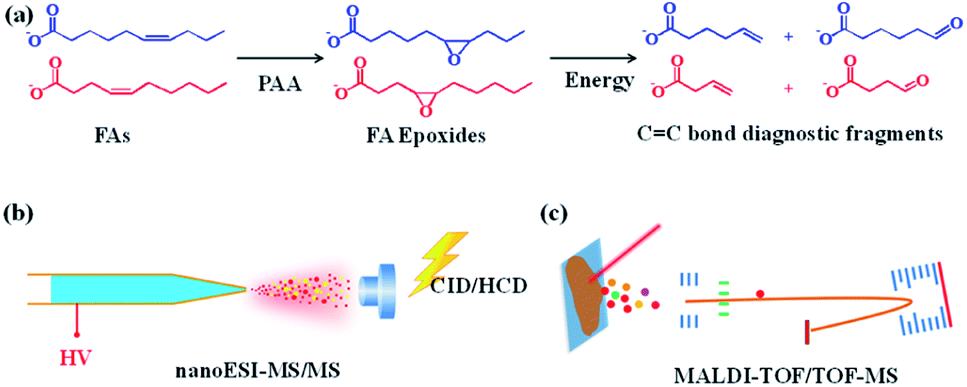 | ||
| Fig. 1 PAA epoxidation used in conjunction with MS for pinpointing CC bond in FAs. (a) Schematic illustration of pinpointing CC position in unsaturated FAs using PAA epoxidation coupled with tandem MS. MS interrogation method for (b) bulk samples and (c) MS imaging of tissue sections. | ||
To test the feasibility of this approach, in-solution epoxidation was conducted using unsaturated FA standards upon reaction with PAA and the epoxide products were subjected to nanoESI-MS/MS analysis. The unsaturated FAs were completely converted into their epoxy forms after the PAA treatment (Fig. S2†). To further confirm the structure of these epoxide products, tandem MS experiments were performed. As shown in Fig. 2, abundant diagnostic fragment ion pairs indicative of the CC positions were obtained in the CID MS/MS. Additionally, HCD tandem MS was performed for the interrogation of these epoxide products using varied normalized collision energy (NCE) (Fig. S3 and S4†). Although diagnostic fragment ions indicating the CC were observed using both fragmentation methods, diagnostic fragment ions with relatively higher abundance, as well as cleaner mass spectra, were obtained using CID mode at NCE of 35%, presumably due to preferential cleavage of the three-membered oxirane ring structure. Furthermore, polyunsaturated FAs including FA 18:3 (6Z, 9Z, 12Z) and FA 20:4 (5Z, 8Z, 12Z, 14Z) were analyzed with their CC bond positions deduced from their diagnostic fragments, despite having more complex MS/MS spectra (Fig. S5†).
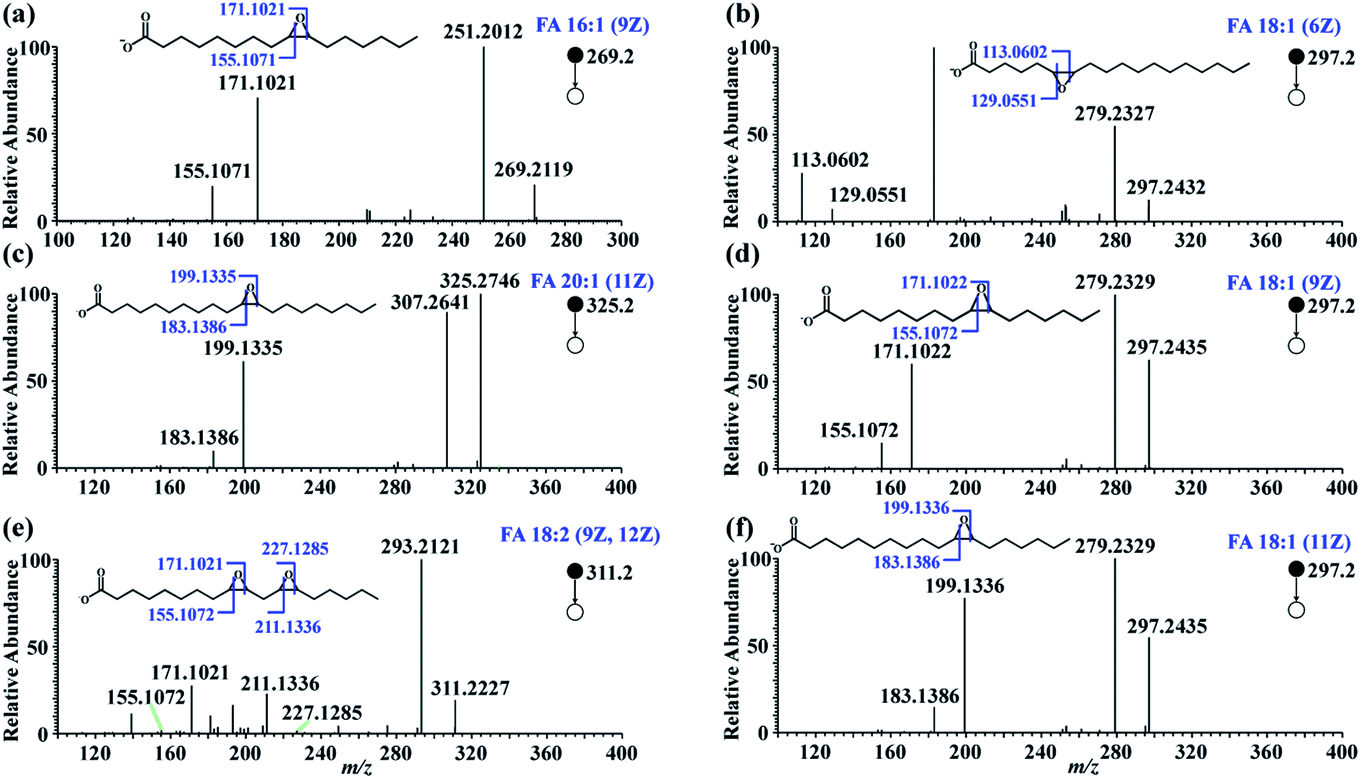 | ||
| Fig. 2 MS/MS spectra of FA epoxides after the in-solution epoxidation of unsaturated FAs: (a) FA 16:1 (9Z), (b) FA 18:1 (6Z), (c) FA 20:1 (11Z), (d) FA 18:1 (9Z), (e) FA 18:2 (9Z, 12Z), (f) FA 18:1 (11Z). | ||
Quantitative analysis of CC bond positional FA isomers
The analytical performance of the proposed method was further examined by quantitatively investigating FAs with different CC bond locations. As shown in Fig. S6,† satisfactory linearity was attained for FA 16:1 (9Z), FA 18:1 (9Z), and FA 18:1 (11Z) over a dynamic range of 0.05–10.0 µM, with R2 values > 0.9989. The limit of detection (LOD), defined by S/N = 3, was estimated to be as low as 4.4 nM for FA 16:1 (9Z), 5.6 nM for FA 18:1 (9Z), and 4.2 nM for FA 18:1 (11Z). The relative standard deviations (RSDs) (n = 3) from all the examined samples were below 5.5%, 3.4%, and 8.9% for FA 16:1 (9Z), FA 18:1 (9Z), and FA 18:1 (11Z), respectively. These data show that the proposed method exhibits higher sensitivity for the detection of FA isomers than recently reported methods,24,32,33 which attributes to the high labeling efficiency and high yield of diagnostic fragment ions.
Relative quantification of FA isomers was also performed by analyzing a series of mixture solutions of FA 18:1 (9Z) and FA 18:1 (11Z), with varied molar ratios from 0.001![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 10:1, at a total concentration of 35 µM. Diagnostic fragment ion pairs from FA 18:1 (9Z) and FA 18:1 (11Z) were observed from the MS/MS spectrum of the isomer mixture (Fig. S7†). As illustrated in Fig. S6d,† satisfactory linearity with R2 = 0.997 was achieved over a dynamic range of molar ratios from 0.001:1 to 10:1. The RSDs of three replicates for all the examined samples were consistently below 6%.
:1 to 10:1, at a total concentration of 35 µM. Diagnostic fragment ion pairs from FA 18:1 (9Z) and FA 18:1 (11Z) were observed from the MS/MS spectrum of the isomer mixture (Fig. S7†). As illustrated in Fig. S6d,† satisfactory linearity with R2 = 0.997 was achieved over a dynamic range of molar ratios from 0.001:1 to 10:1. The RSDs of three replicates for all the examined samples were consistently below 6%.
Analysis of unsaturated FAs in cell-line samples
To test the applicability of the proposed method in complex biological samples, FAs were extracted from human pancreatic duct epithelial cells (HPDE cells), pancreatic cancer cells (PANC-1 cells), and PANC-1 cells treated with stearoyl-CoA desaturase 1 (SCD1) inhibitor A93957238 – a drug that inhibits conversion of saturated fatty acyl-CoA to monounsaturated fatty acyl-CoAs. Following PAA epoxidation, FA samples were subjected to nanoESI-MS/MS analysis. A total of 37 monounsaturated FAs with definitive CC bond positions were identified (Table S1†), showing a high heterogeneity of the FA isomeric composition in these pancreatic cell samples. Interestingly, the proportions of different FA isomers exhibited significant variation between the HPDE and PANC-1 cells (Fig. 3). FA 16:1 (Δ9) was observed as the major isomer for FA 16:1 in both the HPDE and PANC-1 cells, while its proportion decreased from 80.7 ± 1.9% for HPDE cells to 33.1 ± 6.5% for PANC-1 cells (Fig. 3a). A similar decrease in proportion was found for the major isomers of FA 18:1 (Δ9) and FA 18:1 (Δ11) in PANC-1 cells (Fig. 3b). In contrast, increased proportions of some low abundance isomers were observed for FA 16: 1, FA 18:1 and FA 20:1 in the PANC-1 cells. These findings suggest that important FA metabolic pathway alterations may exist in the cancer cell line compared to its normal counterpart. In our proof-of-principle study, the FAs isomers from cell-line samples were tentatively assigned based on high resolution tandem MS data (with tight mass error within 5 ppm). Note that the confidence of isomeric identification of the low abundance FA isomers could be further increased by combining with other lipid CC double bond characterization methods such as PB reactions,17,24 covalent adduct chemical ionization (CACI) GC MS/MS,34 ozone-induced dissociation.19 Unfortunately, these techniques usually require specific MS instrument modifications or specialized instrumentation, which are not directly available on our current hi-res MS instrument. Nonetheless, these methods can be exploited for more confident FAs isomeric identification in future studies.
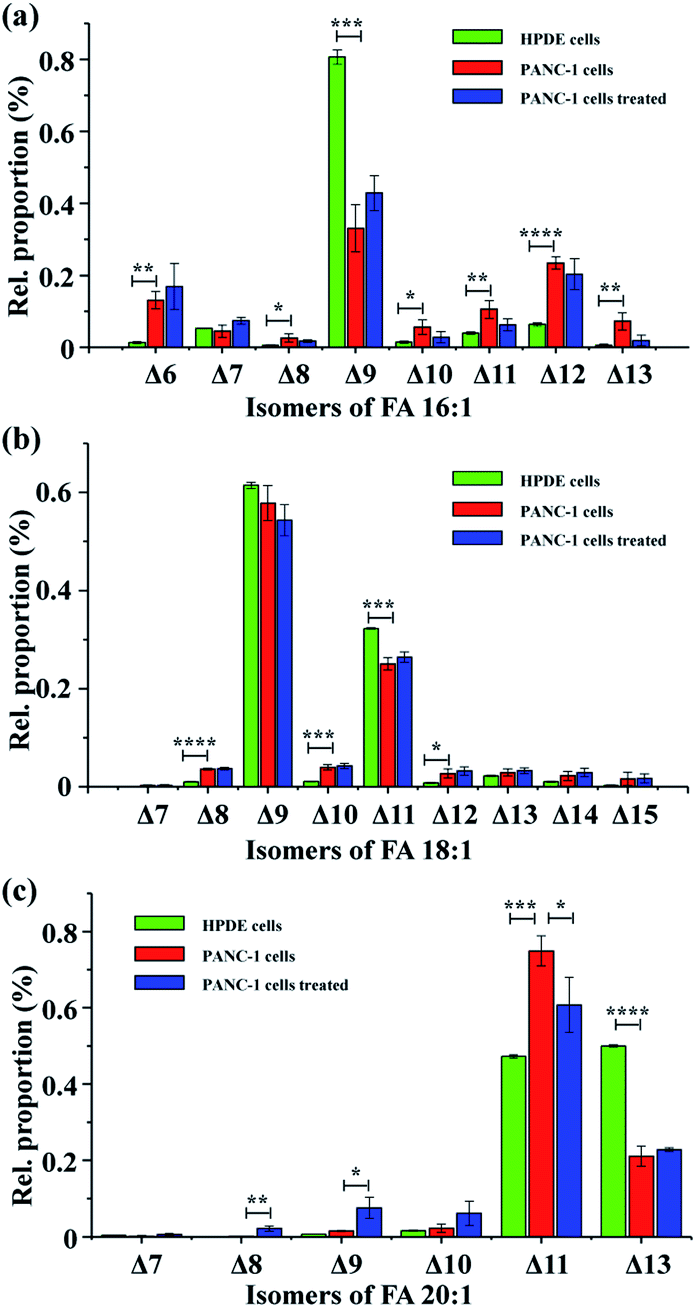 | ||
| Fig. 3 Analysis of CC positional isomers for three major mono-unsaturated FA in pancreatic cells. (a) FA 16:1, (b) FA 18:1, (c) FA 20:1. The green, red, and blue bars represent data obtained from HPDE cells, PANC-1 cells, and PANC-1 cells treated with SCD1 inhibitor A939572, respectively. Differences between the two groups were evaluated using the two-tailed Student's t tests, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. n = 3. | ||
Further, PANC-1 cells dosed with 200 nM SCD1 inhibitor A93957238 were tentatively investigated. No significant change was observed in the isomeric compositions for FA 16:1 and FA 18:1, while several isomers from FA 20:1 were notably different after the 24 hour drug treatment.38 Inhibition of SCD1 has been found to effectively suppress proliferation and induce apoptosis of tumor cells.38 The limited FA metabolic change in PANC-1 cells after the drug treatment in the current case might be due to an insufficient drug treatment period, as the 24 hour treatment did not yield a significant change in cell density (data not shown). It is worth mentioning that although the CC bond positions could be obtained for polyunsaturated FA standards (Fig. S5†), the in-depth analysis of polyunsaturated FAs in complex biological samples would benefit from additional chromatographic separation29,33 to achieve more accurate identification of CC bond position. A related study is currently underway in our laboratory.
Spatial characterization of CC positional isomers using MALDI-TOF/TOF-MS/MS
We further applied the PAA epoxidation strategy to map the spatial distribution of FA isomers within tissue samples using a matrix-assisted laser desorption/ionization time-of-flight tandem mass spectrometry (MALDI-TOF/TOF MS) platform. MALDI-TOF/TOF MS imaging was performed on tumor tissue sections from a murine melanoma model. The PAA incubation time was set to 2 h to ensure complete epoxidation of unsaturated FAs (Fig. S8†). As shown in Fig. 4, the spatial distribution of FA 18:0 at m/z 283.2, and epoxy-FA 18:1 at m/z 297.2 obtained from a tissue section pretreated with PAA epoxidation (Fig. 4 c and d) were in line with that of FA 18:0 at m/z 283.2, and FA 18:1 at m/z 281.2 from an adjacent underivatized tissue section (Fig. 4 a and b). These results show that the on-tissue epoxidation reaction process has neither dislocated the unreacted saturated FAs (such as FA 18:0), nor altered the distribution of reacted FA species, providing accurate spatial distribution of the target analytes. The untreated control (left side section) and the radiation-treated tissue (right side) exhibit the same spatial pattern in that lipid signal intensity (FA 18:1 and FA 18:0) is stronger in the peripheral tissue region rich in cancer cells compared to the necrotic inner tissue region that contains fewer cancer cells (Fig. 4 a and b). Receiver Operating Characteristic (ROC) tests of the intensity of FA 18:1 between the radiated and control tissues resulted in an area under the curve (AUC) value of 0.647 (Fig. S9†), which indicates a slight decrease in FA 18:1 after radiation treatment. We successfully mapped 5 isomers of FA 18:1 with MALDI-TOF/TOF MS/MS imaging targeted at epoxy-FA 18:1 (Fig. 4 e–i and S10†). One isomer (FA18:1(Δ9)) showed significantly decreased signal intensity in the cancer region after radiation treatment compared to untreated samples (Fig. 4h and S11†). This demonstrates the biological significance of detecting FA isomers on tissue sections.
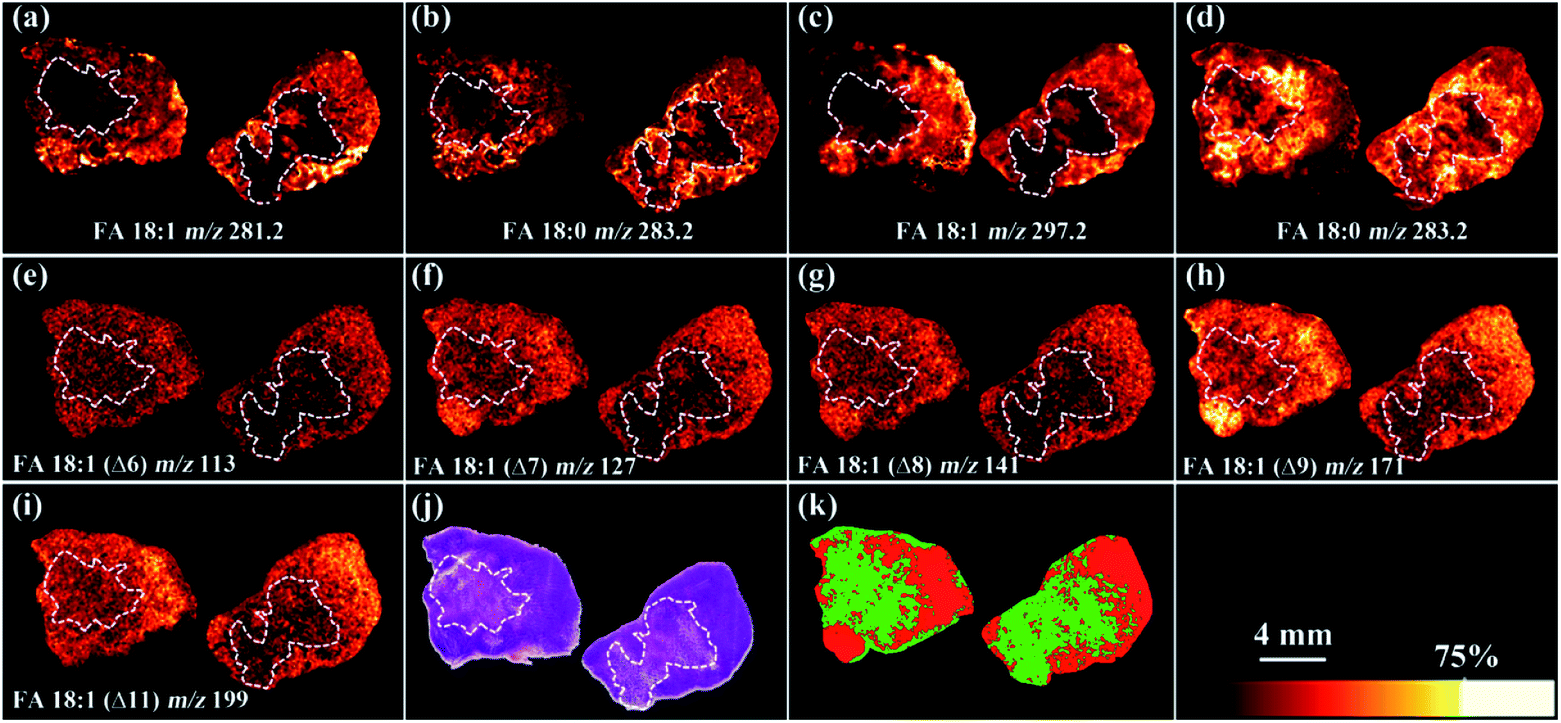 | ||
| Fig. 4 Full-MS images of (a) FA 18:1 and (b) FA 18:0 without epoxidation; full-MS images of (c) epoxy-FA 18:1 and (d) FA 18:0 after on-tissue epoxidation treatment. MALDI-TOF/TOF-tandem MS images (e) FA 18:1 (Δ6), (f) FA 18:1 (Δ7), (g) FA 18:1 (Δ8), (h) FA 18:1 (Δ9), (i) FA 18:1 (Δ11). (j) The H&E stained histological image. (k) Segmentation pipeline analysis of the MS2 data: region rich in cancer cells (red) and region containing few cancer cells with necrosis (green). The left side tissue section was from untreated sample while the right-side tissue section was from sample treated with radiation in all the images. Mass tolerance of all the full-MS images is ±0.05 Da whereas that of ±0.1 Da for MS/MS images. | ||
A segmentation pipeline was employed to segment the MS/MS data into two distinct regions, which were tentatively identified as tumor and necrotic regions after comparing with the histological image (Fig. 4j and k). Based on the segmentation and histology results, we found that downregulation of FA 18:1 isomers was tightly associated with tumor necrosis. Specifically, FA 18:1 (Δ9) yielded AUC value of 0.964 in comparisons between the tumor and necrotic regions (Fig. S12†), suggesting that the concentration of FA 18:1 (Δ9) was notably reduced in necrosis. This result highlights the potential value of FAs as downstream biomarkers that reflect metabolic alterations linked to the progression of tumor necrosis.
It is worth mentioning that m-CPBA epoxidation method was also adopted for pinpointing CC bond positions in unsaturated lipid recently.28,29 Both the m-CPBA and PAA strategies have features such as high sensitivity, high specificity, and simplicity with no need for instrumental modifications. In comparison with the m-CPBA epoxidation, the advances of PAA epoxidation over the m-CPBA treatment are also obvious. (i) For in-solution derivatization, it is much easier for sample clean-up with PAA as the excess PAA in the reaction system could be easily removed through a SpeedVac owing to its high volatility, whereas m-CPBA is a nonvolatile reagent which can be difficult to remove from the sample using a SpeedVac and needs much more tedious sample preparation steps to clean-up excess m-CPBA. (ii) For on-tissue epoxidation, a notable advantage of PAA is minimal chemical contamination. Since PAA rapidly evaporates from the tissue after incubation due to its high volatility, potential side effects such as ion suppression for subsequent MS imaging analysis are greatly minimized. However, a layer of m-CPBA would be coated on the tissue after spraying the m-CPBA reagent onto the tissue (Fig. S13†). The excess m-CPBA on the tissue would oxidize the MALDI matrix and suppress the signal of derivatization products. A comparison between the PAA and m-CPBA methods using MALDI-TOF/TOF MS/MS under consistent instrumental parameters showed that the signal was 6-fold higher in the PAA treated samples compared to the m-CPBA treated samples at the MS2 level (Fig. S14†). (iii) Another advance of PAA over m-CPBA is the on-tissue epoxidation found to be much faster when using PAA. It takes less than 2 hours to reach a nearly 100% epoxidation efficiency for the unsaturated FAs with PAA (Fig. S8†), whereas the m-CPBA strategy needs overnight incubation (ca. 18 h) in a sealed humidity chamber to achieve nearly 100% reaction efficiency.
Conclusions
In summary, a versatile approach for the structural characterization of FA CC bond isomers via PAA epoxidation coupled with tandem MS has been developed in this study, which enables both quantification and spatial mapping of the distribution of FA isomers from biological samples. Accurate localization of CC bond position in FAs has been achieved based on the characteristic diagnostic fragments yielded by the epoxidated FAs under tandem MS conditions such as CID, HCD on an ESI Orbitrap platform and a MALDI-TOF/TOF tandem MS platform. A PAA epoxidation reaction of CC bond from unsaturated FAs can be implemented through either in-solution or on-tissue with close to 100% labeling efficiency. The data show that the proposed method exhibits higher sensitivity than recently reported methods for the detection of FA isomers. Furthermore, our method does not need any MS instrumental modification which offers general accessibility as well as compatibility with commercial mass spectrometers. Overall, the merits of the newly developed method include excellent sensitivity, no MS instrument modification requirements, high reaction efficiency, and the provision of spatially resolved FA isomeric information on tissue samples. These advances will facilitate research endeavors for fine structural elucidation and characterization of FAs in structural lipidomics. It is noted that, although the PAA epoxidation coupled with tandem MS strategy is feasible for the identification of both monounsaturated and polyunsaturated FAs, we note that the PAA epoxidation method provides easier interpretation for monounsaturated FAs than polyunsaturated FAs, as the tandem mass spectra of epoxy-polyunsaturated FAs become more complex. To facilitate the identification process, establishing a spectral library and data searching interface for automated matching and annotation of the diagnostic fragment ions will be explored in the future to further improve the identification accuracy of polyunsaturated FAs.
Experimental
Materials and reagents
For details about the materials and reagents used in the study, refer to the ESI.†Pancreatic cell-lines
The commercial pancreatic cancer cell line PANC-1 (ATCC® CRL-1469) obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) was cultured and maintained in DMEM:F12 (Hyclone, VWR International, Radnor, PA, USA) containing 10% fetal bovine serum (FBS) (Gibco, Thermo Fischer, Waltham, MA, USA), 1% penicillin–streptomycin solution (Gibco, Thermo Fischer Scientific, Waltham, MA, USA). The HPV16-E6E7 immortalized human pancreatic duct epithelial (HPDE/E6E7) cell line obtained from Applied Biological Materials (Richmond, BC, Canada) was maintained in DMEM:F12 containing 10% FBS, 1% penicillin–streptomycin solution. All cells were cultured in a 37 °C moisture incubator supplied with 5% CO2. The stearoyl-CoA desaturase 1 (SCD1) inhibitor A939572 was dissolved in DMSO (Sigma-Aldrich, St. Louis, MO) at a concentration of 10 mM divided to 100 µL aliquots and stored at −20 °C until use. PANC-1 cells and HPDE cells were seeded to 10 cm cell culture dishes at a density of 1.5 × 106 cells per dish then allowed to attach for 24 hours. After attachment, medium was replaced by serum free DMEM:F12. PANC-1 cells were treated with 200 nM A939572 or an equivalent volume of DMSO (control group, 1% DMSO) in triplicate. HPDE/E6E7 cells were treated with an equivalent volume of DMSO. All the cell samples were harvested after 24 h in culture.Murine tumor models
The murine melanoma B78-D14 (B78) cell line, derived from B16 melanoma as previously described, was obtained from Ralph Reisfeld (Scripps Research Institute) in 2002.39 B78 cells were grown in RPMI-1640 and were supplemented with 10% FBS, 100 U mL−1 penicillin, and 100 µg mL−1 streptomycin. Cells were incubated in a humidified incubator at 37 °C with 5% CO2. Cell line authentication was performed per ATCC guidelines using morphology, growth curves, and mycoplasma testing within 6 months of use.Mice were housed and treated under a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Wisconsin–Madison. Female C57BL/6 mice were purchased at age 6 to 8 weeks from Taconic Biosciences, Inc. (Rensselaer, NY, USA.). B78 tumors were engrafted by subcutaneous flank injection of 2 × 106 tumor cells. Tumor size was determined using calipers and volume approximated as (width2 × length)/2. Treatment began when tumors were well-established (∼500 mm3), which occurred approximately 5 weeks following the engraftment. Before treatment initiation, mice were randomized to receive either external beam radiation (EBRT) or no treatment.
Delivery of EBRT in vivo was performed using an X-ray biological cabinet irradiator X-RAD 320 (Precision X-ray, Inc., North Branford, CT, USA). EBRT was prescribed to 20 Gy. The dose rate for EBRT delivery in all experiments was approximately 2 Gy min−1. Dosimetric calibration and monthly quality assurance checks were performed on these irradiators by University of Wisconsin Medical Physics Staff. The tumors where harvested 24 hours after EBRT treatment and stored under −80 °C before use.
To prepare the tissue section for MS imaging, the tumor tissue was embedded in gelatin aqueous solution (100 mg mL−1), followed by snap frozen on dry ice. The tumor tissue was sectioned at 12 µm thickness using a cryostat (ThermoFisher Scientific, San Jose, CA, USA) set at −20 °C and the tissue sections were mounted on the ITO-coated glass microscope slides. The tissue section samples were dried in a desiccator and stored at −80 °C until analysis.
In-solution epoxidation of unsaturated FAs for shotgun lipidomics
Unsaturated FA standards including FA 16:1 (9Z), FA 18:1 (6Z), FA 18:1 (9Z), FA 18:1 (11Z), FA 20:1 (11Z), FA 18:2 (9Z, 12Z), γ-linolenic acid FA 18:3 (6Z, 9Z, 12Z), and arachidonic acid FA 18:4 (5Z, 8Z, 11Z, 14Z) were dissolved in 2-propanol (IPA) at a concentration of 2 mg mL−1 as stock solutions, respectively. The FAs stock solutions were stored under −20 °C before use. PAA solution was also prepared in IPA to a concentration of 500 mM. For derivatization of FA standards, an aliquot of 20 µL FA standard solution (10 mM) and 20 µL PAA solution (500 mM) were mixed in a 0.6 mL tube, and then the mixture was shaken at 300 rpm at room temperature for 1 h. Completed epoxidation of unsaturated FAs was readily achieved by adding excessed amount of PAA. Afterwards, 360 µL IPA was added to dilute the reaction system. The extraction of fatty acids from the cell samples used a modified Folch protocol. Briefly, cell suspensions were centrifuged at 800g for 2 min, after which the upper aqueous layer was discarded. Then, the cells were washed with 2 mL dPBS for three times (800g and 2 min centrifugation) and the cell pellets were obtained. Afterwards, 0.5 mL water, 0.5 mL methanol and 0.5 mL chloroform were added into the cell tube. The mixture was sonicated in a water bath for 15 min and vigorous vortexing for 2 min before centrifugation (12000g) at 4 °C for 10 min. The chloroform layer was transferred to a screw-capped glass tube. The extraction repeated once and the combined chloroform layers were dried under a nitrogen stream. All the lipid extracts were stored at −20 °C before analysis. The FAs extract total was reconstituted in 40 µL IPA. The epoxidation was carried out with 20 µL FAs extract and 20 µL PAA (500 mM), after 1 h shaking (300 rpm) at room temperature, the reaction system was diluted with 360 µL IPA. All the derivatized samples were further diluted to target concentration using IPA for nanoESI-MS/MS interrogation without any other pretreatment.
Quantitative analysis
For absolute quantification of FA isomers, FA 18:1 (6Z) was used as internal standard (IS) for quantitative analysis of FA 18:1 (9Z), FA 18:1 (11Z) and FA 16:1 (9Z). To plot the calibration curves, a series of working solutions at 0.01–40.00 µM of FA 18:1 (9Z), FA 18:1 (11Z) and FA 16:1 (9Z) containing a constant 5 µM FA 18:1 (6Z) internal standard were subjected to nanoESI-MS/MS analysis, respectively. The signal intensities of diagnostic ion pairs were summed for each specific FAs, and the ratios of the summed signal intensities between the FA and the IS were plotted against the concentrations of FA. Relative quantification of FA isomers was also demonstrated by analyzing a series of mixture solution of FA 18:1 (9Z) and FA 18:1 (11Z) with varied molar ratios from 0.001:1 to 10:1, at a total concentration of 35 µM. To plot the relative quantification calibration curve, the signal intensities of diagnostic fragment ions were summed for FA 18:1 (9Z) and FA 18:1 (11Z), respectively, and the summed signal intensity ratios between FA 18:1 (9Z) and FA 18:1 (11Z) were plotted against their molar ratios.
On-tissue epoxidation of unsaturated FAs for MALDI-TOF/TOF MSI
The schematic analytical workflow of on-tissue epoxidation of FAs from tissue section samples for MALDI-TOF-MSI was shown in Fig. S1.† The tissue sections were thawed in a desiccator for 10 min at room temperature. After drying, the tissue sections were incubated in a sealed chamber with PAA vapor (PAA, 10% (v/v) aqueous solution) for 2 h. Afterwards, 1,5-diaminonaphthalene (DAN) matrix in acetonitrile/water (v/v, 70:30) solution at concentration of 10 mg mL−1 was sprayed onto the tissue section using a M5-Sprayer (HTX Technologies, Carrboro, NC, USA) at a flow rate of 50 µL min−1 with tracking space of 2 mm for 2 passes. The drying time between each pass was set to 30 s. The nozzle temperature was set to 50 °C, the nozzle nitrogen gas pressure was 10 psi, and the moving velocity of the nozzle was 1000 mm min−1. For samples without on-tissue epoxidation, the PAA incubation was skipped from the workflow aforementioned. The tissue section samples were dried inside a desiccator for 20 min before MALDI-TOF-MS imaging. In a comparison experiment of using m-CPBA for on-tissue epoxidation, m-CPBA in methanol/water (v/v, 50:50) solution at concentration of 10 mg mL−1 was sprayed onto the tissue section using a M5-Sprayer at a flow rate of 100 µL min−1 with tracking space of 2 mm for 4 passes. The drying time between each pass was set to 10 s. The nozzle temperature was set to 30 °C, the nozzle nitrogen gas pressure was 10 psi, and the moving velocity of the nozzle was 600 mm min−1. Then, the m-CPBA coated tissue section (Fig. S13†) was incubated in a humidity chamber (water in the bottom) under 37 °C for 18 h to reach a high epoxidation efficiency. The application of DAN matrix and MALDI-TOF-MS parameters were kept consistent with the scenario of using the PAA reagent.
Data acquisition and analysis
The shotgun nanoESI-MS analysis was carried out on Orbitrap Fusion™ Lumos™ Tribrid™ mass spectrometer (Thermo Scientific, San Jose, CA, USA). Mass spectra were collected in a mass range of m/z 100–600 in negative ion detection mode. The ionization voltage was set to −2.0 kV. The heated ion transportation capillary was maintained at 320 °C. For tandem MS analysis, either collision-induce dissociation (CID) or high-energy collisional dissociation (HCD) were performed. The precursor ions were isolated with a mass window width of 0.8 Da, and normalized collision energy (NCE) was set to 20–45%. Other MS instrumental parameters were set to default values without any further optimization. Three replicates were run to collect nanoESI MS data.For molecular imaging of FAs, tissue section samples were analyzed using a Bruker rapifleX MALDI Tissuetyper TOF mass spectrometer (Bruker Scientific, LLC, Bremen, Germany) fitted with a Smartbeam 3D Nd:YAG (355 mm) laser. Smartbeam laser settings used were 20 µm diameter circular spot size, with 100 shots per pixel and a raster step size of 50 µm. The Smartbeam laser was set to 40% and 70% power for MS1 and MS2 scan, respectively. The MS imaging data were acquired in negative ion detection mode over a mass range of 100–600 Da with a sampling rate of 1.25 GS s−1, and baseline subtraction was performed during the MS imaging data acquisition. TOF/TOF fragmentation was performed for MS2 imaging with precursor ions isolated using a mass window of 1 Da. For each measurement, the instrument was externally calibrated using the red phosphorus according to literature.40 Images were visualized using FlexImaging v5.0 and SCiLS Lab 2020b Pro (Bruker Scientific, LLC, Bremen, Germany) with data normalized to total ion count (TIC). Six replicates were run to obtain MALDI TOF MS data.
Histology staining
H&E staining was performed according to literature.41 Briefly, the slides were rinsed in water, followed by consecutive rinsing in hematoxylin, 1% acid alcohol and eosin, respectively. Afterwards, the slides were passed through ethanol to dehydrate the tissue and rinsed in xylene to render it completely transparent. A thin layer of mounting medium was applied before mounted with a glass cover slip. Images were obtained using a Nikon Eclipse TE2000-inverted microscope (Minato, Tokyo, Japan).Author contributions
HZ and LL designed the study. HZ, MX and ZL performed the experiments and analyzed the data. XS and NVW provided tissue samples for the MS imaging method development. YL and MM prepared the pancreatic cell-line samples. JCJ and ZSM contributed tissue samples from murine tumor models. HZ and LL prepared the manuscript and all authors provided editorial feedback. All authors discussed the results and commented on the paper.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Aspects of this work were supported by the NIH grants R01DK071801, R56MH110215, RF1AG052324, U01CA231081, R01DC010777, P50DE026787, P30CA014520, P01CA250972, T32GM008692, F30CA250263, and TL1TR002375. The MS instruments were supported by grants NIH-NCRR S10RR029531 and S10OD025084. MX acknowledges NSF Graduate Research Fellowship Program (DGE-1747503). LL acknowledges a Vilas Distinguished Achievement Professorship and Charles Melbourne Johnson Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin-Madison School of Pharmacy.References
- S. Y. Zhang and X. B. Lin, J. Am. Chem. Soc., 2019, 141, 15884–15890 CrossRef CAS PubMed.
- S. Vanni, L. Riccardi, G. Palermo and M. De Vivo, Acc. Chem. Res., 2019, 52, 3087–3096 CrossRef CAS PubMed.
- B. Yang, H. Q. Chen, C. Stanton, R. P. Ross, H. Zhang, Y. Q. Chen and W. Chen, J. Funct. Foods, 2015, 15, 314–325 CrossRef CAS.
- F. Rohrig and A. Schulze, Nat. Rev. Cancer, 2016, 16, 732–749 CrossRef PubMed.
- L. Hodson and P. J. Gunn, Nat. Rev. Endocrinol., 2019, 15, 689–700 CrossRef CAS PubMed.
- R. P. Bazinet and S. Laye, Nat. Rev. Neurosci., 2014, 15, 771–785 CrossRef CAS PubMed.
- M. R. Wenk, Cell, 2010, 143, 888–895 CrossRef CAS PubMed.
- X. Han, Nat. Rev. Endocrinol., 2016, 12, 668–679 CrossRef CAS PubMed.
- S. N. Nguyen, J. E. Kyle, S. E. Dautel, R. Sontag, T. Luders, R. Corley, C. Ansong, J. Carson and J. Laskin, Anal. Chem., 2019, 91, 11629–11635 CrossRef CAS PubMed.
- M. Wang, C. Wang, R. H. Han and X. Han, Prog. Lipid Res., 2016, 61, 83–108 CrossRef CAS PubMed.
- Y. H. Rustam and G. E. Reid, Anal. Chem., 2018, 90, 374–397 CrossRef CAS PubMed.
- T. P. Siegel, K. Ekroos and S. R. Ellis, Angew. Chem., Int. Ed., 2019, 58, 6492–6501 CrossRef PubMed.
- X. Ma, L. Chong, R. Tian, R. Shi, T. Y. Hu, Z. Ouyang and Y. Xia, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2573–2578 CrossRef CAS PubMed.
- P. E. Williams, D. R. Klein, S. M. Greer and J. S. Brodbelt, J. Am. Chem. Soc., 2017, 139, 15681–15690 CrossRef CAS PubMed.
- F. Waldchen, B. Spengler and S. Heiles, J. Am. Chem. Soc., 2019, 141, 11816–11820 CrossRef PubMed.
- M. R. L. Paine, B. L. J. Poad, G. B. Eijkel, D. L. Marshall, S. J. Blanksby, R. M. A. Heeren and S. R. Ellis, Angew. Chem., Int. Ed., 2018, 57, 10530–10534 CrossRef CAS PubMed.
- A. Bednarik, S. Bolsker, J. Soltwisch and K. Dreisewerd, Angew. Chem., Int. Ed., 2018, 57, 12092–12096 CrossRef CAS PubMed.
- C. L. Feider, L. A. Macias, J. S. Brodbelt and L. S. Eberlin, Anal. Chem., 2020, 92, 8386–8395 CrossRef CAS PubMed.
- M. C. Thomas, T. W. Mitchell, D. G. Harman, J. M. Deeley, J. R. Nealon and S. J. Blanksby, Anal. Chem., 2008, 80, 303–311 CrossRef CAS PubMed.
- H. J. Yoo and K. Hakansson, Anal. Chem., 2010, 82, 6940–6946 CrossRef CAS PubMed.
- H. T. Pham, T. Ly, A. J. Trevitt, T. W. Mitchell and S. J. Blanksby, Anal. Chem., 2012, 84, 7525–7532 CrossRef CAS PubMed.
- T. Baba, J. L. Campbell, J. C. Y. Le Blanc and P. R. S. Baker, Anal. Chem., 2017, 89, 7307–7315 CrossRef CAS PubMed.
- M.-E. N. Born and B. M. Prentice, Int. J. Mass Spectrom., 2020, 452 Search PubMed.
- G. Feng, Y. Hao, L. Wu and S. Chen, Chem. Sci., 2020, 11, 7244–7251 RSC.
- W. B. Cao, S. M. Cheng, J. Yang, J. X. Feng, W. P. Zhang, Z. S. Li, Q. H. Chen, Y. Xia, Z. Ouyang and X. X. Ma, Nat. Commun., 2020, 11, 11 CrossRef PubMed.
- R. A. Harris, J. C. May, C. A. Stinson, Y. Xia and J. A. McLean, Anal. Chem., 2018, 90, 1915–1924 CrossRef CAS PubMed.
- M. C. Thomas, T. W. Mitchell and S. J. Blanksby, J. Am. Chem. Soc., 2006, 128, 58–59 CrossRef CAS PubMed.
- Y. Feng, B. M. Chen, Q. Y. Yu and L. J. Li, Anal. Chem., 2019, 91, 1791–1795 CrossRef CAS PubMed.
- T. H. Kuo, H. H. Chung, H. Y. Chang, C. W. Lin, M. Y. Wang, T. L. Shen and C. C. Hsu, Anal. Chem., 2019, 91, 11905–11915 CrossRef CAS PubMed.
- J. I. Zhang, W. A. Tao and R. G. Cooks, Anal. Chem., 2011, 83, 4738–4744 CrossRef CAS PubMed.
- S. L. Tang, H. Y. Cheng and X. Yan, Angew. Chem., Int. Ed., 2020, 59, 209–214 CrossRef CAS PubMed.
- K. Chintalapudi and A. K. Badu-Tawiah, Chem. Sci., 2020, 11, 9891–9897 RSC.
- S. Takashima, K. Toyoshi, T. Yamamoto and N. Shimozawa, Sci. Rep., 2020, 10, 12988 CrossRef CAS PubMed.
- D. H. Wang, Z. Wang, J. R. Cortright, K. P. Le, L. Liu, K. S. D. Kothapalli and J. T. Brenna, Anal. Chem., 2020, 92, 8209–8217 CrossRef CAS PubMed.
- C. E. Randolph, D. J. Foreman, S. J. Blanksby and S. A. McLuckey, Anal. Chem., 2019, 91, 9032–9040 CrossRef CAS PubMed.
- F. Waldchen, F. Mohr, A. H. Wagner and S. Heiles, Anal. Chem., 2020, 92, 14130–14138 CrossRef PubMed.
- A. Bednarik, J. Preisler, D. Bezdekova, M. Machalkova, M. Hendrych, J. Navratilova, L. Knopfova, E. Moskovets, J. Soltwisch and K. Dreisewerd, Anal. Chem., 2020, 92, 6245–6250 CrossRef CAS PubMed.
- Z. Tracz-Gaszewska and P. Dobrzyn, Cancers, 2019, 11, 25 CrossRef PubMed.
- M. Haraguchi, S. Yamashiro, A. Yamamoto, K. Furukawa, K. Takamiya, K. O. Lloyd and H. Shiku, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 10455–10459 CrossRef CAS PubMed.
- K. Sladkova, J. Houska and J. Havel, Rapid Commun. Mass Spectrom., 2009, 23, 3114–3118 CrossRef CAS PubMed.
- A. Ly, A. Buck, B. Balluff, N. Sun, K. Gorzolka, A. Feuchtinger, K.-P. Janssen, P. J. K. Kuppen, C. J. H. van de Velde, G. Weirich, F. Erlmeier, R. Langer, M. Aubele, H. Zitzelsberger, L. McDonnell, M. Aichler and A. Walch, Nat. Protoc., 2016, 11, 1428–1443 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01614h |
| This journal is © The Royal Society of Chemistry 2021 |