
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The origin of unidirectional charge separation in photosynthetic reaction centers: nonadiabatic quantum dynamics of exciton and charge in pigment–protein complexes†
Hiroyuki
Tamura
 *ab,
Keisuke
Saito
ab and
Hiroshi
Ishikita
ab
*ab,
Keisuke
Saito
ab and
Hiroshi
Ishikita
ab
aDepartment of Applied Chemistry, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8654, Japan. E-mail: tamura@protein.rcast.u-tokyo.ac.jp
bResearch Center for Advanced Science and Technology, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8904, Japan
First published on 5th May 2021
Abstract
Exciton charge separation in photosynthetic reaction centers from purple bacteria (PbRC) and photosystem II (PSII) occurs exclusively along one of the two pseudo-symmetric branches (active branch) of pigment–protein complexes. The microscopic origin of unidirectional charge separation in photosynthesis remains controversial. Here we elucidate the essential factors leading to unidirectional charge separation in PbRC and PSII, using nonadiabatic quantum dynamics calculations in conjunction with time-dependent density functional theory (TDDFT) with the quantum mechanics/molecular mechanics/polarizable continuum model (QM/MM/PCM) method. This approach accounts for energetics, electronic coupling, and vibronic coupling of the pigment excited states under electrostatic interactions and polarization of whole protein environments. The calculated time constants of charge separation along the active branches of PbRC and PSII are similar to those observed in time-resolved spectroscopic experiments. In PbRC, Tyr-M210 near the accessary bacteriochlorophyll reduces the energy of the intermediate state and drastically accelerates charge separation overcoming the electron–hole interaction. Remarkably, even though both the active and inactive branches in PSII can accept excitons from light-harvesting complexes, charge separation in the inactive branch is prevented by a weak electronic coupling due to symmetry-breaking of the chlorophyll configurations. The exciton in the inactive branch in PSII can be transferred to the active branch via direct and indirect pathways. Subsequently, the ultrafast electron transfer to pheophytin in the active branch prevents exciton back transfer to the inactive branch, thereby achieving unidirectional charge separation.
1. Introduction
Light reactions of photosynthesis achieve an extremely high internal quantum efficiency from photoabsorption to separated electrons and holes1 through ingeniously regulated pathways of energy and charge transfers in pigment–protein complexes. Light-harvesting (antenna) complexes, which contain a number of pigments, absorb a photon to create an electronically excited state characterized as a bound electron–hole pair, i.e. exciton.1–7 Exciton charge separation necessitates a sufficient potential difference between the donor and acceptor of electrons for overcoming the electron–hole Coulomb binding energy.Photosystem II (PSII) consists of core antenna complexes (CP43 and CP47) and a reaction center (RC).1–4 Chlorophyll a (Chl) molecules in CP43 and CP47 mediate exciton transfers to the RC consisting of Chls (PD1, PD2, ChlD1, and ChlD2), pheophytin a (PheoD1 and PheoD2), and plastoquinone (QA and QB) (Fig. 1).8–38 Charge separation occurs in the RC, where the electron reduces plastoquinone and the hole eventually oxidizes water at the Mn4CaO5 cluster.11–13 Similarly, bacteriochlorophyll a (BChl) molecules in the light harvesting complex I (LHI) from purple bacteria, Rhodobacter sphaeroides, transfer an exciton to the RC (PbRC) consisting of BChl (PL, PM, BL, and BM), bacteriopheophytin a (BPheo, HL and HM), and ubiquinone (QA and QB) (Fig. 1).11
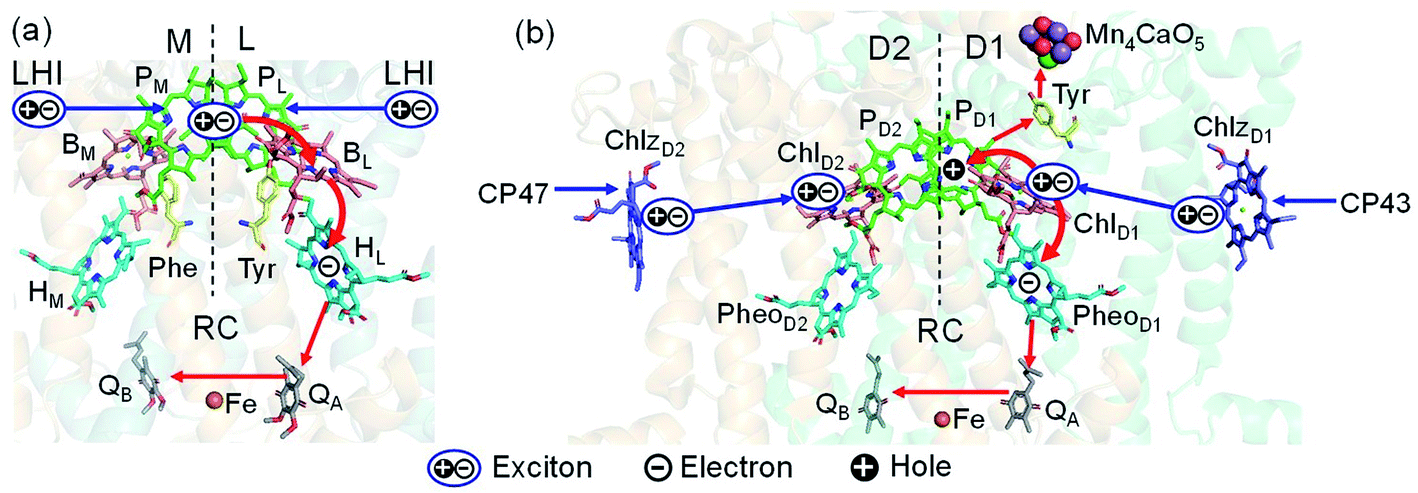 | ||
| Fig. 1 Exciton transfer5–7,9,10 and charge separation11 pathways in (a) PbRC and (b) PSII. Dashed lines indicate the rotation axis of pseudo-C2 symmetry. Blue and red arrows indicate exciton and charge transfer pathways, respectively. Phe and Tyr denote phenylalanine and tyrosine. | ||
Charge separation in PSII and PbRC occurs only along the active branch of the pseudo-C2 symmetric pigment–protein complexes, i.e., D1- and L-branches, respectively (Fig. 1).11 The D2- and M-branches are referred to as inactive branches. PSII and PbRC may have been evolved from a common ancestor and classified as type II RCs.11–13 In type II RCs, QB in the inactive branch accepts an electron from QA in the active branch while it does not directly accept an electron from (B) Pheo in the inactive branch (Fig. 1).
In PbRC, the strong electronic coupling between the special pair BChls, PL and PM, leads to stabilization of the delocalized exciton, (PLPM)*.11,34 The PLPM can accept an exciton from LHI, which absorbs a near infrared photon.5–7 Time-resolved spectroscopic measurements indicated that the excited electron in (PLPM)* is transferred to HLvia BL along the L-branch on a time scale of a few ps.39–50 Despite the pseudo-C2 symmetric cofactor arrangement, the difference in the amino acid sequences between the L- and M-branches leads to the difference in the redox potentials of the pigments via electrostatic interactions and polarization.51–53
A previous study using time-dependent density functional theory (TDDFT) with the quantum mechanics/molecular mechanics/polarizable continuum model (QM/MM/PCM) method indicated that the intermediate states of charge separation along the L- and M-branches, i.e., [PLPM]˙+BL˙− and [PLPM]˙+BM˙−, are lower and higher in energy than that of (PLPM)*, respectively.34
In contrast to PbRC, the excitation energies of PD1 and PD2 in PSII are higher than those of ChlD1 and ChlD2,9,34 where the exciton tends to be localized on a single pigment owing to a weak excitonic coupling. Charge separation in PSII creates a hole localized on PD1˙+, which is the nearest pigment to the Mn4CaO5 cluster located on the D1 side.54–57 The localized nature of a hole on PD1˙+ is important for PSII to keep a high oxidation potential.58
In PSII, CP43 and CP47 transfer an exciton to the RC, presumably, via the peripheral Chls on the D1 (ChlzD1) and D2 (ChlzD2) sides.3,9,10 Time-resolved spectroscopic measurements on PSII suggested that the primary electron transfer occurs from an exciton on 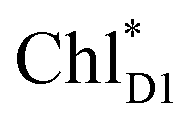 to PheoD1 on a time scale of a few hundred fs.14,22 The hole on ChlD1˙+ is, in turn, transferred to PD1 on a time scale of a few ps.14
to PheoD1 on a time scale of a few hundred fs.14,22 The hole on ChlD1˙+ is, in turn, transferred to PD1 on a time scale of a few ps.14
Because the potential for electron transfer is energetically downhill along both the D1- and D2-branches toward PheoD1 and PheoD2, respectively,33,34 the energetics alone cannot explain unidirectional charge separation in PSII. Given that both ChlD1 and ChlD2 can accept an exciton from the core antenna complexes, the mechanism that leads to charge separation exclusively along the D1-branch is of particular interest. The charge separation pathways in pigment–protein complexes can be determined by various factors including energetics, electronic coupling, vibronic coupling, and quantum effects.2–4,18–21,31,44–46
In this study, we address the long-standing open question as to how PbRC and PSII achieve unidirectional charge separation exclusively along the active branch, by means of nonadiabatic quantum dynamics calculations59–62 parametrized on the basis of TDDFT in the framework of the QM/MM/PCM method.63,64 First, we show that the experimentally observed kinetics of charge separation along the active branches of PbRC and PSII are fairly well reproduced by nonadiabatic quantum dynamics calculations, which is based on the energetics and electronic coupling of the pigments, accounting for electrostatic interactions and polarization of whole protein environments from the X-ray crystal structures. On this basis, we clarify the essential factors which regulate the charge separation pathways in the reaction centers.
2. Methods
The energetics and electronic couplings in PbRC and PSII are analyzed by means of the polarizable QM/MM/PCM method, using the QuanPol code63 implemented in the GAMESS code.65 The electronic states in the QM regions are calculated using DFT and TDDFT with the CAMB3LYP functional66 with the range separation parameter μ of 0.14, α of 0.19, and β of 0.46, which is well suited for the present systems including charge separated states.34 The quantitative values of excitation energies may depend on functionals and parameters.32,38,67 The 6-31G(d) basis set is used for all the QM calculations.The QM region comprises pigments, ligands, hydrogen bonded water, and residues which interact directly with pigments as detailed in a previous report.34 A polarizable amber-02 force field68 is applied for proteins in the MM region, where induced dipoles of the MM atoms are taken into account to reproduce the dielectric screening. The PCM with a dielectric constant of 80 is applied to reproduce the polarization of water, which surrounds the proteins and fills the cavities. The PCM in the QuanPol code is based on a conductor-like screening model,63,64 where the polarization points are put on spheres of radius 3.0 Å from the atom positions.34 All atoms from the X-ray crystal structures are explicitly considered, where each MM atom contains an induced dipole in addition to the permanent charge. The induced dipole of each MM atom is determined iteratively together with the self-consistent field calculation of electronic states, considering the electrostatic interactions with the electrons and nuclei in the QM region as well as the permanent charges and induced dipoles of other MM atoms.63 The molecular orbital levels of the cofactors calculated using QM/MM reproduce the redox potential values calculated solving the Poisson–Boltzmann equation.33,34,57,69 While the dielectric constant for the membrane region may be lower than 80 (e.g. 20),70 a small dielectric constant makes the electrostatic interactions with the charged groups in the membrane-extrinsic region overestimated for membrane proteins. The optimal values for the dielectric constant depend on the protein model used.71,72 The dielectric constant of 80 for the bulk region appears to be optimal for the present models, as suggested previously.33,34
The atomic coordinates of PSII and PbRC are obtained from the X-ray crystal structures from Thermosynechococcus vulcanus at 1.9 Å resolution (PDB code, 3ARC)73 and from Rhodobacter sphaeroides at 2.01 Å resolution (PDB code, 3I4D),74 respectively. The intramolecular reorganization energies of pigments are calculated through geometry optimization with QM/MM, where DFT with the CAMB3LYP functional plus Grimme's dispersion correction75 is used for the QM region. The atomic coordinates of the MM region are fixed to the X-ray crystal structures. The reorganization energy of the MM region is not taken into account.
The electronic coupling between excited states is evaluated on the basis of the diabatization scheme for TDDFT76 in the framework of the QM/MM/PCM method.34,77 The protocol of diabatization is summarized below.
(1) We prepare a set of reference wavefunctions, ΦI, that possess pure characters of the excited states such as an exciton on a single molecule (i.e., Frenkel exciton) and charge separated states for decoupled molecules.
(2) We calculate adiabatic electronic states in the pigment–protein complexes using TDDFT-QM/MM/PCM.
(3) The diabatic wavefunctions are expressed as a linear combination of the adiabatic wavefunctions, ΨJ, by evaluating the overlap integrals between the reference and adiabatic wavefunctions:
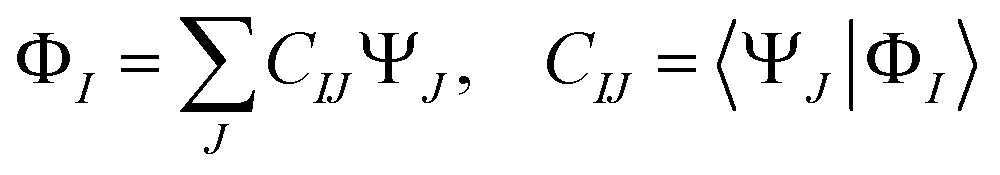 | (1) |
That is, the adiabatic states from the TDDFT-QM/MM/PCM calculations are considered as basis functions for expanding the diabatic states. We consider 10 adiabatic states for expanding the diabatic states. The diabatic coupling is then evaluated as follows:
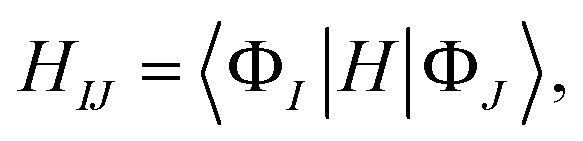 | (2) |
For the nonadiabatic quantum dynamics calculations, we consider the following linear vibronic coupling Hamiltonian in the diabatic representation:
 | (3) |
 | (4) |
H IJ is the diabatic coupling (electronic coupling) between the states I and J. HII is the vertical excitation energy of the Ith electronic states. ωi, xi, and pi are the frequency, position, and momentum of the ith vibrational mode (harmonic oscillator) in the dimensionless coordinate. κiI is the vibronic coupling of the ith vibrational mode in the Ith electronic state.
The exciton on the special pair, (PLPM)*, is considered for the initial conditions of the quantum dynamics calculations of charge separation in PbRC. For PSII, in addition to the exciton localized on 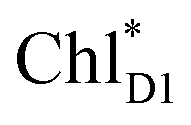 in the D1-branch,
in the D1-branch,  in the D2-branch is also considered for the initial conditions of the quantum dynamics calculations of charge separation. For the exciton transfer between
in the D2-branch is also considered for the initial conditions of the quantum dynamics calculations of charge separation. For the exciton transfer between  and
and  , the direct pathway and the indirect pathway via
, the direct pathway and the indirect pathway via and
and 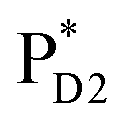 are considered, where the quantum dynamics calculations account for the interference of the phase factors from several pathways. The initial vibrational wave packet is put on the Franck–Condon region of the initial electronic state.
are considered, where the quantum dynamics calculations account for the interference of the phase factors from several pathways. The initial vibrational wave packet is put on the Franck–Condon region of the initial electronic state.
ω
i
and κiI in eqn (4), i.e., spectral density, are determined on the basis of the normal mode analysis and the geometry optimization of the pigments using the QM/MM/PCM method, where the atomic displacements from the Franck–Condon region to the potential bottom on the respective electronic states are projected onto the normal modes. The present model explicitly considers the vibronic couplings of the pigments and axial ligands, which are relevant to the dynamics of charge separation on a time scale of a few ps, whereas slow vibrational modes from surrounding proteins are neglected. The vibrational modes are reduced to a limited number of effective modes which reproduce the short-time dynamics and the reorganization energy of the system (see ESI†).60–62 We consider 25 effective modes for each pigment, unless otherwise noted. For charge separation in PSII via indirect exciton transfer from  to
to  , 10 effective modes are considered for the respective intermediate states,
, 10 effective modes are considered for the respective intermediate states,  and
and  . The multi-configuration time-dependent Hartree (MCTDH) method59 is used for the nonadiabatic quantum dynamics calculations, which properly consider correlations among the nuclear degrees of freedom, the Franck–Condon factor of vibrational wavefunctions, and vibrational energy redistribution along with electronic state transitions.
. The multi-configuration time-dependent Hartree (MCTDH) method59 is used for the nonadiabatic quantum dynamics calculations, which properly consider correlations among the nuclear degrees of freedom, the Franck–Condon factor of vibrational wavefunctions, and vibrational energy redistribution along with electronic state transitions.
For analyzing the time constants of the first (τ1) and second (τ2) charge transfers along the active branches, τ1 and τ2 in the following rate equations are determined via curve fitting against the populations of the exciton (PEX), and the first (PCS1) and second (PCS2) charge separated states in the quantum dynamics calculations:
 | (5) |
 | (6) |
3. Results and discussion
3.1. Charge separation in PbRC
(PLPM) in PbRC can be regarded as a single molecular site owing to the strong electronic coupling.34 The electron transfers from (PLPM)* to (PLPM)˙+BL˙− and (PLPM)˙+BM˙− are exothermic (downhill) and endothermic (uphill), respectively (Fig. 2a).34 As a benchmark, we first compare the calculated time constants of charge separation along the L-branch with the corresponding experimental values. The quantum dynamics calculations indicate that (PLPM)* initially transfers the excited electron to BL on a time scale of τ1 ≈ 3.2 ps. BL˙−, in turn, transfers the electron to HL on a time scale of τ2 ≈ 1.8 ps (Fig. 2c and f). A similar order of time constants was observed in time-resolved spectroscopic measurements on charge separation in PbRC (τ1 = 3.5 ± 0.4 ps and τ2 = 1.2 ± 0.3 ps).41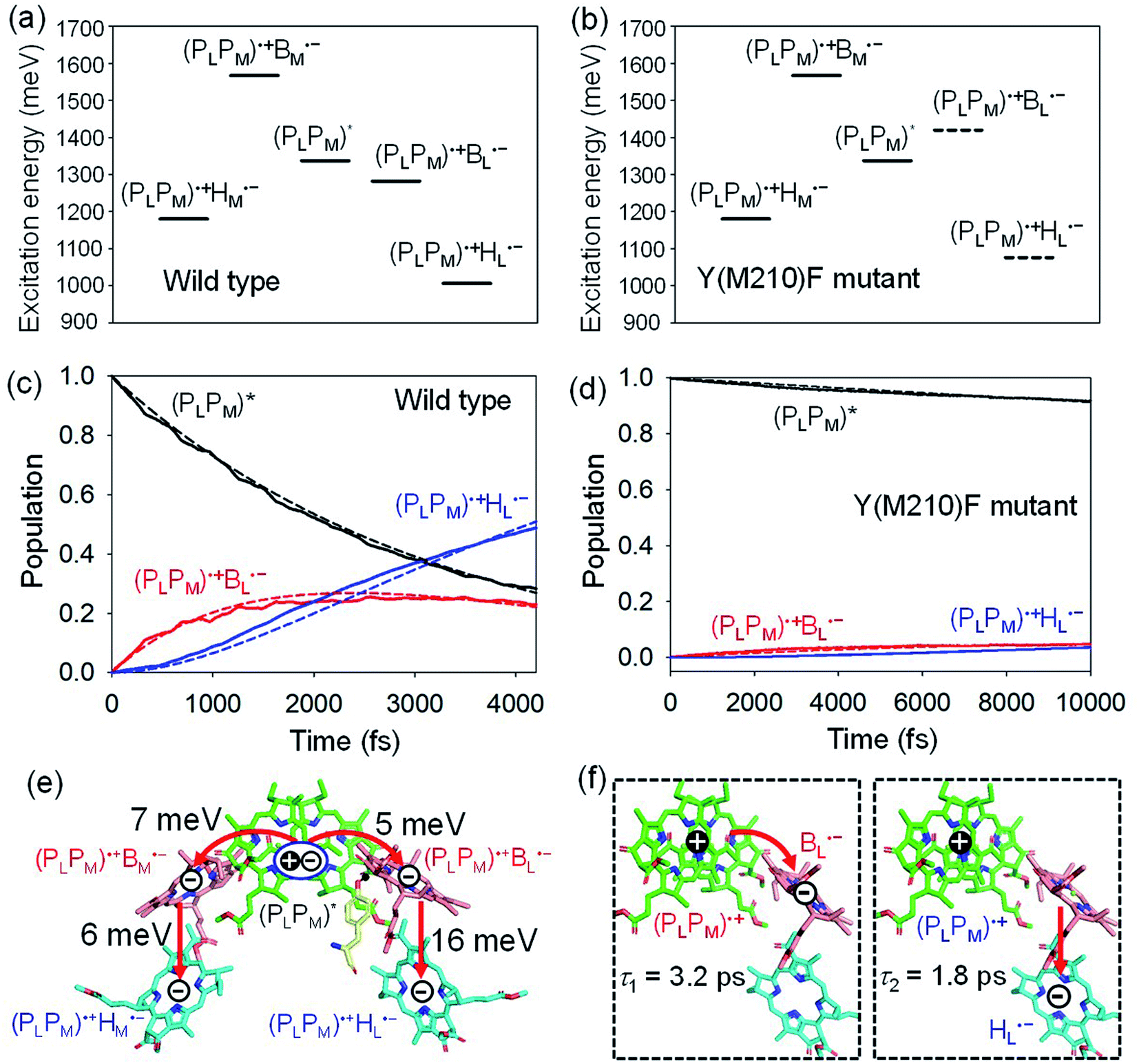 | ||
| Fig. 2 Bottom-to-bottom (adiabatic) excitation energies of the electronic states considering the intramolecular reorganization energies in (a) wild type and (b) Y(M210)F mutant PbRC. Dotted lines indicate the destabilized charge separated states in Y(M210)F mutant PbRC. Population of electronic states during quantum dynamics calculations of charge separation in (c) wild type and (d) Y(M210)F mutant PbRC, where the (PLPM)*, (PLPM)˙+BL˙−, and (PLPM)˙+HL˙− states along the L-branch are considered. Dotted lines indicate curve fitting by using eqn (5). (e) Diagram of charge transfer pathways (red lines) with electronic coupling (meV). Tyr-M210 is shown in yellow. (f) Diagram of the electron and hole locations in the (PLPM)˙+BL˙− and (PLPM)˙+HL˙− states with τ1 and τ2 (ps) for wild type PbRC (∼5 ps in total). τ1 and τ2 for the mutant PbRC are 110 and 8 ps, respectively (∼118 ps in total). | ||
The electronic coupling of the BL˙− → HL˙− transfer (16 meV) is stronger than that of the (PLPM)* → (PLPM)˙+BL˙− transfer (5 meV) (Fig. 2e). Thus, the population of the intermediate (PLPM)˙+BL˙− state is kept small (Fig. 2c). The fast electron transfer from BL˙− to HL is advantageous for preventing charge recombination, because (PLPM)˙+HL˙− is difficult to decay to the ground state owing to a negligibly small orbital overlap between (PLPM)˙+ and HL˙−. Charge separation along the M-branch is negligibly slow, because the intermediate (PLPM)˙+BM˙− state is substantially higher in energy than (PLPM)*, even though (PLPM)˙+HM˙− is lower in energy than (PLPM)* (Fig. 2a).
The previous time-resolved spectroscopic measurements of mutant PbRC suggested that some specific residues especially contribute to unidirectional charge separation.47–50,52 We have extensively analyzed the contribution of each residue to the potential shift on the pigments one by one, and concluded that Tyr-M210 near BL has the largest contribution to the stabilization of BL˙−,33,34 where Phe-L181 is located at the counterpart position near BM.
To verify the essential role of Tyr-M210, we consider the mutation of Tyr-M210 to phenylalanine, Y(M210)F, and investigate charge separation in the mutant PbRC by means of quantum dynamics calculations. The present TDDFT-QM/MM/PCM calculations indicate that the Y(M210)F mutation, in which the hydroxyl group is replaced with hydrogen, makes the intermediate (PLPM)˙+BL˙− state energetically uphill with respect to (PLPM)*, even though the final (PLPM)˙+HL˙− state remains downhill (Fig. 2b). The quantum dynamics calculation indicates that the destabilization of the intermediate (PLPM)˙+BL˙− state drastically slows charge separation along the L-branch through the superexchange mechanism (Fig. 2d). This trend is qualitatively consistent with the experimental observations for the mutant PbRC,47–50,52 where the calculated time constant (∼118 ps) is quantitatively larger than the experimental values (∼16 ps).47 Here, only the local geometry of Phe-M210 was optimized in QM/MM, fixing surrounding proteins at the original geometry of wild type, although the mutation may also affect the surrounding geometry. Overall, the present analysis highlights the impact of the electrostatic interaction of Tyr-M210 on the efficient charge separation along the L-branch.
3.2. Charge separation in PSII
In PSII, ChlD1 and ChlD2 are supposed to accept an exciton from CP43 and CP47 via and
and  , respectively (Fig. 3a).9,10 The present calculations indicate that the bottom-to-bottom excitation energy of
, respectively (Fig. 3a).9,10 The present calculations indicate that the bottom-to-bottom excitation energy of 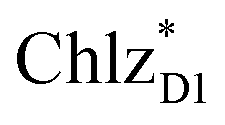 (1991 meV) lies between those of
(1991 meV) lies between those of  (1965 meV) and
(1965 meV) and 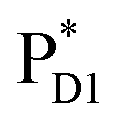 (2032 meV, Fig. 3b). Similarly, the
(2032 meV, Fig. 3b). Similarly, the  energy (2015 meV) lies between those of
energy (2015 meV) lies between those of  (1992 meV) and
(1992 meV) and  (2038 meV, Fig. 3b). Thus,
(2038 meV, Fig. 3b). Thus,  and
and  can accept an exciton from
can accept an exciton from 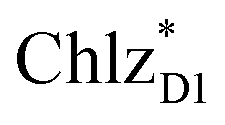 and
and 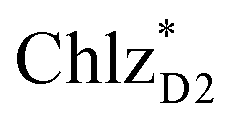 , respectively, in terms of energetics.
, respectively, in terms of energetics.
 | ||
| Fig. 3 (a) Arrangement of Chl molecules in PSII. Calculated bottom-to-bottom (adiabatic) excitation energies considering the intramolecular reorganization energies of the (b) exciton and (c) exciton and charge separated states in PSII. The intermediate and final charge separated states are indicated in red and blue, respectively. | ||
The absolute values of the calculated excitonic couplings in PSII are in the range of 7 to 15 meV (Table 1). The lowest and second lowest excitons obtained by diagonalizing the coupling matrix are localized on  and
and  , respectively (Fig. S2†), which can be regarded as Frenkel excitons. The quantitative values of the exciton energies in PSII calculated using TDDFT-QM/MM/PCM with the CAMB3LYP functional tend to be blue-shifted as compared to the experimental values,25–27 where the calculated lowest exciton energy of 632 nm is blue-shifted as compared to the experimental value of 680 nm.25–27
, respectively (Fig. S2†), which can be regarded as Frenkel excitons. The quantitative values of the exciton energies in PSII calculated using TDDFT-QM/MM/PCM with the CAMB3LYP functional tend to be blue-shifted as compared to the experimental values,25–27 where the calculated lowest exciton energy of 632 nm is blue-shifted as compared to the experimental value of 680 nm.25–27
|
|
|
|
|
ChlD1˙+PheoD1˙− | |
|---|---|---|---|---|---|
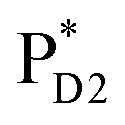
|
−10.1 | ||||
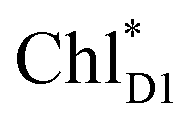
|
7.4 | −13.6 | |||
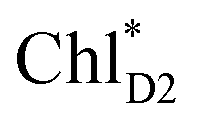
|
−14.4 | 7.0 | −1.8 | ||
| ChlD1˙+PheoD1˙− | −21.6 | ||||
| PD1˙+PheoD1˙− | 6.4 | ||||
| PD1˙+ChlD1˙− | −6.5 | 5.4 | |||
| PD1˙+ChlD2˙− | 0.7 | −0.3 | |||
| PD2˙+ChlD1˙− | −0.5 | −5.4 |

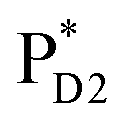

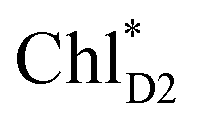
We analyze charge separation from an exciton on  by means of quantum dynamics calculations. The initial electron transfer from
by means of quantum dynamics calculations. The initial electron transfer from  to ChlD1˙+PheoD1˙− occurs on an ultrafast time scale (τ1 ≈ 0.15 ps) (Fig. 4a and c) owing to a strong electronic coupling (∼22 meV, Fig. 4c and Table 1). The subsequent hole transfer to PD1˙+PheoD1˙− occurs on a time scale of τ2 ≈ 3.7 ps (Fig. 4a and c). Thus, once
to ChlD1˙+PheoD1˙− occurs on an ultrafast time scale (τ1 ≈ 0.15 ps) (Fig. 4a and c) owing to a strong electronic coupling (∼22 meV, Fig. 4c and Table 1). The subsequent hole transfer to PD1˙+PheoD1˙− occurs on a time scale of τ2 ≈ 3.7 ps (Fig. 4a and c). Thus, once  accepts an exciton, charge separation occurs efficiently along the D1-branch. Similar time constants of charge separation in PSII were observed in the time-resolved spectroscopy measurements.14,22 Another charge separation pathway,
accepts an exciton, charge separation occurs efficiently along the D1-branch. Similar time constants of charge separation in PSII were observed in the time-resolved spectroscopy measurements.14,22 Another charge separation pathway, 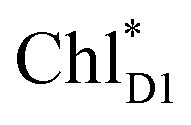 → PD1˙+ChlD1˙−, is endothermic (Fig. 3c) and thus cannot compete with
→ PD1˙+ChlD1˙−, is endothermic (Fig. 3c) and thus cannot compete with 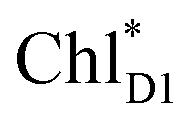 → ChlD1˙+PheoD1˙−. Although other charge separation pathways from an exciton on
→ ChlD1˙+PheoD1˙−. Although other charge separation pathways from an exciton on  and
and 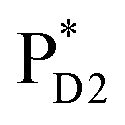 were also proposed,23,24 the quantum dynamical analysis for these pathways is beyond the scope of the present study. Overall, we can conclude that charge separation along the D1-branch proceeds via two-step
were also proposed,23,24 the quantum dynamical analysis for these pathways is beyond the scope of the present study. Overall, we can conclude that charge separation along the D1-branch proceeds via two-step  → ChlD1˙+PheoD1˙− and ChlD1˙+ → PD1˙+ transfers, considering the quantum dynamical analysis based on the energetics and electronic couplings from the QM/MM/PCM method.
→ ChlD1˙+PheoD1˙− and ChlD1˙+ → PD1˙+ transfers, considering the quantum dynamical analysis based on the energetics and electronic couplings from the QM/MM/PCM method.
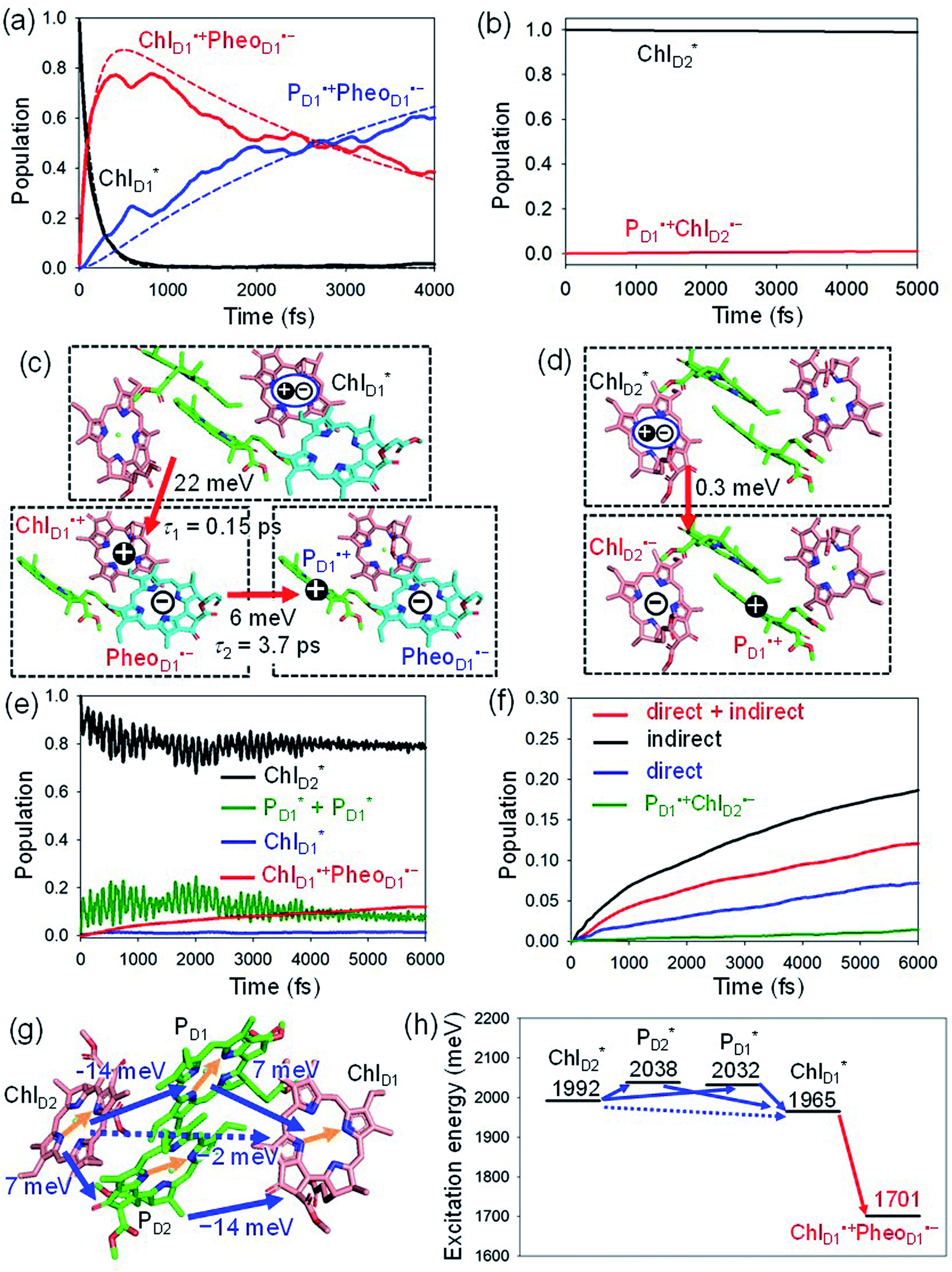 | ||
Fig. 4 Population of excited states during quantum dynamics calculations of charge separation in PSII: (a)  , ChlD1˙+PheoD1˙−, and PD1˙+PheoD1˙− along the D1-branch and (b) from , ChlD1˙+PheoD1˙−, and PD1˙+PheoD1˙− along the D1-branch and (b) from  to PD1˙+ChlD2˙−. Dotted lines indicate curve fitting by using eqn (5). Diagram of charge separation with the absolute value of electronic coupling (meV): (c) from to PD1˙+ChlD2˙−. Dotted lines indicate curve fitting by using eqn (5). Diagram of charge separation with the absolute value of electronic coupling (meV): (c) from  and (d) from and (d) from  . Population of excited states during quantum dynamics calculations from . Population of excited states during quantum dynamics calculations from  : (e) simultaneously considering indirect : (e) simultaneously considering indirect 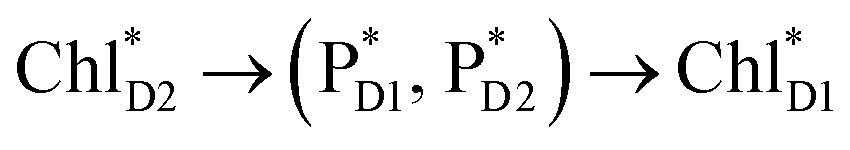 → ChlD1˙+PheoD1˙− (τ ≈ 50 ps) and direct → ChlD1˙+PheoD1˙− (τ ≈ 50 ps) and direct  → ChlD1˙+PheoD1˙− pathways, and (f) ChlD1˙+PheoD1˙− population considering only the indirect (black) or the direct (blue) pathway. The PD1˙+ChlD2˙− population (green) is shown again for comparison. (g) Exciton transfer pathways (blue arrows) with the excitonic coupling (meV). Orange arrows indicate the direction of transition dipole moment. Solid and dotted arrows indicate indirect and direct exciton transfers, respectively. (h) Exciton transfer and charge separation pathways with the excitation energy. → ChlD1˙+PheoD1˙− pathways, and (f) ChlD1˙+PheoD1˙− population considering only the indirect (black) or the direct (blue) pathway. The PD1˙+ChlD2˙− population (green) is shown again for comparison. (g) Exciton transfer pathways (blue arrows) with the excitonic coupling (meV). Orange arrows indicate the direction of transition dipole moment. Solid and dotted arrows indicate indirect and direct exciton transfers, respectively. (h) Exciton transfer and charge separation pathways with the excitation energy. | ||
The electronic coupling of the  → ChlD1˙+PheoD1˙− transfer (∼22 meV) is stronger than that of the ChlD1˙+ → PD1˙+ transfer (∼6 meV, Fig. 4c and Table 1). The strong electronic coupling between the accessary Chl/BChl and the Pheo/BPheo is a common feature of PSII/PbRC. Nevertheless, the BL˙− → HL˙− electron transfer (∼1.8 ps) in PbRC is slower than the
→ ChlD1˙+PheoD1˙− transfer (∼22 meV) is stronger than that of the ChlD1˙+ → PD1˙+ transfer (∼6 meV, Fig. 4c and Table 1). The strong electronic coupling between the accessary Chl/BChl and the Pheo/BPheo is a common feature of PSII/PbRC. Nevertheless, the BL˙− → HL˙− electron transfer (∼1.8 ps) in PbRC is slower than the  → ChlD1˙+PheoD1˙− transfer (∼0.15 ps) in PSII, because the population of the (PLPM)˙+BL˙− intermediate state in PbRC is kept small.
→ ChlD1˙+PheoD1˙− transfer (∼0.15 ps) in PSII, because the population of the (PLPM)˙+BL˙− intermediate state in PbRC is kept small.
Because ChlD2 can also accept an exciton form CP47 on the D2 side,3,9,10 the question arises as to how the exciton on 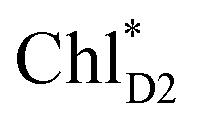 eventually undergoes charge separation in the D1-branch. To analyze charge separation mechanisms from an exciton in the D2-branch, we carried out quantum dynamics calculations considering the initial exciton localized on
eventually undergoes charge separation in the D1-branch. To analyze charge separation mechanisms from an exciton in the D2-branch, we carried out quantum dynamics calculations considering the initial exciton localized on  . The charge separated state in the D2-branch, PD1˙+PheoD2˙−, is less stable than that in the D1-branch, PD1˙+PheoD1˙− (Fig. 3c), owing mainly to a difference in the potentials between PheoD1˙− and PheoD2˙−.33,34,36
. The charge separated state in the D2-branch, PD1˙+PheoD2˙−, is less stable than that in the D1-branch, PD1˙+PheoD1˙− (Fig. 3c), owing mainly to a difference in the potentials between PheoD1˙− and PheoD2˙−.33,34,36
The most stable charge separated state in the D2-branch is PD1˙+ChlD2˙− (Fig. 3c). However, PSII can avoid charge separation from 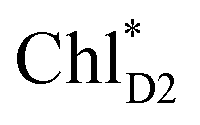 to PD1˙+ChlD2˙− (Fig. 4b) because of a weak electronic coupling (∼0.3 meV, Fig. 4d and Table 1), which is significantly weaker than that between
to PD1˙+ChlD2˙− (Fig. 4b) because of a weak electronic coupling (∼0.3 meV, Fig. 4d and Table 1), which is significantly weaker than that between  and PD2˙+ChlD1˙− (∼5.4 meV) on the counterpart side (Fig. 5a and Table 1). The difference originates from the difference in the vinyl-group orientation between PD1 and PD2 (Fig. 5). The vinyl group is rather in plane for PD1 and out of plane for PD2 (Fig. 6).57 The present results indicate that the in-plane PD1 vinyl group interferes with the π–π interaction between PD1 and ChlD2 (Fig. 5b), thereby preventing the charge transfer to form PD1˙+ChlD2˙−.
and PD2˙+ChlD1˙− (∼5.4 meV) on the counterpart side (Fig. 5a and Table 1). The difference originates from the difference in the vinyl-group orientation between PD1 and PD2 (Fig. 5). The vinyl group is rather in plane for PD1 and out of plane for PD2 (Fig. 6).57 The present results indicate that the in-plane PD1 vinyl group interferes with the π–π interaction between PD1 and ChlD2 (Fig. 5b), thereby preventing the charge transfer to form PD1˙+ChlD2˙−.
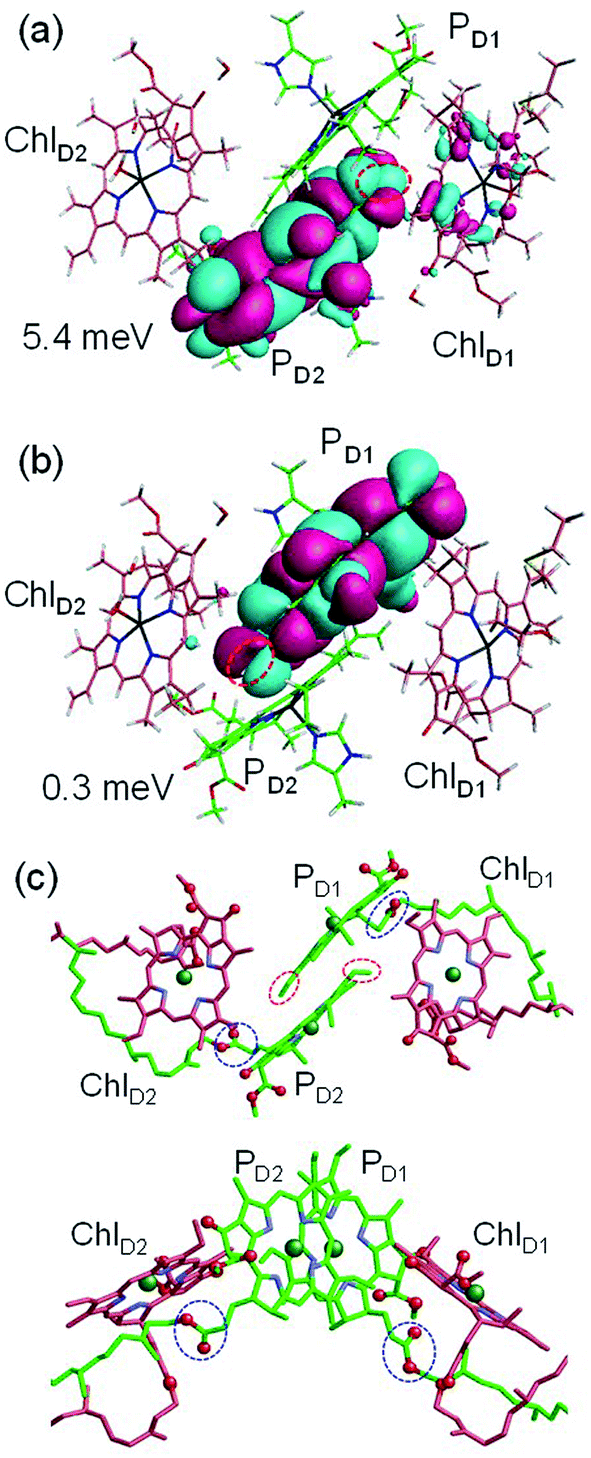 | ||
Fig. 5 Equilibrium geometry and the highest occupied molecular orbital (HOMO) of the (a) PD2ChlD1 dimer and (b) PD1ChlD2 dimer. Red circles indicate the vinyl groups. Electronic coupling (meV) between  and PD2˙+ChlD1˙− and that between and PD2˙+ChlD1˙− and that between  and PD1˙+ChlD2˙− are shown. (c) Configuration of the vinyl (red circles) and phytol (blue circles) groups of PD1 and PD2. and PD1˙+ChlD2˙− are shown. (c) Configuration of the vinyl (red circles) and phytol (blue circles) groups of PD1 and PD2. | ||
 | ||
Fig. 6 Potential energy curve as a function of the C–C–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C torsion angle (degree) of the vinyl group of (a) PD1 and (b) PD2 in PSII, where the geometry of chlorophyll is optimized by QM/MM, fixing the torsion angle. The vinyl group is in the same plane as the chlorin ring at 0 degree. The black arrows indicate the equilibrium angle. C torsion angle (degree) of the vinyl group of (a) PD1 and (b) PD2 in PSII, where the geometry of chlorophyll is optimized by QM/MM, fixing the torsion angle. The vinyl group is in the same plane as the chlorin ring at 0 degree. The black arrows indicate the equilibrium angle. | ||
The out-of-plane orientation of the PD2 vinyl group is caused by the relatively large steric hindrance from the PD1 phytol chain as compared with that between the PD1 vinyl group and the PD2 phytol chain (Fig. 5c and 6). The potential curves calculated using QM/MM indicate that the rotations of the PD1 and PD2 vinyl groups are hindered in the protein environments (Fig. 6 and S1†). Umena et al. reported that the conformations of vinyl groups were determined unambiguously from the corresponding electron density distributions and most of the vinyl groups are located in or near the same plane of the chlorine ring,73 which suggests that the out-of-plane vinyl orientation for PD2 is exceptional. The same conformations of the PD1 and PD2 vinyl groups have also been observed in the X-ray free electron laser (XFEL) structure.78 Thus, the observed vinyl orientations of PD1 and PD2 are considered to be robust in the protein environments. The phytol chains of PD1 and PD2 are less flexible due to the presence of the highly packed protein environment of D1/D2/CP43/CP47, as compared to those exposed to the protein surface in antenna proteins (e.g., LH1 and the Fenna–Matthews–Olson protein). Umena et al. also confirmed that all of the C8 and C13 positions in the phytol chains have a (R,R) configuration as indicated by the low B-factor values.73 Notably, the difference in the phytol-chain conformation also contributes to the asymmetric hole distribution on PD1 and PD2, i.e., PD1˙+ > PD2˙+.57 These results suggest that the symmetry-breaking of the PD1PD2 geometry not only increases the PD1˙+ population, which facilitates water oxidation at the Mn4CaO5 moiety on the D1 side, but also prevents charge separation along the D2-branch via a weak electronic coupling between  and PD1˙+ChlD2˙−.
and PD1˙+ChlD2˙−.
The quantum dynamics calculations indicate that the exciton on 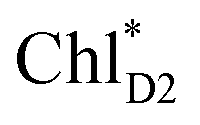 can be transferred to ChlD1via the direct pathway and the indirect pathway mediated by
can be transferred to ChlD1via the direct pathway and the indirect pathway mediated by 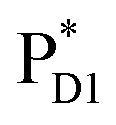 and
and  owing to adequately strong excitonic couplings (Table 1 and Fig. 4g) and small energy differences (Fig. 4h). Note that among the residues near ChlD1 and ChlD2, D1-Met172 adjacent to ChlD1 contributes to the difference in excitation energy between
owing to adequately strong excitonic couplings (Table 1 and Fig. 4g) and small energy differences (Fig. 4h). Note that among the residues near ChlD1 and ChlD2, D1-Met172 adjacent to ChlD1 contributes to the difference in excitation energy between  and
and  .34 Previous calculations by Sirohiwal et al.38 using other DFT functionals and an equation-of-motion coupled cluster method also indicated that ChlD1 exhibits the lowest excitation energy in the protein environment of PSII. Once the exciton is transferred to ChlD1, subsequent charge separation to ChlD1˙+PheoD1˙− occurs rapidly. The present analysis indicates that an overall time scale of charge separation from
.34 Previous calculations by Sirohiwal et al.38 using other DFT functionals and an equation-of-motion coupled cluster method also indicated that ChlD1 exhibits the lowest excitation energy in the protein environment of PSII. Once the exciton is transferred to ChlD1, subsequent charge separation to ChlD1˙+PheoD1˙− occurs rapidly. The present analysis indicates that an overall time scale of charge separation from  to ChlD1˙+PheoD1˙− is in the order of a few tens ps (Fig. 4e and f), where exponential fitting indicates a τ of ∼50 ps. It is highly likely that the exciton on
to ChlD1˙+PheoD1˙− is in the order of a few tens ps (Fig. 4e and f), where exponential fitting indicates a τ of ∼50 ps. It is highly likely that the exciton on 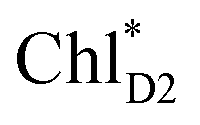 can eventually undergo charge separation in the D1-branch without charge separation in the D2-branch. Even though the energy difference between
can eventually undergo charge separation in the D1-branch without charge separation in the D2-branch. Even though the energy difference between  and
and 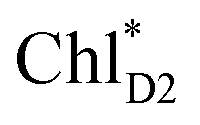 is small (Fig. 4h), the ultrafast charge separation from
is small (Fig. 4h), the ultrafast charge separation from 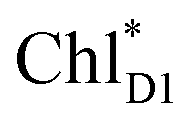 to ChlD1˙+PheoD1˙− prevents exciton back transfer to ChlD2, enhancing the robustness of unidirectional charge separation along the D1-branch. The charge separation pathway via the exciton transfer from the D2- to D1-branches may correspond to the delayed component observed in the time-resolved spectroscopic measurements apart from the ultrafast
to ChlD1˙+PheoD1˙− prevents exciton back transfer to ChlD2, enhancing the robustness of unidirectional charge separation along the D1-branch. The charge separation pathway via the exciton transfer from the D2- to D1-branches may correspond to the delayed component observed in the time-resolved spectroscopic measurements apart from the ultrafast 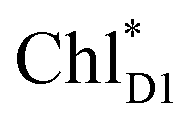 → ChlD1˙+PheoD1˙− charge separation within the D1-branch.14
→ ChlD1˙+PheoD1˙− charge separation within the D1-branch.14
The direct excitonic coupling between  and
and 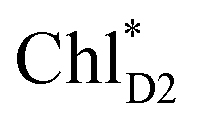 is relatively weak (∼2 meV) owing to the long distance (∼20 Å) as compared with the coupling between neighboring Chls, i.e.,
is relatively weak (∼2 meV) owing to the long distance (∼20 Å) as compared with the coupling between neighboring Chls, i.e.,  pairs (Fig. 4g and Table 1). Consequently, charge separation considering only the direct
pairs (Fig. 4g and Table 1). Consequently, charge separation considering only the direct 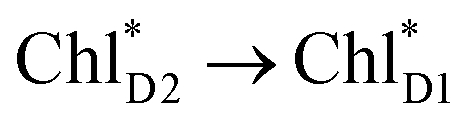 → ChlD1˙+PheoD1˙− pathway is slower than charge separation considering only the indirect
→ ChlD1˙+PheoD1˙− pathway is slower than charge separation considering only the indirect  → ChlD1˙+PheoD1˙− pathway in the quantum dynamics calculations (Fig. 4f). The excitonic coupling between
→ ChlD1˙+PheoD1˙− pathway in the quantum dynamics calculations (Fig. 4f). The excitonic coupling between  and
and  (−14 meV) is stronger than that between
(−14 meV) is stronger than that between  and
and  (7 meV, Table 1). The former and latter are characterized as J- and H-aggregates (minus and plus signs), respectively, considering the directions of the transition dipole moments (Fig. 4g). The excitonic coupling is relatively insensitive to the orbital overlap as compared with the case of the charge transfer coupling. The direct and indirect
(7 meV, Table 1). The former and latter are characterized as J- and H-aggregates (minus and plus signs), respectively, considering the directions of the transition dipole moments (Fig. 4g). The excitonic coupling is relatively insensitive to the orbital overlap as compared with the case of the charge transfer coupling. The direct and indirect  exciton transfers exhibit the destructive interference of the quantum phase factor, which is dictated by the signs of excitonic couplings, i.e., relative orientation of the transition dipole moments. Consequently, the exciton transfer rate considering all pathways is slower than the rate considering only the indirect pathway (Fig. 4f). Thus, in terms of the phase factor, the configuration of Chls in PSII is not necessarily optimal for accelerating the
exciton transfers exhibit the destructive interference of the quantum phase factor, which is dictated by the signs of excitonic couplings, i.e., relative orientation of the transition dipole moments. Consequently, the exciton transfer rate considering all pathways is slower than the rate considering only the indirect pathway (Fig. 4f). Thus, in terms of the phase factor, the configuration of Chls in PSII is not necessarily optimal for accelerating the 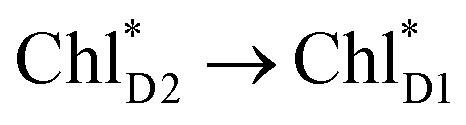 transfer, while the configuration is optimal for charge separation along the D1-branch.
transfer, while the configuration is optimal for charge separation along the D1-branch.
Overall, it can be concluded that the  exciton transfer followed by charge separation to ChlD1˙+PheoD1˙− in the D1-branch is overwhelmingly faster than charge separation in the D2-branch (Fig. 4f). The irreversible
exciton transfer followed by charge separation to ChlD1˙+PheoD1˙− in the D1-branch is overwhelmingly faster than charge separation in the D2-branch (Fig. 4f). The irreversible  exciton transfer allows PSII to utilize the excitation energy from both the CP43 and CP47 antenna complexes for charge separation in the active branch.
exciton transfer allows PSII to utilize the excitation energy from both the CP43 and CP47 antenna complexes for charge separation in the active branch.
3.3. Role of Mn4CaO5 in the charge separation pathway in PSII
The localized electronic states on PD1 in PSII are advantageous to maintain a high oxidation potential for water splitting in contrast to the strongly coupled (PLPM)* and (PLPM)˙+ in PbRC.34 The hole on PD1˙+ is largely stabilized by acidic residues near the Mn4CaO5 cluster, namely D1-Asp61, D1-Glu189, and D1-Asp170.34,57 This may explain why the Mn4CaO5 cluster is located on the D1 side, because the electrostatic potential, which attracts a hole toward the D1 side, also enhances charge separation to PD1˙+PheoD1˙−.33,34,58 Because the difference in the redox potential between PD1 and ChlD1 is small,34 PD1˙+ChlD1˙− is substantially higher in energy than ChlD1˙+PheoD1˙− (Fig. 3c). Thus, the exciton funneling to rather than
rather than  is a reasonable design principle for efficient charge separation to use excitons from the antenna complexes in PSII.
is a reasonable design principle for efficient charge separation to use excitons from the antenna complexes in PSII.
4. Conclusion
Quantum dynamics calculations indicated that two-step (PLPM)* → (PLPM)˙+BL˙− and BL˙− → HL˙− electron transfers occur on a time scale of ∼3.2 and ∼1.8 ps, respectively (Fig. 2c). The population of the intermediate (PLPM)˙+BL˙− state is kept small, owing to a strong BL˙− → HL˙− coupling (∼16 meV, Fig. 2e). The rapid electron transfer to HL is advantageous for preventing charge recombination, because the orbital overlap between (PLPM)˙+ and HL˙− is negligibly small owing to a long molecular distance. The electrostatic interaction with the hydroxyl group of Tyr-M210 near BL stabilizes the intermediate (PLPM)˙+BL˙− state and accelerates charge separation along the L-branch, highlighting the essential role of Tyr-M210 in efficient unidirectional charge separation.In PSII, both ChlD1 and ChlD2 can accept an exciton from CP43 and CP47, respectively. The 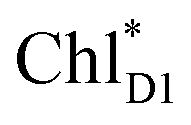 → ChlD1˙+PheoD1˙− electron transfer occurs on an ultrafast time scale (∼0.15 ps), followed by the ChlD1˙+ → PD1˙+ hole transfer on a time scale of ∼3.7 ps (Fig. 4a), as suggested by time-resolved spectroscopic measurements.14 Charge separation in the D2-branch is unlikely to occur despite the relatively stable PD1˙+ChlD2˙− state, because the in-plane PD1 vinyl group interferes with the π–π interaction between PD1 and ChlD2, thereby weakening the electronic coupling. The exciton on
→ ChlD1˙+PheoD1˙− electron transfer occurs on an ultrafast time scale (∼0.15 ps), followed by the ChlD1˙+ → PD1˙+ hole transfer on a time scale of ∼3.7 ps (Fig. 4a), as suggested by time-resolved spectroscopic measurements.14 Charge separation in the D2-branch is unlikely to occur despite the relatively stable PD1˙+ChlD2˙− state, because the in-plane PD1 vinyl group interferes with the π–π interaction between PD1 and ChlD2, thereby weakening the electronic coupling. The exciton on  can be transferred to ChlD1via the direct and indirect pathways. Subsequently, the ultrafast
can be transferred to ChlD1via the direct and indirect pathways. Subsequently, the ultrafast  → ChlD1˙+PheoD1˙− charge separation prevents exciton back transfer to ChlD2, thereby enhancing the robustness of unidirectional charge separation in the D1-branch. Thus, PSII efficiently utilizes excitons not only from CP43 (D1 side) but also from CP47 (D2 side) for charge separation in the D1-branch, which leads to electron transfer to QBvia QA and hole transfer to the Mn4CaO5 cluster on the D1 side.
→ ChlD1˙+PheoD1˙− charge separation prevents exciton back transfer to ChlD2, thereby enhancing the robustness of unidirectional charge separation in the D1-branch. Thus, PSII efficiently utilizes excitons not only from CP43 (D1 side) but also from CP47 (D2 side) for charge separation in the D1-branch, which leads to electron transfer to QBvia QA and hole transfer to the Mn4CaO5 cluster on the D1 side.
Author contributions
H. T. designed the research. H. T., K. S., and H. I. performed the research. H. T. wrote the main part of the manuscript. All the authors were involved in the discussion of the results and contributed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by JST CREST (JPMJCR1656 to H. I), JSPS KAKENHI (JP18H01937 to H. T. and H. I., JP18H05155, JP20H03217, and JP20H05090 to H. I., JP18H01186 to K. S., and JP16H06560 to K. S.), and the Interdisciplinary Computational Science Program in CCS, University of Tsukuba. The authors acknowledge valuable discussions with prof. Akihiko Ishizaki.References
- E. Wientjes, H. van Amerongen and R. Croce, J. Phys. Chem. B, 2013, 117, 11200–11208 CrossRef CAS PubMed.
- G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. van Grondelle, Nat. Chem., 2011, 3, 763–774 CrossRef CAS PubMed.
- C. Kreisbeck and A. Aspuru-Guzik, Chem. Sci., 2016, 7, 4174–4183 RSC.
- J. Pan, A. Gelzinis, V. Chorošajev, M. Vengris, S. S. Senlik, J.-R. Shen, L. Valkunas, D. Abramavicius and J. P. Ogilvie, Phys. Chem. Chem. Phys., 2017, 19, 15356–15367 RSC.
- R. van Grondelle and V. I. Novoderezhkin, Phys. Chem. Chem. Phys., 2006, 8, 793–807 RSC.
- F. Ma, L.-J. Yu, Z.-Y. Wang-Otomo and R. van Grondelle, Biochim. Biophys. Acta Bioenerg., 2016, 1857, 408–414 CrossRef CAS PubMed.
- L.-M. Tan, J. Yu, T. Kawakami, M. Kobayashi, P. Wang, Z.-Y. Wang-Otomo and J.-P. Zhang, J. Phys. Chem. Lett., 2018, 9, 3278–3284 CrossRef CAS PubMed.
- M. Germano, A. Y. Shkuropatov, H. Permentier, R. de Wijn, A. J. Hoff, V. A. Shuvalov and H. J. van Gorkom, Biochemistry, 2001, 40, 11472–11482 CrossRef CAS PubMed.
- B. A. Diner and F. Rappaport, Annu. Rev. Plant Biol., 2002, 53, 551–580 CrossRef CAS PubMed.
- S. Vasil’ev, J.-R. Shen, N. Kamiya and D. Bruce, FEBS Lett., 2004, 561, 111–116 CrossRef.
- T. Cardona, A. Sedoud, N. Cox and A. W. Rutherford, Biochim. Biophys. Acta Bioenerg., 2012, 1817, 26–43 CrossRef CAS PubMed.
- T. Cardona and A. W. Rutherford, Trends Plant Sci., 2019, 24, 1008–1021 CrossRef CAS PubMed.
- T. Cardona, J. W. Murray and A. W. Rutherford, Mol. Biol. Evol., 2015, 32, 1310–1328 CrossRef CAS PubMed.
- M. L. Groot, N. P. Pawlowicz, L. J. G. W. van Wilderen, J. Breton, I. H. M. van Stokkum and R. van Grondelle, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13087 CrossRef CAS PubMed.
- M.-L. Groot, F. van Mourik, C. Eijckelhoff, I. H. M. van Stokkum, J. P. Dekker and R. van Grondelle, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4389 CrossRef CAS PubMed.
- A. R. Holzwarth, M. G. Müller, M. Reus, M. Nowaczyk, J. Sander and M. Rögner, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6895 CrossRef CAS PubMed.
- M. Hasegawa, H. Nagashima, R. Minobe, T. Tachikawa, H. Mino and Y. Kobori, J. Phys. Chem. Lett., 2017, 8, 1179–1184 CrossRef CAS PubMed.
- J. A. Myers, K. L. M. Lewis, F. D. Fuller, P. F. Tekavec, C. F. Yocum and J. P. Ogilvie, J. Phys. Chem. Lett., 2010, 1, 2774–2780 CrossRef CAS.
- F. D. Fuller, J. Pan, A. Gelzinis, V. Butkus, S. S. Senlik, D. E. Wilcox, C. F. Yocum, L. Valkunas, D. Abramavicius and J. P. Ogilvie, Nat. Chem., 2014, 6, 706–711 CrossRef CAS PubMed.
- E. Romero, R. Augulis, V. I. Novoderezhkin, M. Ferretti, J. Thieme, D. Zigmantas and R. van Grondelle, Nat. Phys., 2014, 10, 676–682 Search PubMed.
- E. Romero, V. I. Novoderezhkin and R. van Grondelle, Nature, 2017, 543, 355–365 CrossRef CAS PubMed.
- G. Raszewski and T. Renger, J. Am. Chem. Soc., 2008, 130, 4431–4446 CrossRef CAS PubMed.
- E. Romero, I. H. M. van Stokkum, V. I. Novoderezhkin, J. P. Dekker and R. van Grondelle, Biochemistry, 2010, 49, 4300–4307 CrossRef CAS PubMed.
- V. I. Novoderezhkin, E. Romero, J. P. Dekker and R. van Grondelle, ChemPhysChem, 2011, 12, 681–688 CrossRef CAS PubMed.
- G. Raszewski, W. Saenger and T. Renger, Biophys. J., 2005, 88, 986–998 CrossRef CAS PubMed.
- V. I. Novoderezhkin, J. P. Dekker and R. van Grondelle, Biophys. J., 2007, 93, 1293–1311 CrossRef CAS PubMed.
- K. Acharya, B. Neupane, V. Zazubovich, R. T. Sayre, R. Picorel, M. Seibert and R. Jankowiak, J. Phys. Chem. B, 2012, 116, 3890–3899 CrossRef CAS PubMed.
- G. Raszewski, B. A. Diner, E. Schlodder and T. Renger, Biophys. J., 2008, 95, 105–119 CrossRef CAS PubMed.
- F. Müh, M. Plöckinger and T. Renger, J. Phys. Chem. Lett., 2017, 8, 850–858 CrossRef PubMed.
- A. Gelzinis, L. Valkunas, F. D. Fuller, J. P. Ogilvie, S. Mukamel and D. Abramavicius, New J. Phys., 2013, 15, 075013 CrossRef CAS.
- Y. Fujihashi, M. Higashi and A. Ishizaki, J. Phys. Chem. Lett., 2018, 9, 4921–4929 CrossRef CAS PubMed.
- M. A. Kavanagh, J. K. G. Karlsson, J. D. Colburn, L. M. C. Barter and I. R. Gould, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 19705 CrossRef CAS PubMed.
- K. Kawashima and H. Ishikita, Chem. Sci., 2018, 9, 4083–4092 RSC.
- H. Tamura, K. Saito and H. Ishikita, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 16373 CrossRef CAS PubMed.
- M. Mandal, K. Kawashima, K. Saito and H. Ishikita, J. Phys. Chem. Lett., 2020, 11, 249–255 CrossRef CAS PubMed.
- H. Ishikita, J. Biesiadka, B. Loll, W. Saenger and E.-W. Knapp, Angew. Chem., Int. Ed., 2006, 45, 1964–1965 CrossRef CAS PubMed.
- L. Zhang, D.-A. Silva, H. Zhang, A. Yue, Y. Yan and X. Huang, Nat. Commun., 2014, 5, 4170 CrossRef CAS PubMed.
- A. Sirohiwal, F. Neese and D. A. Pantazis, J. Am. Chem. Soc., 2020, 142, 18174–18190 CrossRef CAS PubMed.
- M. Ziolek, N. Pawlowicz, R. Naskrecki and A. Dobek, J. Phys. Chem. B, 2005, 109, 18171–18176 CrossRef CAS PubMed.
- H. Wang, S. Lin and N. W. Woodbury, J. Phys. Chem. B, 2008, 112, 14296–14301 CrossRef CAS PubMed.
- C. Lauterwasser, U. Finkele, H. Scheer and W. Zinth, Chem. Phys. Lett., 1991, 183, 471–477 CrossRef CAS.
- W. Zinth and J. Wachtveitl, ChemPhysChem, 2005, 6, 871–880 CrossRef CAS PubMed.
- A. Niedringhaus, V. R. Policht, R. Sechrist, A. Konar, P. D. Laible, D. F. Bocian, D. Holten, C. Kirmaier and J. P. Ogilvie, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3563 CrossRef CAS PubMed.
- P. Huppman, T. Arlt, H. Penzkofer, S. Schmidt, M. Bibikova, B. Dohse, D. Oesterhelt, J. Wachtveit and W. Zinth, Biophys. J., 2002, 82, 3186–3197 CrossRef CAS PubMed.
- F. Ma, E. Romero, M. R. Jones, V. I. Novoderezhkin and R. van Grondelle, J. Phys. Chem. Lett., 2018, 9, 1827–1832 CrossRef CAS PubMed.
- F. Ma, E. Romero, M. R. Jones, V. I. Novoderezhkin and R. van Grondelle, Nat. Commun., 2019, 10, 933 CrossRef PubMed.
- U. Finkele, C. Lauterwasser, W. Zinth, K. A. Gray and D. Oesterhelt, Biochemistry, 1990, 29, 8517–8521 CrossRef CAS PubMed.
- H. Wang, Y. Hao, Y. Jiang, S. Lin and N. W. Woodbury, J. Phys. Chem. B, 2012, 116, 711–717 CrossRef CAS PubMed.
- C. Kirmaier, C. He and D. Holten, Biochemistry, 2001, 40, 12132–12139 CrossRef CAS PubMed.
- P. D. Laible, D. K. Hanson, J. C. Buhrmaster, G. A. Tira, K. M. Faries, D. Holten and C. Kirmaier, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 865 CrossRef CAS PubMed.
- M. A. Steffen, K. Lao and S. G. Boxer, Science, 1994, 264, 810 CrossRef CAS PubMed.
- T. P. Treynor, C. Yoshina-Ishii and S. G. Boxer, J. Phys. Chem. B, 2004, 108, 13523–13535 CrossRef CAS.
- M. Saggu, S. D. Fried and S. G. Boxer, J. Phys. Chem. B, 2019, 123, 1527–1536 CrossRef CAS PubMed.
- S. E. J. Rigby, J. H. A. Nugent and P. J. O'Malley, Biochemistry, 1994, 33, 10043–10050 CrossRef CAS PubMed.
- B. A. Diner, E. Schlodder, P. J. Nixon, W. J. Coleman, F. Rappaport, J. Lavergne, W. F. J. Vermaas and D. A. Chisholm, Biochemistry, 2001, 40, 9265–9281 CrossRef CAS PubMed.
- T. Okubo, T. Tomo, M. Sugiura and T. Noguchi, Biochemistry, 2007, 46, 4390–4397 CrossRef CAS PubMed.
- K. Saito, T. Ishida, M. Sugiura, K. Kawakami, Y. Umena, N. Kamiya, J. R. Shen and H. Ishikita, J. Am. Chem. Soc., 2011, 133, 14379–14388 CrossRef CAS PubMed.
- H. Ishikita, W. Saenger, J. Biesiadka, B. Loll and E.-W. Knapp, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9855 CrossRef CAS PubMed.
- M. H. Beck, A. Jäckle, G. A. Worth and H. D. Meyer, Phys. Rep., 2000, 324, 1–105 CrossRef CAS.
- H. Tamura, E. R. Bittner and I. Burghardt, J. Chem. Phys., 2007, 127, 034706 CrossRef PubMed.
- H. Tamura, J. G. S. Ramon, E. R. Bittner and I. Burghardt, Phys. Rev. Lett., 2008, 100, 107402 CrossRef.
- H. Tamura, J. Chem. Phys., 2009, 130, 214705 CrossRef PubMed.
- N. M. Thellamurege, D. Si, F. Cui, H. Zhu, R. Lai and H. Li, J. Comput. Chem., 2013, 34, 2816–2833 CrossRef CAS PubMed.
- N. M. Thellamurege and H. Li, J. Chem. Phys., 2012, 137, 246101 CrossRef PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery Jr, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- L. Cupellini, S. Caprasecca, C. A. Guido, F. Müh, T. Renger and B. Mennucci, J. Phys. Chem. Lett., 2018, 9, 6892–6899 CrossRef CAS PubMed.
- P. Cieplak, J. Caldwell and P. Kollman, J. Comput. Chem., 2001, 22, 1048–1057 CrossRef CAS.
- K. Saito and H. Ishikita, Biophys. J., 2011, 101, 2018–2025 CrossRef CAS PubMed.
- G. M. Ullmann and E.-W. Knapp, Eur. Biophys. J., 1999, 28, 533–551 CrossRef CAS PubMed.
- C. N. Schutz and A. Warshel, Proteins, 2001, 44, 400–417 CrossRef CAS PubMed.
- A. Warshel, P. K. Sharma, M. Kato and W. W. Parson, Biochim. Biophys. Acta, 2006, 1764, 1647–1676 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- A. W. Roszak, V. Moulisová, A. D. P. Reksodipuro, A. T. Gardiner, R. Fujii, H. Hashimoto, N. W. Isaacs and R. J. Cogdell, Biochem. J., 2012, 442, 27–37 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- H. Tamura, J. Phys. Chem. A, 2016, 120, 9341–9347 CrossRef CAS PubMed.
- K. Mitsuhashi, H. Tamura, K. Saito and H. Ishikita, J. Phys. Chem. B, 2021, 125, 2879–2885 CrossRef CAS PubMed.
- J. Kern, R. Chatterjee, I. D. Young, F. D. Fuller, L. Lassalle, M. Ibrahim, S. Gul, T. Fransson, A. S. Brewster, R. Alonso-Mori, R. Hussein, M. Zhang, L. Douthit, C. de Lichtenberg, M. H. Cheah, D. Shevela, J. Wersig, I. Seuffert, D. Sokaras, E. Pastor, C. Weninger, T. Kroll, R. G. Sierra, P. Aller, A. Butryn, A. M. Orville, M. Liang, A. Batyuk, J. E. Koglin, S. Carbajo, S. Boutet, N. W. Moriarty, J. M. Holton, H. Dobbek, P. D. Adams, U. Bergmann, N. K. Sauter, A. Zouni, J. Messinger, J. Yano and V. K. Yachandra, Nature, 2018, 563, 421–425 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01497h |
| This journal is © The Royal Society of Chemistry 2021 |