
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptoid-based reprogrammable template for cell-permeable inhibitors of protein–protein interactions†
Yasuhiro
Fukuda
 a,
Marin
Yokomine
a,
Daisuke
Kuroda
ab,
Kouhei
Tsumoto
abc,
Jumpei
Morimoto
*a and
Shinsuke
Sando
*ab
a,
Marin
Yokomine
a,
Daisuke
Kuroda
ab,
Kouhei
Tsumoto
abc,
Jumpei
Morimoto
*a and
Shinsuke
Sando
*ab
aDepartment of Chemistry and Biotechnology, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, 113-8656, Japan. E-mail: jmorimoto@chembio.t.u-tokyo.ac.jp; ssando@chembio.t.u-tokyo.ac.jp
bDepartment of Bioengineering, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, 113-8656, Japan
cInstitute of Medical Science, The University of Tokyo, 4-6-1, Shirokanedai, Minato-ku, Tokyo, 108-8639, Japan
First published on 3rd August 2021
Abstract
The development of inhibitors of intracellular protein–protein interactions (PPIs) is of great significance for drug discovery, but the generation of a cell-permeable molecule with high affinity to protein is challenging. Oligo(N-substituted glycines) (oligo-NSGs), referred to as peptoids, are attractive as potential intracellular PPI inhibitors owing to their high membrane permeability. However, their intrinsically flexible backbones make the rational design of inhibitors difficult. Here, we propose a peptoid-based rational approach to develop cell-permeable PPI inhibitors using oligo(N-substituted alanines) (oligo-NSAs). The rigid structures of oligo-NSAs enable independent optimization of each N-substituent to improve binding affinity and membrane permeability, while preserving the backbone shape. A molecule with optimized N-substituents inhibited a target PPI in cells, which demonstrated the utility of oligo-NSA as a reprogrammable template to develop intracellular PPI inhibitors.
Introduction
Protein–protein interactions (PPIs) play fundamental roles in diverse biological processes and are involved in the pathogenesis of various diseases.1 Extensive efforts have been devoted to developing PPI inhibitors.2,3 However, it remains challenging to generate inhibitors of intracellular PPIs because it is difficult to produce a molecule having a large recognition surface area and high membrane permeability.4Peptidomimetic synthetic oligomers, such as β-peptides,5,6 peptoids,7,8 oligoureas,9,10 and γ-AApeptides,11,12 are attractive PPI inhibitor candidates because oligomers with desirable sequences can be prepared using well-established modular synthetic methods. Among them, peptoids, usually referring to oligo(N-substituted glycines) (oligo-NSGs), are advantageous in that diverse functional substituents can be introduced during oligomer synthesis using a submonomer synthetic method,13 and they have high membrane permeability14 owing to their N-substituted amide structure. Peptoids that inhibit intracellular PPIs, e.g., the interaction between Skp2 and p300,15 have been reported. However, the reported peptoids showed relatively weak binding to proteins and their KD values remain in the μM order. The reason for their low affinity has been considered to be the entropic penalty upon binding due to their intrinsically flexible backbone.
To overcome this limitation of oligo-NSGs, attempts have been made to realize conformationally constrained peptoids, for example, by the introduction of bulky substituents16–23 or N-aryl-substituted glycine residue24 which respectively cause steric or electronic repulsions, and head-to-tail macrocyclization.25–27 Recently, a peptoid that inhibits the interaction between β-catenin and T-cell factor 3 in cells was elegantly developed using conformation-constraining strategies.28 The peptoid showed inhibitory activity in vivo, and this work demonstrated the utility of peptoids for the development of intracellular PPI inhibitors. However, this peptoid-based approach requires the introduction of specific types of bulky N-substituents to achieve a constrained conformation. As a result, the choice of functional groups that can be introduced as N-substituents in ligand design is restricted.
Recently, we demonstrated that peptoids can be conformationally constrained by backbone modifications without introducing bulky N-substituents or N-aryl-substituted glycine residue and macrocyclization. More specifically, the introduction of methyl groups on the backbone α-carbons of oligo-NSG was shown to conformationally constrain the peptoid, and the resulting oligo(N-substituted alanine) (oligo-NSA, Fig. 1a) stably formed an extended shape.29 The methyl groups on the α-carbons introduce steric repulsions on the backbone, known as pseudo-1,3-allylic strains, with the carbonyl oxygen in the preceding residue and the N-substituent of the following residue, and the bond rotation about the φ and ψ angles is restricted (Fig. 1a).30 As a result, the oligo-NSA structure is controlled on a per-residue basis by steric effects on the backbone and independently from N-substituents. Therefore, oligo-NSA can serve as a peptoid-based scaffold for PPI inhibitors with little restriction on the choice of functional groups introduced as N-substituents.
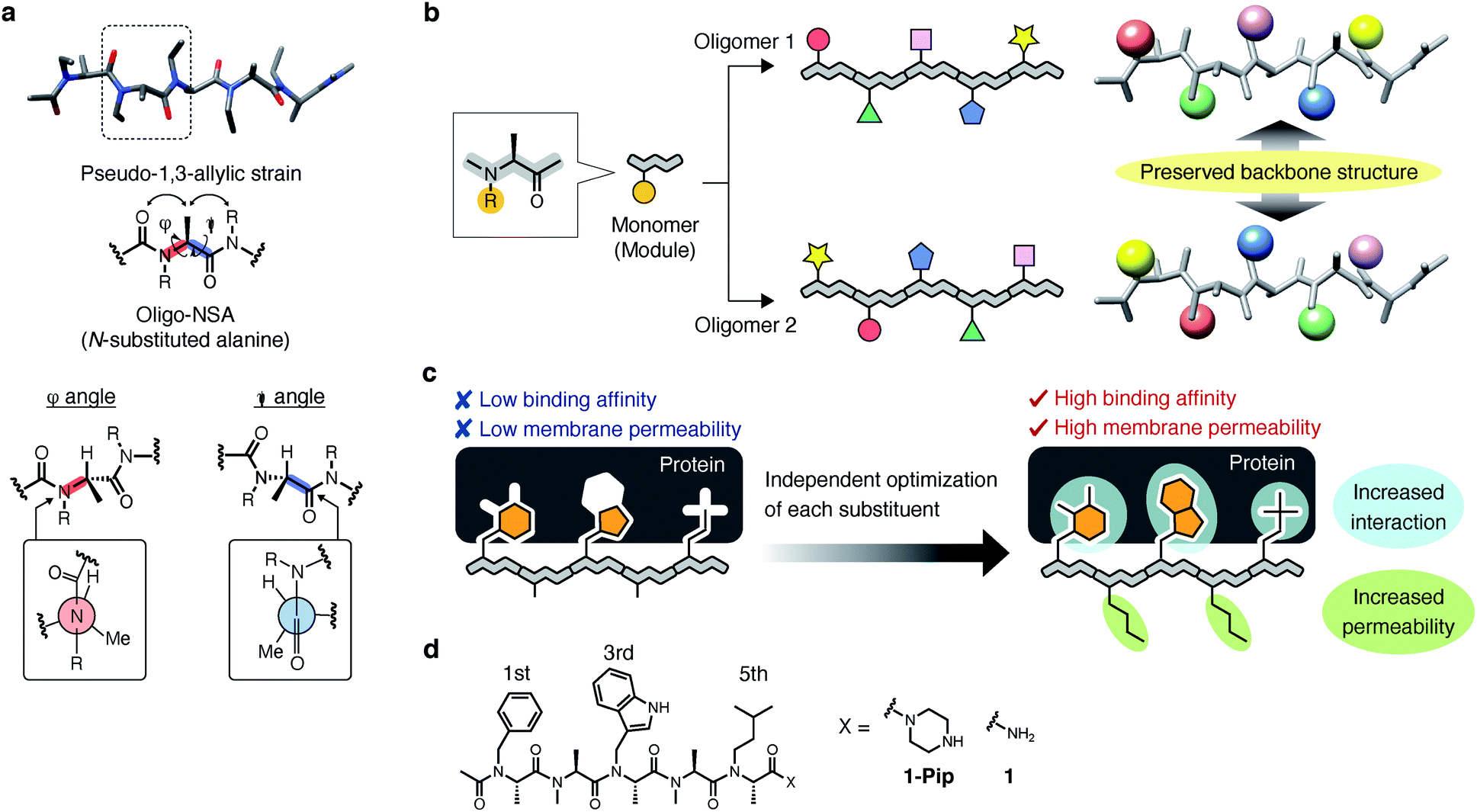 | ||
| Fig. 1 Design strategy for development of inhibitors of intracellular protein–protein interactions (PPIs) using oligo-NSA. (a) Schematic illustration of the rotational restrictions on the backbone about the dihedral angles φ and ψ in oligo-NSA exerted by pseudo-1,3-allylic strains. (b) The oligo-NSA structure is preorganized on a per-monomer basis. Oligo-NSAs with different functional groups as N-substituents with preserved backbone structures can be prepared by modularly replacing each monomer. (c) Design strategy for the development of cell-permeable PPI inhibitors. The rigid structure of oligo-NSA enables independent optimization of N-substituents to improve the binding affinity and membrane permeability, while preserving an extended backbone structure. (d) Chemical structures of 1-Pip and 1 that inhibit PPI between MDM2 and p53. | ||
Here, we propose oligo-NSA as a peptoid-based modularly reprogrammable template for the development of intracellular PPI inhibitors. Because the oligo-NSA structure is preorganized on a per-monomer basis irrespective of the structures of N-substituents, each N-substituent is modularly replaceable to optimize the binding affinity and membrane permeability, while preserving the backbone structure (Fig. 1b). More specifically, the monomers displaying N-substituents toward the target protein can be optimized to improve the affinity (highlighted in cyan in Fig. 1c right), while the monomers displaying N-substituents not facing the target protein can be optimized to improve membrane permeability without disturbing the interaction with the target protein (highlighted in green in Fig. 1c right).
In the previous study, we reported the oligo-NSA 1-Pip as a ligand of MDM2 (Fig. 1d).291-Pip was designed by mimicking the alignment of hot-spot residues in p53, the natural ligand of MDM2. p53 recognizes MDM2 with three hot-spot residues, Phe19, Trp23, and Leu26, displayed on the p53 transactivation domain. Therefore, we designed the oligo-NSA 1-Pip, bearing hot spots of p53, and the oligo-NSA exhibited binding ability to MDM2 in vitro.
In this study, we demonstrated the utility of oligo-NSA as a modularly reprogrammable peptoid for the development of intracellular PPI inhibitors by generating an inhibitor targeting the interaction between cancer-related MDM2 and p53.
Results and discussion
Initial design of inhibitors of the interaction between MDM2 and p53
The ligand designed in the previous study did not inhibit MDM2–p53 interaction in cellulo. The inhibitory activity of 1-Pip against intracellular MDM2–p53 interaction was examined in MDM2-overexpressing SJSA-1 cells. In MDM2-overexpressing cells, the ubiquitination and successive degradation of p53 or masking of the p53 transactivation domain by MDM2 negatively regulate p53 functions. If 1-Pip would inhibit the intracellular PPI, p53 degradation would be suppressed and the intracellular levels of p53 and p21, a transcriptional target of p53, would be increased (Fig. 2a). However, no detectable p53 or p21 were observed after treatment of SJSA-1 cells with 1-Pip even at 20 μM (Fig. 2b and S1†). We initially assumed that the C-terminal cationic secondary amine adversely affected membrane permeability. Thus, we synthesized an oligo-NSA with the same sequence, but without the C-terminal piperazine (1, Fig. 1d), and evaluated its inhibitory activity. 1 showed in vitro inhibitory activity comparable to that of 1-Pip in a competitive fluorescence polarization (FP) assay using a previously reported MDM2 ligand, PMI peptide,31 as a fluorescent probe (Fig. 2c and Table S1†). However, 1 also did not inhibit MDM2–p53 interaction in cellulo (Fig. 2b). The poor inhibitory activity of these initially designed inhibitors in cultured cells was considered to be due to their low affinity for MDM2 and their low membrane permeability. | ||
| Fig. 2 Initial design of oligo-NSA-based inhibitors of MDM2–p53 interaction. (a) Schematic illustration of the negative regulation of p53 function by MDM2 and activation of the p53 pathway via the inhibition of MDM2–p53 interaction. (b) Intracellular protein levels of p53, p21, and β-actin. SJSA-1 cells were treated with 20 μM oligo-NSA for 8 h and cell lysates were analyzed by western blotting. Lysate of SJSA-1 cells treated with a known inhibitor of the MDM2–p53 interaction, Nutlin-3a, was analyzed as a positive control. Representative results of three independent experiments are shown. Other results are shown in Fig. S1.† (c) Inhibitory curves of 1-Pip and 1 against the interaction between fluorescently labeled PMI peptide and MDM2 from a competitive FP assay. Y-axis indicates change in fluorescence anisotropy (ΔFA). Error bars represent standard deviations of triplicates. | ||
Design strategy for improved binding affinity and membrane permeability
The high modularity of oligo-NSAs was expected to allow independently optimizing each N-substituent to improve the binding affinity or membrane permeability, while preserving the backbone structure. We assumed that the binding affinity for MDM2 can be improved by optimizing the structures of the 1st, 3rd, and 5th N-substituents that face MDM2, while the membrane permeability can be improved by introducing lipophilic structures on the 2nd and 4th N-substituents that do not face MDM2. To support this assumption, we performed molecular dynamics (MD) simulations. First, MD simulations of 1 alone were performed using a CHARMM36m/CGenFF force field. From the obtained frames, the structure with the best geometrical match of N and Nα atoms of every other residue with the Cα and Cβ atoms of the three hot-spot residues in p53-TAD was selected. The structure was placed on the predicted binding site of MDM2, and the complex structure was subjected to MD simulations. A representative structure of the complex during the simulations is shown in Fig. 3a. In the complex, the 1st, 3rd, and 5th N-substituents on 1 were buried in the cleft of MDM2, and the 2nd and 4th N-substituents did not face MDM2. To analyze the engagement of each substituent in protein binding more quantitatively, the solvent accessible surface area (SASA) of each N-substituent during the simulations was calculated (Fig. S2†). While the ratio of SASA to the whole surface area of the 1st, 3rd, and 5th N-substituents was below 0.1 on average, the ratios of SASA of the 2nd and 4th N-substituents remained high (0.7–0.9 on average). Thus, throughout the simulation time, the 1st, 3rd, and 5th N-substituents remained associated with the MDM2 surface, and the 2nd and 4th N-substituents were not facing MDM2, which supported our assumption that the preserved backbone structure of oligo-NSAs will allow independently optimizing each N-substituent to improve binding affinity and membrane permeability.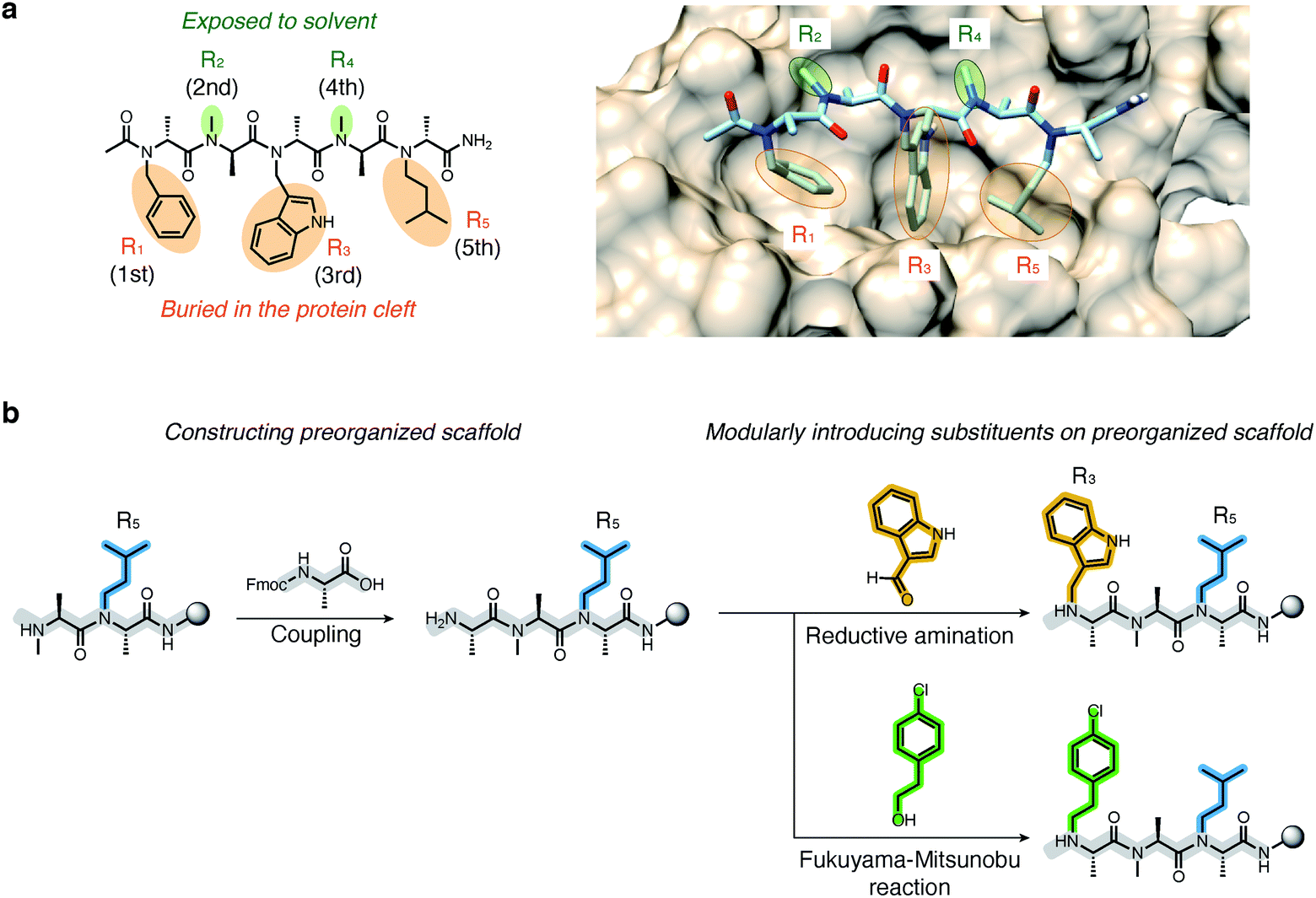 | ||
| Fig. 3 Strategy for structural optimization to improve binding affinity and membrane permeability using oligo-NSA as a modularly reprogrammable template. (a) Molecular dynamics (MD) simulations of oligo-NSA 1 in complex with MDM2. A representative structure of the complex during the simulations is shown. (b) Submonomer synthesis of oligo-NSA. Each N-substituent is modularly introduced using aldehydes or alcohols as submonomers. | ||
Directions of N-substituents must be critical to the binding affinity. Therefore, we investigated distributions of the dihedral angles (χ angles) of N-substituents at R1, R3 and R5 during the MD simulations (Fig. S3†). As shown in Fig. S3,† oligo-NSA 1 has two preferred directions for N-substituents around χ = 90° and −90°. The χ angles of N-isopentyl group (χ = 87°), N-benzyl (χ = −78°) and N-indolylmethyl groups (χ = −117°) of oligo-NSA binding to MDM2 in Fig. 3a are within one of these preferred directions. Therefore, the conformer of oligo-NSA 1 that is preferable for binding to MDM2 is expected to be stochastically formed in solution.
Structural optimization to improve binding affinity
We first derivatized the 1st, 3rd, and 5th N-substituents to improve the binding affinity of the oligo-NSA 1. N-Substituents were introduced by previously reported reductive amination29,32 or Fukuyama–Mitsunobu reaction33 using commercially available aldehydes or alcohols as submonomers (Fig. 3b). This submonomer synthetic method allows the modular replacement of each N-substituent. We independently optimized the 1st, 3rd, and 5th substituents, and several of the tested modifications showed improved inhibitory activity against the interaction between MDM2 and PMI peptide in a competitive FP assay (Fig. 4a, S4, S5 and Table S2†). Based on the previously reported chemical structures of MDM2 inhibitors, we introduced halogens to the phenyl group at R1 and the indole ring at R3.34,35 However, modifications of the R1 substituent did not improve affinity (compounds S1–S3). Introduction of chloride to the indole group at R3, especially, substitution to a 6-chloroindolylmethyl group, improved the binding affinity by 2–11-fold (compounds 2 and S4). Compound S5, in which the indolylmethyl group at R3 in 1 is replaced with a 4-chlorophenethyl group, also exhibited improved binding affinity. We assumed increased hydrophobicity of the N-substituent at R5 will improve the binding affinity to MDM2 according to the literature,36,37 and introduced N-substituents with higher hydrophobicity at R5. The neohexyl group (compound 3) was preferred over the isopentyl group, n-butyl group (compound S6), and cyclohexylmethyl group (compound S7). Based on these results, we synthesized and evaluated the binding affinity of compound 4 bearing benzyl, 6-chloroindolylmethyl, and neohexyl as the 1st, 3rd, and 5th N-substituent, respectively. The Ki value of 4 was 0.34 ± 0.06 μM, which is 15-fold stronger than that of the original ligand 1. These results demonstrated that the rigid backbone of oligo-NSA enables independent optimization of each substituent to improve the binding affinity.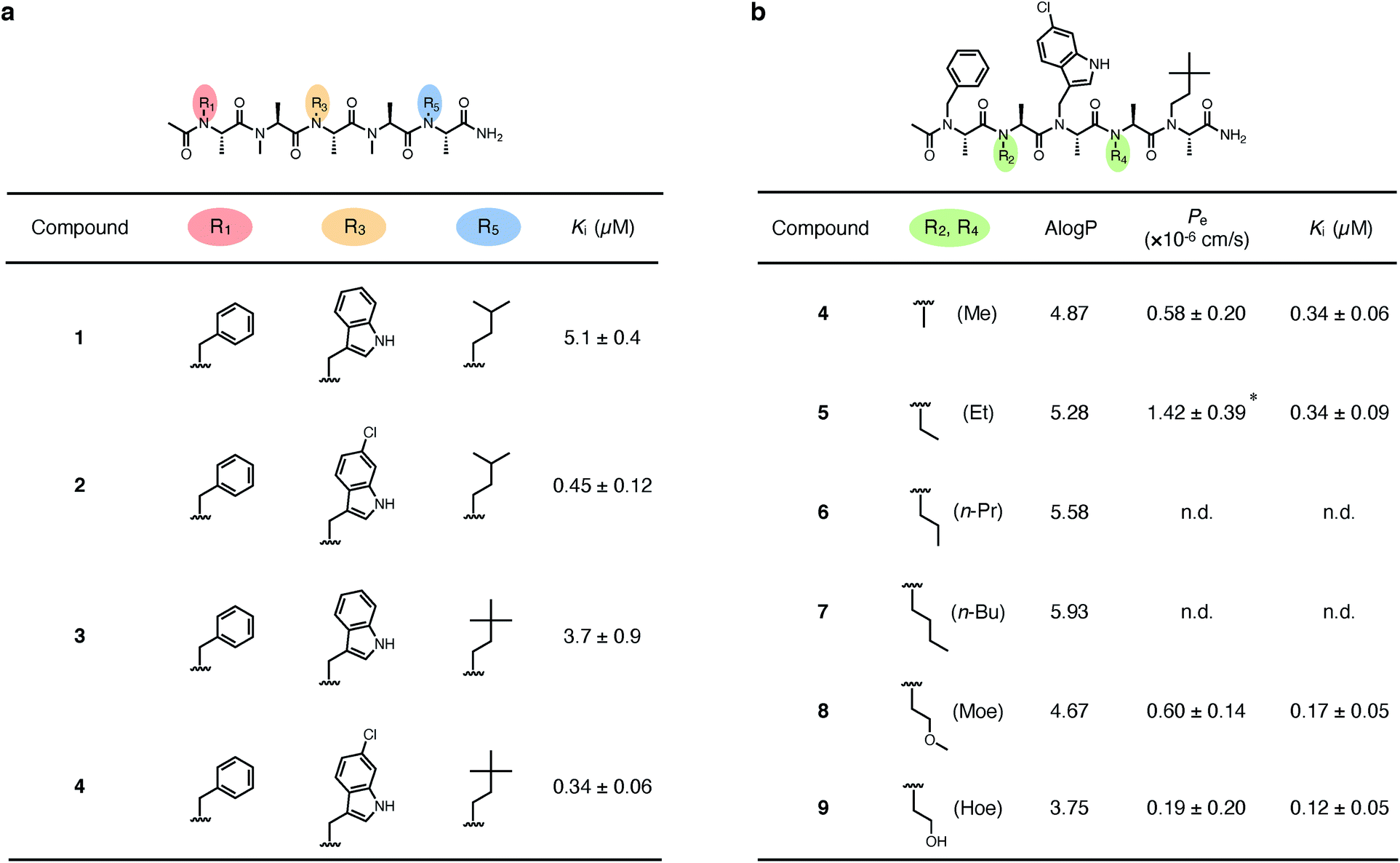 | ||
Fig. 4 Structural optimization of oligo-NSA to improve binding affinity and membrane permeability. (a) Chemical structures of substituents at R1, R3, and R5 and Ki values of compounds 1–4. Ki values were determined by a competitive FP assay using fluorescently labeled PMI peptide and MDM2. (b) Chemical structures of substituents at R2 and R4, A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP values, Pe values, and Ki values of compounds 4–9. AlogP values are indices of lipophilicity of compounds. Pe values were determined by a Caco-2 assay. Ki values were determined by a competitive FP assay using fluorescently labeled PMI peptide and MDM2. n.d.: not determined due to low water solubility in aqueous solution. *p < 0.05 compared with compound 4. logP values, Pe values, and Ki values of compounds 4–9. AlogP values are indices of lipophilicity of compounds. Pe values were determined by a Caco-2 assay. Ki values were determined by a competitive FP assay using fluorescently labeled PMI peptide and MDM2. n.d.: not determined due to low water solubility in aqueous solution. *p < 0.05 compared with compound 4. | ||
Structural optimization to improve membrane permeability
We next designed derivatives of 4 to improve membrane permeability by modifying the substituents R2 and R4. First, the membrane permeability of 4 was measured by a Caco-2 assay (Fig. 4b). 4 showed only modest membrane permeability, with an effective permeability constant (Pe) of 0.58 ± 0.20 × 10−6 cm s−1. To adjust the lipophilicity to a range preferable for membrane permeability, we synthesized compounds 5–9 bearing substituents with different lipophilicity, ethyl (Et), n-propyl (n-Pr), n-butyl (n-Bu), 2-methoxyethyl (Moe), or 2-hydroxyethyl (Hoe) groups, at the R2 and R4 positions (Fig. 4b), and evaluated their membrane permeability. Differences in the Pe value were observed in the Caco-2 assay, and introduction of the Et groups (compound 5) enhanced the Pe value by 2.4-fold. 6 and 7, with more lipophilic substituents, were too hydrophobic and thus, not soluble in aqueous buffer, and 8 and 9, with more hydrophilic substituents, showed lower Pe values. These results are consistent with the fact that moderate lipophilicity is important for high membrane permeability and too high lipophilicity results in low water solubility.38,39 We also evaluated the permeability of 4–9 by a parallel artificial membrane permeability assay (PAMPA) (Table S3†). Pe values showed the same tendencies as in the Caco-2 assay, and oligo-NSA 5, bearing Et groups, showed the highest membrane permeability. These results demonstrated that the membrane permeability of oligo-NSA can be improved by appropriately modulating the lipophilicity via modification of the N-substituents.We confirmed that the derivatization of R2 and R4 did not interrupt the interactions of 4, 5, 8, and 9 with MDM2 in a competitive FP assay (Fig. 4b and S6†).
The results of the competitive binding assay and the permeability assay suggested that the membrane permeability of oligo-NSA can be improved without adversely affecting the binding ability by introducing substituents with appropriate lipophilicity at R2 and R4 that are not facing the interaction surface.
Evaluation of backbone preservation upon modification of the N-substituents
To validate the assumption that the N-substituents of oligo-NSA can be replaced without disturbing the extended shape of the backbone, we conducted structural analysis.The structural similarity of 1–5, 8 and 9 was first evaluated by circular dichroism (CD) measurements (Fig. S7†). All oligo-NSAs exhibited similar spectral shapes; all spectra had a maximum around 195 nm and a minimum around 225 nm. These spectral shapes also resemble reported spectra of oligo-NSAs forming β-strand structures,29 which suggested that all the oligo-NSAs maintained the extended backbone shape.
To obtain more detailed structural information, we conducted NMR studies of the original oligo-NSA, 1, and the most potent oligo-NSA, 5. To facilitate obtaining the compounds at the multi-milligram scale, which is required for NMR analysis, these oligo-NSAs were synthesized with C-terminal piperazine (1-Pip and 5-Pip), which gives higher yields than the oligo-NSA 1 and 5 with C-terminal amide. Spatially proximal protons on the backbones of 1-Pip and 5-Pip were determined by ROESY measurements (Fig. S8 and S9†). The α-protons, β-protons, and Nα protons of the compounds on the 1H NMR spectra were assigned with the aid of COSY, 13C NMR, HMQC, and HMBC spectra (Fig. S10–S19†). For both compounds, NOE signals indicating rotational restrictions about φ and ψ via pseudo-1,3-allylic strains were observed (Fig. 5a–c). These results supported the preservation of the extended backbone structure before and after modification of the N-substituents.
 | ||
| Fig. 5 Structural analysis of MDM2-binding oligo-NSAs. (a) Schematic illustration of the preferred conformations via pseudo-1,3-allylic strains. Red and blue arrows indicate spatially proximal protons via rotational restrictions of the backbone dihedral angles. Part of the three-dimensional model structure is shown next to each Newman diagram to visualize the proximities of the protons. (b and c) Summary of spatially proximal protons in 1-Pip (b) and 5-Pip (c) obtained from ROESY measurements. Protons with NOEs that suggest rotational restrictions about φ (red arrows) and ψ (blue arrows) are indicated. (d and e) Overlay of five representative structures of 1 (d) and 5 (e) during MD simulations. Dominant conformations were determined by clustering analysis, and the backbones of the representative structures from each of the top five clusters were overlaid. (f and g) Representative structure of 1 (shown in cyan in (f)) and 5 (shown in pink in (g)) in complex with MDM2 during the MD simulations. The representative structures were determined by clustering analysis. | ||
To assess the dynamics of each oligomer structure, we conducted MD simulations. First, simulations of oligo-NSA 5 alone were conducted, and the results were evaluated together with the MD simulations of oligo-NSA 1 performed to prepare the model complex structure of 1 and MDM2 shown in Fig. 3a. Representative structures of 1 and 5 during MD simulations were generated by clustering the trajectories (Fig. S20†), and the representative structures were superimposed over each other (Fig. 5d and e). The overall backbone structures were maintained in the extended shape throughout the simulation period, although some degree of structural fluctuation was observed for both oligomers (Fig. S21†). The maintenance of the backbone structures was also suggested from distributions of the backbone dihedral angles, φ and ψ (Fig. S22†). The φ and ψ angles were restricted around the region with low energy on the previously reported energy diagram during the majority of the simulation time.
Next, we conducted MD simulations of 5 in complex with MDM2, and the results were compared with the MD simulations of 1 in complex with MDM2 shown in Fig. 3a to assess the effect of the modification on the binding mode (Fig. 5f, g and S23–S25†). Throughout the simulations, the N-substituents of the 1st, 3rd, and 5th residues of both oligomers were nearly completely buried in the p53-binding cleft of MDM2, while the N-substituents of the 2nd and 4th residues were not associated with MDM2 and exposed to the solvent as shown by the SASA plot (Fig. S26†).
These results supported the validity of our design principle, in which each N-substituent is independently optimized to improve the binding affinity and membrane permeability.
Evaluation of the selectivity of oligo-NSA 5 against MDM2–p53 interaction
The MDM2–p53 interaction is mediated by α-helix. It is important that the designed oligo-NSA selectively inhibits MDM2–p53 interaction and does not interrupt other similar α-helix mediated PPIs toward application of the compound in cells. We chose the interaction between MCL-1 and an α-helical BH3 peptide as an example of another α-helix mediated PPI, and evaluated an inhibitory activity of compound 5 against the interaction by a competitive FP assay (Fig. S27†). Compound 5 did not show inhibitory activity against the PPI, which supported the selective inhibition of the oligo-NSA against MDM2–p53 interaction.Evaluation of intracellular activities
Finally, we evaluated the intracellular activity of the optimized oligo-NSA with improved binding affinity and membrane permeability. Intracellular levels of p53 and p21 in SJSA-1 cells with and without oligo-NSA 1 and 5 were analyzed by western blotting. Treatment of SJSA-1 cells with 5 for 8 h led to a dose-dependent increase in the levels of p53 and p21, whereas treatment with 1, the initial oligo-NSA before optimization of the N-substituents, showed no effects on the p53 and p21 levels (Fig. 6a, S28 and S29†). This result indicated that optimization of the N-substituents led to improved intracellular activity. Furthermore, we evaluated the activity of 5-Rv, which is the reverse sequence of 5. 5-Rv showed a substantially lower inhibitory activity than 5in vitro (Fig. S30†) and had no effects on intracellular p53 and p21 levels (Fig. 6a, S28 and S29†). This result suggested that p53 pathway activation by 5 is due to selective interaction of 5 with MDM2.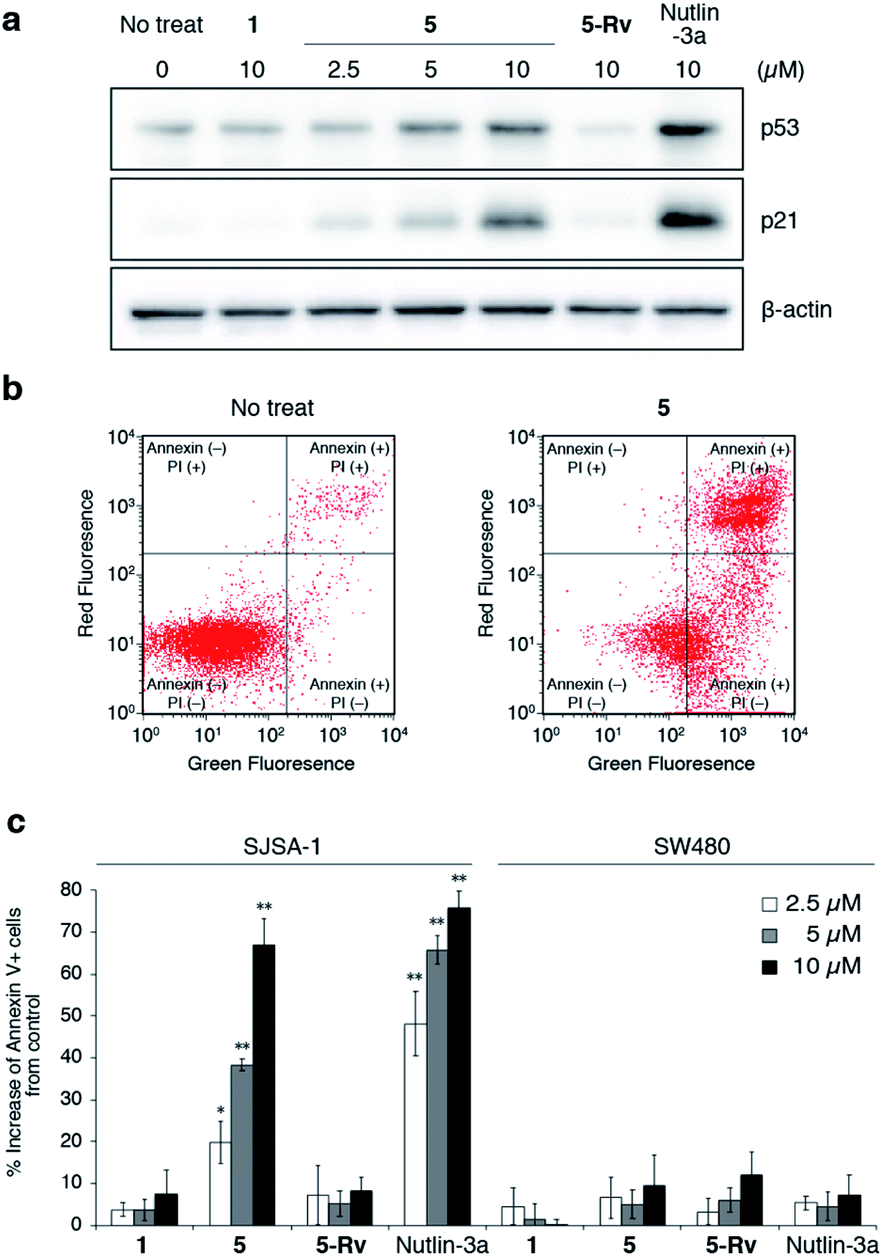 | ||
| Fig. 6 Evaluation of inhibitory activity of oligo-NSAs in cellulo. (a) Intracellular protein levels of p53, p21, and β-actin in MDM2-overexpressing SJSA-1 cells treated with oligo-NSAs. SJSA-1 cells were incubated with 0, 2.5, 5, or 10 μM 5, 10 μM 1, 10 μM 5-Rv, or 10 μM Nutlin-3a for 8 h and cell lysates were analyzed by western blotting. Representative results of three independent experiments are shown. Other results are shown in Fig. S28.† (b) Measurements of apoptotic cells by staining with Annexin V-FITC and propidium iodide (PI). After treatment with 10 μM 5 for 48 h, SJSA-1 cells were labeled with Annexin-V and PI and analyzed by flow cytometry. Representative results of three independent experiments are shown. Other results are shown in Fig. S31–S33.† (c) Apoptotic responses of SJSA-1 cells (left) and p53-mutated SW480 cells (right) to 1, 5, 5-Rv, and Nutlin-3a. Error bars represent standard deviations of three independent experiments. *p < 0.05 and **p < 0.01 from t test between each sample and the vehicle control. | ||
Next, we evaluated the apoptosis-inducing ability of 5 by staining SJSA-1 cells treated with 5 with Annexin V-FITC and propidium iodide (PI) (Fig. 6b and S31–S33†). Treatment with 5 increased the ratios of Annexin V-positive cells and Annexin V- and PI-double positive cells, which indicates the induction of apoptosis. The degree of apoptosis induction increased in a dose-dependent manner (Fig. 6c). Consistent with the western blotting results, little apoptosis induction was observed in cells treated with 5-Rv. This result suggested that cell apoptosis induced by 5 is due to its interaction with MDM2, and not due to non-specific toxicity of the oligo-NSA. Furthermore, treatment with 5 had little effect on SW480 cells, which are mutant p53-bearing cancer cells (Fig. 6c and S34–S36†), although high basal p53 levels were observed irrespective of the compounds used for treatments (Fig. S37 and S38†). Thus, cell apoptosis induced by 5 was suggested to depend on p53 function. Therefore, 5 was suggested to trigger p53 pathway-dependent apoptosis, further supporting the selective PPI inhibitory effect of the oligo-NSA.
The contribution of increased cell permeability to the improved intracellular activity of oligo-NSA was assessed by comparing the intracellular activities of 5 with those of 4 by Annexin assay (Fig. S39 and S40†). The increase of the ratio of Annexin V-positive cells and Annexin V- and PI-double positive cells was significantly larger after the treatment with 5 than that of 4 at 10 μM. This result indicated that the increased cell permeability was an important factor for the improved intracellular activity of oligo-NSA 5.
Altogether, we successfully developed a peptoid active in cells by using oligo-NSA as a modularly reprogrammable template and independently optimizing each N-substituent.
Conclusions
In this study, we demonstrated that the rigid structure of oligo-NSA enables independent optimization of N-substituents for improving the binding ability and membrane permeability. Specific substituents are not required to achieve a constrained scaffold structure and thus, each substituent can be optimized without restriction on the choice of N-substituents. This modular approach using oligo-NSA as a reprogrammable template enabled peptoid-based facile development of an intracellular PPI inhibitor that induces cell apoptosis.Peptoid-based approaches for developing PPI inhibitors have been faced with the challenge that the intrinsically flexible backbone of oligo-NSG often leads to low protein affinity. The introduction of bulky N-substituents is effective to constrain the oligo-NSG conformation, but it hampers structural optimization for affinity maturation due to the requirements of introducing the conformationally constraining elements on the N-substituents.40 The rigid backbone of oligo-NSA resolved the problem and liberated the N-substituents from structural ordering. Thus, rapid optimization of the N-substituents for high affinity and high membrane permeability was realized using oligo-NSA as a template structure.
Low-molecular weight compounds with constrained conformations, such as terphenyl,41 oligobenzamide,42 pyrrolopyrimidine,43 triazine-piperazine-triazines44 and oligooxopiperazine,45 have been also reported to serve as template structures for PPI inhibitors.46 Some of the compounds and oligo-NSAs have high modularity. We here demonstrated that the modularity of these template structures can be utilized for facile optimization of not only binding affinity to a target protein but also membrane permeability.
Furthermore, considering oligo-NSAs form a β-strand-like extended structure, oligo-NSA might be also useful to mimic the orientation of hot-spot residues on β-strand structures in proteins. Therefore, oligo-NSA may serve as templates of inhibitors targeting not only α-helix mediated PPIs but also β-strand and β-hairpin mediated PPIs.47
In conclusion, the peptoid-based modularly reprogrammable template will facilitate the development of inhibitors of diverse intracellular PPIs and expand the utility of peptoids in biomedical applications.
Data availability
Experimental data is provided in the manuscript and the ESI.†Author contributions
J. M. and S. S. conceived and directed the study. Y. F. contributed to the execution of this study. Y. F. and M. Y. synthesized compounds and conducted in vitro assays, cellular assays, and structural analysis. M. Y., D. K., and K. T. organized and/or conducted computational studies. Y. F., J. M., and S. S. wrote the manuscript and all authors approved the manuscript.Conflicts of interest
The authors declare the following competing financial interest: J. M., Y. F., and S. S. have filed a patent application (PCT/JP2020/27010).Acknowledgements
This work was supported by JSPS KAKENHI (JP19K15692, JP19H04202, JP18H02082, JP20H02531), AMED (JP20ak0101117, JP20wm0325002, JP20ak0101139, JP18am0101094, JP18fm0208030, JP18fk0108073), and partly by CREST (JPMJCR13L4). The computations were performed at the Research Center for Computational Science, Okazaki, Japan. We thank Drs Y. Goto and H. Suga at the University of Tokyo for the use of a CD spectrometer. This work was partly supported by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under grant numbers JP19am0101094 and JP20am0101087 (support number 2605). This work was partly supported by the World-leading Innovative Graduate Study Program for Life Science and Technology, The University of Tokyo, as part of the WISE Program (Doctoral Program for World-leading Innovative & Smart Education), MEXT, Japan. pGEX6P2-MDM2 (17-125) was a gift from Gary Daughdrill (#62063, Addgene plasmid). Molecular graphics and clustering analyses of MD simulations were performed using UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.Notes and references
- A. A. Ivanov, F. R. Khuri and H. Fu, Trends Pharmacol. Sci., 2013, 34, 393–400 CrossRef CAS PubMed.
- M. R. Arkin, Y. Tang and J. A. Wells, Chem. Biol., 2014, 21, 1102–1114 CrossRef CAS PubMed.
- P. Wójcik and Ł. Berlicki, Bioorg. Med. Chem. Lett., 2016, 26, 707–713 CrossRef.
- X. Ran and J. E. Gestwicki, Curr. Opin. Chem. Biol., 2018, 44, 75–86 CrossRef CAS.
- R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219–3232 CrossRef CAS PubMed.
- D. Seebach and J. Gardiner, Acc. Chem. Res., 2008, 41, 1366–1375 CrossRef CAS.
- R. J. Simon, R. S. Kania, R. N. Zuckermann, V. D. Huebner, D. A. Jewell, S. Banville, S. Ng, L. Wang, S. Rosenberg and C. K. Marlowe, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9367–9371 CrossRef CAS.
- P. G. Alluri, M. M. Reddy, K. Bachhawat-Sikder, H. J. Olivos and T. Kodadek, J. Am. Chem. Soc., 2003, 125, 13995–14004 CrossRef CAS.
- L. Fischer, P. Claudon, N. Pendem, E. Miclet, C. Didierjean, E. Ennifar and G. Guichard, Angew. Chem., Int. Ed., 2010, 49, 1067–1070 CrossRef CAS PubMed.
- J. Fremaux, C. Venin, L. Mauran, R. H. Zimmer, G. Guichard and S. R. Goudreau, Nat. Commun., 2019, 10, 924 CrossRef PubMed.
- Y. Niu, Y. Hu, X. Li, J. Chen and J. Cai, New J. Chem., 2011, 35, 542–545 RSC.
- P. Sang, Z. Zhou, Y. Shi, C. Lee, Z. Amso, D. Huang, T. Odom, V. T. B. Nguyen-tran, W. Shen and J. Cai, Sci. Adv., 2020, 6, eaaz4988 CrossRef CAS.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- P. Yu, B. Liu and T. Kodadek, Nat. Biotechnol., 2005, 23, 746–751 CrossRef CAS PubMed.
- M. Oh, J. H. Lee, H. Moon, Y. J. Hyun and H. S. Lim, Angew. Chem., Int. Ed., 2016, 55, 602–606 CrossRef CAS.
- C. W. Wu, T. J. Sanborn, K. Huang, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2001, 123, 6778–6784 CrossRef CAS PubMed.
- G. L. Butterfoss, P. D. Renfrew, B. Kuhlman, K. Kirshenbaum and R. Bonneau, J. Am. Chem. Soc., 2009, 131, 16798–16807 CrossRef CAS PubMed.
- J. R. Stringer, J. A. Crapster, I. A. Guzei and H. E. Blackwell, J. Am. Chem. Soc., 2011, 133, 15559–15567 CrossRef CAS.
- G. L. Butterfoss, B. Yoo, J. N. Jaworski, I. Chorny, K. A. Dill, R. N. Zuckermann, R. Bonneau, K. Kirshenbaum and V. A. Voelz, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14320–14325 CrossRef CAS.
- J. S. Laursen, J. Engel-Andreasen, P. Fristrup, P. Harris and C. A. Olsen, J. Am. Chem. Soc., 2013, 135, 2835–2844 CrossRef CAS PubMed.
- O. Roy, G. Dumonteil, S. Faure, L. Jouffret, A. Kriznik and C. Taillefumier, J. Am. Chem. Soc., 2017, 139, 13533–13540 CrossRef CAS.
- B. C. Gorske, E. M. Mumford, C. G. Gerrity and I. Ko, J. Am. Chem. Soc., 2017, 139, 8070–8073 CrossRef CAS.
- D. Gimenez, G. Zhou, M. F. D. Hurley, J. A. Aguilar, V. A. Voelz and S. L. Cobb, J. Am. Chem. Soc., 2019, 141, 3430–3434 CrossRef CAS.
- N. H. Shah, G. L. Butterfoss, K. Nguyen, B. Yoo, R. Bonneau, D. L. Rabenstein and K. Kirshenbaum, J. Am. Chem. Soc., 2008, 130, 16622–16632 CrossRef CAS.
- S. B. Y. Shin, B. Yoo, L. J. Todaro and K. Kirshenbaum, J. Am. Chem. Soc., 2007, 129, 3218–3225 CrossRef CAS PubMed.
- B. Yoo, S. B. Y. Shin, M. L. Huang and K. Kirshenbaum, Chem.–Eur. J., 2010, 16, 5528–5537 CrossRef CAS.
- P. D. Renfrew, T. W. Craven, G. L. Butterfoss, K. Kirshenbaum and R. Bonneau, J. Am. Chem. Soc., 2014, 136, 8772–8782 CrossRef CAS PubMed.
- J. A. Schneider, T. W. Craven, A. C. Kasper, C. Yun, M. Haugbro, E. M. Briggs, V. Svetlov, E. Nudler, H. Knaut, R. Bonneau, M. J. Garabedian, K. Kirshenbaum and S. K. Logan, Nat. Commun., 2018, 9, 4396 CrossRef PubMed.
- J. Morimoto, Y. Fukuda, D. Kuroda, T. Watanabe, F. Yoshida, M. Asada, T. Nakamura, A. Senoo, S. Nagatoishi, K. Tsumoto and S. Sando, J. Am. Chem. Soc., 2019, 141, 14612–14623 CrossRef CAS PubMed.
- Y. Gao and T. Kodadek, Chem. Biol., 2013, 20, 360–369 CrossRef CAS.
- M. Pazgier, M. Liu, G. Zou, W. Yuan, C. Li, C. Li, J. Li, J. Monbo, D. Zella, S. G. Tarasov and W. Lu, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4665–4670 CrossRef CAS.
- K. Pels and T. Kodadek, ACS Comb. Sci., 2015, 17, 152–155 CrossRef CAS.
- G. A. Sable, K. J. Lee, M. K. Shin and H. S. Lim, Org. Lett., 2018, 20, 2526–2529 CrossRef CAS.
- B. L. Grasberger, T. Lu, C. Schubert, D. J. Parks, T. E. Carver, H. K. Koblish, M. D. Cummings, L. V. LaFrance, L. Milkiewicz, R. R. Calvo, D. Maguire, J. Lattanze, C. F. Franks, S. Zhao, K. Ramachandren, G. R. Bylebyl, M. Zhang, C. L. Manthey, E. C. Petrella, M. W. Pantoliano, I. C. Deckman, J. C. Spurlino, A. C. Maroney, B. E. Tomczuk, C. J. Molloy and R. F. Bone, J. Med. Chem., 2005, 48, 909–912 CrossRef CAS.
- E. A. Harker, D. S. Daniels, D. A. Guarracino and A. Schepartz, Bioorg. Med. Chem., 2009, 17, 2038–2046 CrossRef CAS.
- Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K. H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. Cai, P. Berry, E. Feyfant, X. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3445–3454 CrossRef.
- F. Touti, Z. P. Gates, A. Bandyopadhyay, G. Lautrette and B. L. Pentelute, Nat. Chem. Biol., 2019, 15, 410–418 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Deliv. Rev., 2001, 46, 3–26 CrossRef CAS.
- A. Furukawa, C. E. Townsend, J. Schwochert, C. R. Pye, M. A. Bednarek and R. S. Lokey, J. Med. Chem., 2016, 59, 9503–9512 CrossRef CAS PubMed.
- T. Hara, S. R. Durell, M. C. Myers and D. H. Appella, J. Am. Chem. Soc., 2006, 128, 1995–2004 CrossRef CAS PubMed.
- B. P. Orner, J. T. Ernst and A. D. Hamilton, J. Am. Chem. Soc., 2001, 123, 5382–5383 CrossRef CAS.
- J. P. Plante, T. Burnley, B. Malkova, M. E. Webb, S. L. Warriner, T. A. Edwards and A. J. Wilson, Chem. Commun., 2009, 5091–5093 RSC.
- J. H. Lee, Q. Zhang, S. Jo, S. C. Chai, M. Oh, W. Im, H. Lu and H. S. Lim, J. Am. Chem. Soc., 2011, 133, 676–679 CrossRef CAS.
- M. Oh, J. H. Lee, W. Wang, H. S. Lee, W. S. Lee, C. Burlak, W. Im, Q. Q. Hoang and H. S. Lim, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11007–11012 CrossRef CAS.
- B. B. Lao, K. Drew, D. A. Guarracino, T. F. Brewer, D. W. Heindel, R. Bonneau and P. S. Arora, J. Am. Chem. Soc., 2014, 136, 7877–7888 CrossRef CAS PubMed.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- A. M. Watkins and P. S. Arora, ACS Chem. Biol., 2014, 9, 1747–1754 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01560e |
| This journal is © The Royal Society of Chemistry 2021 |