
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirecting transition metal-based oxygen-functionalization catalysis
Gracita M.
Tomboc
 ,
Yeji
Park
,
Kwangyeol
Lee
* and
Kyoungsuk
Jin
*
,
Yeji
Park
,
Kwangyeol
Lee
* and
Kyoungsuk
Jin
*
Department of Chemistry and Research Institute for Natural Sciences, Korea University, Seoul 02841, Republic of Korea. E-mail: kylee1@korea.ac.kr; kysjin@korea.ac.kr
First published on 23rd June 2021
Abstract
This review presents the recent progress of oxygen functionalization reactions based on non-electrochemical (conventional organic synthesis) and electrochemical methods. Although both methods have their advantages and limitations, the former approach has been used to synthesize a broader range of organic substances as the latter is limited by several factors, such as poor selectivity and high energy cost. However, because electrochemical methods can replace harmful terminal oxidizers with external voltage, organic electrosynthesis has emerged as greener and more eco-friendly compared to conventional organic synthesis. The progress of electrochemical methods toward oxygen functionalization is presented by an in-depth discussion of different types of electrically driven-chemical organic synthesis, with particular attention to recently developed electrochemical systems and catalyst designs. We hope to direct the attention of readers to the latest breakthroughs of traditional oxygen functionalization reactions and to the potential of electrochemistry for the transformation of organic substrates to useful end products.
 Gracita M. Tomboc | Dr Gracita M. Tomboc received her Bachelor's degree in Chemical Engineering from Adamson University, Manila Philippines in 2012. She became a registered Chemical Engineer in 2013. She received her PhD degree in Energy Science and Technology from Myongji University, Republic of Korea in 2019. She is currently working as postdoctoral researcher at Korea University. Her research interests include the advanced materials processing of ideal electrode materials for electrochemical energy storage system applications such as supercapacitor, water splitting process, direct methanol fuel cells, and hydrogen generation. |
 Kwangyeol Lee | Professor Kwangyeol Lee obtained his PhD degree (1997) in Chemistry from the University of Illinois at Urbana-Champaign. After fulfilling his military obligation, he joined Korea University in 2003 as a chemistry faculty member, before being appointed as a professor. He is the recipient of the Wiley-KCS Young Scholar Award (2009, Korean Chemical Society) and the Excellent Research Award (2019, KCS Inorganic Chemistry Division). His current interests include the development of synthetic methodologies for nanoscale materials and the development of nanotechnologies to support the environment by creating sustainable energy. |
 Kyoungsuk Jin | Professor Kyoungsuk Jin is an assistant professor in the department of chemistry at Korea University. He received his PhD degree (2016) in Material Science from Seoul National University. During his postdoctoral research at the Massachusetts Institute of Technology (MIT), he studied the electrochemical oxygen functionalization reactions. He joined the department of chemistry at Korea University to begin his independent career in 2020. His research focuses on the fundamental understanding of electrochemical reactions and organic electrosynthesis including various oxygen-atom functionalization catalysis, asymmetric synthesis, biochemical reactions. |
1. Introduction
Oxygen atom functionalization has played an indispensable role in industrial applications and biological processes. For example, epoxides are versatile chemical intermediates that have been mainly produced from the oxygen functionalization of olefin substrates. Indeed, ethylene oxide and propylene oxide are among the highest volume products in our industry, with annual production rates of 15 and 3 Mt per year, respectively. These epoxides are widely used to manufacture diverse products, including ethylene glycol, ethylene carbonate, polyester, polyethylene glycol, and polyurethane. Representative examples of oxygen functionalization other than epoxidation are C–H bond oxidation, sulfoxidation, phosphorous oxidation, allylic oxidation, lactonization, Baeyer–Villiger oxidation, etc.In nature, there exist various enzymes that are capable of oxidizing target substrates with high selectivity. For example, methane monooxidase (MMO) in methanotrophic bacteria oxidizes methane into methanol under ambient conditions.1,2 Interestingly, the active site of the MMO enzyme is comprised of copper-based particulate MMO (pMMO) and iron-based soluble MMO (sMMO) that selectively capture the methane substrate.3 In addition to MMO, several transition metal-based enzymes are known to catalyze several oxygen functionalization reactions.4,5 Inspired by nature, various synthetic transition metal-based complexes have been investigated to mimic biological systems.6,7 In line with this, oxygen-atom functionalization generally follows the stepwise process shown below:
| MxOyL + oxidant → Mx+2Oy+1L |
| Mx+2Oy+1L + subs → MxOyL + prods, |
Apart from this route, an oxygen-functionalization reaction can be conducted via electrochemical methods, where an external voltage is applied to drive the reaction instead of oxidants. Although relatively low catalytic performance and limited reaction scope have been reported compared to the one by the non-electrochemical pathways, it has been regarded as a promising alternative as electrification itself does not require any additives and is ideally free of greenhouse gas (GHG, CO/CO2) emission.
This review aims to provide an overview of previous oxygen functionalization studies, which are divided into two parts: non-electrochemical and electrochemical methods. We categorized non-electrochemical methods according to their synthetic routes, namely, chemical route, photochemical route, and heterogeneous catalysis. We also discussed various catalytic systems, focusing on reaction mechanism, high-valent metal–oxo complexes, heterogeneous catalysts, and oxidants. The second part of this work featured recent studies and research directions of electrochemical oxygen functionalization. We divided electrochemical oxygen functionalization into three groups: (1) direct oxidation via outer-sphere electron transfer, (2) indirect oxidation, and (3) direct oxidation via inner-sphere electron transfer. In addition, recent progress in electrochemical oxygen functionalization was classified according to their synthetic strategies and interfacial kinetics. Lastly, the future direction of electrochemical oxygen functionalization will be described based on current understanding.
2. Non-electrochemical oxygen functionalization
For many years, oxidative reaction mostly followed stoichiometric oxidations, which consume toxic and corrosive organic reagents with high oxidizing capacity, such as bromine (Br2),8 chromium trioxide (CrO3),9 and potassium permanganate (KMnO4).10 This process could cause severe damage to the environment associated with the production of vast amounts of heavy-metal waste. As a result, many new works leaned towards the development of greener catalytic oxidations catalyzed by various metals and metal complex catalysts under mild conditions. The progress of catalytic processes directs the exploration of oxidants that give no waste as by-products and the use of inexpensive catalysts that are easy to prepare, handle, regenerate, and are stable and reactive under mild reaction conditions.11,12The oxidation of organic molecules primarily represents either the elimination of hydrogen, as in the sequential dehydrogenations of ethane: CH3CH3 → CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 → HC
CH2 → HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, or the replacement of a hydrogen atom bonded to carbon with a more electronegative element like oxygen, as in the series of oxidative transformations of methane: CH4 → CH3OH → CH2O → HCO2H → CO2.13 Selective oxidation reactions of alcohols, alkanes, and alkenes are some of the most important transformations in chemical synthesis that directly produce essential chemicals and intermediates, such as aldehydes, acids, alcohols, epoxides, and ketones.14 Here, we categorize non-electrochemical oxygen functionalization according to its synthetic routes and highlight the reaction mechanisms, generation of high-valent metal oxo species, and different catalysts and chemical oxidants typically considered for the oxidative transformation of organic substrates.
CH, or the replacement of a hydrogen atom bonded to carbon with a more electronegative element like oxygen, as in the series of oxidative transformations of methane: CH4 → CH3OH → CH2O → HCO2H → CO2.13 Selective oxidation reactions of alcohols, alkanes, and alkenes are some of the most important transformations in chemical synthesis that directly produce essential chemicals and intermediates, such as aldehydes, acids, alcohols, epoxides, and ketones.14 Here, we categorize non-electrochemical oxygen functionalization according to its synthetic routes and highlight the reaction mechanisms, generation of high-valent metal oxo species, and different catalysts and chemical oxidants typically considered for the oxidative transformation of organic substrates.
2.1. Oxygen atom sources
Chemical oxidants play an important role in the formation of metal–oxo complexes as they function as both an oxygen source and activating catalysts in oxygen functionalization.15 Molecular O2, hydrogen peroxide (H2O2), potassium bisulfate (KHSO4), chromate (CrO4), sodium hypochlorite (NaOCl), tert-butyl hydroperoxide (TBHP), iodosylbenzene (PhIO), and peroxyacid (RCO3H) have been widely employed to assist the formation of metal–oxo species under various reaction conditions.16 Among these, molecular O2 has been broadly investigated since it is abundant, cheap, and only produces water as a by-product.17 The activation of molecular O2 on the surface of heterogeneous catalysts is the key step in different oxidation reactions, such as epoxidation and CO-, hydrocarbon-, alcohol-, and glucose- oxidations.18 However, oxidation reactions using molecular O2 oxidant often have several challenges: (1) the direct oxidation of organic substrates using molecular O2 typically occurs at harsh conditions (toxic solvents, high temperature, and high pressure); (2) a co-reductant is normally required to complete the oxidation of transition metal-based catalysis; and (3) product selectivity is relatively moderate.18,19 Likewise, hydrogen peroxide has been studied as it also generates water as its by-product. Fe-, Mn-, Rh-, W-, and Ti-based catalytic systems are known for employing H2O2 as the terminal oxidant.20–24 The major limitations of this oxidant are the hazards associated with the storage and transportation of H2O2 and the requirement of toxic organic solvents for the recovery of catalysts used during its production.14,25,26On the other hand, water is perhaps the most environmentally benign oxygen source for different organic oxidation reactions. However, the problem of using water for organic oxidations is correlated to its stoichiometric catalytic activation. The reaction usually requires strong oxidants, like CeIV and ammonium nitrate, to remove electrons and protons. e.g.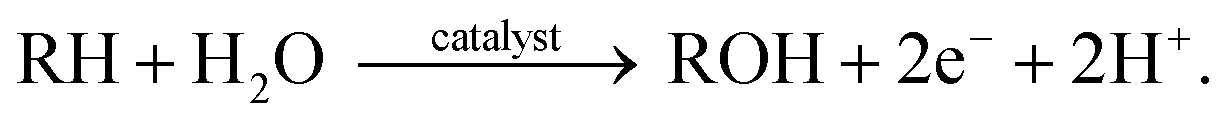 27–30 In line with this, Nam et al. recently suggested that much weaker oxidants can replace these strong one-electron oxidants if the reaction is light-driven (see Section 2.2 for details).6,30,31
27–30 In line with this, Nam et al. recently suggested that much weaker oxidants can replace these strong one-electron oxidants if the reaction is light-driven (see Section 2.2 for details).6,30,31
2.2. Metal–oxo species
High-valent metal oxo species, Mn+O or Mn+–OH, are highly reactive intermediates that are widely utilized in various oxidation processes in biological systems and chemical industries. In enzymatic and synthetic oxidation catalysis, high-valent transition metal–oxo intermediates are generally produced and serve as the active oxygen atom transfer (OAT) species. Transition metal-based high-valent metal–oxo complexes, such as Fe-, Mn-, Ru-, Cr-, Mo-, V-, and Os–oxo complexes, have been investigated.32–35 The oxidizing power of the high-valent oxo-form of these metal atoms is sufficient for the oxidation of target substrates. The structures, properties, and reactivities of these complexes in organic oxidation reactions have been well documented in the past.7,36Fig. 1 presents some of the representative metal–oxo species employed for oxygen functionalization reactions, including Fe-, Mn-, and Ru–oxo complexes.
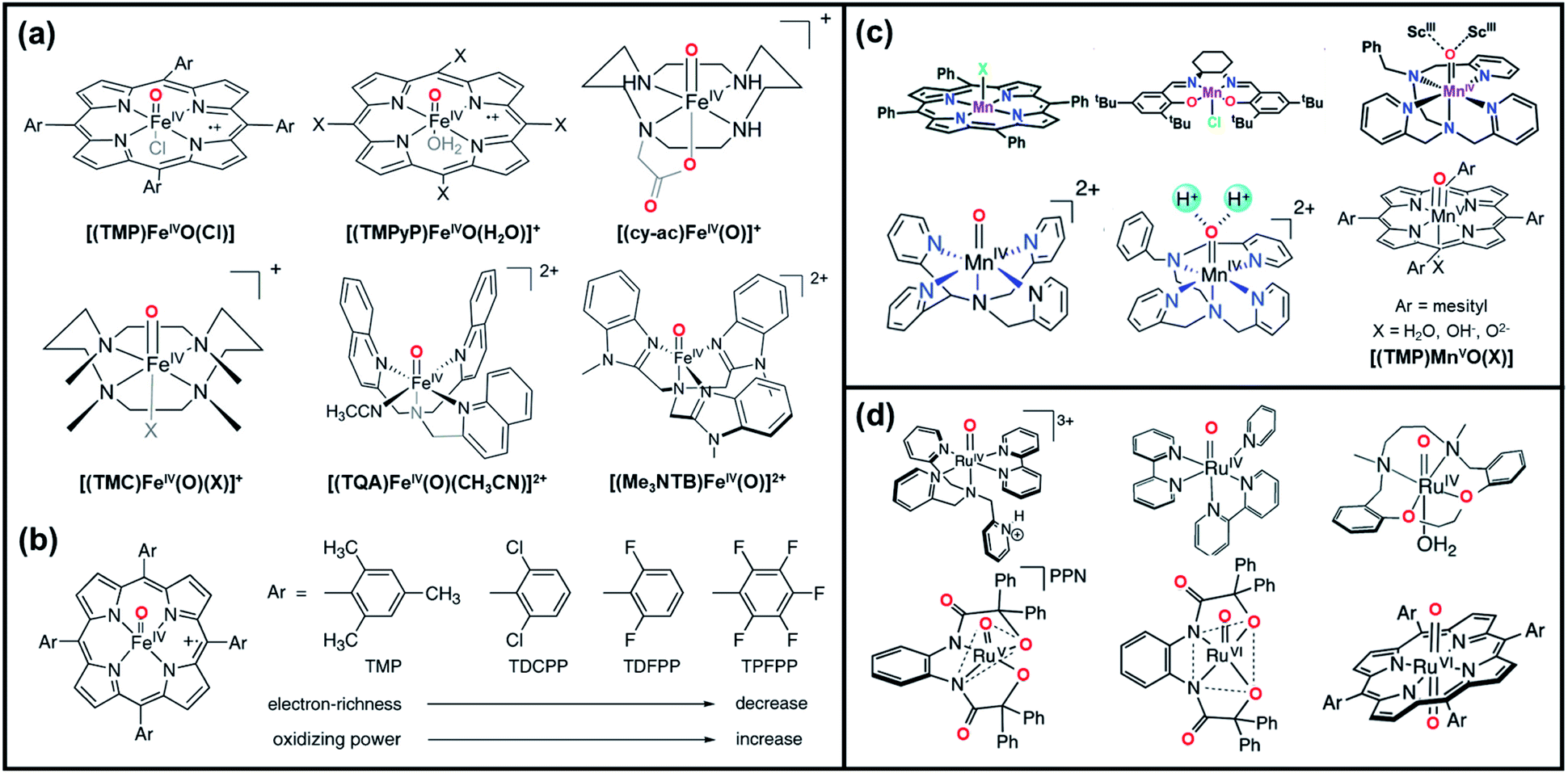 | ||
| Fig. 1 (a) Examples of high-valent Fe–oxo porphyrin complex. Reproduced with permission.33 Copyright 2018, American Chemical Society. (b) Correlation between FeIV–oxo species paired with different axial ligands and their reactivities. Reproduced with permission.193 Copyright 2007, American Chemical Society. (c) Representatives of high-valent Mn–oxo porphyrin and salen complex. Reproduced with permission.33,53,194 Copyright 2020, The Royal Society of Chemistry; Copyright 2016, American Chemical Society; and Copyright 2018, American Chemical Society. (d) Examples of high-valent Ru–oxo porphyrin complex. Reproduced with permission.54 Copyright 2016, The Royal Society of Chemistry. | ||
Oxidation reactions such as alkane hydroxylation, olefin epoxidation, and olefin cis-dihydroxylation catalyzed by iron centers in enzymes have been a subject of interest for many years. High-valent porphyrin Fe–oxo intermediates are central oxidizing species in heme and non-heme iron enzymes and synthetic oxidation catalysts.37–41Fig. 1a displays the examples of synthetic model systems of high-valent Fe–oxo porphyrin complex. Among these, FeIV–oxo porphyrin π-cation radical [(P˙+)FeIV(O)] is a well-known intermediate formed during the reaction of Fe(TMP)Cl (TMP = meso-tetramesitylporphyrin) and m-chlorobenzoic acid (m-CPBA) in presence of dichloromethane and methanol. The formed Fe–oxo species was found to be a competent oxidant in olefin epoxidation.4,5,42–45 Likewise, FeIV–oxo tetraphenylporphyrin [(TPP)FeIV(O)]˙+ produced via the oxidation of FeIII tetraphenylporphyrin chloride [(TPP)FeIII(Cl)] and PhIO with cytochrome P450 catalyst are discovered as reactive oxo species for driving alkane hydroxylation.4,5,43,44 Since then, FeIV–oxo species with electron-rich/deficient porphyrins paired with different axial ligands (Fig. 1b) have been designed and investigated towards various oxidation reactions in an effort to understand the electronic effects of porphyrin and axial ligands on the chemical properties of the Fe–oxo intermediates.40,41,46
High-valent Mn–oxo species have also received a great deal of attention for oxygen functionalization reactions. The model complexes of manganese are categorized into two groups based on the ligand systems: (1) Mn–porphyrin based systems and (2) Mn–salen based systems. Groves et al. synthesized the three kinds of MnO complexes namely, (TMP)Mn(O), (TMP)Mn(O)(OH), and (TPP)Mn(O) (TMP = 5, 10, 15, 20-tetramesitylporphyrin), by facile reaction of MnIII complex with m-CPBA as oxidants. The generated transient species were assigned to MnVO species, which showed high reactivity towards olefin substrates, thus forming epoxide products.47 Similarly, high-valent MnIV–oxo species generated from the oxidation of tetra-N-((methylpyridyl)-porphyrinato)MnIII-complex [MnIII(TMPyP)] are identified as active species for olefin epoxidation and alkane hydroxylation catalysis.37,48–50 Moreover, manganese–salen complexes (Mn–salen) are well-known catalysts in the epoxidation of alkenes and other olefinic materials. Mn–salen complexes with PhIO as terminal oxidant are typically oxidized to MnV(salen)O, which are considered as the active species responsible for the epoxidation of various substrates.51 In particular, the Jacobsen's catalyst, a chiral salen based MnIII complex, successfully transformed wide range of substrates including conjugated Z-enynes, acyclic conjugated polyenes, cyclic dienes, enol ethers, and some tetra-substituted conjugated alkenes.52 The combination of this catalyst with iodosyl benzene and triethylamine trihydrofluoride as fluoride donor showed selective fluorination of a large variety of substrates containing benzylic C–H bond. The representative examples of Mn–oxo species are shown in Fig. 1c.53
Lastly, high-valent Ru–oxo species, such as RuIVO, RuVO, and RuVIO, have emerged as reactive species for water oxidation, epoxidation, and C–H oxidation.54 Ruthenium imine complexes, like Ru(bpy)2Cl2 (bpy = 2,2′-bipyridine), are among the most distinguished ruthenium complexes oxidized to generate the RuIVO intermediate.55 Interestingly, the catalytic epoxidation efficiency of Ru(bpy)2Cl2 was increased upon the incorporation of Lewis acid like Al(OTf)3 (OTF: trifluoromethanesulfonate), which led to the formation of more active species, the RuIVO/AlIII intermediate. Such results have highlighted the roles of non-redox metal ions, like Lewis acids, in regulating the reactivity of high-valent metal–oxo complexes.56 On the other hand, RuV–oxo complexes, like [RuV(O)(N4O)2+] (N4O = bis (2-(2-pyridyl)-ethyl)(2-hydroxy-2-(2 pyridyl)ethyl)amine), have been reported to proceed with the oxidative CC bond cleavage of styrene and trans- and cis-stilbene in the presence of acetonitrile, leading to the formation of benzaldehyde products. Additionally, trans-dioxo-RuVI–porphyrin complexes [(RuVI(TMP)O2)] are well-known intermediates for aerobic asymmetric epoxidation of olefins.57,58Fig. 1d displays some examples of high-valent Ru–oxo porphyrin complex.
The details of the electron transfer mechanisms and catalytic performances of metal–oxo species towards various oxygen functionalization reactions are discussed in the succeeding sections.
2.3. Proposed non-electrochemical oxygen functionalization mechanism
In this section, we present the recent progress of non-electrochemical methods for oxygen functionalization. The non-electrochemical methods for the transformation of organic molecules are categorized based on synthetic routes, namely, the (1) chemical route, (2) photochemical route, and (3) heterogeneous catalysis. Scheme 1 displays a schematic illustration of non-electrochemical methods for oxidation functionalization.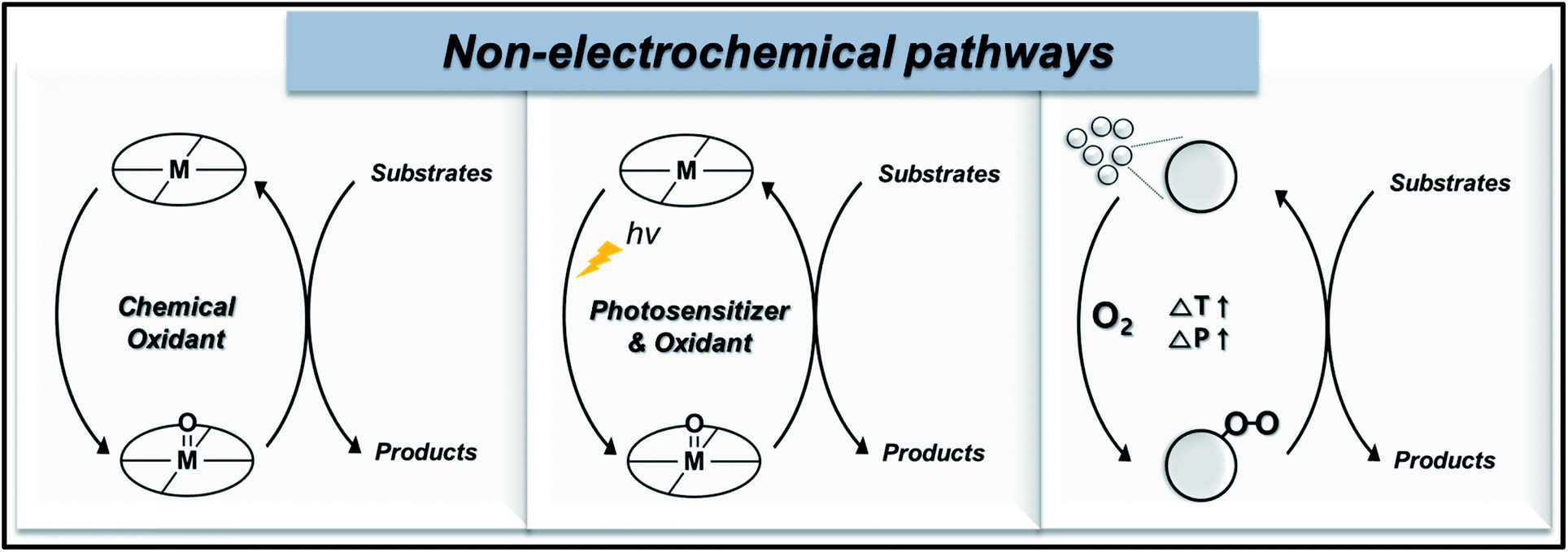 | ||
| Scheme 1 Schematic illustration of non-electrochemical pathways: (from left to right) chemical route, photochemical route, and heterogeneous catalysis. | ||
An oxygen functionalization reaction via chemical route is generally catalyzed by high-valent metal–oxo complexes in presence of chemical oxidants, like H2O2 or molecular O2, under high temperature and pressure. The photochemical route undergoes a similar reaction mechanism as that of the chemical route, except for the utilization of solar radiant energy as the driving force of the organic transformations, which thus enables the oxidation reaction under milder conditions. A typical photochemical oxygen functionalization reaction uses a photosensitizer, chemical oxidant, homogenous catalyst, oxygen source, and substrate. Conversely, oxygen functionalization via heterogeneous catalysis proceeds via the adsorption of reactant molecules on the catalyst surface, which are converted into surface intermediates, followed by the formation and desorption of the final product. High temperature and pressure are the usual activation conditions of this pathway, while the reaction kinetics are highly dependent on the activation energies (Ea) of adsorbed reactants, adsorbed intermediates, and desorption of products. The activity, selectivity, resistance to deactivation, and regeneration of catalyst are vital properties in heterogeneous catalysis, especially at the industrial level.22,59,60
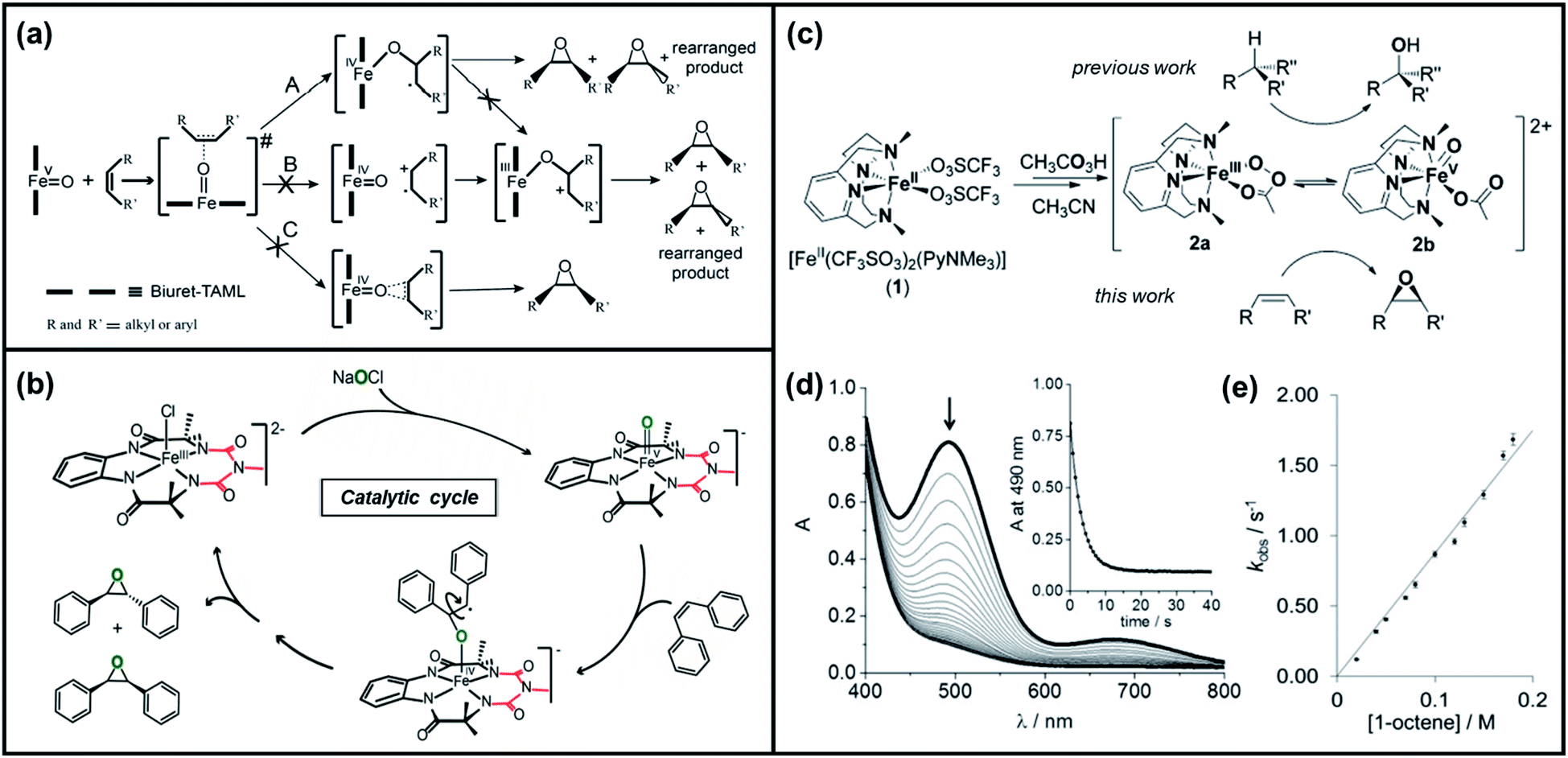 | ||
| Fig. 2 (a) Three possible mechanisms of epoxidation of alkenes by [FeV(O)–(biuret-TAML)] based on the initial attack of “O” into the substrate during the oxidation reaction and (b) the suggested catalytic cycle for epoxidation of olefins by FeV(O) and NaOCl. Reproduced with permission.67 Copyright 2015, American Chemical society. (c) Formation of species 2a and 2b upon the reaction of [FeII(CF3SO3)2(PyNMe3)] with peracetic acid in acetonitrile, (d) kinetics monitoring of the reaction of 2 with 1-octene in acetonitrile/acetone via UV/vis spectroscopy (inset displays the decay of the 490 nm chromophore fitted to a single exponential), and (e) plot of rate constants kobs against the 1-octene concentration for the reaction of 2 with 1-octene. Reproduced with permission.72 Copyright 2016, Wiley-VCH. | ||
O species, and RuIVO intermediate with Sc3+, respectively
| Entry | Catalyst | Oxidant | Substrate | Product | % yield | % conversion |
|---|---|---|---|---|---|---|
a [FeV(O)–(biuret-TAML)]− complex 2.67
b Mn(TPA)Cl2 (TPA = tris(pyridin-2-ylmethyl)amine).
c 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 ratio MnII:Al3+.
d 1:0 ratio MnII:Al3+.74
e Ru(bpy)2Cl2 (bpy = 2,2′ -bipyridine).55 :0 ratio MnII:Al3+.
d 1:0 ratio MnII:Al3+.74
e Ru(bpy)2Cl2 (bpy = 2,2′ -bipyridine).55
|
||||||
| 1 | FeV(O)a | NaOCl/m-CPBA |
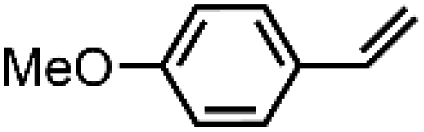
|
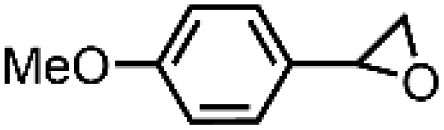
|
74 ± 6 | N.D. |
| 2 | FeV(O)a | NaOCl/m-CPBA |
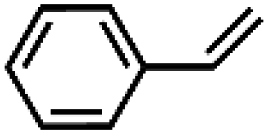
|
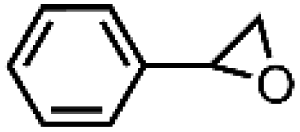
|
71 ± 6 | N.D. |
| 3 | FeV(O)a | NaOCl/m-CPBA |
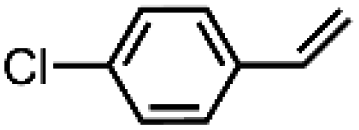
|
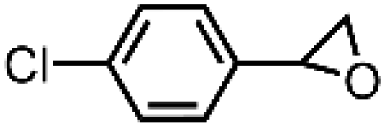
|
70 ± 6 | N.D. |
| 4 | FeV(O)a | NaOCl/m-CPBA |
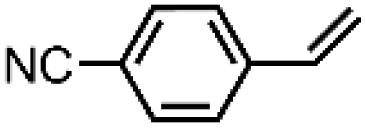
|
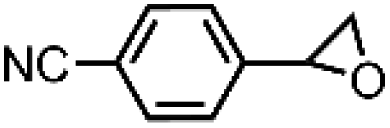
|
62 ± 5 | N.D. |
| 5 | FeV(O)a | NaOCl/m-CPBA |
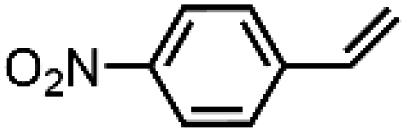
|
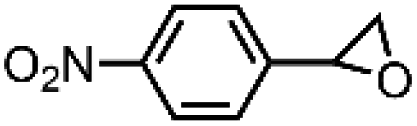
|
66 ± 5 | N.D. |
| 6 | FeV(O)a | NaOCl/m-CPBA |
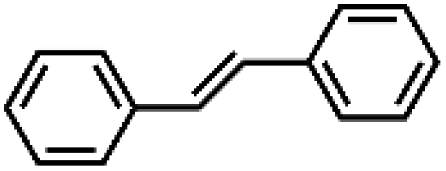
|
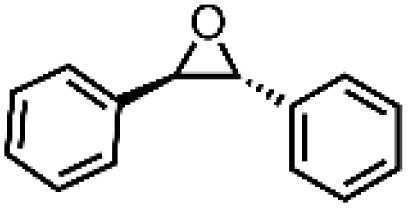
|
81 ± 4 | N.D. |
| 7 | FeV(O)a | NaOCl/m-CPBA |
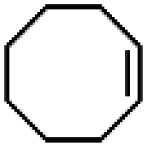
|
|
65 ± 5 | N.D. |
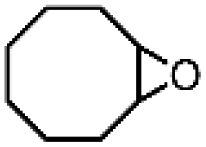
| 8 | FeV(O)a | NaOCl/m-CPBA |
|

|
86 ± 6 | N.D. |
| 9 | FeV(O)a | NaOCl/m-CPBA |
|

|
78 ± 7 | N.D. |
| 10 | Mn(TPA)Cl2b | Phl(OAc)2 |
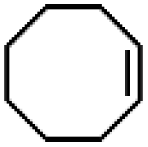
|
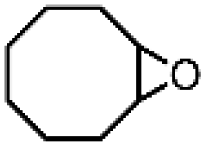
|
4.1c | 9.9c |
| 91.4d | 97.8d | |||||
| 11 | Mn(TPA)Cl2b | Phl(OAc)2 |
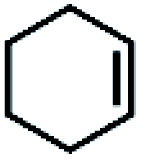
|
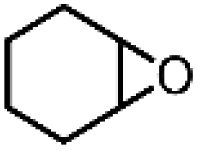
|
6.7c | 9.9c |
| 69.0d | 91.3d | |||||
| 12 | Mn(TPA)Cl2b | Phl(OAc)2 |
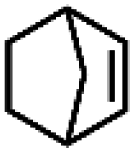
|
|
5.1c | 11.1c |
| 47.4d | 84.3d | |||||
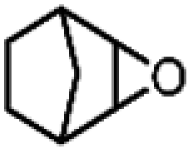
| 13 | Mn(TPA)Cl2b | Phl(OAc)2 |
|
|
4.9c | 9.1c |
| 45.1d | 71.9d | |||||
| 14 | Mn(TPA)Cl2b | Phl(OAc)2 |

|

|
3.7c | 9.3c |
| 51.7d | 75.1d | |||||
| 15 | Mn(TPA)Cl2b | Phl(OAc)2 |

|

|
2.5c | 13.5c |
| 45.1d | 90.3d | |||||
| 16 | Ru(bby)2Cl2e | Phl(OAc)2 |
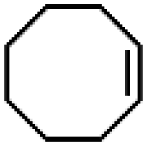
|
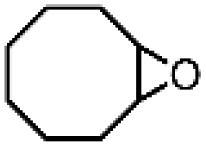
|
89.9 | 100 |
| 17 | Ru(bby)2Cl2e | Phl(OAc)2 |
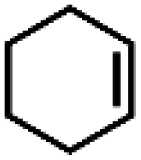
|
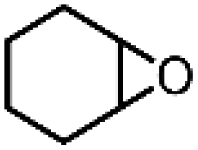
|
82.2 | 100 |
| 18 | Ru(bby)2Cl2e | Phl(OAc)2 |
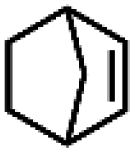
|
|
33.6 | 98.9 |
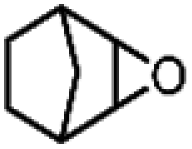
| 19 | Ru(bby)2Cl2e | Phl(OAc)2 |
|

|
30.0 | 10 |
| 20 | Ru(bby)2Cl2e | Phl(OAc)2 |

|

|
74.6 | 94.6 |
| 21 | Ru(bby)2Cl2e | Phl(OAc)2 |

|

|
67.3 | 100 |
Costas et al. further scrutinized the reactivity of FeV(O) intermediate for catalytic epoxidation reactions.72 As shown in Fig. 2c, the reaction of [FeII(CF3SO3)2(PyNMe3)] (compound 1) with peracetic acid and acetonitrile at −41 °C led to the formation of compound 2, which is made up of two thermally unstable components: [FeIII(OOAc)(PyNMe3)]2+(2a) and [FeV(O)(OAc)(PyNMe3)]2+ (2b). The group investigated the reaction kinetics of 2b with cyclooctene substrate by conducting a series of ultraviolet/visible (UV/vis) stopped-flow experiments. The rapid disappearance of the absorbance band at 490 nm following the addition of excess olefin was primarily used as an evaluation point (Fig. 2d). Three crucial observations stemmed from these results. First, based on the plot of the rate constant versus substrate concentration (Fig. 2e), it was determined that the second-order reaction rates of FeV(O)+ are independent of the peracid concentration. Next, the oxidation rate of 2b towards olefins was directly proportional to the degree of olefin substitution. Such observation implied that rather than the steric factors, the electronic effects actually dominated the reactivity of the FeV(O) intermediate towards olefins. Finally, the higher reactivity of 2b compared to synthetic FeIV–oxo porphyrin radical species supported the proposal of FeV(O) intermediates as plausible oxygen functionalization agents for high selective epoxidations.
Moreover, the catalytic oxidation of various alkanes catalyzed by manganese complex [LMnIV(O)3MnIVL](PF6)2 (L = 1,4,7-trimethyl-1,4,7-triazacyclononane) with peroxyacetic acid or H2O2 in acetonitrile was first reported as viable by Shul'pin et al. in the late 1990s.73 The reactions catalyzed by [LMnIV(O)3MnIVL](PF6)2 were rationalized by assuming the formation of high-valent metal–oxo intermediates, possibly MnVO, in the presence of H2O2 oxidant. More interestingly, the stereoselective oxygenation of hexane, cyclohexane, and cycloheptane was dramatically accelerated upon the addition of acetic acid in the H2O2/[LMnIV(O)3MnIVL](PF6)2 system. This result suggested the critical role of carboxylic acid additives in improving the product yield and selectivity of asymmetric oxidation reactions. Likewise, the addition of redox-inactive metal ions, like Al3+ as Lewis acids during the generation of MnIVO species has shown positive effects on the epoxidation efficiency of non-heme manganese complex, as investigated by Yin et al.74 The effect of adding Al(OTf)3 in the epoxidation of cyclo-olefins and terminal linear olefins catalyzed by Mn(TPA)Cl2 complex (TPA = tris(pyridin-2-ylmethyl)amine) is presented in Table 1 (entry 10–15). For example, the epoxidation of cyclooctene to cyclooctene oxide catalyzed by Mn(TPA)Cl2 catalyst alone has demonstrated sluggish activity, obtaining low product yield and conversion (4.1% and 9.9%, respectively). Under identical conditions, the addition of Al3+ generated a much higher oxidation activity with 97.8% conversion and 91.4% yield of epoxide.
Subsequent studies have reported the use of related manganese complexes to catalyze the oxidation of alkene, alcohols, and phenols in both aqueous and organic solutions. Collectively, the factors affecting the generation of active high-valent Mn–oxo intermediates were narrowed down to three points: (1) addition of additives (i.e., carboxylic acid, sulfuric acid, non-redox metal ions as Lewis acids), (2) the number of ligands or cis-binding sites available in the structure of the manganese complex, which can bind both of the oxidant and additives, and (3) the nature of reactive intermediates responsible for the catalytic oxidation reaction.37–39,75 In line with this, Nam et al. revealed the role of additives and the nature of active intermediates in Mn complex-catalyzed enantioselective epoxidation of olefins.76 They employed non-heme Mn catalysts with tetradendate N4 ligand (LN4) (1, Fig. 3a) and pentadentate N5 ligand (LN5) (2, Fig. 3b), together with H2O2, alkyl hydroperoxides, and iodosylbenzene (PhIO) as oxidants, respectively. Fig. 3c and d illustrate the “carboxylic acid-assisted” mechanisms of olefin epoxidation catalyzed by 1 and 2, respectively. On one hand, it was perceived that when using H2O2 and alkyl hydroperoxides as oxidants, only the Mn complex bearing a LN4 ligand can bind the carboxylic acid at the manganese center (Fig. 3c, pathway A), which promoted heterolytic O–O bond cleavage (Fig. 3c, pathway C) followed by the formation of highly valent manganese–oxo species. On the other hand, using PhIO as an oxidant, both 1 and 2 catalysts supported the formation of the active manganese–oxo species during asymmetric epoxidation of olefins. Both catalysts obtained high product yields with high enantioselectivity (Fig. 3d). Based on these observations, Nam et al. concluded that irrespective of the oxidants in carboxylic acid, the Mn complex with two cis-binding sites is the more efficient catalyst. Moreover, the results of their mechanistic studies in olefin epoxidation suggested that a common intermediate (either MnIV(O) or MnV(O)) was generated as an active species when the reactions using different oxidants were carried out in the presence of carboxylic acid. By conducting a number of stoichiometric and catalytic reactions, it was implicated that MnV(O) species are more plausible active oxidants in the catalytic oxidation reaction than MnIV(O) species (Fig. 3e), which is similar to previous proposals.
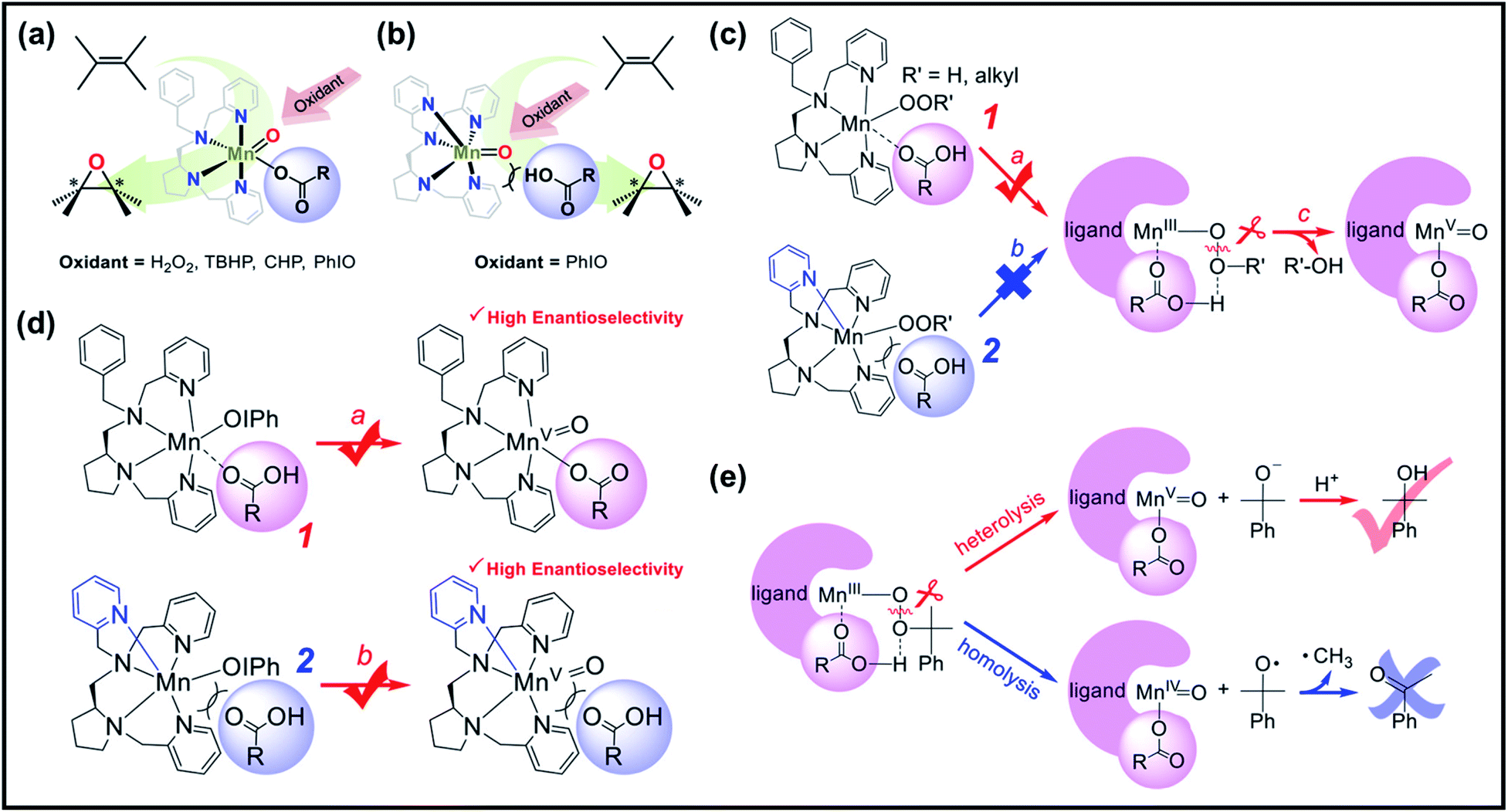 | ||
| Fig. 3 Asymmetric epoxidation reactions by manganese complexes bearing (a) tetradentate N4 ligands (1) and (b) tetradentate N5 ligands (2). (c) Proposed carboxylic acid-assisted mechanism involving the formation of active manganese–oxo species in the reactions of H2O2 and alkyl hydroperoxides. (d) Formation of highly reactive and enantioselective manganese–oxo intermediates during the reactions of 1 and 2 with PhIO in the presence of carboxylic acid. (e) Identification of the mode of O–O bond cleavage of the Mn–(hydro)alkylperoxo intermediate: heterolytic vs. homolytic O–O bond cleavage mechanisms. Reproduced with permission.76 Copyright 2018, American Chemical Society. | ||
Furthermore, ruthenium complexes, like ruthenium porphyrin complexes, have also been regarded as efficient catalysts for the epoxidation of olefins. The interest in ruthenium is attributed to its proximity to iron in the periodic table and its ability to generate various high-valent metal–oxo species. RuIVO to RuVIIO species have been postulated to be the most likely active intermediates when olefins are selectively oxidized to the corresponding epoxides via oxygen functionalization.77Table 1 (entry 16–21) presents some examples of the epoxidation of different alkenes catalyzed by the RuIVO intermediate. In this regard, Sheldon et al. conducted a comparison study to investigate the reactivity of cis and trans-RuVI-dioxo complexes towards the epoxidation of a trans-stilbene substrate with sodium periodate (NaIO4) as oxygen donor and ruthenium bis[2-(phenyl azo)pyridine]Cl2 complex [(Ru(pap)2Cl2)] as a catalyst.78 The employed catalyst could be transformed into five isomeric forms, including trans–trans–trans (γ), trans–cis–cis (α), and cis–cis–cis (β) isomers. Both cis-β- and trans-γ-isomers showed remarkable selectivity for epoxide (47% and 75%, respectively), while the most stable cis-α-isomer gave inferior results (34%). The different results observed for the two cis-isomers (α and β) were correlated to the steric effect, in which the α-isomer appeared to be more sterically hindered than the β-isomer.
Kojima et al. have recently reported a comprehensive review of the characteristics and reactivity of ruthenium oxo complexes towards the oxidation of various organic compounds, including alcohols, alkenes, and alkanes.54 The authors categorized the synthetic procedures of high-valent ruthenium–oxo complexes into three groups, as shown in Fig. 4a. The formation of the Ru–oxo species can follow either of the following: (1) introduction of a high-valent oxo source into a ligand, (2) employing of an oxo-transfer reagent to oxidize a low-valent Ru complex, and (3) proton-coupled electron transfer (PCET) oxidation of a low-valent Ru–OH2 complex. As an alternative, Zhang et al. recently investigated the potential of dendritic ruthenium porphyrins as a new class of highly selective catalysts for epoxidation and cyclopropanation of alkenes.79 In their work, poly(benzyl ether) dendritic wedge dendrons (denoted as [G-n]) were attached to carbonyl rutheniumII–meso-tetraphenylporphyrin (abbreviated as 5) to form dendritic ruthenium porphyrins (5–[G-n]m), where n is the generation number of the dendrons (n = 0, 1, 2) and m is the total number of dendrons attached to 5 (m = 4, 8), as displayed in Fig. 4b. It was discovered that the attachment of [G-n] to complex 5 dramatically increased the epoxide yield during the catalytic styrene oxidation reaction. Also, the chemoselectivity of the prepared catalyst significantly increased as the number of attached dendrons increased (Fig. 4c). Notably, prior to this study, there was no inclusive report regarding the dendritic metalloporphyrin catalyzed epoxidation of steroids.
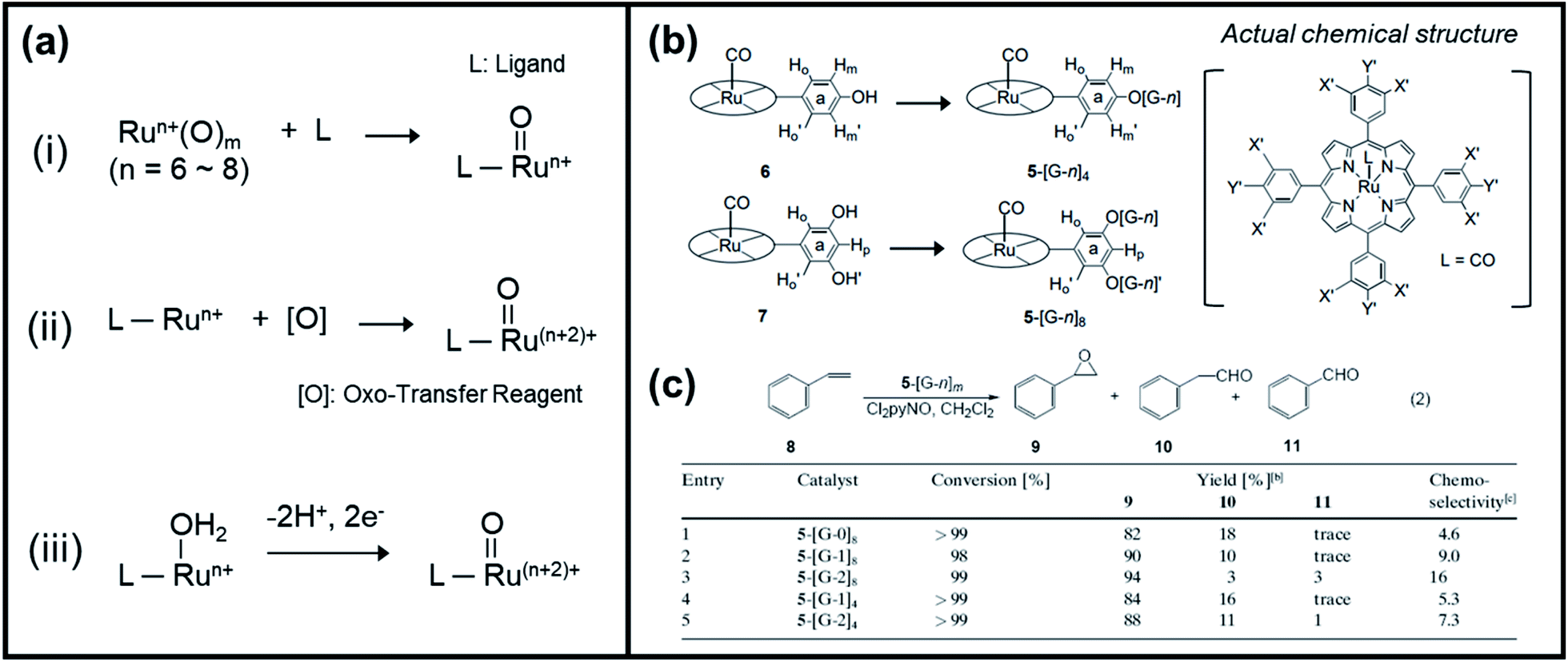 | ||
| Fig. 4 (a) Categorized synthetic procedures of high-valent ruthenium–oxo complexes. Reproduced with permission.54 Copyright 2016, The Royal Society of Chemistry. (b) Formation of dendritic ruthenium porphyrins (5–[G-n]4 (n = 1, 2) and 5–[G-n]8 (n = 0, 1, 2)) from complex 6 and 7, respectively, and its actual chemical structure. (c) Catalytic behavior of the dendritic ruthenium porphyrins 5–[G-n]m toward the epoxidation of styrene (8) with excess Cl2pyNO. Reproduced with permission.79 Copyright 2002, Wiley-VCH. | ||
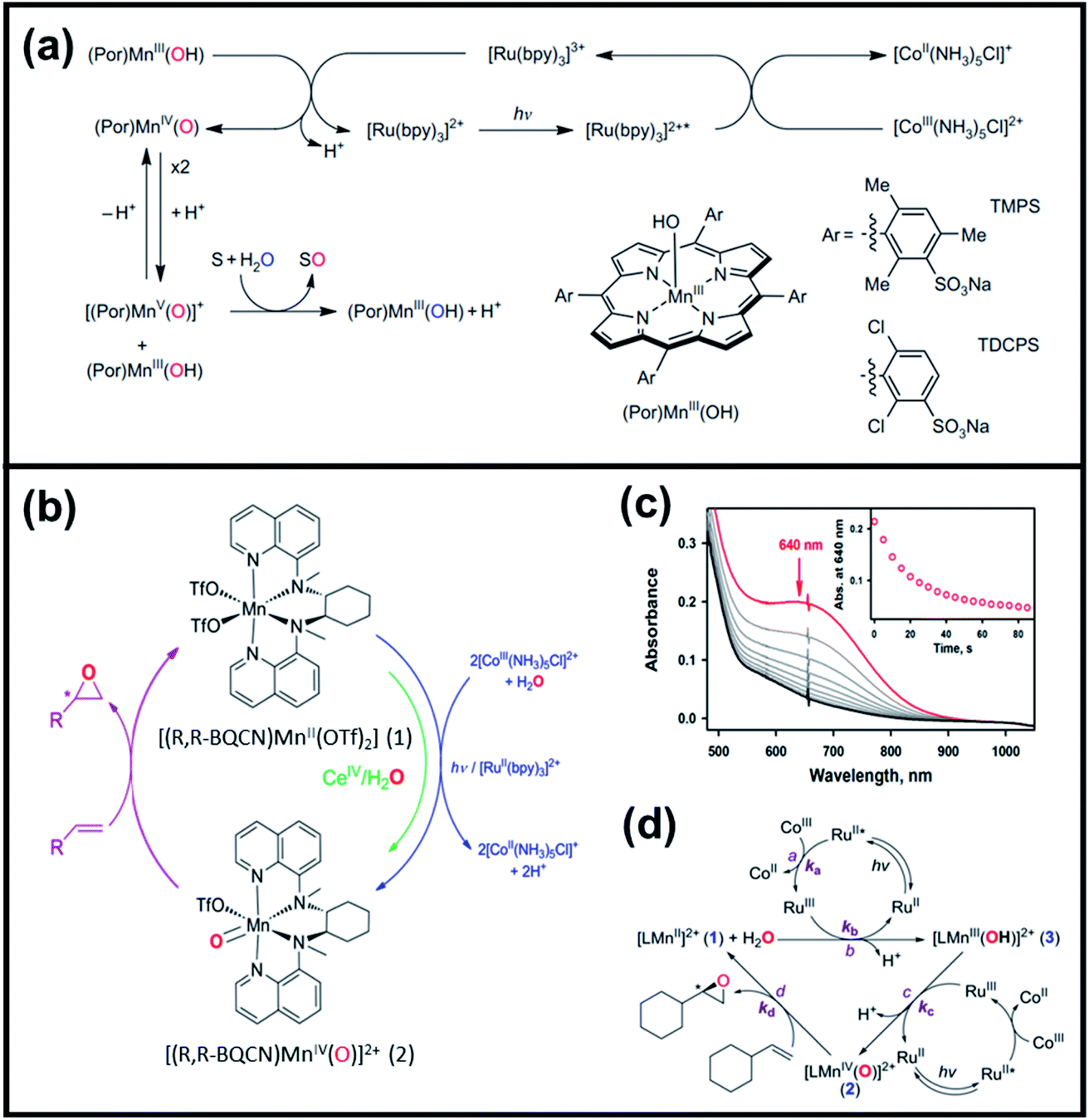 | ||
| Fig. 5 (a) Proposed reaction mechanism of photocatalytic oxygenation reactions of organic substrates using water, (Por)MnIII(OH), and [RuII(bpy)3]2+ as the oxygen source, catalyst, and photocatalyst, respectively. Reproduced with permission.30 Copyright 2011, Macmillan Publishers Limited. (b) Schematic diagram of photocatalytic and stoichiometric enantioselective epoxidation of terminal olefins using MnIV–oxo complex, (c) UV/vis monitoring of [(R,R-BQCN)MnIV(O)2+] reacted with vinylcyclohexane (50 mM) in CH3CN/H2O solvent (inset shows the decay of the adsorption band at 640 nm), and (d) proposed mechanism of photoinduced generation of non-heme MnIV–oxo complex and its reaction with olefin. Reproduced with permission.31 Copyright 2016, American Chemical Society. | ||
| Entry | Catalyst | Oxygen source | Substrate | Product | % yield | % conversion c |
|---|---|---|---|---|---|---|
| a [(R,RBQCN)MnII(OTf)2] (BQCN = N,N′-dimethyl-N,N′-bis(8-quinolyl)cyclohexanediamine and OTf = CF3SO3−).31 b [Et4N][(bTAML) FeIII(OH2)] (bTAML = biuret-modified tetraamidomacrocyclic ligand).80 | ||||||
| 1 | (R,R-BQCN)MnII(OTf)2a | Water |
|
|
59 ± 4 | 53 ± 2 |
| 2 | (R,R-BQCN)MnII(OTf)2a | Water |
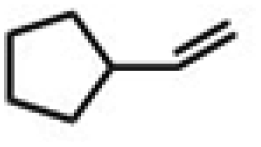
|
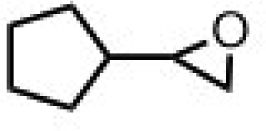
|
68 ± 5 | 50 ± 2 |
| 3 | (R,R-BQCN)MnII(OTf)2a | Water |

|

|
24 ± 3 | 57 ± 1 |
| 4 | (R,R-BQCN)MnII(OTf)2a | Water |

|
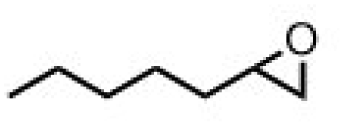
|
26 ± 3 | 60 ± 2 |
| 5 | (R,R-BQCN)MnII(OTf)2a | Water |

|
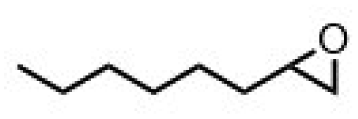
|
34 ± 4 | 56 ± 2 |
| 6 | (R,R-BQCN)MnII(OTf)2a | Water |
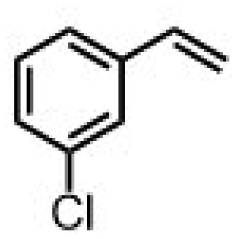
|
|
65 ± 5 | 43 ± 2 |
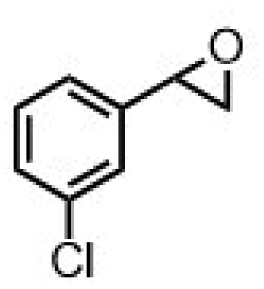
| 7 | [Et4N][(bTAML)FeIII(OH2)]b | Water |
|
|
90 | 72 |
| 8 | [Et4N][(bTAML)FeIII(OH2)]b | Water |
|
|
84 | 50 |
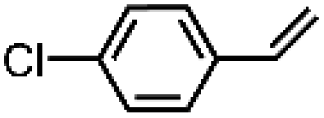
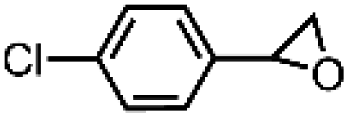
| 9 | [Et4N][(bTAML)FeIII(OH2)]b | Water |
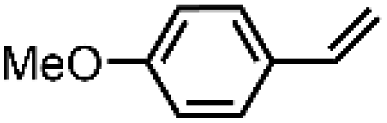
|
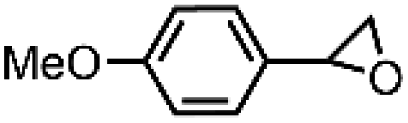
|
92 | 95 |
| 10 | [Et4N][(bTAML)FeIII(OH2)]b | Water |
|
|
79 | 58 |
| 11 | [Et4N][(bTAML)FeIII(OH2)]b | Water |
|
|
94 | 51 |
| 12 | [Et4N][(bTAML)FeIII(OH2)]b | Water |
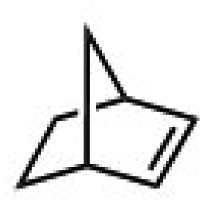
|
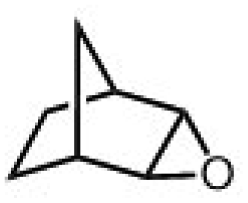
|
92 | 64 |
Subsequently, Gupta et al. investigated the selective photocatalytic alkane hydroxylation and olefin epoxidation using high-valent Fe–oxo complex, [Et4N][(bTAML) FeIII(OH2)] (1) as a catalyst, assisted by [RuII(bpy)3]Cl2, [CoIII(NH3)5Cl]Cl2, and water as a photosensitizer, weak one-electron acceptor, and oxygen atom sources, respectively.80Fig. 6a displays the proposed catalytic cycle for the photochemical oxygenation reaction catalyzed by the Fe–oxo complex. Upon irradiation with light (3 W blue LED, 440 nm), the iron–oxo complex FeIII–bTAML (1) was oxidized to [(bTAML) FeIV–OH]− which was immediately converted into [{(bTAML)FeIV}2(μ-O)]2− (2) dimer. After the addition of a substrate (alkenes or alkanes) to this solution, the dimer (2) reacted with the substrate to form FeV(O) and regenerate the compound (1). The formed [{(bTAML)FeIV}2(μ-O)]2- (2) dimer was revealed as the active oxidant, allowing the hydroxylation of alkanes and epoxidation of alkenes with high selectivity. The photocatalytic hydroxylation of different alkenes by [Et4N][(bTAML)FeIII(OH2)] in the presence of water as the oxygen source is summarized in Table 2 (entries 7–12).
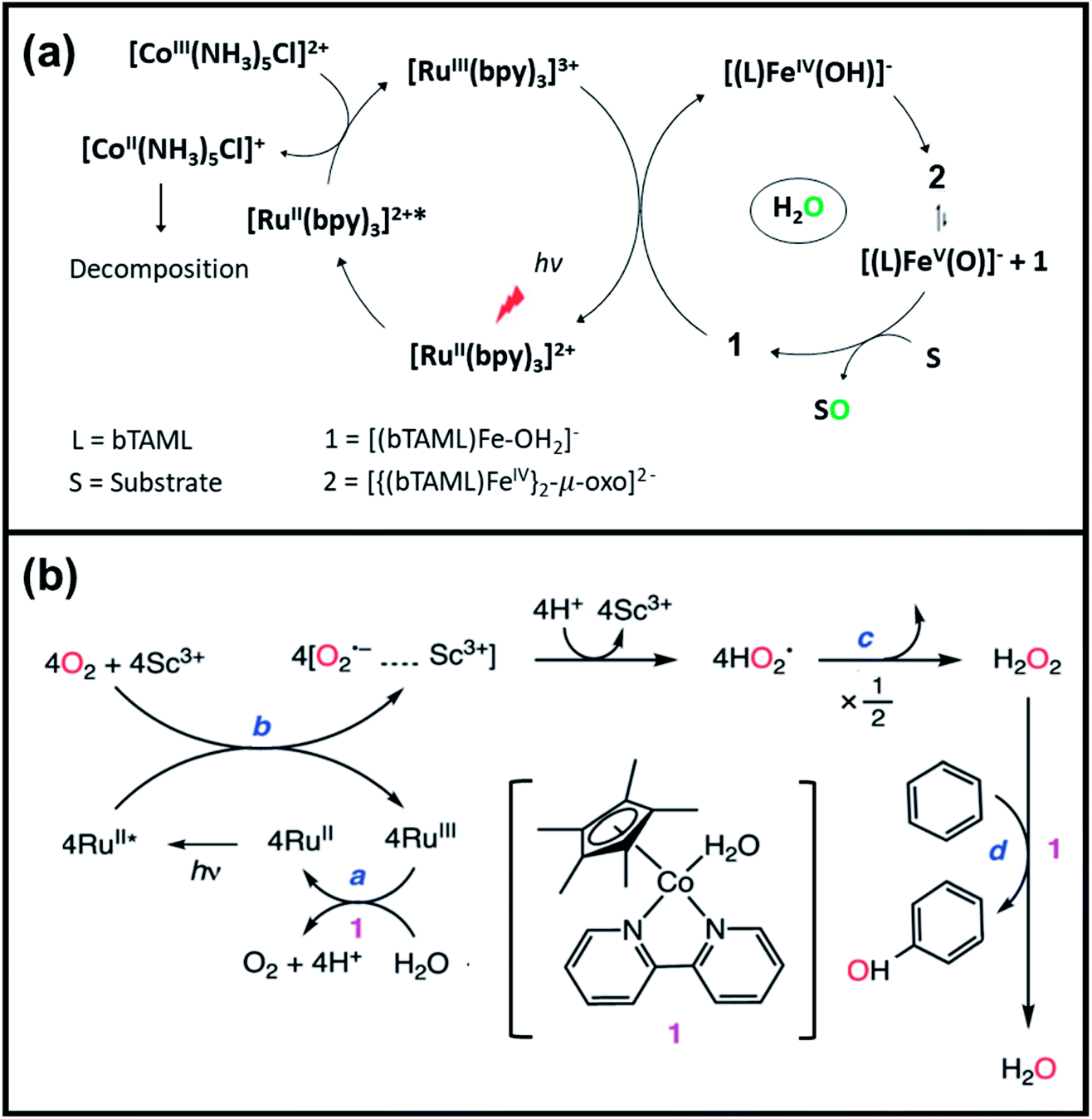 | ||
| Fig. 6 (a) Proposed reaction mechanism of the selective photocatalytic hydroxylation and olefin epoxidation using [Et4N][(bTAML)FeIII(OH)2], [RuII(bpy)3]Cl2, [CoIII(NH3)5Cl]Cl2, and water as catalyst, photosensitizer, one-electron acceptor, and oxygen atom source, respectively. Reproduced with permission.80 Copyright 2017, The Royal Society of Chemistry. (b) Mechanism of the photocatalytic oxidation of benzene to phenol using dioxygen and water as oxygen and electron source, respectively, in the presence of a cobalt catalyst. Reproduced with permission.85 Copyright 2017, The Royal Society of Chemistry. | ||
The selectivity of the photochemical oxygenation reaction of cyclohexene was considerably improved upon the introduction of homogeneous RuII porphyrin as a photocatalyst, with water functioning as an oxygen donor.81–83 Takagi et al. recently proposed that the selectivity for epoxidation of alkenes can be further improved using a RuII–porphyrin–clay hybrid photocatalyst.84 The authors prepared two types of +2-charged RuII–carbonyl porphyrin clay-hybrids as sensitizers for the photochemical oxygenation of cyclohexene: (1) RuIIDMPyP2+ (5,10-diphenyl-15,20-di(N-methylpyridinium-4-yl)porphyrinato rutheniumII-carbonyl dichloride) and (2) RuIIDMPyMP2+ (5,10-di[2,6-dimethyl-4-(N-methylpyridinium-3-yl)phenyl]-15,20-di(2,4,6trimethylphenyl)porphyrinato rutheniumII-carbonyl dichloride). Note that the carbonyl group coordinated to ruthenium may function as an axial ligand to enhance triplet excited lifetime, which is conducive to photoinduced electron-transfer reaction. By performing a photochemical reaction using visible light (λirr. = 420 nm, LED light source), the RuIIDMPyMP2+–clay complex was discovered to work as a longer-lived photocatalyst, attributed to the steric and electronic protection of porphyrin chromophore by 2,6-dimethyl substituents of the phenyl group. The photochemical epoxidation of cyclohexene-sensitive by RuII–porphyrin-clay hybrid achieved a high selectivity of 96%.
Aside from photocatalytic alkane hydroxylation and olefin epoxidation, photocatalytic oxygenation of benzene and ring-substituted toluene in the presence of O2 oxidant and water have also shown progress. For example, Fukuzumi et al. reported for the first time the photocatalytic hydroxylation of benzene to phenol in the presence of Sc(NO3)3 under visible light irradiation.85 They used [RuII(Me2phen)3]2+ (Me2phen = 4,7-dimethyl-1,10-phenanthroline) as photocatalyst and [CoIII(Cp*)(bpy)(H2O)]2+ (Cp = η5 – penta methyl cyclo penta dienyl) as catalyst for both water oxidation and benzene hydroxylation. Fig. 6b illustrates the mechanism of photocatalytic hydroxylation of benzene to phenol. Specifically, the visible light irradiation of [RuII(Me2phen)3]2+ resulted in the formation of H2O2 due to photoinduced electron transfer from the excited state of [RuII(Me2phen)3]2+ to O2 oxidant in the presence of Sc3+, as shown in the following equation: 2[RuII(Me2phen)3]2+ + O2 + 2H+ → 2[RuIII(Me2phen)3]3+ + H2O2. PhOH was then produced via the catalytic oxidation of H2O2 catalyzed by [CoIII(Cp*)(bpy)(H2O)]2+, in which p-benzoquinone product was also detected.
Thus, pure Ag metal catalyst can only obtain EO selectivity up to 40–50%.87,88 The addition of alkali metals like Cs and Re or atomic Cl as additives or inhibitors, to Ag catalyst was found to increase the epoxidation selectivity up to 80%.89 The selectivity enhancement is attributed to the partially reversible poisoning of Ag metal surface that hinders the atomic chemisorption of O2 and therefore impedes the oxidation of ethylene to CO2 and H2O.89 Moreover, optimization of size, shape, and composition of heterogenous Ag-based catalysts have shown promising results.90–93 For example, Linic et al. compared the catalytic performance of Ag nanocubes, pentagonal nanowires, and spherical particles of varying size to demonstrate the impact of chemical and physical factors on the reaction selectivity.94–96Fig. 7a–c display high-resolution transmission electron microscopy (HR-TEM) images of an Ag nanowire, nanocube, and the EO selectivity as a function of size and shape of Ag catalysts, respectively. The authors reported that for a given set of external conditions, Ag nanocubes are superior to both nanowires and nanosphere particles and, for a specific particle shape, those with larger sizes have obtained higher EO selectivity. DFT calculations revealed that the difference in the activation barriers is associated with the formation of acetaldehyde and ethylene oxide during epoxidation and is 0.1 eV larger on Ag(100) than on Ag(111). This result suggests that the variation of results among particles of different shapes may be correlated to the exposed crystal facet. Ag(100) facets present in nanocubes intrinsically display higher selectivity towards ethylene oxide compared to Ag(111) facets.
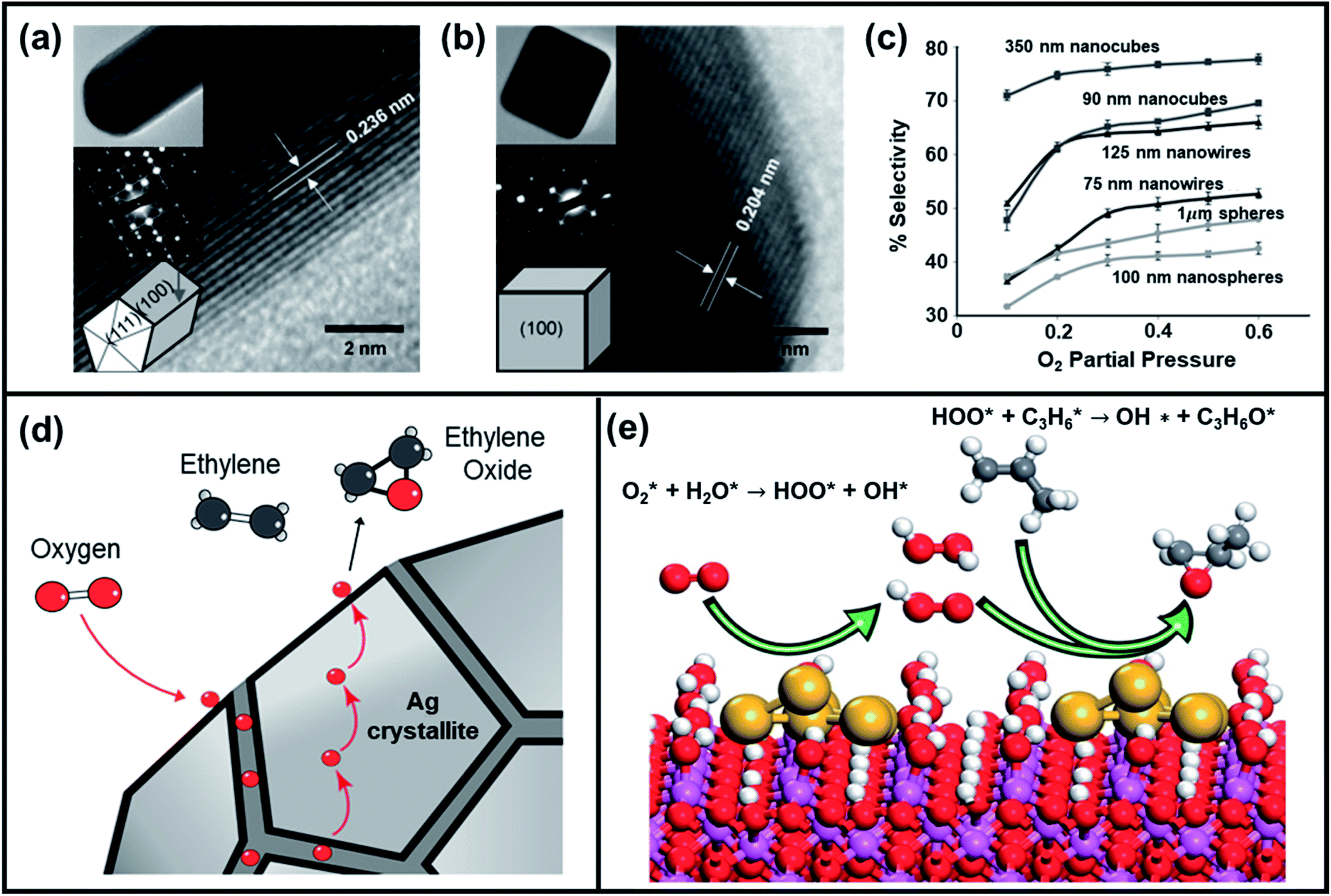 | ||
| Fig. 7 HRTEM images of a (a) Ag nanowire and (b) Ag nanocube (insets show the zoomed-out TEM images of the nanostructures, selected area electron diffraction patterns, and model structures). (c) Ethylene oxide selectivity as a function of the size and shape of Ag catalytic particles and O2 partial pressure at 510 K reaction temperature. Reproduced with permission.95 Copyright 2010, Wiley-VCH. (d) Representative model of Ag catalyst showing the role of grain boundaries for ethylene epoxidation. Reproduced with permission.90 Copyright 2019, American Chemical Society. (e) Illustration of reaction mechanism of Au-based catalyst (Au/α-Al2O3) towards propene epoxidation with O2–H2O mixture. Reproduced with permission.118 Copyright 2016, American Chemical Society. | ||
The size effect of Ag catalysts was further investigated by Hensen et al. They found that particles larger than 50 nm are composed of multiple Ag crystallites with much smaller domain sizes between 25 and 30 nm.90 The epoxidation selectivity increased when larger Ag particles exhibited grain boundaries between smaller Ag crystallites, which played a vital role in supplying oxygen atoms to the external surface. Fig. 7d showed the proposed reaction mechanism of Ag-catalyzed ethylene epoxidation. Hensen et al. suggested that oxygen atoms from grain boundaries of Ag crystallites slowly dissolve in the bulk Ag and diffuse to the external surface of the catalyst, where they react with ethylene to form ethylene oxide. Thus, the presence of oxygen on the catalyst surface may have stabilized the Ag(111), Ag(110), and Ag(100) facets, and may have induced surface reconstruction.88,97,98 It was reasoned that the transformation rate of the ethylene is not exactly structure-sensitive, but dependent on the oxygen content of bulk Ag.
Similar to ethylene oxide, propene epoxide (PO) is an versatile chemical intermediates in various chemical industries, including the production of polyether polyols, polyurethane, and propene glycol. Today, the industrial processes for producing PO are the chlorohydrin and the hydroperoxidation (Halcon) processes.99–103 The major drawback of the former process is the generated chlorinated by-products, which not only impose serious environmental hazards but also destroy equipment due to corrosion.104–106 Likewise, Halcon process is greatly hindered by the high cost associated with the multiple steps required and low efficiency due to the formation of unwanted by-products, like styrene and tert-butyl alcohol. Ideally, propene could be directly oxidized using molecular oxygen assisted by a suitable catalyst. Total and partial oxidation of propene generates propylene oxides with multiple C3 oxygenates, like acrolein, propanal, 1-propanol, isopropanol, and acetone.107 However, to date, no reported catalyst is being used industrially. Haruta et al. were the first to report the potential of nanometer-sized gold clusters dispersed on TiO2 as an active and highly selective catalyst for direct epoxidation of propene with O2.108–110 Since then, the control of Au particle size, support materials, and catalyst preparation methods have been the focus of many studies.111,112 For example, it was observed that propene molecules adsorbed on adjacent Ti sites generate CO2 as by-product, and thus, TiO2 was replaced with titanium silicate (TS-1) as support. Later on, various metal oxides, such as titania, ceria, and iron oxides have also been explored as supports for nanosized Au catalysts.113–116 In view of synthetic methods for Au catalysts, the best activity was observed for catalysts prepared by the deposition–precipitation method, which gave propene epoxidation selectivity of over 90%.108,117 Moreover, mechanistic studies showed that the molecular adsorption of O2 and H2O on Au clusters could lead to O2 activation via a hydroperoxyl (*OOH) intermediate. To further explicate the mechanism of Au-based catalyst (Au/α-Al2O3) towards propene epoxidation with an O2–H2O mixture, Li et al. applied DFT and ab initio molecular dynamics (AIMD) methods.118 The reaction mechanism is displayed in Fig. 7e. The calculation results demonstrated that *OOH species are readily formed from co-adsorbed intermediates (*O2 and *H2O) at the catalyst interface, in which the oxygen molecules prefer the edge of Au clusters while hydrogen actively moves between *OH, *OOH, and *H2O. The resulting *OOH intermediate was determined to be the key oxidative species, which can be dissociated into atomic oxygen species to produce PO or combined with propene to generate *C3H6OOH species that could be easily transformed to epoxypropane.
Silica-supported Au catalysts were then discovered to be capable of catalyzing the selective oxidation of primary and secondary alcohols to aldehydes or ketones using molecular O2 as the oxidant at atmospheric pressure.119 In the case of a primary alcohol, 1% Au on the silica support functioned as a surface modifier, which strongly favored the selectivity of aldehydes. However, for the oxidation of secondary alcohols, the Au catalyst acted as a strong activator of organic molecules since the silica support is almost inactive at low temperatures, resulting in a very low conversion rate.
2.4. Overview of non-electrochemical oxygen functionalization
In section 2, we have categorized the non-electrochemical methods for oxygen functionalization into three synthetic routes, namely, chemical routes, photochemical routes, and heterogeneous catalysis. Notably, these non-electrochemical routes have generally exhibited high selectivity and yield. As we discussed in the previous section, the metal–oxo species generated from transition-metal catalysts are considered key intermediate species for most chemical and photochemical studies. In order to generate oxo-species from the initial (resting) state of metal-complex, chemical oxidants or chemical oxidants with photosensitizer are typically added. The appropriate combination of transition metal-based catalysts and chemical oxidants is a vital requirement not only to achieve high selectivity and yield but also to avoid undesirable oxidation reactions such as over-oxidation or oxidation of other functional groups.In addition, controlling the difference between the redox potential of substrates and metal–oxo species may favor the reaction energetically, hence improve the overall reactivity. Indeed, Nam et al. found that the addition of redox-inactive metals to a transition metal complex could modulate the redox potential of metal–oxo species and affected the reactivity.120 Interestingly, similar phenomena could be found in a biological system, the CaMn4 cluster, an oxygen-evolving complex (OEC) in photosystem II. Agapie et al. found that the redox-inactive calcium in the cluster allows a buildup of localized positive charge, modulating the reduction potentials of the Mn in the OEC.121,122 In this vein, the design of a metal complex with additional hetero atoms or other functional groups could be a prerequisite to expand the scope of oxygen functionalization by a non-electrochemical method.
3. Electrochemical oxygen functionalization
The large energy consumption of modern society produces a significant carbon footprint. This is attributed to the fact that most end products or precursors are formed via thermochemical routes. For example, ammonia, a high-volume commodity chemical, is currently produced using the Haber–Bosch process, which proceeds to the reaction of hydrogen (H2) and nitrogen (N2) gas under high temperature and high pressure (∼500 °C, 150–300 bar). Although it has been regarded as one of the most successful commercialized technologies, it still has major issues, particularly the massive carbon footprint that can reach up to 1.87 tons per ton of ammonia produced.Electrochemical organic synthesis (also denoted as electro-organic synthesis or organic electrosynthesis) has been studied extensively ever since the first experiments were carried out by Faraday in the 18th century.123 It has been considered a straightforward and powerful approach for the transformation of organic molecules. Electrochemical organic synthesis is green and environmentally benign, considering external voltage is used as a substitute for toxic redox reagents under mild conditions.124,125 Unlike the conventional thermochemical route or chemical/photochemical routes, organic electrosynthesis normally does not require any oxidants nor high temperature/pressure. Despite these strong merits, however, only a limited number of commodity chemicals have been produced via electrochemical routes, including anthraquinone, perfluorinated hydrocarbons (PFCs), and adiponitrile.126,127 Herein, the general aspects of electrochemical organic synthesis are discussed and followed by the developed approaches for this field. The progress of electrochemical oxygen functionalization is shown by the critical assessment of various oxidative electrochemical processes, including the simple ethylene epoxidation reaction and some complex organic substrate functionalization.
3.1. Electrochemical system
 | ||
| Scheme 2 Schematic diagram of representative electron transfer mechanisms. Briefly, no chemical bonds are formed between reactant/product and electrode surface in the case of outer-sphere electron transfer, thus, electron transfer event occurs outside of electric double layer. On the other hand, inner-sphere electron transfer is accompanied by direct bond formation between reactant/product and electrode surface. Thus, tuning of reactivity can be attained via changing the binding affinity of electrode surface toward reactant/product. | ||
Inner-sphere electron transfer events are always accompanied by the bond formation between the reactant and the electrode surface (catalytic process) within the electrical double layer. This mechanism utilizes a redox-active catalyst, which changes its oxidation state upon the application of an external potential. The oxidized form of this species reacts with the other reactants or substrates to form the desired products. Therefore, the selectivity is largely affected by the chemical reactivity between the reactant and catalyst. Modification of the catalyst surface and properties has been attempted to improve electrochemical performance via this mechanism. Conversely, the electron transfer from the electrode to the reactant species is often accompanied by an outer-sphere mechanism, which is simply driven by the applied potential, and no bond formation occurs between the two species (non-catalytic process). The selectivity can only be determined by the potential difference between the electrode and the substrate, in which a redox mediator may help reduce the required overpotential for the process to proceed. It is important to clarify that this type of mechanism may or may not require a redox mediator to facilitate the electron transfer and that the demand for such components solely depends on the electrochemical reaction pathways. The details of this matter are discussed in section 3.2.
Electrochemical oxygen functionalization reactions require the following components: organic substrates, oxygen atom sources, redox mediators (or electrocatalysts), and reaction media (solvents and supporting electrolytes). Redox mediators (also referred to as redox catalysts, electron transfer mediators, or just mediators) are species soluble in electrolytes that have redox activity in a typical working potential window. Redox mediators act as charge carriers, they are reduced/oxidized at the electrode surface and are then transferred from the reaction site and chemically react with O2.133 From a mechanistic point of view, redox mediators play three key roles in electrochemical reaction systems: (1) facilitating the transfer of electrons between the anode and organic substrate to improve electro-kinetics, (2) lowering the overpotential needed for radical generation, and (3) controlling the selectivity of the chemical steps that occur after electron transfer.134 An activated form of the mediator is generated (Medox) when the applied potential reaches the redox potential of the mediator (Emed). A charge transfer process occurs between Medox and the substrate to generate highly reactive radicals or cations as intermediate species, followed by the formation of desired products. Based on this, several criteria of the appropriate mediator were recently suggested.124,135 For example, the redox potential of the mediator must be lower than that of the substrate so as to not allow the substrate to be oxidized or reduced. However, the potential difference between the two components should not be large lest the reaction rates become impractically slow. More importantly, both the reduced and oxidized form of the mediator should be inert to all processes other than electron transfer since irreversible side reactions dramatically reduce the catalytic activity. Lastly, the solubility of these transformed species must be high enough to ensure homogeneity throughout the electrolysis period.
Reaction media such as solvents and supporting electrolytes play significant roles in the control of electrochemical reactions. These components serve as an environment for molecular events, such as electron transfer and subsequent chemical processes, including the separation and extraction of products from reaction mixtures. Acetonitrile (ACN), dimethylformamide (DMF), and tetrahydrofuran (THF) are generally used with a supporting electrolyte.136–139 However, these compounds are destructive to both human health and the environment. In this vein, ionic liquids and supercritical fluids are being regarded as alternative reaction media for organic electrochemical reactions.140–146 Their primary advantages include low vapor pressure, nonflammability, high polarity, and relative inertness, whereas these reaction media are costly although they can be recovered and reused after reactions.
3.2. Proposed electrochemical pathways
The progress of electrochemical oxygen-atom functionalization has led to the development of three routes, as follows: (1) direct electro-oxidation via outer-sphere electron transfer, in which reagent-electron transfer is conducted on the electrode surface; (2) indirect electro-oxidation in presence of redox mediator to facilitate the activation of the substrate; and (3) direct electro-oxidation via inner-sphere electron transfer, which employs an electrocatalyst to mediate the transformation of the organic substrate. Scheme 3 shows schematic diagrams and simplified energy profiles of the three electro-organic synthesis pathways to demonstrate their differences.125,147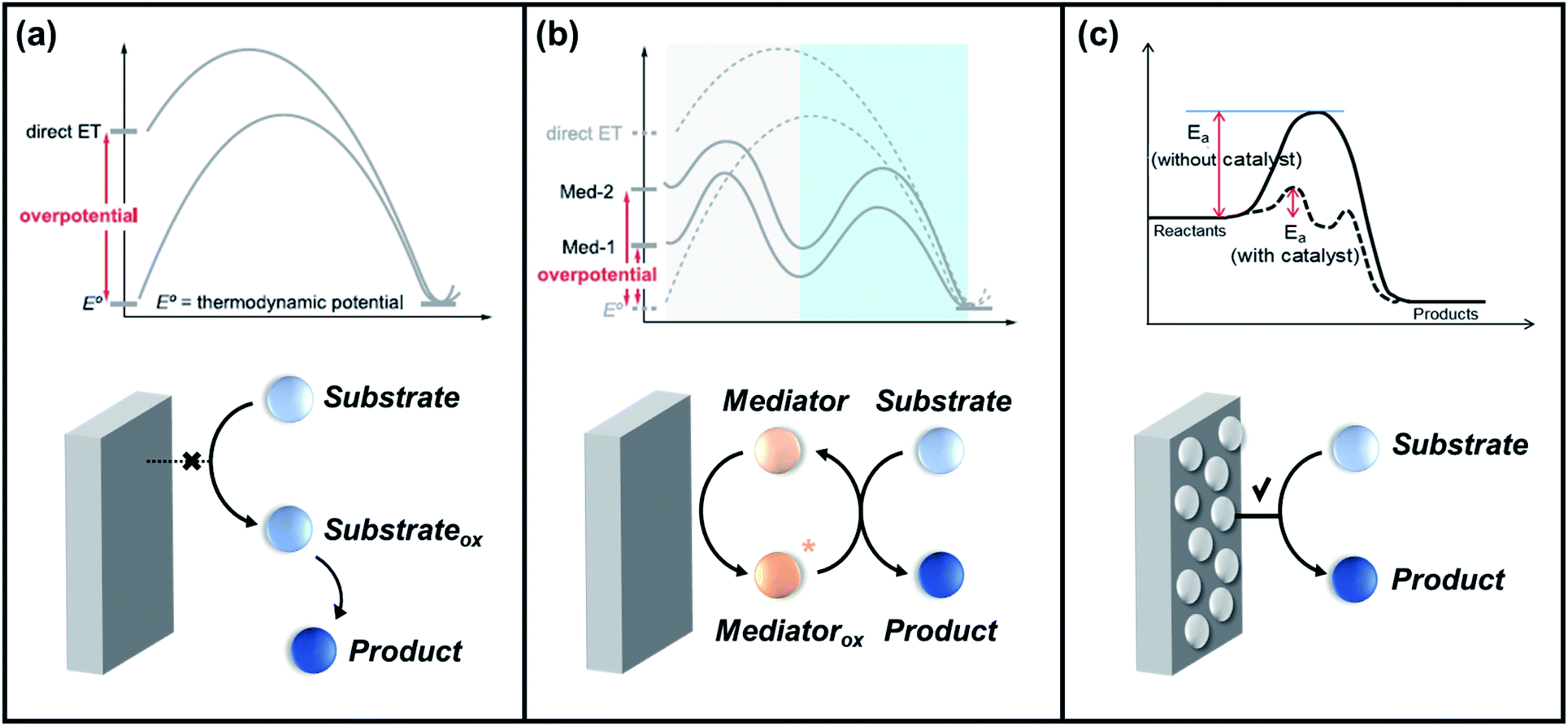 | ||
| Scheme 3 Simplified energy profiles and schematic diagrams for (a) direct electro-oxidation via the outer sphere mechanism, (b) indirect electro-oxidation with mediators, and (c) direct electro-oxidation via the inner sphere mechanism. Reproduced with permission.125,147 Copyright 2020, American Chemical society and Copyright, 2020 MDPI. | ||
In search for efficient electrochemical approaches where the substrate and electrode could interact directly to generate reactive and valuable species, Patureau et al. designed a metal- and chemical oxidants-free electro-oxidation process to produce aromatic esters.150 Initially, the group investigated 4-methylanisole as the model substrate reacting with methanol in an undivided cell, composed of carbon as anode and nickel as cathode. Upon the application of potential, three compounds were formed including the target oxidative coupling product ester, aldehyde, and acetal. Fig. 8a shows the proposed mechanism of the production of ester products. By passing an electric current to the substrate (a), an aryl radical cation (a′) is formed at the anode followed by deprotonation and oxidation, which eventually forms a quinone methide derivative (a′′). Further anodic oxidation events lead to the formation of acetal intermediate (e). The direct electro-oxidation of this intermediate species at the anode side would then generate the ester product (c). Importantly, the proposed electro-oxidative method to construct esters and acetals from methylarenes and benzaldehydes offers a new approach in synthetic chemistry. Likewise, Chiba et al. reported a selective functionalization of styrenes with O2 into carbonyls products.151 The group suggested the plausible mechanisms of olefin cleavage via direct anodic oxidation of styrene, as shown in Fig. 8b. The production of carbonyl products via the olefin cleavage of styrene substrate (2a) may follow two pathways. Initially, a styrene radical cation intermediate (2a˙+) with trapped O2˙− species would be formed upon the application of potential. Subsequently, dioxetane intermediate (major pathway) and epoxide intermediate (minor pathway) are formed, whereas the former species is immediately decomposed into the target carbonyl compounds.
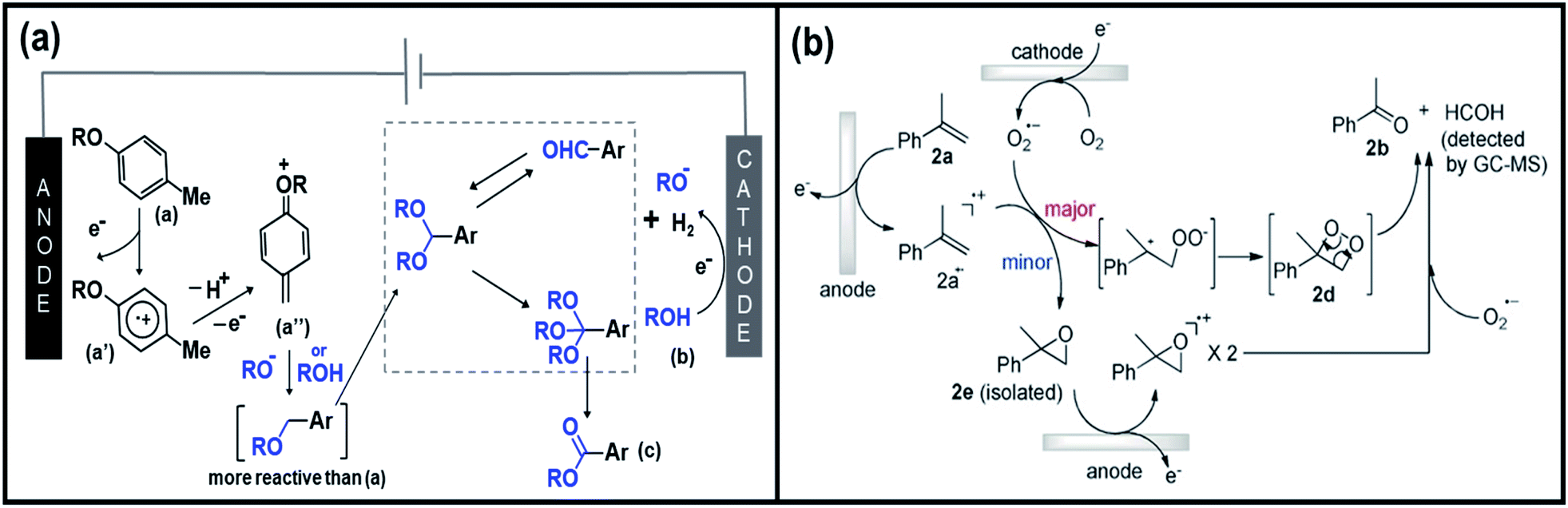 | ||
| Fig. 8 (a) Illustration of the proposed mechanism of direct electro-oxidation of methylarenes to ester products. Reproduced with permission.150 Copyright 2020, Wiley-VCH. (b) Plausible mechanisms of olefin cleavage via direct anodic oxidation of styrene. Reproduced with permission.151 Copyright 2019, Wiley-VCH. | ||
O complex, as shown in Fig. 9a. The overall electrochemical process has achieved yields and % ee of epoxides that were almost comparable to those obtained from chemical epoxidation, as shown in Table 3. Subsequently, the production of epoxides has followed the halogen-mediated route for a long time due to high efficiency. However, the overall process is not economically viable because of the formation of by-products.
 | ||
| Fig. 9 (a) Possible reaction mechanism of Mn–salen complex mediated electro-epoxidation of olefins water/CH2Cl2 two-phase system. Reproduced with permission.157 Copyright 2001, Elsevier Science B.V. (b) Coupling of the electrogeneration of H2O2 in [bdmim][BF4] ionic liquid with the manganese/bicarbonate-activated epoxidation system. Reproduced with permission.160 Copyright 2006, Elsevier Ltd. | ||
| Entry | Catalyst | Oxidant/electrolyte | Substrate | Product | % yield | % conversionb |
|---|---|---|---|---|---|---|
| a (S,S)-N,N-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediaminomanganeseIII chloride.157 b % enantioselective epoxide. | ||||||
| 1 | Mn-salen3a | CH2Cl2/NaCl |
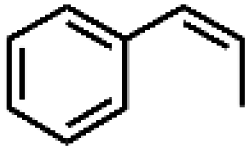
|
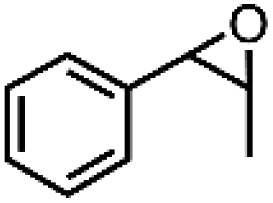
|
93 | 87b |
| 2 | Mn-salen3a | CH2Cl2/NaCl |
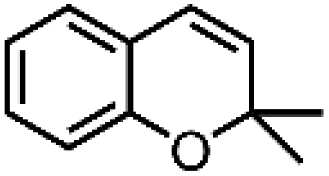
|
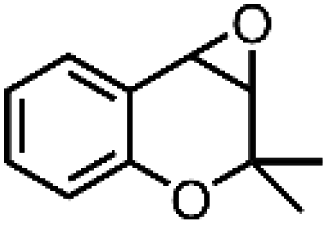
|
80 | 81b |
| 3 | Mn-salen3a | CH2Cl2/NaCl |
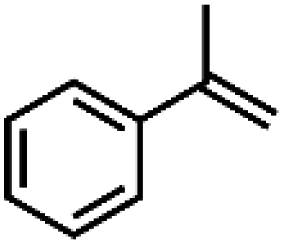
|
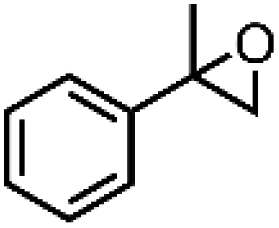
|
69 | 43b |
| 4 | Mn-salen3a | CH2Cl2/NaCl |
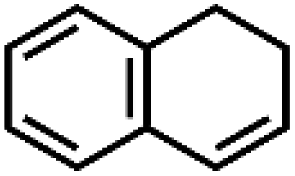
|
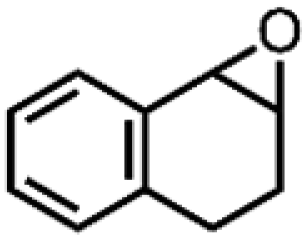
|
47 | 40b |
| 5 | Mn-salen3a | CH2Cl2/NaCl |
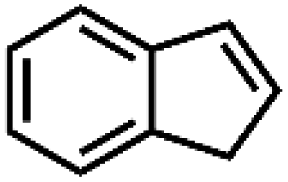
|
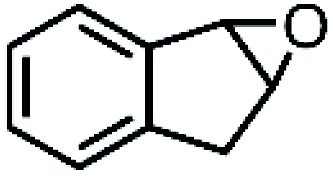
|
30 | 26b |
Alternatively, hydrogen peroxide (H2O2) has been considered one of the environmentally benign oxidants for electro-organic synthesis since the by-product being produced is only water.158,159 However, H2O2 is usually produced through the anthraquinone process, which requires toxic solvents to regenerate the employed catalysts, contradictory to the originally intended function of H2O2. Also, the hazards associated with the storage and transportation of concentrated H2O2 have been a hindrance to the progress of this approach. One solution proposed is to produce low strength H2O2 by electrochemical reduction of O2, which offers an alternative way to utilize this oxidant. A particular example is the investigation of Ho et al. regarding the electro generation of H2O2 in [bdmim][BF4] IL (ionic liquid), followed by the in situ epoxidation of electrophilic alkenes in the H2O2/HCO3−/MnII system, represented in Fig. 9b.160 Notably, the addition of water in the [bdmim][BF4]/NaOH mixture resulted in the efficient production of H2O2, while the introduction of CO2 in the H2O2/[bdmim][BF4]/NaOH(aq) with MnII catalyst generated the peroxymonocarbonate anion, HCO4−, which was suggested to be the active species corresponding to the epoxidation. The ionic liquid was recovered and reused multiple times without showing significant changes in electrochemical performance, which is significant since the IL is the most expensive component in the proposed system. Table 4 summarizes the epoxidation of various lipophilic alkenes in H2O2/[bdmim][BF4]/NaOH(aq) reaction media and MnII catalyst.
| Entry | Catalyst | Oxidant/electrolyte | Substrate | Product | % yield | % conversion |
|---|---|---|---|---|---|---|
| a Ionic liquid, [bdmim][BF4].160 | ||||||
| 1 | MnsO4/HCO3 | H2O2/IL/NaOHa |
|

|
83 | >99 |
| 2 | MnsO4/HCO3 | H2O2/IL/NaOHa |
|
|
84 | >99 |
| 3 | MnsO4/HCO3 | H2O2/IL/NaOHa |
|
|
80 | >99 |
| 4 | MnsO4/HCO3 | H2O2/IL/NaOHa |
|
|
90 | >99 |
| 5 | MnsO4/HCO3 | H2O2/IL/NaOHa |
|
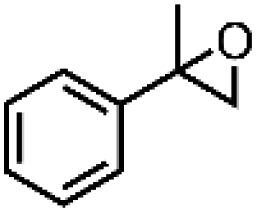
|
83 | 95 |
| 6 | MnsO4/HCO3 | H2O2/IL/NaOHa |
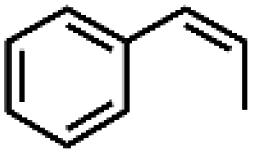
|
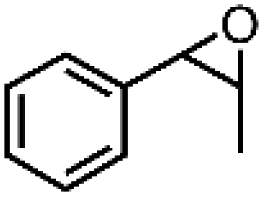
|
76 | 87 |
| 7 | MnsO4/HCO3 | H2O2/IL/NaOHa |
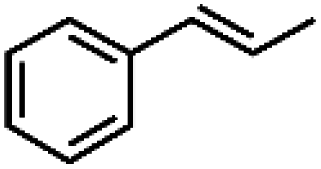
|
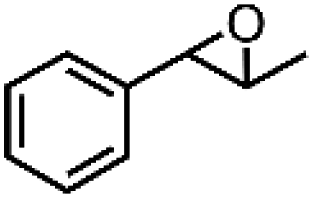
|
71 | 81 |
| 8 | MnsO4/HCO3 | H2O2/IL/NaOHa |
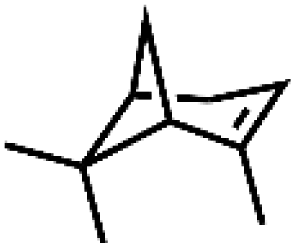
|
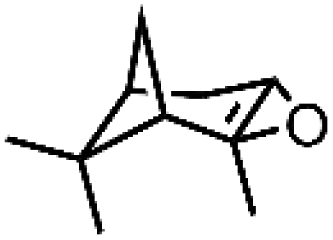
|
62 | 75 |
| 9 | MnsO4/HCO3 | H2O2/IL/NaOHa |
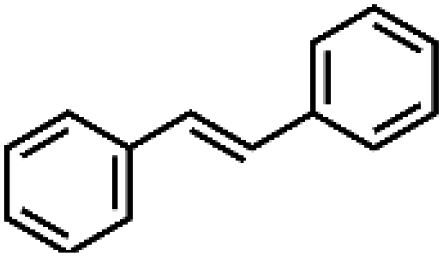
|
|
42 | 44 |
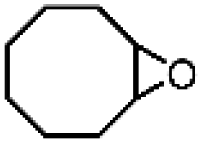
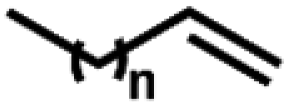
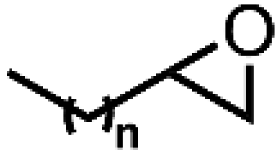
O species (resting state of Mn3O4 NPs), cyclooctene oxides are formed together with MnII-vacant sites. Accordingly, the nucleophilic attack of water to the available MnII sites is suggested as the rate-determining step of the reaction, which induced the formation of MnIII–OH2 accompanied by a one-electron transfer. The suggested alternative electro-oxidation route not only anticipated the utilization of water for the electrochemical epoxidation reaction at ambient conditions, but also projected a lower kinetic barrier associated with the use of suitable electrocatalysts. However, low conversion efficiencies (30% of the maximum theoretical efficiency), shown in Table 5 (entries 1 and 2), were initially achieved and more efforts are still necessary to fully unfold the potential of this innovative direct electro-oxidation.
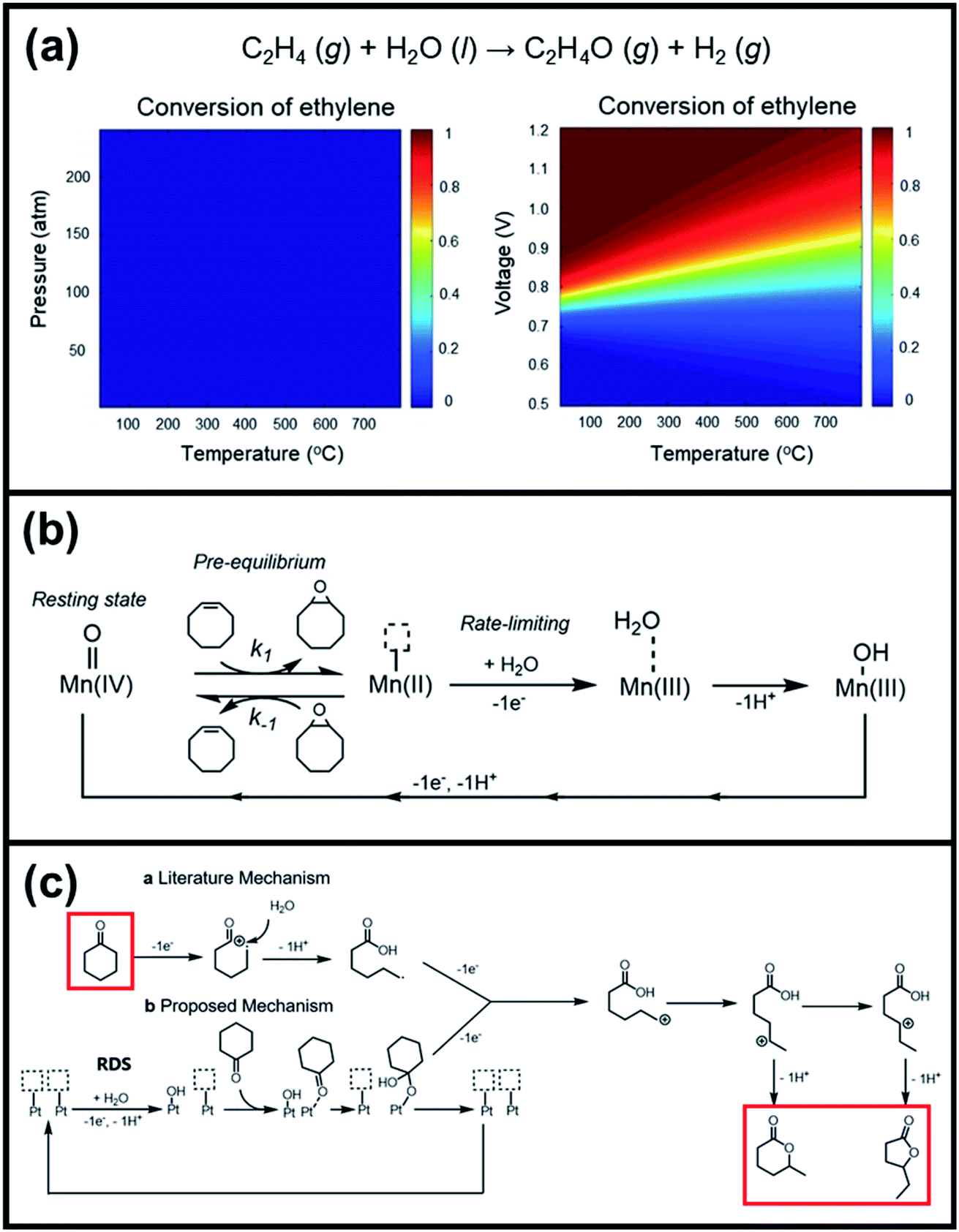 | ||
| Fig. 10 (a) Analyses of the conversion of ethylene to ethylene-oxides through the thermochemical route and electrochemical route. (b) Suggested mechanism for electrochemical epoxidation of cyclooctene using Mn3O4 nanoparticles as oxidation catalysts and water as an oxygen atom source. Reproduced with permission.162 Copyright 2019, American Chemical Society. (c) Proposed mechanism for electrochemical lactonization of cyclic ketones using Pt foil electrodes and water as an oxygen atom source. The rate-determining step involves the activation of water molecules by Pt in an electron transfer to form Pt–OH. Reproduced with permission.163 Copyright 2020, American Chemical Society. | ||
| Entry | Catalyst | Oxidant/electrolyte | Substrate | Product | % yield | % conversion |
|---|---|---|---|---|---|---|
| a n = 8. b n = 7.162 c Rh2(OAC)4 with anhydrous TBHP (tert-butyl hydroperoxide).174 | ||||||
| 1 | Mn3O4 NPs | Water |
|
|
28.3a | 55.1a |
| 2 | Mn3O4 NPs | Water |
|
|
18.9b | 30.2b |
| 3 | Rh2(cap)4c | CH2Cl2/tBuOOH |
|
|
94 | N.D. |
| 4 | Rh2(cap)4c | CH2Cl2/tBuOOH |
|
|
77 | N.D. |
| 5 | Rh2(cap)4c | CH2Cl2/tBuOOH |
|

|
92 | N.D. |
| 6 | Rh2(cap)4c | CH2Cl2/tBuOOH |
|
|
75 | N.D. |
| 7 | Rh2(cap)4c | CH2Cl2/tBuOOH |
|
|
86 | N.D. |
| 8 | Rh2(cap)4c | CH2Cl2/tBuOOH |
|
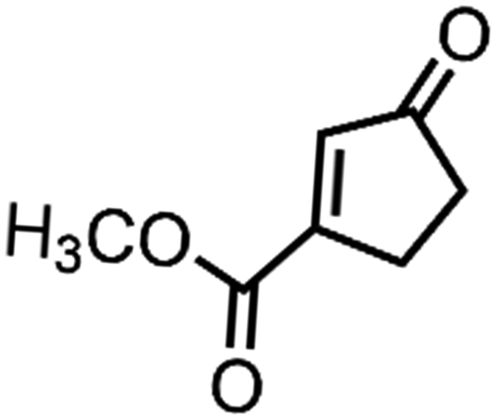
|
79 | N.D. |
Manthiram et al. then investigated the possibility of electrochemical lactone formation by following the same concept of their previous study.163 In their proposed reaction mechanism (Fig. 10c), the water molecule is initially activated on a Pt electrode surface following an inner-sphere electron transfer to form Pt–OH. The adsorbed OH species then reacts with the adsorbed cyclohexanone to form an active intermediate, which can proceed to the formation of carboxylic acid cation via ring-opening electron transfer. By extending the electrochemical oxidation, the formed cation can carry on to ring-closure, thus forming lactone products. Isotope labeling experiments verified that the oxygen in the lactone products is inserted into cyclohexanone from the water in the electrolyte, with hydrogen gas co-produced at the cathode. With the use of water as the sole oxygen atom source, the authors were able to produce lactones from cyclohexanone with faradaic efficiencies of approximately 20%.
However, aside from the low conversion efficiencies obtained from initial experiments, the identification of conditions under which the desired reaction proceeds selectively imposes a major technical challenge for this electrochemical approach. Specifically, the lack of profound comprehension regarding the involved reaction, including the electron and proton movement, rate of oxygen functionalization from the reactant to the surface of the catalyst, and the role of the catalysts, is a major concern of direct electrooxidation following an inner-sphere mechanism. It is then imperative to address these issues to optimize the overall performance of direct electrochemical oxidation in terms of conversion, faradaic efficiency, yield, and selectivity.
3.3. Oxygen functionalization by electrochemical methods
C bond cleavage and addition of oxygen atoms can give rise to valuable products, including epoxides, ketones, 1,2-diols, aldehydes, and carboxylic acids. Notably, the anodic oxidation of these low-cost unsaturated hydrocarbons can proceed with or without a redox mediator. Metal–oxo catalyst systems employing iron, ruthenium, manganese, chromium, and osmium metals are commonly used mediators in the electrochemical oxygenation of olefins. The chemistry of metal–oxo complexes has been extensively studied for non-electrochemical methods, as these species are capable of initializing several types of transformations, such as oxygen functionalization and hydrogen abstraction of strong C–H, N–H, and O–H bonds. This section of the review presents the promising transformation of olefin substrates via electrochemical oxygen functionalization.
The direct chemical reaction between oxygen and olefins is the conventional method to synthesize various epoxides, however, this process is economically challenged by the harsh reaction conditions and formation of unwanted by-products. Driven by these limitations, Van der Eijk et al. established the first non-conventional route involving the electrochemical oxidation of higher olefins into their respective epoxides.164 The challenge of this approach is the identification of conditions under which the desired reaction proceeds selectively. As a response, an electrochemically generated AgII–pyridine (Ag(Py)2+) was employed as a mediator to selectively oxidized propene and four isomeric butenes. Taking propene saturated in a silver acetate solution as an example, they demonstrated that upon electrolysis, AgI was oxidized to a AgII complex, then this complex was readily reacted with propene to give AgI and propene oxide. Eqn (1)–(3) summarize the involved reaction mechanism.
| Ag(Py)2+ + 2Py → Ag(Py)42+ + e− | (1) |
| 2Ag(Py)42+ + H2O ↔ Ag(Py)2+ + Ag(Py)2O+ + 4Py + 2H+ | (2) |
| Ag(Py)2O+ + CnH2n → Ag (Py)2+ + CnH2nO | (3) |
Van der Eijk et al. confirmed that the rate-determining step in the electrochemical epoxidation of higher olefins is the transfer of an electrophilic oxygen atom from the mediator to the homogeneously dissolved substrates. Importantly, their results brought out the potential of AgIII oxides as a competent selective epoxidation agent for higher olefin substrates.165,166
Moreover, ruthenium complexes, such as ruthenium porphyrin complexes, have been regarded as efficient electrocatalysts for the epoxidation of olefins due to the accessibility of high-valent ruthenium–oxo species (RuIVO to RuVIIO). These species were presumed to be the most likely active intermediates when olefins are selectively oxidized to the corresponding epoxides via oxygen functionalization.77,78 Krtil et al. recently reported regarding the electrocatalytic ethene epoxidation on nanocrystalline RuO2 interesting to discuss in detail.167 The electrocatalytic properties of the Ru-based oxides were tuned by controlling the particle size and by doping with Co. However, they realized that CO2 is the main ethene oxidation reaction product on nanocrystalline RuO2 and Ru0.8Co0.2O2 electrodes in acid media. To shift the reaction pathway, they employed the Cl− ion as a surface blocking agent, which effectively cut-off the CO2-reaction channel due to the formation of Ru–O–Cl surface species. Such hindrance to the formation of CO2 allowed the production of ethylene oxide in acid media. The sudden appearance of this product above the potential of approximately 1.2 V upon the addition of 0.3 M Cl, was explained through DFT calculations, which showed that the lone surface oxygen sites (Ru–O) surrounded by Cl− ions are the active species responsible for the epoxidation process. Accordingly, Cl was confirmed as an efficient epoxidation promoter that switches off the combustion pathway toward CO2 and enables the epoxidation reaction channel by surface reactive site blocking.
Sargent et al. recently reported chloride-mediated selective electrosynthesis of ethylene and propylene oxides at high current density under ambient conditions.168 They postulated that chloride is likely an efficient redox mediator at the anode that facilitates the selective partial oxidation of ethylene to ethylene oxide. As shown in Fig. 11a, the extended heterogeneous/homogeneous interface induced by the Cl− ions can buffer ethylene from uncontrolled oxidation, leading to the production of ethylene oxide. Using platinum (Pt) foil as the working electrode (anode), nickel (Ni) foam, and Ag/AgCl (3.0 M KCl) as the counter electrode and reference electrode, respectively, the authors tested their idea in a flow-cell setup with 1.0 M potassium chloride (KCl) electrolyte, in which they mixed the catholyte and anolyte output streams as displayed in Fig. 11b. Briefly, Cl− is oxidized to Cl2 at the Pt anode, which bonds with H2O to form hypochlorous and hydrochloric acid (HOCl and HCl, respectively). HOCl then reacted with ethylene dissolved in the electrolyte to form ethylene chlorohydrin (HOCH2CH2Cl). Upon mixture of hydroxide (OH−, from the catholyte stream) and HOCH2CH2Cl (anolyte stream), ethylene oxide was generated, and Cl− were regenerated. Through this modified system, they achieved a faradaic efficiency of ∼70% and product specificities of ∼97%.
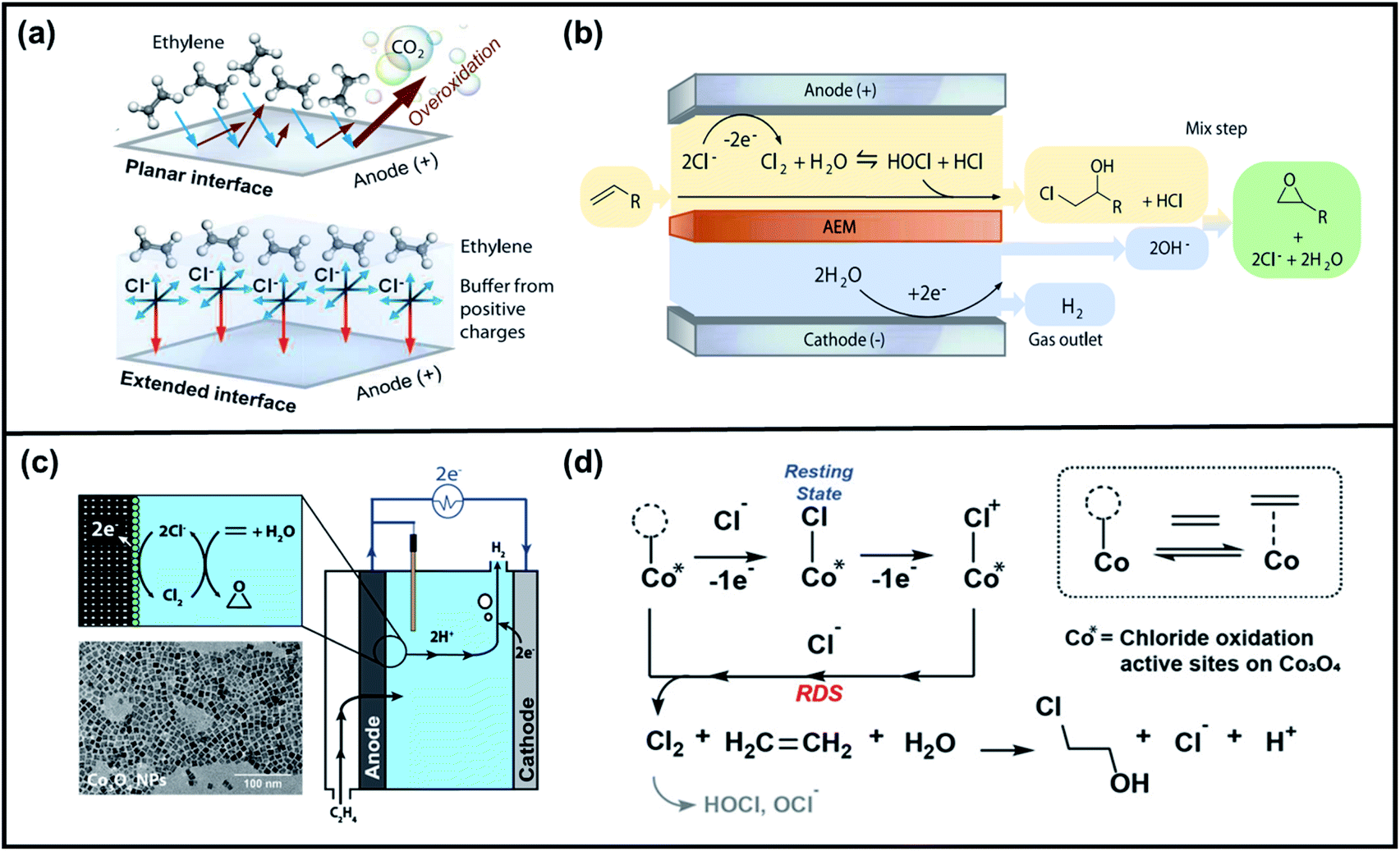 | ||
| Fig. 11 (a) Schematic diagram illustrating ethylene oxidation at a planar interface (leading to overoxidation) and the proposed chloride-mediated selective electrosynthesis that presents an extended interface and (b) the chloride-mediated electrochemical system of ethylene to ethylene-oxide transformation, in which the formation of ethylene chlorohydrin in the anolyte and the generation of ethylene oxide in the mixing step are proposed. Reproduced with permission.168 Copyright 2020, American Association for the Advancement of Science. (c) Schematic of an electrochemical ethylene oxidation cell (inset shows the mechanism of electrochemical epoxidation and TEM image of Co3O4 nanoparticles) and (d) the proposed mechanism for chlorine-mediated electrochemical ethylene oxidation in saline water. Reproduced with permission.169 Copyright 2020, American Chemical Society. | ||
Manthiram et al. conducted a series of electrochemical kinetic studies to understand the mechanism of ethylene oxidation in saline water using cobalt oxide nanoparticle catalysts and chlorine as a redox mediator.169Fig. 11c illustrates the electrochemical cell set-up used under simulated seawater electrolyte. Ethylene gas was introduced through the porous carbon paper anode. Given that 2-choloroethanol is expected to be formed as a product in the chlorohydrin process, it was hypothesized that the chloride ions in the electrolyte are likely to be oxidized to active chlorine species (i.e., Cl2, HOCl, and OCl−) at the anode surface, which act as a redox mediator to oxidize ethylene. Among the suspected chlorine species, molecular chlorine was determined as most active for oxidizing ethylene to 2-choloroethanol and further aging of the as-electrolyzed solution converted 2-choloroethanol into ethylene oxide. Generally, chlorine evolution reaction (CER) under neutral pH follows the Krishtalik mechanism where the second electron-transfer step is the rate-determining step, that forms the adsorbed Cl+ species. The authors found that in the presence of ethylene, the resting state and rate-determining step (RDS) of the chlorine-mediated ethylene oxidation reaction are different from that of CER.170,171 The group suggested a new pathway where adsorbed chlorine exist as a resting state and one electron oxidation occurs to generate Cl+ species, followed by the chemical rate-determining step to produce Cl2 (Fig. 11d).
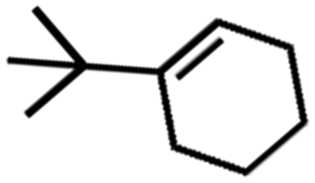
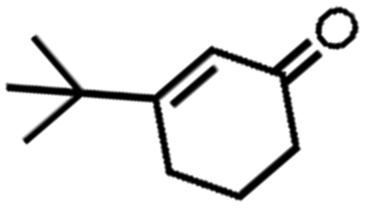
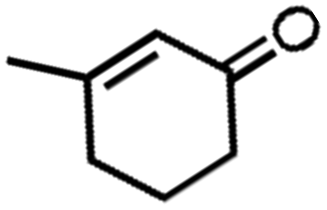
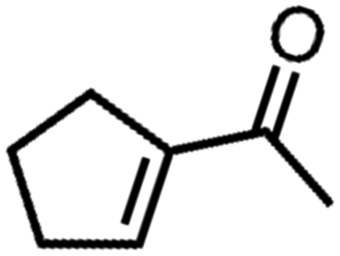
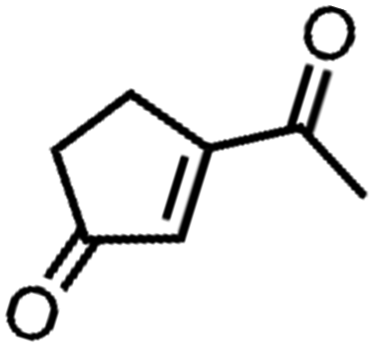
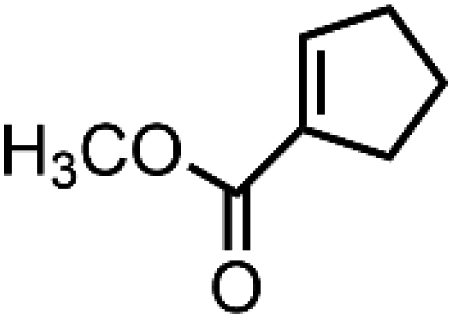
![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) Fe3+, Cu1+ Cu2+, and Co2+ Co3+. Bearing this in mind, Doyle et al. developed a novel catalytic allylic oxidation protocol based on dirhodium(II) caprolactamate (denoted as Rh2(cap)4).174 The unique reactivity of Rh2(cap)4 emanates from its ability to undergo facile redox chemistry, namely Rh24+/Rh25+. A minimum catalyst loading of 0.1 mol% Rh2(cap)4 was able to produce enedieones with impressive yield in just 1 h (Table 5, entries 3–8). However, toxic oxidizers and large solvent quantities are required for allylic oxidation via organic synthesis, which limits their application.
Fe3+, Cu1+ Cu2+, and Co2+ Co3+. Bearing this in mind, Doyle et al. developed a novel catalytic allylic oxidation protocol based on dirhodium(II) caprolactamate (denoted as Rh2(cap)4).174 The unique reactivity of Rh2(cap)4 emanates from its ability to undergo facile redox chemistry, namely Rh24+/Rh25+. A minimum catalyst loading of 0.1 mol% Rh2(cap)4 was able to produce enedieones with impressive yield in just 1 h (Table 5, entries 3–8). However, toxic oxidizers and large solvent quantities are required for allylic oxidation via organic synthesis, which limits their application.
Consequently, electrochemistry was introduced to allylic oxidation to realize a robust, easy to use, and scalable electro-organic process. Masui et al. were the first to conduct anodic oxidation of olefins to α, β unsaturated ketones using N-hydroxyphthalimide (NHPI) as a redox mediator and atmospheric oxygen as an oxygen source.175 However, unsatisfactory yield and selectivity were obtained and there was a limited scope in terms of accessible substrates. In response, Baran et al. improved the original scheme by employing a tetrachloro-derivative to enhance the reactivity of NHPI and tert-butylhydroperoxide (tBuOOH) as co-oxidant (Fig. 12a).176 They then proposed an optimized mechanism for electrochemical allylic oxidation using the modified mediator (Cl4NHPI), as displayed in Fig. 12b. Briefly, the transformation of olefinic substrate 1 initially involves the deprotonation of Cl4NHPI in the presence of excess pyridine, followed by the anodic oxidation, which generated tetra-chlorophthalimido N-oxyl radical species. Subsequently, olefin 1 proceeded to hydrogen atom abstraction, which induced the regeneration of the redox mediator and formation of allylic radical species 2. Finally, the reaction between the radical species and electrochemically generated tBuOO˙ produced allylic peroxide 3, which, upon the removal of tBuOH, yielded enone 4. Notably, the optimized method was successfully applied to more than 30 substrates with good yield and selectivity, thus demonstrating its practical utility.
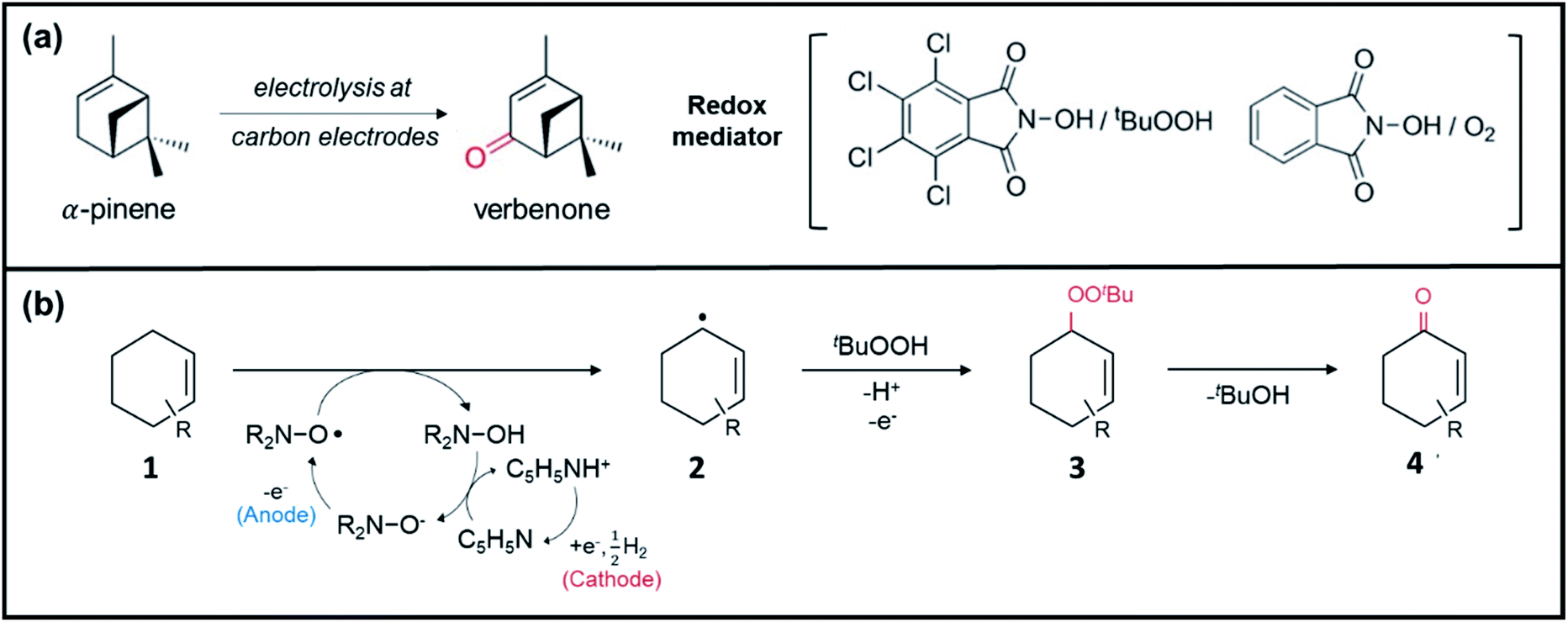 | ||
| Fig. 12 (a) Anodic oxidation of α-pinene using the original and modified NHPI redox mediator. Reproduced with permission.173 Copyright 2020, American Chemical Society. (b) Proposed mechanism for electrochemical allylic oxidation of olefins using the modified NHPI redox mediator. Reproduced with permission.176 Copyright 2016, Macmillan Publishers Limited. | ||
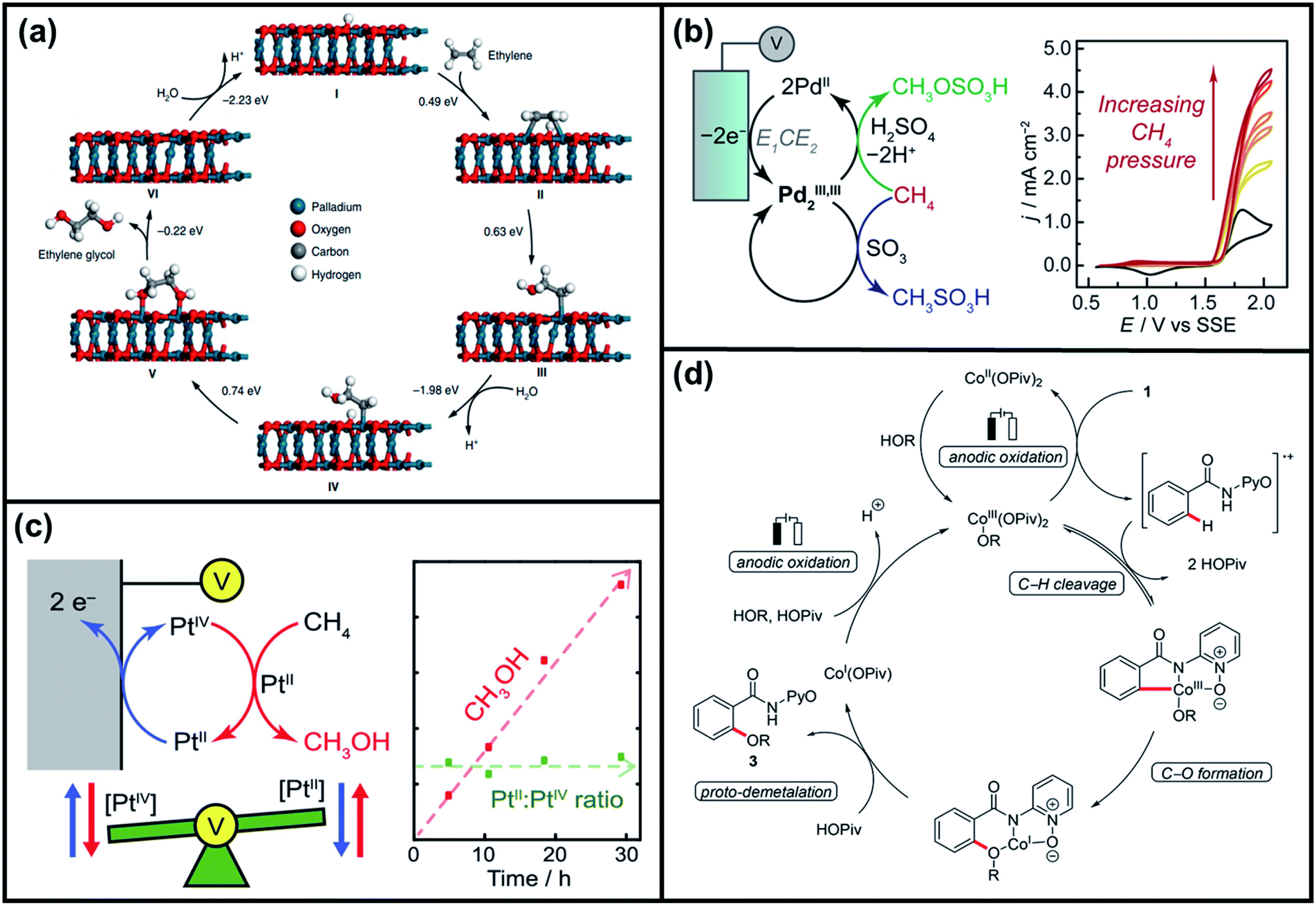 | ||
| Fig. 13 (a) Proposed reaction mechanism of electrochemical oxidation of ethylene to ethylene glycols, in which DFT calculations implied the involvement of the lattice OH in PdO as a direct reaction intermediate. Reproduced with permission.177 Copyright 2020, Springer Nature Limited. (b) Proposed mechanism for electrochemical methane functionalization by a PdIII,III2 intermediate. Green and blue arrows indicate faradaic and non-faradaic reaction pathways, respectively. Inset displays the methane functionalization by an electrogenerated PdIII,III2 species with varying CH4 species. Reproduced with permission.187 Copyright 2017, American Chemical Society. (c) Schematic of methane to methanol catalysis via the electrochemical regeneration of PtIV. Inset shows the effect of a constant PtII:PtIV ratio to methanol production with respect to reaction time. Reproduced with permission.188 Copyright 2019, American Chemical Society. (d) Proposed catalytic cycle for electrochemical cobalt-catalyzed C–H oxygenation at room temperature. Reproduced with permission.189 Copyright 2017, American Chemical Society. | ||
Particular examples to discuss are the recent works of Surendranath et al., which tackled new electrochemical approaches for promoting rapid methane monofunctionalization.187Fig. 13b displays their proposed mechanism for electrochemical methane functionalization driven by PdIII,III2 intermediate species. Specifically, PdIISO4 was electrooxidized in concentrated sulfuric acid to form the PdIII,III2 species, which then reacts with methane. The activation of methane by the electrogenerated Pd species proceeds with low activation barrier energy (25.9 ± 2.6 kcal mol−1), generating methyl bisulfate (CH3OSO3H) and methanesulfonic acid (CH3SO3H) as methanol precursors. Both of these methanol precursors can be thermally and hydrolytically converted to methanol via faradaic and non-faradaic pathways (green and blue arrows in Fig. 13b), respectively, which significantly increases the electrical energy efficiency of the reaction while maintaining high rates of catalysis. Afterward, an electrochemical approach for continuous, steady-state methane to methanol conversion was established in the presence of stoichiometric PtIV oxidants in PtII chloride salt aqueous solution (PtIICl42−) at a low reaction temperature (Fig. 13c).188 They emphasized that, while the Cl-adsorbed Pt surfaces were active for the inner-sphere two-electron transfer of PtII to PtIV, this electrode is inert toward the simultaneous oxidation of methanol products. By careful optimization and maintenance of the PtIV:PtII ratio (4.3:1.4), it was possible to carry out steady-state and continuous methane to methane catalysis up to 30 h with 70% methanol selectivity.
Meanwhile, cobalt complexes have been recently identified as cost-effective electrochemical catalysts. Ackermann et al. reported for the first time the successful silver-free electrochemical cobalt-catalyzed C–H activation of arenes and alkenes via C–H oxygenation under mild conditions at 23 °C.189 They identified Co(OAc)2 as an optimal catalyst among a representative set of cobalt precursors, while NaOPiv showed better performance as a base than NaOAc. Also, control experiments showed that electrochemical C–H functionalization was not feasible without the presence of a base, electric current, and/or cobalt catalyst. On the basis of their mechanistic studies, they proposed a catalytic cycle of the cobalt-catalyzed C–H activation. Fig. 13d shows that the reaction mechanism is possibly initiated by a facile electrochemical catalyst generation, followed by electrochemical single electron transfer of the aromatic benzamide via anodic oxidation. Subsequently, C–O bond formation occurs by chelation assistance, securing excellent positional selectivity. Then, the desired product is released while the catalytically active CoIII species is regenerated by means of anodic oxidation.
3.4. Overview of electrochemical oxygen functionalization
In section 3, we have discussed the great potential of electrochemical methods towards oxygen functionalization reactions in terms of (1) electro-oxidation pathways and (2) synthetic strategies. The first part of this section presented the growth of electrochemical method based on the electron transfer mechanisms, namely, direct electro-oxidation via outer-sphere electron transfer, indirect electro-oxidation with mediators, and direct electro-oxidation via inner-sphere electron transfer. Subsequently, we assessed the progress of this advanced method by presenting various electrically driven chemical-organic syntheses, in which we discussed the advancement of well-known electrochemical systems and catalyst designs. Fig. 14 represents the different types of electrochemical oxygen functionalization reactions discussed in this review.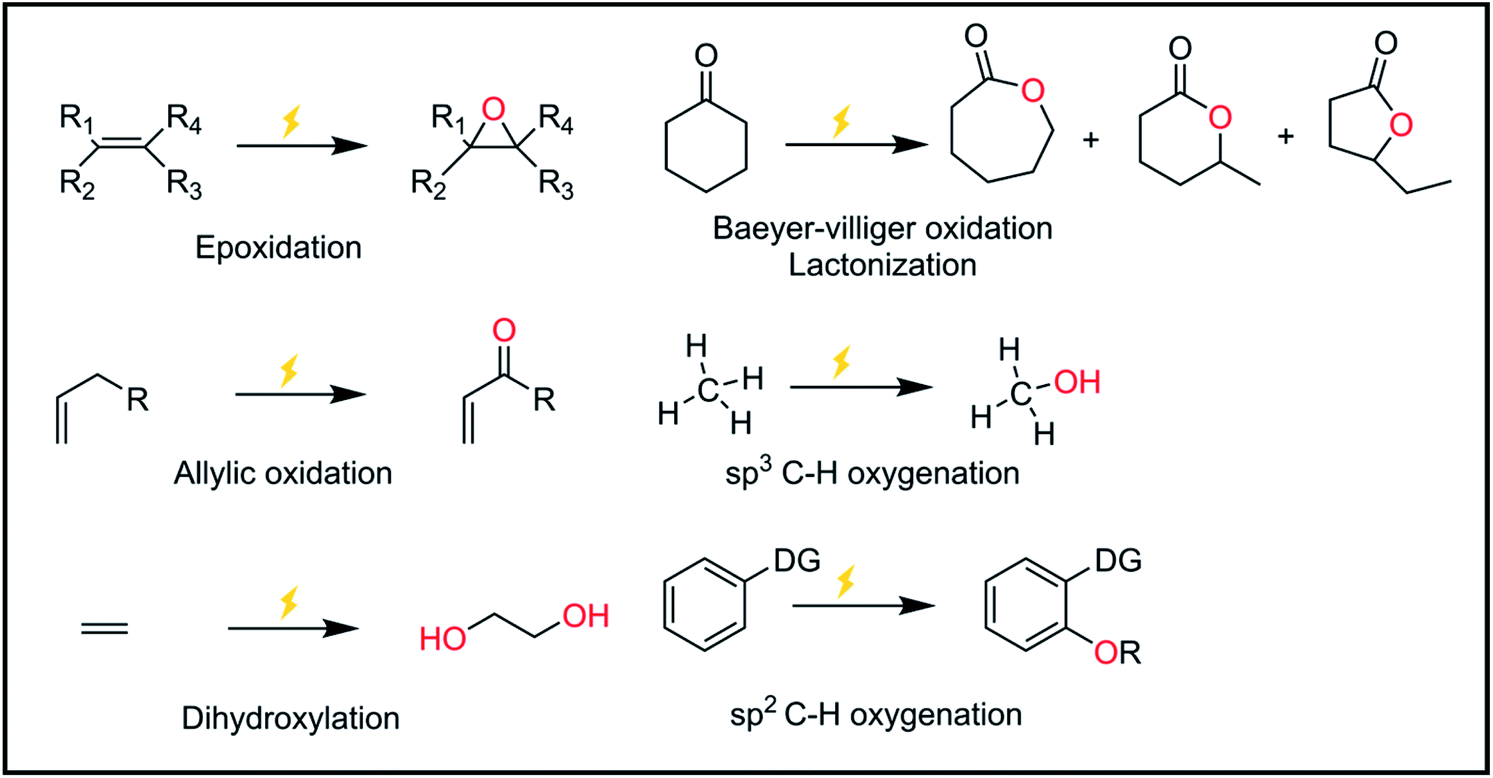 | ||
| Fig. 14 Summary of previously studied electrochemical oxygen functionalization reactions. | ||
As already stated, electrochemical methods can proceed with various oxygen functionalization reactions under milder conditions and with total elimination of unwanted by-products. More importantly, the exploitation of electricity as sole oxidant to drive the oxidation reactions has prevented the excessive use of harmful and toxic chemical oxidants, leading to a greener chemistry. However, the major obstacle for the commercialization of electro-organic synthesis is the required high energy cost correlated to the high overpotentials. The poor selectivity towards desired products and the difficulty to identify the suitable conditions under which the reaction could proceed selectively with high yield also raise a major obstruction for the proposed electrochemical approach. In addition, supporting electrolytes and additives are mostly required to reduce the ohmic resistance inflicted by the poor conductivity of organic solvents, which also poses a challenge in scaling up.
Because of the above-mentioned difficulties, immense efforts have been made to expand the application of electro-organic synthesis. For example, the use of water as an oxygen atom source is difficult (thermochemically), as it requires unreasonably high temperatures and pressures. However, the idea to apply an electrical potential instead of pressure, to release the oxygen from water and to eventually drive the chemical reaction between water and the olefin substrate, made it possible to use water as an oxygen atom source without any strong chemical oxidants. Significantly, the epoxidation of ethylene with water proceeded under room temperature and ambient pressure without the production of carbon dioxide or carbon monoxide. In this regard, we believe that electrochemical oxygen functionalization utilizing water can become an alternative methodology for the decarbonization of the chemical industry. Additionally, continuous efforts are given to the modification of the structure and composition of various electrocatalysts, search of suitable reaction media (solvent, additives, supporting electrolytes), and enhancement of the different electrochemical systems to fully optimize the overall performance of electrochemical oxygen functionalization in terms of selectivity, conversion, faradaic efficiency, and yield.
4. Summary and outlook
This Review article collectively highlights the recent development of oxygen functionalization reactions based on non-electrochemical and electrochemical methods. While the non-electrochemical methods may be considered as the conventional approach for the oxidation of organic substrates, the electrochemical process offers a greener chemistry as it can proceed the oxidation reaction under milder conditions with zero waste/unwanted by-products. However, the tangible progress of the latter progress is just beginning to blossom even though it has been studied for several decades. Hence, in the last part of this review, we hope to briefly discuss the future directions of electrochemistry towards the transformation of organic substrates into useful end products.Various strategies have been exploited in an attempt to achieve selective electrochemical oxygen functionalization, but further breakthroughs must be made to realize its commercialization. To fully improve the performance of this innovative approach, it is imperative to understand the interfacial kinetics at the electrode. Oxidation at the anode always competes with other side reactions, such as oxygen evolution, solvent oxidation, oxidation of auxiliary functional groups, etc. Thus, we categorized the electrochemical methods with respect to their mechanisms: indirect electro-oxidation with mediators and direct oxidation with inner or outer sphere electron transfer mechanism, respectively. Notably, indirect oxidation in presence of a mediator has provided an efficient strategy to selectively oxidize target substrates using the redox potential of the redox mediator. Since the redox potential of the mediator is typically sufficiently lower than that of the substrates, the direct oxidation of the substrate at the electrode is avoided.
Tuning the binding affinity of reactants toward the electrode (catalyst) surface has been previously regarded as a vital technique in the field of electrochemical water splitting, oxygen reduction reactions, and carbon dioxide. Also, it was verified that selectivity and overpotential values are largely affected by the types of catalysts on the electrode. However, most studied schemes, including the so-called volcano plot based on the Sabatier principle, present the oxygen functionalization via an outer-sphere pathway, which limits the role of catalysts. Because the electrode surface is polarized and an electric double layer is formed when an external voltage is applied, it became necessary to examine the solvation phenomena of reactant species in blended electrolytes in order to boost the interaction between electrode materials and reactant species. In line with this, a direct electro-oxidation reaction following an inner-sphere mechanism is considered as an alternative way of directing selectivity correlated to its capacity to control the interfacial kinetics happening on the electrode surface. We introduced in Section 3 the recent attempts to conduct oxygen functionalization reactions with inner-sphere fashion. In particular, Manthiram et al. mediated direct bond formation via pre-activation of water molecules (MO or M–OH) as an oxygen source.162
Relevant to the aforementioned issues, deeper studies of interfacial kinetics in the electrochemical system should be explored. Because polarized electrode surfaces normally disfavor non-polar organic reactants, covalent bonding between the electrode surface and reactant species is hardly formed. To overcome this, solvation of solvent and electrolyte molecules may play a key role in enhancing the solubility and facilitating the approach of the reactant species. Kamlet–Taft parameters are solvochromic properties that contain polarizability, hydrogen bond donating, and accepting abilities of solvent molecules.190,191 It was first suggested by Taft and Kamlet and has been widely utilized to predict the effect of solvent molecules on reaction kinetics, such as catalytic hydrogenation and cross-coupling reactions. Considering most oxygen-atom functionalization requires hydrogen/proton transfer, it is expected that an appropriate choice of solvent molecules based on K–T parameters might enhance the reaction efficiency.192 We believe that this review provides the valuable insights for transition metal-based oxygen functionalization catalysis.
Author contributions
K. J. conceptualized the paper. G. M. T., Y. P., K. L., and K. J. prepared the original draft of the manuscript. G. M. T., K. L., and K. J. reviewed and edited its contents. K. L. and K. J. supervised the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. M. T., Y. P., and K. L. were supported by National Research Foundation of Korea (NRF-2020R1A2B5B03002475 and NRF-2019R1A6A1A11044070), the Korea Basic Science Institute under the R&D program (Project No. C38530) supervised by the Ministry of Science, and the Hydrogen Energy Innovation Technology Development Program of the National Research Foundation of Korea funded by the Korean government (Ministry of Science and ICT (MSIT)) (No. NRF-2019M3E6A1064709). K. J. was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. NRF-2021R1C1C1004264) and a Korea University Grant.Notes and references
- R. L. Lieberman and A. C. Rosenzweig, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 147–164 CrossRef CAS PubMed.
- J. C. Murreil, B. Gilbert and I. R. McDonald, Arch. Microbiol., 2000, 173, 325–332 CrossRef PubMed.
- R. Banerjee, Y. Proshlyakov, J. D. Lipscomb and D. A. Proshlyakov, Nature, 2015, 518, 431–434 CrossRef CAS PubMed.
- J. T. Groves, T. E. Nemo and R. S. Myers, J. Am. Chem. Soc., 1979, 101, 1032–1033 CrossRef CAS.
- J. T. Groves, J. Chem. Educ., 1985, 62, 924–927 CrossRef.
- S. Fukuzumi, T. Kojima, Y. M. Lee and W. Nam, Coord. Chem. Rev., 2017, 333, 44–56 CrossRef CAS.
- V. A. Larson, B. Battistella, K. Ray, N. Lehnert and W. Nam, Nat. Rev. Chem., 2020, 4, 404–419 CrossRef CAS.
- I. Saikia, A. J. Borah and P. Phukan, Chem. Rev., 2016, 116, 6837–7042 CrossRef CAS PubMed.
- J. D. Lou and Z. N. Xu, Tetrahedron Lett., 2002, 43, 6095–6097 CrossRef CAS.
- C. Wang, L. Zong and C. H. Tan, J. Am. Chem. Soc., 2015, 137, 10677–10682 CrossRef CAS PubMed.
- J. Y. Li, Y. H. Li, M. Y. Qi, Q. Lin, Z. R. Tang and Y. J. Xu, ACS Catal., 2020, 10, 6262–6280 CrossRef CAS.
- C. Dai, J. Zhang, C. Huang and Z. Lei, Chem. Rev., 2017, 117, 6929–6983 CrossRef CAS PubMed.
- R. A. Sheldon, J. Mol. Catal., 1983, 20, 1–26 CrossRef CAS.
- Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480–3524 RSC.
- R. H. Holm, Chem. Rev., 1987, 87, 1401–1449 CrossRef CAS.
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, WILEY-VCH, 4th edn, 2003, vol. 53 Search PubMed.
- Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC.
- S. Hu, W. Xiao, W. Yang, J. Yang, Y. Fang, J. Xiong, Z. Luo, H. Deng, Y. Guo, L. Zhang and J. Ding, ACS Appl. Mater. Interfaces, 2018, 10, 17167–17174 CrossRef CAS PubMed.
- B. Yang and T. A. Manz, RSC Adv., 2016, 6, 88189–88215 RSC.
- H. T. Li, Q. Gao, B. Han, Z. H. Ren, K. S. Xia and C. G. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 371–380 CrossRef CAS PubMed.
- Y. Feng, J. England and L. Que, ACS Catal., 2011, 1, 1035–1042 CrossRef CAS.
- B. Wang, C. Li, K. Bin Cho, W. Nam and S. Shaik, J. Chem. Theory Comput., 2013, 9, 2519–2525 CrossRef CAS PubMed.
- K. P. Bryliakov, Chem. Rev., 2017, 117, 11406–11459 CrossRef CAS PubMed.
- M. Ciclosi, C. Dinoi, L. Gonsalvi, M. Peruzzini, E. Manoury and R. Poli, Organometallics, 2008, 27, 2281–2286 CrossRef CAS.
- Y. Yi, L. Wang, G. Li and H. Guo, Catal. Sci. Technol., 2016, 6, 1593–1610 RSC.
- S. P. Teong, X. Li and Y. Zhang, Green Chem., 2019, 21, 5753–5780 RSC.
- D. W. Low, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 1996, 118, 117–120 CrossRef CAS.
- Y. M. Lee, S. N. Dhuri, S. C. Sawant, J. Cho, M. Kubo, T. Ogura, S. Fukuzumi and W. Nam, Angew. Chem., Int. Ed., 2009, 48, 1803–1806 CrossRef CAS.
- M. D. Kärkäs and B. Åkermark, Dalton Trans., 2016, 45, 14421–14461 RSC.
- S. Fukuzumi, T. Kishi, H. Kotani, Y. M. Lee and W. Nam, Nat. Chem., 2011, 3, 38–41 CrossRef CAS PubMed.
- D. Shen, C. Saracini, Y. M. Lee, W. Sun, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2016, 138, 15857–15860 CrossRef CAS PubMed.
- X. Engelmann, I. Monte-Pérez and K. Ray, Angew. Chem., Int. Ed., 2016, 55, 7632–7649 CrossRef CAS PubMed.
- M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2019, 5, 13–28 CrossRef CAS PubMed.
- S. Sahu and D. P. Goldberg, J. Am. Chem. Soc., 2016, 138, 11410–11428 CrossRef CAS PubMed.
- K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942–13958 CrossRef CAS PubMed.
- Z. Chen and G. Yin, Chem. Soc. Rev., 2015, 44, 1083–1100 RSC.
- E. P. Talsi and K. P. Bryliakov, Coord. Chem. Rev., 2012, 256, 1418–1434 CrossRef CAS.
- K. P. Bryliakov and E. P. Talsi, Coord. Chem. Rev., 2014, 276, 73–96 CrossRef CAS.
- R. V. Ottenbacher, E. P. Talsi and K. P. Bryliakov, Molecules, 2016, 21, 1454–1470 CrossRef PubMed.
- A. Bassan, M. R. A. Blomberg, T. Borowski and P. E. M. Siegbahn, J. Phys. Chem. B, 2004, 108, 13031–13041 CrossRef CAS.
- M. M. Abu-Omar, A. Loaiza and N. Hontzeas, Chem. Rev., 2005, 105, 2227–2252 CrossRef CAS PubMed.
- K. Chen, M. Costas and L. Que, Dalton Trans., 2002, 672–679 RSC.
- J. T. Groves, R. C. Haushalter, M. Nakamura, T. E. Nemo and B. J. Evans, J. Am. Chem. Soc., 1981, 103, 2884–2886 CrossRef CAS.
- J. T. Groves and Y. Watanabe, J. Am. Chem. Soc., 1988, 110, 8443–8452 CrossRef CAS.
- R. Andreu, E. E. Ferapontova, L. Gorton and J. J. Calvente, J. Phys. Chem. B, 2007, 111, 469–477 CrossRef CAS PubMed.
- M. B. Neibergall, A. Stubna, Y. Mekmouche, E. Münck and J. D. Lipscomb, Biochemistry, 2007, 46, 8004–8016 CrossRef CAS.
- J. T. Groves, J. Lee and S. S. Marla, J. Am. Chem. Soc., 1997, 119, 6269–6273 CrossRef CAS.
- J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175–4205 CrossRef CAS PubMed.
- K. J. Young, B. J. Brennan, R. Tagore and G. W. Brudvig, Acc. Chem. Res., 2015, 48, 567–574 CrossRef CAS.
- X. X. Li, M. Guo, B. Qiu, K. Bin Cho, W. Sun and W. Nam, Inorg. Chem., 2019, 58, 14842–14852 CrossRef CAS PubMed.
- F. Teixeira, R. A. Mosquera, A. Melo, C. Freire and M. N. D. S. Cordeiro, Phys. Chem. Chem. Phys., 2014, 16, 25364–25376 RSC.
- E. M. McGarrigle and D. G. Gilheany, Chromium— and Manganese—Salen Promoted Epoxidation of Alkenes, 2005, vol. 36 Search PubMed.
- N. Kannan, A. R. Patil and A. Sinha, Dalton Trans., 2020, 49, 14344–14360 RSC.
- T. Ishizuka, H. Kotani and T. Kojima, Dalton Trans., 2016, 45, 16727–16750 RSC.
- Z. Lv, W. Zheng, Z. Chen, Z. Tang, W. Mo and G. Yin, Dalton Trans., 2016, 45, 11369–11383 RSC.
- Y. Liu and T. C. Lau, J. Am. Chem. Soc., 2019, 141, 3755–3766 CrossRef CAS PubMed.
- J. T. Groves and R. Quinn, Inorg. Chem., 1984, 23, 3844–3846 CrossRef CAS.
- J. T. Groves and R. Quinn, J. Am. Chem. Soc., 1985, 107, 5790–5792 CrossRef CAS.
- F. Zaera, J. Phys. Chem. B, 2002, 106, 4043–4052 CrossRef CAS.
- C. M. Friend and B. Xu, Acc. Chem. Res., 2017, 50, 517–521 CrossRef CAS PubMed.
- W. Nam, Y. M. Lee and S. Fukuzumi, Acc. Chem. Res., 2014, 47, 1146–1154 CrossRef CAS PubMed.
- C. C. Phifer and D. R. McMillin, Inorg. Chem., 1986, 25, 1329–1333 CrossRef CAS.
- W. Nam, S. Y. Oh, Y. J. Sun, J. Kim, W. K. Kim, S. K. Woo and W. Shin, J. Org. Chem., 2003, 68, 7903–7906 CrossRef CAS PubMed.
- C. M. Elliott, J. R. Dunkle and S. C. Paulson, Langmuir, 2005, 21, 8605–8608 CrossRef CAS PubMed.
- T. Niwa and M. Nakada, J. Am. Chem. Soc., 2012, 134, 13538–13541 CrossRef CAS PubMed.
- B. Mondal, F. Neese, E. Bill and S. Ye, J. Am. Chem. Soc., 2018, 140, 9531–9544 CrossRef CAS PubMed.
- K. K. Singh, M. K. Tiwari, B. B. Dhar, K. Vanka and S. Sen Gupta, Inorg. Chem., 2015, 54, 6112–6121 CrossRef CAS PubMed.
- C. Krebs, D. G. Fujimori, C. T. Walsh and J. M. Bollinger, Acc. Chem. Res., 2007, 40, 484–492 CrossRef CAS.
- H. Miyake, K. Chen, S. J. Lange and L. Que, Inorg. Chem., 2001, 40, 3534–3538 CrossRef CAS PubMed.
- M. A. Ehudin, L. B. Gee, S. Sabuncu, A. Braun, P. Moënne-Loccoz, B. Hedman, K. O. Hodgson, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2019, 141, 5942–5960 CrossRef CAS PubMed.
- M. J. Park, J. Lee, Y. Sun, J. Kim and W. Nam, J. Am. Chem. Soc., 2006, 128, 2630–2634 CrossRef CAS PubMed.
- J. Serrano-Plana, A. Aguinaco, R. Belda, E. García-España, M. G. Basallote, A. Company and M. Costas, Angew. Chem., 2016, 128, 6418–6422 CrossRef.
- J. R. Lindsay Smith and G. B. Shul'pin, Tetrahedron Lett., 1998, 39, 4909–4912 CrossRef CAS.
- C. Choe, L. Yang, Z. Lv, W. Mo, Z. Chen, G. Li and G. Yin, Dalton Trans., 2015, 44, 9182–9192 RSC.
- G. B. Shul'pin, G. Süss-Fink and L. S. Shul'pina, J. Mol. Catal. A: Chem., 2001, 170, 17–34 CrossRef.
- J. Du, C. Miao, C. Xia, Y. M. Lee, W. Nam and W. Sun, ACS Catal., 2018, 8, 4528–4538 CrossRef CAS.
- R. Zong and R. P. Thummel, J. Am. Chem. Soc., 2005, 127, 12802–12803 CrossRef CAS PubMed.
- G. A. Barf and R. A. Sheldon, J. Mol. Catal. A, 1995, 98, 143–146 CrossRef CAS.
- J. L. Zhang, H. B. Zhou, J. S. Huang and C. M. Che, Chem.–Eur. J., 2002, 8, 1554–1562 CrossRef CAS.
- B. Chandra, K. K. Singh and S. Sen Gupta, Chem. Sci., 2017, 8, 7545–7551 RSC.
- T. Shiragami, K. Kubomura, D. Ishibashi and H. Inoue, J. Am. Chem. Soc., 1996, 118, 6311–6312 CrossRef CAS.
- S. Funyu, T. Isobe, S. Takagi, D. A. Tryk and H. Inoue, J. Am. Chem. Soc., 2003, 125, 5734–5740 CrossRef CAS PubMed.
- K. Kurimoto, T. Yamazaki, Y. Suzuri, Y. Nabetani, S. Onuki, S. Takagi, T. Shimada, H. Tachibana and H. Inoue, Photochem. Photobiol. Sci., 2014, 13, 154–156 CrossRef CAS PubMed.
- D. Tatsumi, T. Tsukamoto, R. Honna, S. Hoshino, T. Shimada and S. Takagi, Chem. Lett., 2017, 46, 1311–1314 CrossRef CAS.
- J. W. Han, J. Jung, Y. M. Lee, W. Nam and S. Fukuzumi, Chem. Sci., 2017, 8, 7119–7125 RSC.
- W. Huang, G. Sun and T. Cao, Chem. Soc. Rev., 2017, 46, 1977–2000 RSC.
- T. Pu, H. Tian, M. E. Ford, S. Rangarajan and I. E. Wachs, ACS Catal., 2019, 10727–10750 CrossRef CAS.
- T. E. Jones, R. Wyrwich, S. Böcklein, E. A. Carbonio, M. T. Greiner, A. Y. Klyushin, W. Moritz, A. Locatelli, T. O. Menteş, M. A. Niño, A. Knop-Gericke, R. Schlögl, S. Günther, J. Wintterlin and S. Piccinin, ACS Catal., 2018, 8, 3844–3852 CrossRef CAS.
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, Weinheim, New York, 1993 Search PubMed.
- A. J. F. Van Hoof, E. A. R. Hermans, A. P. Van Bavel, H. Friedrich and E. J. M. Hensen, ACS Catal., 2019, 9, 9829–9839 CrossRef CAS.
- S. Anandhakumar, M. Sasidharan, C. W. Tsao and A. M. Raichur, ACS Appl. Mater. Interfaces, 2014, 6, 3275–3281 CrossRef CAS PubMed.
- S. S. Sangaru, H. Zhu, D. C. Rosenfeld, A. K. Samal, D. Anjum and J. M. Basset, ACS Appl. Mater. Interfaces, 2015, 7, 28568–28576 CrossRef PubMed.
- N. L. Nguyen, S. Piccinin and S. De Gironcoli, J. Phys. Chem. C, 2011, 115, 10073–10079 CrossRef CAS.
- P. Christopher and S. Linic, J. Am. Chem. Soc., 2008, 130, 11264–11265 CrossRef CAS.
- P. Christopher and S. Linic, ChemCatChem, 2010, 2, 78–83 CrossRef CAS.
- S. Linic and P. Christopher, ChemCatChem, 2010, 2, 1061–1063 CrossRef CAS.
- M. Huš and A. Hellman, ACS Catal., 2019, 9, 1183–1196 CrossRef.
- T. E. Jones, R. Wyrwich, S. Bocklein, T. C. R. Rocha, E. A. Carbonio, A. Knop-Gericke, R. Schlogl, S. Gunther, J. Wintterlin and S. Piccinin, J. Phys. Chem. C, 2016, 120, 28630–28638 CrossRef CAS.
- C. S. Martín, J. Chem. Eng. Data, 2001, 46, 1149–1152 CrossRef.
- M. Drache, P. Roussel and J. P. Wignacourt, Chem. Rev., 2007, 107, 80–96 CrossRef CAS PubMed.
- C. Noguez, J. Phys. Chem. C, 2007, 111, 3606–3619 CrossRef.
- C. Liu, J. Xin, J. Tan, T. Liu, M. R. Kessler and J. Zhang, ACS Omega, 2018, 3, 8718–8723 CrossRef CAS PubMed.
- H. Mimoun, M. Mignard, P. Brechot and L. Saussine, J. Am. Chem. Soc., 1986, 108, 3711–3718 CrossRef CAS.
- M. G. Clerici, G. Bellussi and U. Romano, J. Catal., 1991, 129, 159–167 CrossRef CAS.
- M. G. Clerici and P. Ingallina, J. Catal., 1993, 140, 71–83 CrossRef CAS.
- T. A. Nijhuis, M. Makkee, J. A. Moulijn and B. M. Weckhuysen, Ind. Eng. Chem. Res., 2006, 45, 3447–3459 CrossRef CAS.
- J. Teržan, M. Huš, B. Likozar and P. Djinović, ACS Catal., 2020, 13415–13436 CrossRef.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178, 566–575 CrossRef CAS.
- B. S. Uphade, Y. Yamada, T. Akita, T. Nakamura and M. Haruta, Appl. Catal., A, 2001, 215, 137–148 CrossRef CAS.
- B. S. Uphade, T. Akita, T. Nakamura and M. Haruta, J. Catal., 2002, 209, 331–340 CrossRef CAS.
- G. J. Hutchings, J. Catal., 1985, 96, 292–295 CrossRef CAS.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS.
- J. Gaudet, K. K. Bando, Z. Song, T. Fujitani, W. Zhang, D. S. Su and S. T. Oyama, J. Catal., 2011, 280, 40–49 CrossRef CAS.
- B. Gómez, N. V. Likhanova, M. A. Domínguez-Aguilar, R. Martínez-Palou, A. Vela and J. L. Gázquez, J. Phys. Chem. B, 2006, 110, 8928–8934 CrossRef.
- D. M. Perez Ferrandez, I. Herguedas Fernandez, M. P. G. Teley, M. H. J. M. De Croon, J. C. Schouten and T. A. Nijhuis, J. Catal., 2015, 330, 396–405 CrossRef CAS.
- X. Feng, N. Sheng, Y. Liu, X. Chen, D. Chen, C. Yang and X. Zhou, ACS Catal., 2017, 7, 2668–2675 CrossRef CAS.
- M. Haruta, B. S. Uphade, S. Tsubota and A. Miyamoto, Res. Chem. Intermed., 1998, 24, 329–336 CrossRef CAS.
- J. C. Liu, Y. Tang, C. R. Chang, Y. G. Wang and J. Li, ACS Catal., 2016, 6, 2525–2535 CrossRef CAS.
- S. Biella and M. Rossi, Chem. Commun., 2003, 3, 378–379 RSC.
- S. Bang, Y. M. Lee, S. Hong, K. Bin Cho, Y. Nishida, M. S. Seo, R. Sarangi, S. Fukuzumi and W. Nam, Nat. Chem., 2014, 6, 934–940 CrossRef CAS PubMed.
- E. Y. Tsui, R. Tran, J. Yano and T. Agapie, Nat. Chem., 2013, 5, 293–299 CrossRef CAS.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2013, 333, 733–737 CrossRef.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- F. Wang and S. S. Stahl, Acc. Chem. Res., 2020, 53, 561–574 CrossRef CAS PubMed.
- D. T. Mah, Electrochem. Soc. Interface, 2014, 23, 47 CrossRef CAS.
- J. H. Wagenknecht, J. Chem. Educ., 1983, 60, 271–273 CrossRef CAS.
- L. M. Torres, A. F. Gil, L. Galicia and I. González, J. Chem. Educ., 1996, 73, 808–810 CrossRef CAS.
- A. J. Bard, J. Am. Chem. Soc., 2010, 132, 7559–7567 CrossRef CAS PubMed.
- S. Tanimoto and A. Ichimura, J. Chem. Educ., 2013, 90, 778–781 CrossRef CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- S. J. Hendel and E. R. Young, J. Chem. Educ., 2016, 93, 1951–1956 CrossRef CAS.
- I. Landa-Medrano, I. Lozano, N. Ortiz-Vitoriano, I. Ruiz De Larramendi and T. Rojo, J. Mater. Chem. A, 2019, 7, 8746–8764 RSC.
- G. S. Sauer and S. Lin, ACS Catal., 2018, 8, 5175–5187 CrossRef CAS.
- N. T. Zhang, C. C. Zeng, C. M. Lam, R. K. Gbur and R. D. Little, J. Org. Chem., 2013, 78, 2104–2110 CrossRef CAS PubMed.
- P. Cao, Y. Sun, J. Yao, B. Ren, R. Gu and Z. Tian, Langmuir, 2002, 18, 2737–2742 CrossRef CAS.
- I. Streeter, A. J. Wain, M. Thompson and R. G. Compton, J. Phys. Chem. B, 2005, 109, 12636–12649 CrossRef CAS PubMed.
- G. V. Ramesh, R. Kodiyath, T. Tanabe, M. Manikandan, T. Fujita, N. Umezawa, S. Ueda, S. Ishihara, K. Ariga and H. Abe, ACS Appl. Mater. Interfaces, 2014, 6, 16124–16130 CrossRef CAS PubMed.
- J. Wu, Y. Zhou, Y. Zhou, C. W. Chiang and A. Lei, ACS Catal., 2017, 7, 8320–8323 CrossRef CAS.
- J. W. Lee, J. Y. Shin, Y. S. Chun, H. Bin Jang, C. E. Song and S. G. Lee, Acc. Chem. Res., 2010, 43, 985–994 CrossRef CAS PubMed.
- I. A. Ansari and R. Gree, Org. Lett., 2002, 4, 1507–1509 CrossRef CAS PubMed.
- W. Zheng, M. Wu, C. Yang, Y. Chen, R. Tan and D. Yin, Mater. Chem. Phys., 2020, 256, 123681 CrossRef CAS.
- J. Ghilane, P. Martin, H. Randriamahazaka and J. C. Lacroix, Electrochem. Commun., 2010, 12, 246–249 CrossRef CAS.
- Y. L. Hu, Q. F. Liu, T. T. Lu and M. Lu, Catal. Commun., 2010, 11, 923–927 CrossRef CAS.
- M. Kathiresan and D. Velayutham, Chem. Commun., 2015, 51, 17499–17516 RSC.
- G. Zhao, T. Jiang, W. Wu, B. Han, Z. Liu and H. Gao, J. Phys. Chem. B, 2004, 108, 13052–13057 CrossRef CAS.
- C. Margarita and H. Lundberg, Catalysts, 2020, 10, 1–24 CrossRef.
- M. Panizza and G. Cerisola, Chem. Rev., 2009, 109, 6541–6569 CrossRef CAS PubMed.
- J. B. Sperr and D. L. Wright, Chem. Soc. Rev., 2006, 35, 605–621 RSC.
- C. Yu, B. Özkaya and F. W. Patureau, Chem.–Eur. J., 2021, 27, 3682–3687 CrossRef CAS PubMed.
- Y. Imada, Y. Okada, K. Noguchi and K. Chiba, Angew. Chem., Int. Ed., 2019, 58, 125–129 CrossRef CAS PubMed.
- K. Liu, C. Song and A. Lei, Org. Biomol. Chem., 2018, 16, 2375–2387 RSC.
- H. T. Tang, J. S. Jia and Y. M. Pan, Org. Biomol. Chem., 2020, 18, 5315–5333 RSC.
- R. Lin, A. P. Amrute and J. Pérez-Ramírez, Chem. Rev., 2017, 117, 4182–4247 CrossRef CAS PubMed.
- S. Torii, K. Uneyama, H. Tanaka, T. Yamanaka, T. Yasuda, M. Ono and Y. Kohmoto, J. Org. Chem., 1981, 46, 3312–3315 CrossRef CAS.
- S. Torii, P. Liu, N. Bhuvaneswari, C. Amatore and A. Jutand, J. Org. Chem., 1996, 61, 3056–3060 CrossRef.
- H. Tanaka, M. Kuroboshi, H. Takeda, H. Kanda and S. Torii, J. Electroanal. Chem., 2001, 507, 75–81 CrossRef CAS.
- H. Nishihara, K. Pressprich, R. W. Murray and J. P. Coliman, Inorg. Chem., 1990, 29, 1000–1006 CrossRef CAS.
- L. Espinal, S. L. Suib and J. F. Rusling, J. Am. Chem. Soc., 2004, 126, 7676–7682 CrossRef CAS PubMed.
- K. P. Ho, K. Y. Wong and T. H. Chan, Tetrahedron, 2006, 62, 6650–6658 CrossRef CAS.
- Z. J. Schiffer and K. Manthiram, Joule, 2017, 1, 10–14 CrossRef.
- K. Jin, J. H. Maalouf, N. Lazouski, N. Corbin, D. Yang and K. Manthiram, J. Am. Chem. Soc., 2019, 141, 6413–6418 CrossRef CAS.
- J. H. Maalouf, K. Jin, D. Yang, A. M. Limaye and K. Manthiram, ACS Catal., 2020, 10, 5750–5756 CrossRef CAS.
- J. M. Van der Eijk, T. H. Peters, N. De Wit and H. A. Colijn, Catal. Today, 1988, 3, 259–266 CrossRef CAS.
- C. G. Vayenas, S. Bebelis and S. Neophytides, J. Phys. Chem., 1988, 92, 5083–5085 CrossRef CAS.
- S. Bebelis and C. G. Vayenas, J. Catal., 1992, 138, 588–610 CrossRef CAS.
- J. S. Jirkovský, M. Busch, E. Ahlberg, I. Panas and P. Krtil, J. Am. Chem. Soc., 2011, 133, 5882–5892 CrossRef.
- W. R. Leow, Y. Lum, A. Ozden, Y. Wang, D. H. Nam, B. Chen, J. Wicks, T. T. Zhuang, F. Li, D. Sinton and E. H. Sargent, Science, 2020, 368, 1228–1233 CrossRef CAS PubMed.
- M. Chung, K. Jin, J. S. Zeng and K. Manthiram, ACS Catal., 2020, 14015–14023 CrossRef CAS.
- L. I. Krishtalik, Electrochim. Acta, 1981, 26, 329–337 CrossRef CAS.
- H. Ha, K. Jin, S. Park, K. G. Lee, K. H. Cho, H. Seo, H. Y. Ahn, Y. H. Lee and K. T. Nam, J. Phys. Chem. Lett., 2019, 10, 1226–1233 CrossRef CAS PubMed.
- W. G. Dauben, M. Lorber and D. S. Fullerton, J. Org. Chem., 1969, 34, 3587–3592 CrossRef CAS.
- S. R. Waldvogel and M. Selt, Angew. Chem., Int. Ed., 2016, 55, 12578–12580 CrossRef CAS PubMed.
- A. J. Catino, R. E. Forslund and M. P. Doyle, J. Am. Chem. Soc., 2004, 126, 13622–13623 CrossRef CAS PubMed.
- M. Masui, S. Hara, T. Ueshima, T. Kawaguchi and S. Ozaki, Chem. Pharm. Bull., 1983, 31, 4209–4211 CrossRef CAS.
- E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 77–81 CrossRef CAS PubMed.
- Y. Lum, J. E. Huang, Z. Wang, M. Luo, D. H. Nam, W. R. Leow, B. Chen, J. Wicks, Y. C. Li, Y. Wang, C. T. Dinh, J. Li, T. T. Zhuang, F. Li, T. K. Sham, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 14–22 CrossRef CAS.
- M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC.
- M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS.
- N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS.
- K. Ogura and K. Takamagari, Nature, 1986, 319, 308 CrossRef CAS.
- M. Ma, B. J. Jin, P. Li, M. S. Jung, J. Il Kim, Y. Cho, S. Kim, J. H. Moon and J. H. Park, Adv. Sci., 2017, 4, 1700379 CrossRef.
- L. Arnarson, P. S. Schmidt, M. Pandey, A. Bagger, K. S. Thygesen, I. E. L. Stephens and J. Rossmeisl, Phys. Chem. Chem. Phys., 2018, 20, 11152–11159 RSC.
- Y. Kawamata, M. Yan, Z. Liu, D. H. Bao, J. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448–7451 CrossRef CAS PubMed.
- M. J. Boyd, A. A. Latimer, C. F. Dickens, A. C. Nielander, C. Hahn, J. K. Nrskov, D. C. Higgins and T. F. Jaramillo, ACS Catal., 2019, 9, 7578–7587 CrossRef CAS.
- M. Stoukides, J. Appl. Electrochem., 1995, 25, 899–912 CrossRef CAS.
- M. E. O'Reilly, R. S. Kim, S. Oh and Y. Surendranath, ACS Cent. Sci., 2017, 3, 1174–1179 CrossRef PubMed.
- R. S. Kim and Y. Surendranath, ACS Cent. Sci., 2019, 5, 1179–1186 CrossRef CAS PubMed.
- N. Sauermann, T. H. Meyer, C. Tian and L. Ackermann, J. Am. Chem. Soc., 2017, 139, 18452–18455 CrossRef CAS.
- R. W. Taft and M. J. Kamlet, J. Am. Chem. Soc., 1976, 98, 2886–2894 CrossRef CAS.
- M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877–2887 CrossRef CAS.
- L. Crowhurst, R. Falcone, N. L. Lancaster, V. Llopis-Mestre and T. Welton, J. Org. Chem., 2006, 71, 8847–8853 CrossRef CAS PubMed.
- W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS.
- S. Kim, K. Bin Cho, Y. M. Lee, J. Chen, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2016, 138, 10654–10663 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |