
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidative additions of alkynyl/vinyl iodides to gold and gold-catalyzed vinylation reactions triggered by the MeDalphos ligand†
Jessica
Rodriguez‡
 a,
Alexis
Tabey‡
a,
Sonia
Mallet-Ladeira
b and
Didier
Bourissou
*a
a,
Alexis
Tabey‡
a,
Sonia
Mallet-Ladeira
b and
Didier
Bourissou
*a
aLaboratoire Hétérochimie Fondamentale et Appliquée (UMR 5069), Université de Toulouse (UPS), CNRS, 118 route de Narbonne, F-31062 Toulouse, France. E-mail: dbouriss@chimie.ups-tlse.fr
bInstitut de Chimie de Toulouse (FR 2599), 118 Route de Narbonne, 31062 Toulouse Cedex 09, France
First published on 28th April 2021
Abstract
The hemilabile Ad2P(o-C6H4)NMe2 ligand promotes fast, quantitative and irreversible oxidative addition of alkynyl and vinyl iodides to gold. The reaction is general. It works with a broad range of substrates of various electronic bias and steric demand, and proceeds with complete retention of stereochemistry from Z and E vinyl iodides. Both alkynyl and vinyl iodides react faster than aryl iodides. The elementary step is amenable to catalysis. Oxidative addition of vinyl iodides to gold and π-activation of alkenols (and N-alkenyl amines) at gold have been combined to achieve hetero-vinylation reactions. A number of functionalized heterocycles, i.e. tetrahydrofuranes, tetrahydropyranes, oxepanes and pyrrolidines were obtained thereby (24 examples, 87% average yield). Taking advantage of the chemoselectivity for vinyl iodides over aryl iodides, sequential transformations involving first a hetero-vinylation step and then a C–N coupling, a C–C coupling or an heteroarylation were achieved from a vinyl/aryl bis-iodide substrate.
Introduction
Judicious ligand design has been recently shown to overcome the reluctance of gold(I) complexes to undergo oxidative addition, providing a valuable mean to achieve Au(I)/Au(III) cycles, including 2-electron redox catalysis.1 Our group first reported a bending strategy. Thanks to their unique chelating properties, o-carboranyl diphosphines promote oxidative addition of aryl iodides and strained C–C bonds to gold under mild conditions (Fig. 1a).2 Then, (P,N) ligands, in particular MeDalphos3 proved to also prepare and activate gold(I) towards oxidative addition due to the hemilabile and hard character of the N center. The (P,N) gold complex reacts readily with a variety of aryl iodides and bromides (Fig. 1b).4 The resulting aryl gold(III) complexes are reactive and open perspectives both in catalysis and (bio)conjugation. The ligand-enabled oxidative addition of aryl iodides has been used as entry point to achieve catalytic C–C and C–N cross-couplings such as the arylation of indoles, of anilines and (sulfon)amides.5a–c It has also been successfully merged with π activation, enabling the difunctionalization of alkenes (heteroarylation, diarylation).5d–g,6 In addition, the (P,N) aryl Au(III) proved very powerful and chemoselective to arylate cysteine residues of peptides and proteins, as well as to derivatize B-cages and access well-defined hybrid nanoclusters.7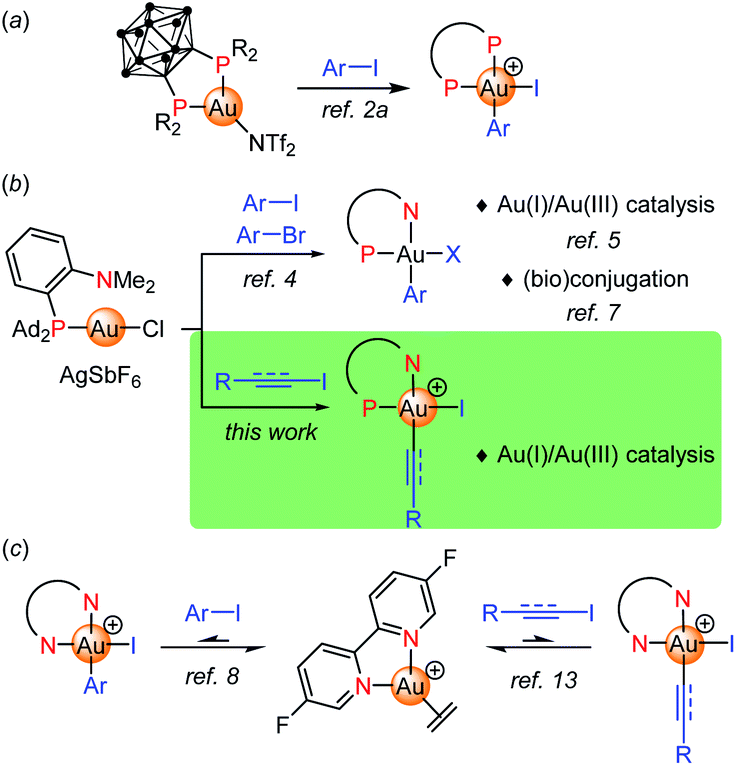 | ||
| Fig. 1 Ligand-enabled oxidative additions of C–I/C–Br bonds to gold. | ||
Apart from P-based ligands, McGrady, Bower and Russell employed a simple bipyridine to trigger oxidative addition to gold.8,9 The corresponding ethylene gold(I) complex reacts with aryl iodides (20 eq.) at 50 °C, quantitative conversions are achieved upon removal of ethylene (Fig. 1c). The oxidative addition is in fact reversible in this case. The feasibility of a Negishi-type biaryl coupling was substantiated by performing the respective elementary steps sequentially in stoichiometric conditions.
Besides aryl substrates, there are also a few precedents of activation and coupling of electrophilic alkynyl and vinyl compounds at gold. Most developed is the use of alkynyl hypervalent iodine reagents, as pioneered by Waser in the late 2000's10 and further extended by Liu, Patil and Hashmi.11 Ethynylbenzoiodoxolone reagents (EBX) proved to be very efficient alkynylating reagents and they were engaged in a number of C(sp2)–C(sp) coupling reactions. Much less is known from alkynyl halides, with only two gold-catalyzed cyclization–alkynylation sequences reported in 2019 by Hashmi with tert-butylallenoates and alkynyl bromides (under thermal conditions),12a and by Ollivier and Fensterbank with o-alkynylphenols and alkynyl iodides (under Ir-photosensitized conditions).12b In addition, Bower and Russell have reported very recently first examples of oxidative addition of alkynyl and vinyl iodides to gold from the (bipyridine)Au(H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)+ complex (Fig. 1c).13 As for aryl iodides, the reaction is reversible and requires an excess of substrate to isolate the respective Au(III) complexes.
CH2)+ complex (Fig. 1c).13 As for aryl iodides, the reaction is reversible and requires an excess of substrate to isolate the respective Au(III) complexes.
Given the high reactivity and versatility that the (P,N) ligand confers to gold in the oxidative addition of aryl halides, we were intrigued about the possibility for the (P,N) gold complex to also activate alkynyl and vinyl iodides. The results we obtained along this line are reported herein. The reaction is shown to proceed readily and quantitatively with a variety of substrates, used in stoichiometric amounts. Chemoselectivity over aryl iodides has been investigated. The elementary step has been leveraged into catalysis. Hetero-vinylation reactions were found to be efficiently catalyzed by the (P,N) gold complex. The scope of the transformation has been explored and it has been applied to the synthesis of various polyfunctional products featuring tetrahydrofurane moieties starting from a vinyl/aryl bis-iodide substrate.
Results and discussion
The ability of the MeDalphos ligand to promote oxidative addition of alkynyl iodides to gold was first investigated (Scheme 1).14 The (P,N)AuCl complex 1 was reacted with iodo phenylacetylene 2a in the presence of silver hexafluoroantimonate. NMR monitoring showed instantaneous and quantitative formation of the Au(III) alkynyl complex 3a. The 31P NMR signal appears at δ 103 ppm, deshielded by 46 ppm with respect to (P,N)AuCl. The 1H NMR signal for the NMe2 group resonates at δ 3.68 ppm, shifted downfield by ca. 1 ppm compared to that of the gold(I) precursor 1. This is reminiscent of that observed for the (P,N)Au(III) aryl complexes deriving from oxidative addition of aryl halides4,5a and suggests coordination of the nitrogen atom to gold. Most diagnostic for the oxidative addition of PhCC–I is the disappareance of the![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–I signal of 2a at δ 6.6 ppm in 13C NMR spectroscopy. In complex 3a, the respective alkynyl carbon atom is found at δ 59.1 ppm. This signal resonates as a doublet with a small JPC coupling constant (6.5 Hz), indicating that the alkynyl sits cis to phosphorus. Crystals of 3a suitable for X-ray diffraction analysis were obtained by vapor diffusion of pentane into a dichloromethane solution at 4 °C (Scheme 1).15 The gold center adopts square-planar geometry, with tight coordination of the NMe2 group (the Au–N distance is short at 2.121(8) Å). The alkynyl group sits indeed cis to phosphorus. The Au–CC–Ph framework only marginally deviates from linearity, despite the proximity of the sterically demanding PAd2 group.
C–I signal of 2a at δ 6.6 ppm in 13C NMR spectroscopy. In complex 3a, the respective alkynyl carbon atom is found at δ 59.1 ppm. This signal resonates as a doublet with a small JPC coupling constant (6.5 Hz), indicating that the alkynyl sits cis to phosphorus. Crystals of 3a suitable for X-ray diffraction analysis were obtained by vapor diffusion of pentane into a dichloromethane solution at 4 °C (Scheme 1).15 The gold center adopts square-planar geometry, with tight coordination of the NMe2 group (the Au–N distance is short at 2.121(8) Å). The alkynyl group sits indeed cis to phosphorus. The Au–CC–Ph framework only marginally deviates from linearity, despite the proximity of the sterically demanding PAd2 group.
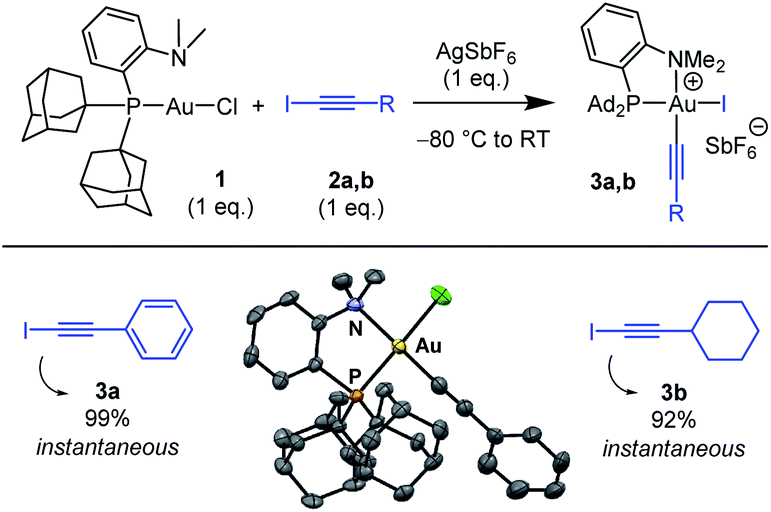 | ||
| Scheme 1 Oxidative addition of alkynyl iodides to the (P,N)AuCl complex 1. Reaction times and spectroscopic yields based on 31P NMR. Molecular structure of the (P,N)Au(III) alkynyl complex 3a (hydrogen atoms and counter anion omitted for clarity, ellipsoids shown at 50% probability). | ||
Oxidative addition of C(sp)–I bonds to gold was then generalized to substrate 2b featuring a cyclohexyl instead of phenyl substituent. Here also the reaction was complete within the time of mixing. The ensuing (P,N)Au(III) alkynyl complex 3b (obtained in 92% yield) was characterized by multi-nuclear NMR spectroscopy and mass spectrometry. Besides extending the reaction to simpler and commercially available Au(I) precursors, the MeDalphos ligand imparts higher reactivity compared to bipyridine. The (P,N)-ligated complex 1 reacts readily with alkynyl iodides 2a,b (instantaneous reactions with only 1 eq. of substrate) and forms monometallic complexes. For comparison, the (bipyridine)Au(I) (H2CCH2)+ complex requires large excess of substrate (20 eq.) along with extended reaction times (1 h), and tends to form Au(I)/Au(III) bimetallic complexes.13
Next, the feasibility of oxidative addition of vinyl iodides to gold was explored (Scheme 2).16 First, the (P,N)AuCl complex 1 was activated by AgSbF6 and reacted with trans iodo styrene 4a. The corresponding (P,N)Au(III) vinyl complex 5a was immediately and cleanly formed, as apparent from NMR spectroscopy. Besides the typical pattern for the MeDalphos ligand chelating a gold(III) center (deshielded 31P NMR signal at δ 75.0 ppm and deshielded 1H NMR signal for the NMe2 group at δ 3.51 ppm), the 1H NMR spectrum shows two new signals characteristic of a CHCH moiety (δ 6.96 and 6.50 ppm), with associated 13C NMR signals at δ 140.7 and 113.2 ppm. The absence of JPC coupling supports cis relationship between the vinyl and PAd2 groups at gold while the large 3JHH coupling constant (14.5 Hz) indicates that the trans geometry of the CC bond is retained. Given that only styrenyl substrates undergo oxidative addition to the (bipyridine)Au(H2CCH2)+ complex,13 we were eager to examine the scope of vinyl iodides prone to react with the (P,N)AuCl complex 1. Gratifyingly, instantaneous and quantitative reactions were observed with the alkyl-substituted substrate 4b, the parent vinyl iodide 4c (oxidative addition takes 3 h in this case), as well as the ester-substituted substrates 4d,e. The transformation thus tolerates substituents with different electronic bias at the CC bond and it works with both Z and E substrates, including trisubstituted ones. In all cases, the Z/E stereochemistry of the substrate is preserved, oxidative addition to gold proceeds with retention.
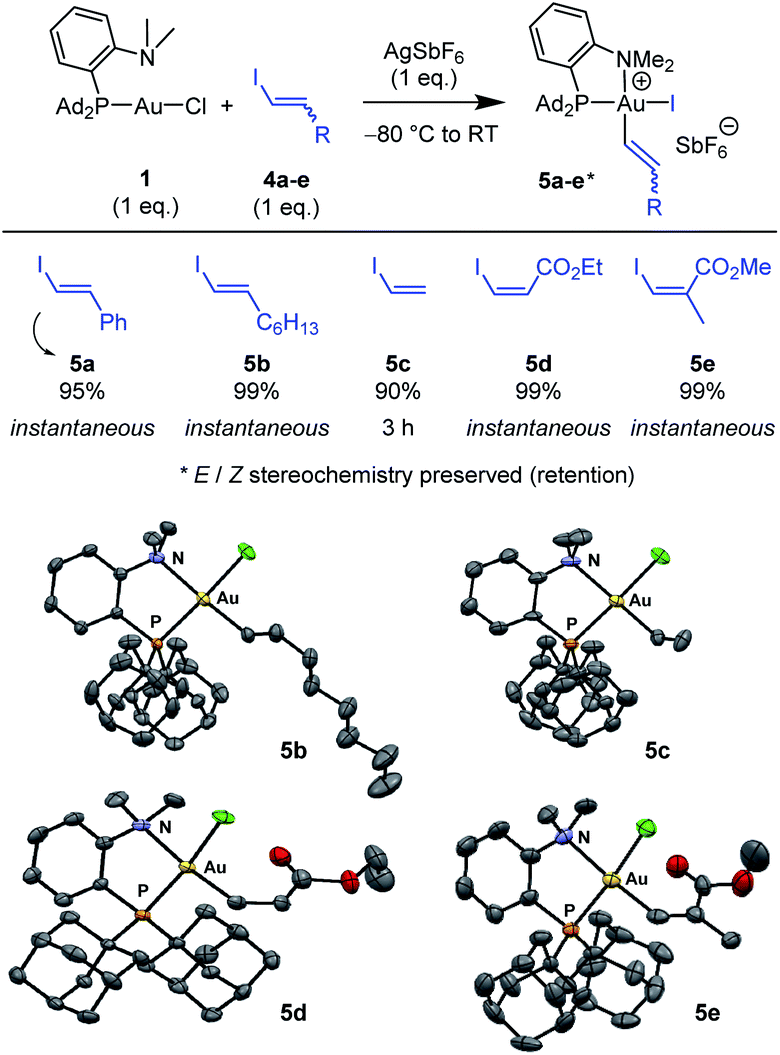 | ||
| Scheme 2 Oxidative addition of vinyl iodides to the (P,N)AuCl complex 1. Reaction times and spectroscopic yields based on 31P NMR. Molecular structures of the (P,N)Au(III) vinyl complexes 5b–e (hydrogen atoms, counter anions and solvate molecules omitted for clarity, ellipsoids shown at 50% probability). | ||
X-ray diffraction analyses were carried out on complexes 5b–e (Scheme 2), confirming the square-planar geometry of gold, the chelation of the (P,N) ligand with strong N → Au coordination, the cis relationship of the vinyl and PAd2 groups and the Z/E stereochemistry of the CC double bond. Of note, the vinyl group is oriented perpendicularly to the gold coordination plane in all complexes to minimize steric constraints.
Having substantiated that oxidative addition of alkynyl and vinyl iodides takes place easily with the (P,N)AuCl complex 1, we were then keen to assess the relative reactivity of these substrates compared to aryl iodides. To this end, competitive experiments were performed between iodo benzene, iodo phenylacetylene 2a and trans iodo styrene 4a (Scheme 3). In all cases, equimolar amounts of the two iodides were combined with 1 eq. of the (P,N)AuCl complex 1 and 1 eq. of AgSbF6. The proportion of the ensuing Au(III) complexes was determined by 31P and 1H NMR spectroscopy at complete consumption of the Au(I) complex.15 The competition between Ph–I and Ph–CC–I revealed strong preference for C(sp)–I oxidative addition, the Au(III) alkynyl and aryl complexes being obtained in 94/6 ratio, while complete selectivity for oxidative addition of the vinyl iodide was observed between Ph–I and Ph–CHCH–I. Thus, both alkynyl and vinyl iodides react faster than aryl iodides with the (P,N)AuCl complex 1.17,18 Furthermore, C(sp)–I prevailed over C(sp2)–I oxidative addition in the competition between Ph–CC–I and Ph–CHCH–I.
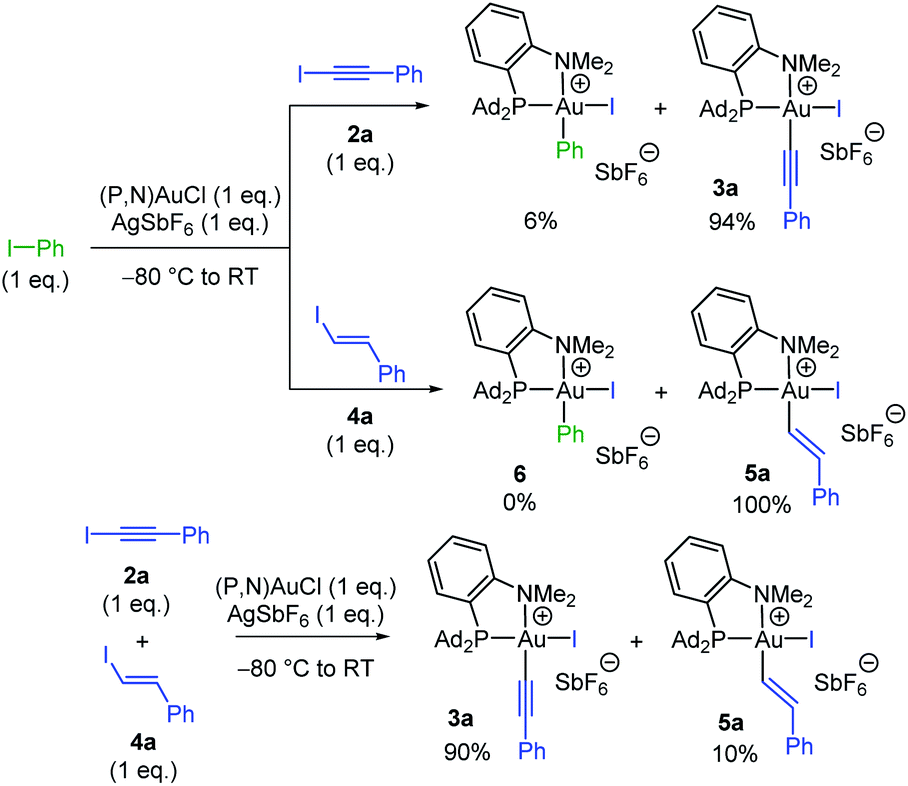 | ||
| Scheme 3 Competitive reactions between iodo benzene, iodo phenylacetylene 2a and trans iodo styrene 4a in the oxidative addition to the (P,N)AuCl complex 1. | ||
Note that the (P,N) ligand and silver salt are needed for the C(sp)/C(sp2)–I oxidative addition to take place at gold. No reaction occurs with the (P,N)AuCl complex alone, with PhCC–I as well as H13C6–CHCH–I. In the case of N-heterocyclic carbene complex (IPr)AuCl, only the π-complex was formed, while (Ph3P)AuCl gave mainly the inactive (Ph3P)2Au+ species.15
Our next goal was to demonstrate that the (P,N) gold complex not only undergoes oxidative addition of alkynyl and vinyl iodides, but also enables catalytic cross-coupling transformations to be achieved.19 Given that the (P,N) ligand triggers fast, quantitative and irreversible addition of C(sp)/C(sp2)–I bonds to gold, we were interested in combining oxidative addition of alkynyl/vinyl iodides and π-activation at gold. To this end, the coupling of alkenols with alkynyl and vinyl iodides was investigated as an attractive extension of the recently reported hetero-arylation reactions of alkenes.20 Unfortunately, no hetero-alkynylation product could be achieved from iodo phenylacetylene and 4-penten-1-ol. In fact, no oxidative addition occurred and only the Au(I) π-complex with 4-penten-1-ol was observed under these conditions. However, positive results were obtained with vinyl iodides, as detailed hereafter. The coupling of trans iodo oct-1-ene with 4-penten-1-ol was used to optimize the reaction conditions. The silver salt, base, solvent, concentration, temperature and catalytic loading were varied (Table 1). The hetero-vinylation product 7a was obtained in 99% yield within 2 h using 5 mol% of (P,N)AuCl, AgSbF6 (1.05 eq.) and K3PO4 (1 eq.) in DCE at 0.4 M and 80 °C (entry 1). The use of other halide scavengers and bases results in lower yields (entries 2–9). The reaction proceeds equally well at 45 °C and can even be performed at room temperature if prolonged for 16 h (entries 10–11). Working in other solvents and lowering the concentration (0.1 M) also results in somewhat decreased efficiency. However, the catalytic loading can be reduced to 1 mol% (entry 17) and the reaction can be performed in air with technical grade solvents15 without noticeable loss of catalytic activity, demonstrating good robustness.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ag salt | Base | Solvent | T (°C) | Yieldb (%) |
| a Reactions performed at 0.4 M concentration with equimolar amounts of alkenol and vinyl iodide (0.4 mmol scale). b Yield determined by 1H NMR using dimethyl terephthalate as internal standard. c 16 h. d Reaction performed at 0.1 M concentration. e Reaction performed with 1 mol% of (P,N)AuCI and 1.01 eq. of AgOTf. DTBP = 2,6-di-tert-butylpyridine. | |||||
| 1 | AgOTf | K 3 PO 4 | DCE | 80 | 99 |
| 2 | AgSbF6 | K3PO4 | DCE | 80 | 50 |
| 3 | AgPF6 | K3PO4 | DCE | 80 | 77 |
| 4 | AgBF4 | K3PO4 | DCE | 80 | 88 |
| 5 | AgOTf | DTBP | DCE | 80 | 92 |
| 6 | AgOTf | Cs2CO3 | DCE | 80 | 58 |
| 7 | AgOTf | NaOAc | DCE | 80 | 50 |
| 8 | AgOTf | DIEA | DCE | 80 | 21 |
| 9 | AgOTf | - | DCE | 80 | 6 |
| 10 | AgOTf | K 3 PO 4 | DCE | 45 | 99 |
| 11c | AgOTf | K3PO4 | DCE | 25 | 84 |
| 12 | AgOTf | K3PO4 | o-DCB | 80 | 95 |
| 13 | AgOTf | K3PO4 | toluene | 80 | 88 |
| 14 | AgOTf | K3PO4 | DCM | 45 | 89 |
| 15 | AgOTf | K3PO4 | chloroform | 45 | 84 |
| 16d | AgOTf | K3PO4 | DCE | 80 | 90 |
| 17 | AgOTf | K 3 PO 4 | DCE | 80 | 98 |
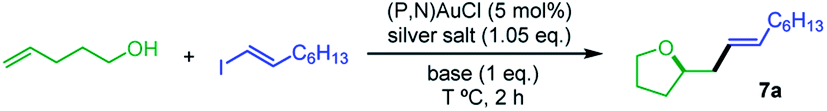
With the optimized conditions in hands, the reaction of 4-penten-1-ol with different vinyl iodides was studied. Good to excellent yields were observed in all cases (Scheme 4), illustrating the generality and efficiency of the transformation. Trans iodo ethyl acrylate gave the corresponding oxy-vinylation product 7b in good yield. The reaction works well and with complete retention of stereochemistry from cis vinyl iodides, as substantiated by the formation of 7c and 7d. Good results were also obtained when preparing 7e–g from the parent, the tBu and the tri-substituted substrates, indicating that the reaction is not very sensitive to the steric demand of the vinyl iodide. With trans iodo styrenes, the transformation was most conveniently achieved at 25 °C over 16 h (to minimize degradation of the vinyl iodide). The corresponding oxy-vinylation products 7h–l were obtained in good yields whatever the steric demand and electronic bias of the substrate. The efficient preparation of 7l is particularly noteworthy, no interference was observed with the Bpin moiety.
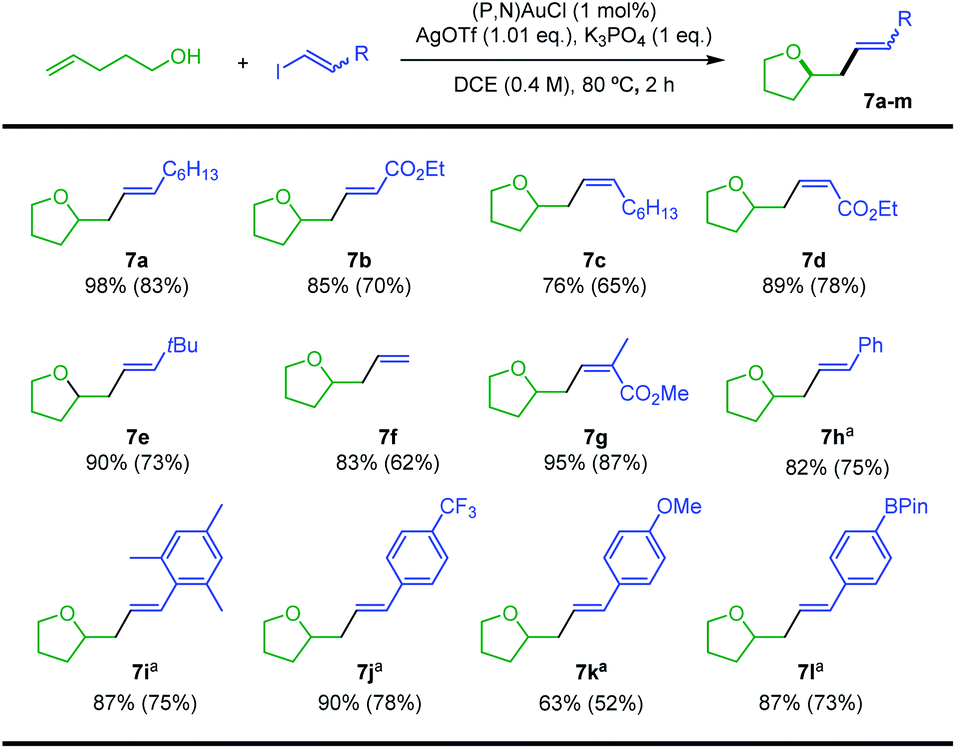 | ||
| Scheme 4 Gold-catalyzed oxy-vinylation, scope of vinyl iodides. Yields determined by 1H NMR spectroscopy with dimethyl terephthalate as internal standard. Isolated yields in parentheses. a25 °C, 16 h. | ||
The scope of alkenols was then assessed (Scheme 5a). The oxy-vinylation tolerates aryl and alkyl substitution on 4-penten-1-ol, as illustrated by the preparation of the tetrahydrofurane derivatives 8a–c. It works well with ortho-allyl phenol to give the benzo-fused product 8d. It is also efficient with gem-disubstituted alkenols and enables the formation of quaternary centers as in 8e. In addition, six and seven-membered ring products 8f–h are formed in high yields from 5-hexen-1-ol and 6-hepten-1-ol. In all cases, the terminal alkenols undergo exo cyclization with complete regioselectivity. Internal alkenols are also suitable substrates, as substantiated by the oxy-vinylation of 4-hexen-1-ols. Of note, the reaction of the Z isomer is fully regioselective for 5-exo cyclization, while 5-exo and 6-endo cyclizations compete for the E isomer. The corresponding tetrahydrofurane/tetrahydropyrane products 8j/8k are obtained in 47 and 33% yields, respectively. A similar tendency was observed for the oxy-arylation, with in the latter case complete switch of regioselectivity from 5-exo to 6-endo between the Z and E isomers of the alkenol.5d The transformation is applicable to N-tosyl alkenamines as well. Amino-vinylation proceeds nicely to give the pyrrolidine products 8l and 8m in >92% yields (Scheme 5b). The feasibility of 3-component oxy-vinylation of alkenes5e was also substantiated by the preparation of product 8n (Scheme 5c).
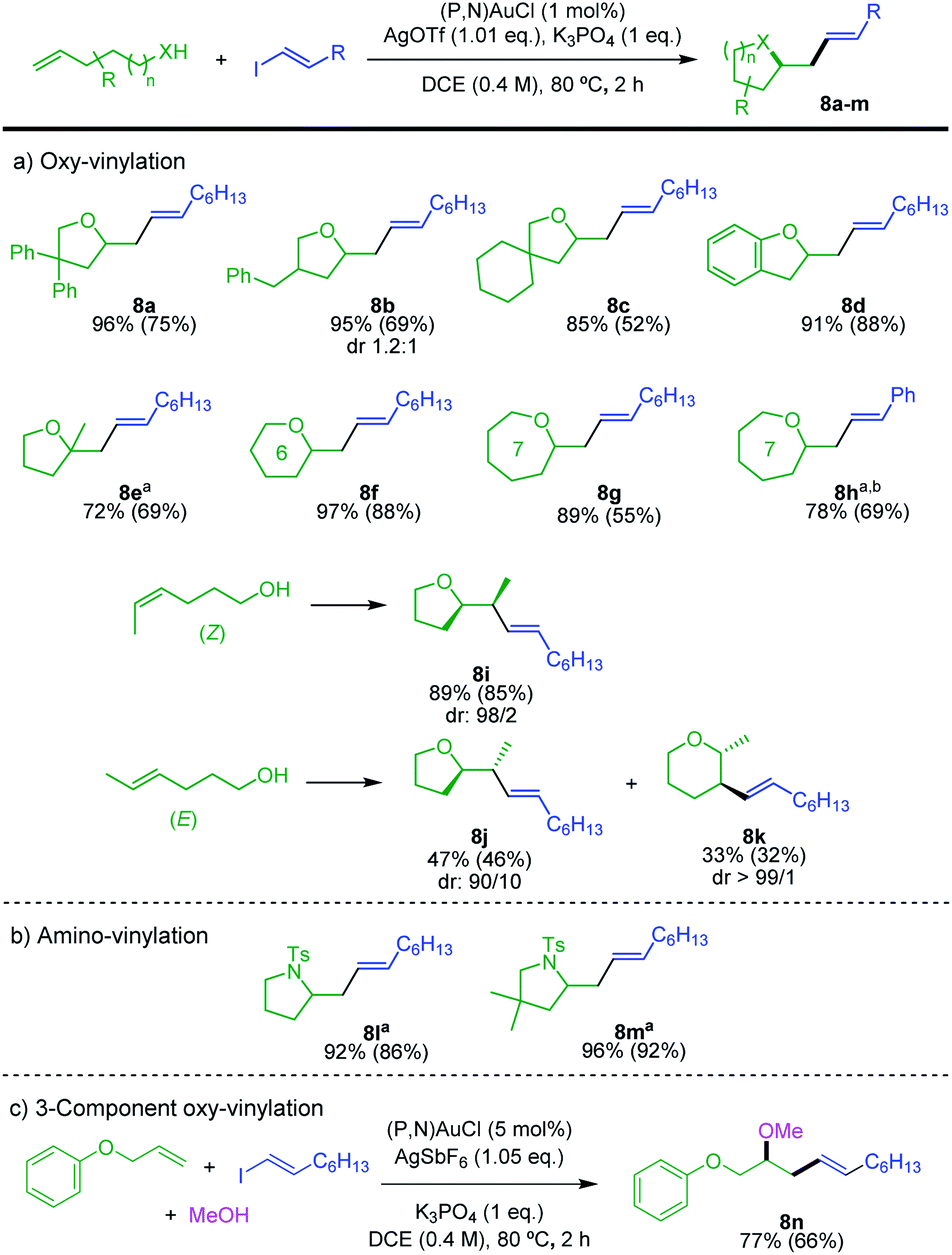 | ||
| Scheme 5 Gold-catalyzed hetero-vinylation, scope of alkenols and N-tosyl alkenamines, as well as 3-component oxy-vinylation. Yields determined by 1H NMR spectroscopy with dimethyl terephthalate as internal standard. Isolated yields in parentheses. a5 mol% (P,N)AuCl, 1.05 eq. AgOTf. b25 °C, 16 h. | ||
Then, the preference observed for oxidative addition of vinyl–I over Ar–I bonds to gold prompted us to explore the possibility to achieve chemoselective catalytic transformation of the difunctional substrate 4f featuring both vinyl–I and aryl–I moieties.21 Pleasingly, using 1 eq. of 4-penten-1-ol or N-tosyl 4-penten-1-amine, the oxy- and amino-vinylations are largely predominant. The tetrahydrofurane and pyrrolidine products 9a,b featuring iodo styryl arms were obtained in good yields (Scheme 6a). 1H NMR analysis of the crude reaction mixture shows no oxy-arylation product and only a small amount of oxy-arylation/oxy-vinylation product resulting from the transformation of both C(sp2)–I bonds of 4f (7% with respect to 9a). The synthesis of 9a was scale up to 4 mmol, demonstrating the robustness and practical interest of the transformation. Further derivatization of 9a was achieved using the ability of the (P,N) gold(I) complex to catalyze C–N and C–C coupling.5a–c Using N-tosyl amine and N-methyl indole, the functionalized tetrahydrofuranes 10a,b were obtained in high yields. In addition, using 3 eq. of 4-penten-1-ol, the oxy-vinylation and oxy-arylation reactions were found to readily proceed giving the styryl-bridged bis-tetrahydrofurane 11a in 85% yield (Scheme 6b).
 | ||
| Scheme 6 Selective and sequential transformation of the bifunctional substrate 4f. Yields determined by 1H NMR spectroscopy with dimethyl terephthalate as internal standard. Isolated yields in parentheses. aGram scale reaction (4 mmol). | ||
Finally, we aimed to take advantage of the reactivity of 4f to achieve sequential hetero-vinylation/hetero-arylation catalytic transformations in one-pot and obtain thereby original heterocyclic products (Scheme 7). To start with, the sequential reaction of 4f with 2 eq. of 4-penten-1-ol was studied. Once the oxy-vinylation reaction achieved with the first eq. of alkenol, another load of gold complex and silver salt was required for the oxy-arylation to proceed when adding the second eq. of 4-penten-1-ol. The bis-tetrahydrofurane 11a was obtained in good yield (63% over two steps) under these conditions. With this procedure in hands, different alkenols were then used for the oxy-vinylation and oxy-arylation steps. Unsymmetrical products 11b–d were thereby obtained in moderate to good yields (38–55% over two steps), combining 4-penten-1-ol with 4-methyl-4-penten-1-ol, ortho-allyl phenol and 5-hexen-1-ol, respectively. This reaction sequence also works with N-tosyl alkenamines, allowing for example the access to 11e and 11f combining tetrahydrofurane and pyrrolidine moieties. Thanks to the vinyl–I/aryl–I chemoselectivity, it is straightforward to obtain these two constitutional isomers by simply changing the order of addition of the alkenol and N-tosyl alkenamine.
 | ||
| Scheme 7 Sequential gold-catalyzed hetero-vinylation/arylation transformations of the bifunctional substrate 4f. Yields over the two steps determined by 1H NMR spectroscopy with dimethyl terephthalate as internal standard. Isolated yields in parentheses. | ||
From a mechanistic viewpoint, the hetero-vinylation reactions catalyzed by the (P,N)Au(I) complex are proposed to occur via the catalytic cycle displayed in Fig. 2. As for the corresponding hetero-arylation, it starts by oxidative addition of the vinyl iodide to gold, then involves π-activation and cyclization of the alkenol at gold, and the product is finally released upon C(sp2)–C(sp3) reductive elimination. In line with this picture, similar results were obtained using the gold(III) vinyl complex 5b resulting from oxidative addition of trans iodo oct-1-ene as catalyst.15 In addition, NMR monitoring of a catalytic run identified the alkenol/gold(I) π-complex as resting state of the transformation.22 The proposed mechanism is consistent with the complete trans selectivity observed experimentally upon addition of the vinyl group and O atom to the CC bond, as apparent in the reaction of the internal alkenols leading to 8i,j,k.
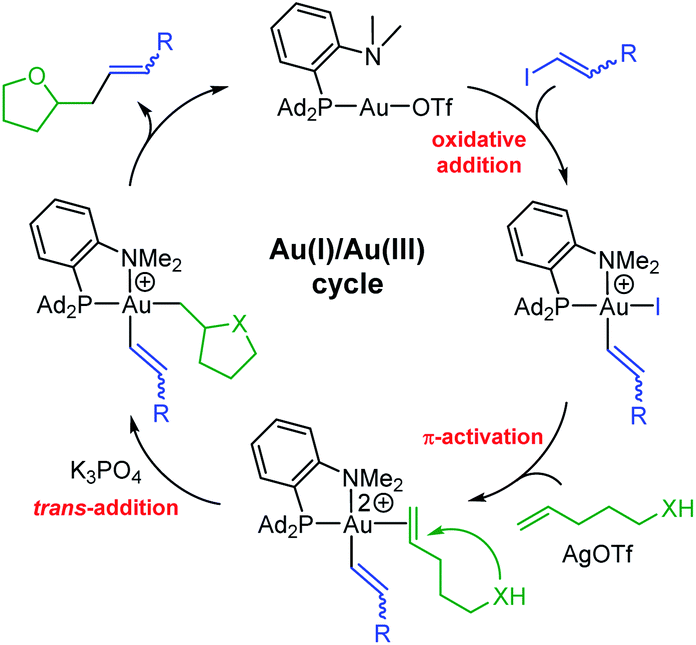 | ||
| Fig. 2 Catalytic cycle proposed to account for oxy-/amino–vinylation reactions catalyzed by the (P,N)Au(I) complex. | ||
Conclusions
The MeDalphos ligand triggers oxidative addition of alkynyl and vinyl iodides to gold under mild conditions. The corresponding (P,N)-chelated organo Au(III) complexes form quickly, quantitatively and irreversibly. They are readily isolated and have been fully characterized. The reaction works with a broad range of substrates of various electronic bias and steric demand, including vinyl iodide itself and iodoacrylates. Z and E vinyl iodides are activated and the oxidative addition proceeds with complete retention of stereochemistry.In the case of vinyl iodides, this elementary step could be used as an entry point for a catalytic transformation merging Au(I)/Au(III) redox cycling and π-activation at gold. The (P,N)Au complex proved efficient and general in promoting the hetero-vinylation of alkenols and N-tosyl alkenamines.
The relative reactivity of vinyl and aryl iodides has been investigated via competitive experiments. This revealed high chemoselectivity of both the oxidative addition and catalytic coupling in favour of the vinyl substrates. This chemoselectivity was exploited synthetically with a vinyl/aryl bis-iodide substrate. Several polyfunctional tetrahydrofurane products were obtained straightforwardly by sequential hetero-vinylation/arylation.
These results further demonstrate the interest and synthetic value of “non-innocent” ligands such as MeDalphos in 2-electron redox gold chemistry. Future work will aim to expand the variety of suitable substrates and achievable catalytic transformations, as well as to develop new ligand frameworks.
Author contributions
J. R. and A. T. performed the experiments; J. R., A. T. and D. B. analyzed the experimental data; and S. M.-L. collected and refined the X-ray diffraction data. All the authors contributed to scientific discussion and participated in writing the manuscript. D. B. guided the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Centre National de la Recherche Scientifique, the Université de Toulouse and the Agence Nationale de la Recherche (ANR-19-CE07-0037) is gratefully acknowledged. J. R. thanks the European Commission for a MCIF (Gold3Cat-799606).Notes and references
- (a) B. Huang, M. Hu and F. D. Toste, Trends Chem., 2020, 2, 707 CrossRef CAS; (b) M. O. Akram, S. Banerjee, S. S. Saswade, V. Bedi and N. T. Patil, Chem. Commun., 2018, 54, 11069 RSC.
- (a) M. Joost, A. Zeineddine, L. Estévez, S. Mallet−Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 14654 CrossRef CAS PubMed; (b) M. Joost, L. Estévez, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 5236 CrossRef CAS PubMed.
- (a) K. D. Hesp and M. Stradiotto, J. Am. Chem. Soc., 2010, 132, 18026 CrossRef CAS PubMed; (b) S. R. J. Lundgren, K. D. Hesp and M. Stradiotto, Synlett, 2011, 17, 2443 Search PubMed.
- A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Nat. Commun., 2017, 8, 565 CrossRef PubMed.
- (a) J. Rodriguez, A. Zeineddine, E. D. Sosa Carrizo, K. Miqueu, N. Saffon-Merceron, A. Amgoune and D. Bourissou, Chem. Sci., 2019, 10, 7183 RSC; (b) J. Rodriguez, N. Adet, N. Saffon-Merceron and D. Bourissou, Chem. Commun., 2020, 56, 94 RSC; (c) M. O. Akram, A. Das, I. Chakrabarty and N. T. Patil, Org. Lett., 2019, 21, 8101 CrossRef CAS PubMed; (d) M. Rigoulet, O. Thillaye du Boullay, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2020, 59, 16625 CrossRef CAS PubMed; (e) A. G. Tathe, C. C. Chintawar, V. W. Bhoyare and N. T. Patil, Chem. Commun., 2020, 56, 9304 RSC; (f) C. C. Chintawar, A. K. Yadav and N. T. Patil, Angew. Chem., Int. Ed., 2020, 59, 11808 CrossRef CAS PubMed; (g) S. Zhang, C. Wang, X. Ye and X. Shi, Angew. Chem., Int. Ed., 2020, 59, 20470 CrossRef CAS PubMed; (h) A. G. Tathe, U. A. K. Yadav, C. C. Chintawar and N. T. Patil, ACS Catal., 2021, 11, 4576 CrossRef CAS.
- For I to F exchange to give (P,N)Au(III) fluoride complexes, see: A. Genoux, M. Biedrzycki, E. Merino, E. Rivera, A. Linden and C. Nevado, Angew. Chem., Int. Ed., 2021, 60, 4161 CrossRef PubMed.
- (a) M. S. Messina, J. M. Stauber, M. A. Waddington, A. L. Rheingold, H. D. Maynard and A. M. Spokoyny, J. Am. Chem. Soc., 2018, 140, 7065 CrossRef CAS PubMed; (b) J. M. Stauber, E. A. Qian, Y. Han, A. L. Rheingold, P. Král, D. Fujita and A. M. Spokoyny, J. Am. Chem. Soc., 2020, 142, 327 CrossRef CAS PubMed; (c) S. L. Zhang and J. J. Dong, Org. Biomol. Chem., 2019, 17, 1245 RSC.
- M. J. Harper, C. J. Arthur, J. Crosby, E. J. Emmett, R. L. Falconer, A. J. Fensham-Smith, P. J. Gates, T. Leman, J. E. McGrady, J. F. Bower and C. A. Russell, J. Am. Chem. Soc., 2018, 140, 4440 CrossRef CAS PubMed.
- For oxidative addition of strained C–C bonds, i.e. biphenylenes, to gold(I) ligated by simple NHCs and a CAAC bearing a pendant imine moiety, see: (a) C.-Y. Wu, T. Horibe, C. Borch Jacobsen and F. D. Toste, Nature, 2015, 517, 449 CrossRef CAS PubMed; (b) J. Chu, D. Munz, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 7884 CrossRef CAS PubMed.
- (a) J. P. Brand, J. Charpentier and J. Waser, Angew. Chem., Int. Ed., 2009, 48, 9346 CrossRef CAS PubMed; (b) J. P. Brand and J. Waser, Angew. Chem., Int. Ed., 2010, 49, 7304 CrossRef CAS PubMed; (c) Y. Li, J. P. Brand and J. Waser, Angew. Chem., Int. Ed., 2013, 52, 6743 CrossRef CAS PubMed; (d) H. Ghari, Y. Li, R. Roohzadeh, P. Caramenti, J. Waser and A. Ariafard, Dalton Trans., 2017, 46, 12257 RSC.
- (a) X. Li, X. Xie, N. Sun and Y. Liu, Angew. Chem., Int. Ed., 2017, 56, 6994 CrossRef CAS PubMed; (b) S. Banerjee and N. T. Patil, Chem. Commun., 2017, 53, 7937 RSC; (c) Y. Yang, P. Antoni, M. Zimmer, K. Sekine, F. F. Mulks, L. Hu, L. Zhang, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2019, 58, 5129 CrossRef CAS PubMed; (d) Y. Yang, L. Eberle, F. F. Mulks, J. F. Wunsch, M. Zimmer, F. Rominger, M. Rudolph and A. S. K. Hashmi, J. Am. Chem. Soc., 2019, 141, 17414 CrossRef CAS PubMed; (e) S. Banerjee, V. W. Bhoyarec and N. T. Patil, Chem. Commun., 2020, 56, 2677 RSC; (f) S. Banerjee, S. B. Ambegave, R. D. Mule, B. Senthilkumar and N. T. Patil, Org. Lett., 2020, 22, 4792 CrossRef CAS PubMed.
- (a) Y. Yang, J. Schießl, S. Zallouz, V. Göker, J. Gross, M. Rudolph, F. Rominger and A. S. K. Hashmi, Chem.–Eur. J., 2019, 25, 9624 CrossRef CAS PubMed; (b) Z. Xia, V. Corcé, F. Zhao, C. Przybylski, A. Espagne, L. Jullien, T. Le Saux, Y. Gimbert, H. Dossmann, V. Mouriès-Mansuy, C. Ollivier and L. Fensterbank, Nat. Chem., 2019, 11, 797 CrossRef CAS PubMed.
- J. A. Cadge, H. A. Sparkes, J. Bower and C. A. Russell, Angew. Chem., Int. Ed., 2020, 59, 6617 CrossRef CAS PubMed.
- For Au(III) alkynyl complexes synthesized by transmetallation with Ag–alkyne, see for example: (a) J. Fernandez-Cestau, B. Bertrand, M. Blaya, G. A. Jones, T. J. Penfold and M. Bochmann, Chem. Commun., 2015, 51, 16629 RSC; (b) L. Rocchigiani, J. Fernandez-Cestau, G. Agonigi, I. Chambrier, P. H. M. Budzelaar and M. Bochmann, Angew. Chem., Int. Ed., 2017, 56, 13861 CrossRef CAS PubMed . For Au(III) alkynyl complexes synthesized by oxidation of Au(I) with alkynyl iodonium salts, see for example ref. 11c and 11d.
- See ESI.†.
- For Au(III) vinyl complexes synthesized by alkyne hydroauration, see for example: (a) A. Pintus, L. Rocchigiani, J. Fernandez-Cestau, P. H. M. Budzelaar and M. Bochmann, Angew. Chem., Int. Ed., 2016, 55, 12321 CrossRef CAS PubMed; (b) L. Rocchigiani, J. Fernandez-Cestau, I. Chambrier, P. Hrobárik and M. Bochmann, J. Am. Chem. Soc., 2018, 140(26), 8287 CrossRef CAS PubMed; (c) R. Kumar, J.-P. Krieger, E. Gómez-Bengoa, T. Fox, A. Linden and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 12862 CrossRef CAS PubMed. For Au(III) vinyl complexes synthesized by nucleophilic addition to π-coordinated alkynes, see for example: (d) M. S. M. Holmsen, A. Nova, D. Balcells, E. Langseth, S. Øien-Ødegaard, R. H. Heyn, M. Tilset and G. Laurenczy, ACS Catal., 2017, 7, 5023 CrossRef CAS. For Au(III) vinyl complexes synthesized by ring-opening of alkylidene cyclopropanes, see: (e) J. A. González, F. Verdugo, J. L. Mascareñas, F. López and C. Nevado, Angew. Chem., Int. Ed., 2020, 59, 20049 CrossRef PubMed.
- For rare studies comparing the oxidative addition of aryl–X and vinyl–X substrates to Pd, see: (a) A. Jutand and S. Négri, Organometallics, 2003, 22, 4229 CrossRef CAS; (b) A. Ariafard and Z. Lin, Organometallics, 2006, 25, 4030 CrossRef CAS.
- The preference for vinyl–X over aryl–X bonds is tentatively attributed to the reduced steric demand around the bond to be activated, resulting in a slightly favoured reaction kinetically.
- For gold-catalyzed alkynylation reactions from electrophilic alkynyl substrates, see ref. 11 and 12.
- For somewhat related hetero-vinylation reactions catalyzed by Cu or Pd, see: (a) D. N. Mai and J. P. Wolfe, J. Am. Chem. Soc., 2010, 132, 12157 CrossRef CAS PubMed; (b) E. Cahard, N. Bremeyer and M. J. Gaunt, Angew. Chem., Int. Ed., 2013, 52, 9284 CrossRef CAS PubMed; (c) D. Holt and M. J. Gaunt, Angew. Chem., Int. Ed., 2015, 54, 7857 CrossRef CAS PubMed; (d) L. Huang, Q. Wang, W. Wu and H. Jiang, J. Org. Chem., 2014, 79, 7734 CrossRef CAS PubMed; (e) J. Li, W. Yang, S. Yang, L. Huang, W. Wu, Y. Sun and H. Jiang, Angew. Chem., Int. Ed., 2014, 53, 7219 CrossRef CAS PubMed; (f) J. Li, W. Hu, C. Li, S. Yang, W. Wu and H. Jiang, Org. Chem. Front., 2017, 4, 373 RSC.
- For rare examples of chemoselective catalytic cross-coupling reactions with pX–C6H4–CHCH–X substrates (X = I, Br), see:
(a) K. J. Powell, L.-C. Han, P. Sharma and J. E. Moses, Org. Lett., 2014, 16, 2158 CrossRef CAS PubMed;
(b) M. Grigalunas, T. Ankner, P. O. Norrby, O. Wiest and P. Helquist, J. Am. Chem. Soc., 2015, 137, 7019 CrossRef CAS PubMed;
(c) R. Dakarapu and J. R. Falck, J. Org. Chem., 2018, 83, 1241 CrossRef CAS PubMed.
- For tricoordinate π-alkene complexes deriving from the (P,N)Au(I) complex, see: (a) M. Navarro, A. Toledo, M. Joost, A. Amgoune, S. Mallet-Ladeira and D. Bourissou, Chem. Commun., 2019, 55, 7974 RSC; (b) M. Navarro, A. Toledo, S. Mallet-Ladeira, E. Daiann Sosa Carrizo, K. Miqueu and D. Bourissou, Chem. Sci., 2020, 11, 2750 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental procedures, characterization data of all new compounds including NMR spectra. CCDC 2069847 (3), 2069844 (5b), 2069846 (5c), 2069845 (5d) and 2069848 (5e). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01483h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |