
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress in the improvement of hydrothermal stability of zeolites
Raquel Simancas
 a,
Anand Chokkalingam
a,
Shanmugam P. Elangovan
a,
Zhendong Liuab,
Tsuneji Sanoa,
Kenta Iyokia,
Toru Wakihara
*ab and
Tatsuya Okubo
*a
a,
Anand Chokkalingam
a,
Shanmugam P. Elangovan
a,
Zhendong Liuab,
Tsuneji Sanoa,
Kenta Iyokia,
Toru Wakihara
*ab and
Tatsuya Okubo
*a
aDepartment of Chemical System Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 13-8656, Japan. E-mail: wakihara@chemsys.t.u-tokyo.ac.jp; okubo@chemsys.t.u-tokyo.ac.jp
bInstitute of Engineering Innovation, The University of Tokyo, 2-11-16 Yayoi, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 17th May 2021
Abstract
Zeolites have been successfully employed in many catalytic reactions of industrial relevance. The severe conditions required in some processes, where high temperatures are frequently combined with the presence of steam, highlight the need of considering the evolution of the catalyst structure during the reaction. This review attempts to summarize the recently developed strategies to improve the hydrothermal framework stability of zeolites.
 Raquel Simancas | Raquel Simancas received her PhD degree in chemistry from the Polytechnic University of Valencia in 2015 under the supervision of professor F. Rey. She moved to Kanagawa University in 2015 for postdoctoral research with professor W. Ueda. In 2018, she moved to the Tokyo Institute of Technology for another postdoctoral research study with Dr T. Yokoi. Since 2020, she has been a project researcher in the Okubo–Wakihara laboratory at The University of Tokyo. Her research interests include the synthesis of zeolites for environmental applications. |
 Anand Chokkalingam | Anand Chokkalingam received his PhD degree in chemistry (supervised by Professor M. Palanichamy) from Anna University, Chennai in January 2012. He carried out postdoctoral research with Professor Ajayan Vinu at the National Institute of Materials Science (NIMS), Japan and The University of Queensland, Australia until December 2013. Since January 2015, he has been a project researcher in the Okubo–Wakihara laboratory at The University of Tokyo. His current research focuses on the synthesis and modification of zeolites for energy and environmental applications. |
 Shanmugam P. Elangovan | Shanmugam P. Elangovan received his PhD degree in Chemistry from Anna University in 1995 under Professor V. Murugesan. After five years of lecturing at the Sri Venkateswara College of Engineering, Chennai, he joined the University of Kaiserslautern in Germany as a post-doctoral researcher with Prof. Martin Hartmann (2001–2003). He then moved to the Professor Okubo group at the University of Tokyo in 2003, holding several collaborative industrial research positions, in between. Since 2018, he has been a project researcher in the Okubo–Wakihara laboratory at The University of Tokyo. His current research focuses on optimizing synthesis conditions for the bulk preparation of zeolites for industries. |
 Toru Wakihara | Toru Wakihara is a professor at the Institute of Engineering Innovation of The University of Tokyo. He received his PhD degree from UTokyo in 2004, and started his academic career at Yokohama National University, where he was promoted to an assistant professor and associate professor in 2007 and 2012, respectively. He moved back to UTokyo as an associate professor in 2013. In 2020, he was promoted to a full professor. His research interests include powder technology, synthesis and application of nanosized zeolites, and synthesis of zeolites under extreme conditions. |
 Tatsuya Okubo | Tatsuya Okubo is a professor in the Department of Chemical System Engineering, and an Executive Vice President of The University of Tokyo. He received his PhD degree from UTokyo and started his academic career at Kyushu University as a research associate in 1988. He moved to UTokyo in 1991, where he was promoted to a lecturer, an associate professor and a full professor in 1994, 1997 and 2006, respectively. He spent one year at the California Institute of Technology (USA) as a visiting associate in 1993–1994. His research interests include the synthesis and application of zeolites. |
1. Introduction
More than 250 different zeolite framework types have been recognized by the International Zeolite Association (IZA).1 Microporous aluminosilicates are the major class of zeolites and their Si/Al ratio is a significant factor that influences properties such as ion-exchange capacity, thermal and hydrothermal stability, hydrophobicity, acidity, and catalytic activity. Most of the industrial applications are limited to less than ten structure types such as FAU, MFI, MOR, FER, LTA, CHA or *BEA.2Zeolites are employed as catalysts in many different processes of industrial relevance, such as fluid catalytic cracking (FCC), selective catalytic reduction (SCR), methanol to hydrocarbons (MTH) reaction, dehydration of alcohols, alkylation, hydroxylation, and epoxidation, among others.3 Hydrothermal stability is an important characteristic of zeolites, which makes them one of the most widely employed materials for automobile applications viz., exhaust purification, hydrocarbon trapping, diesel oxidation catalysis, SCR, etc., where they are repeatedly subjected to processing under a high temperature in the presence of either trapped organics or water.4,5
For instance, a diesel particulate filter (DPF) needs to be treated above 650 °C on a regular basis to burn off all the particulate matter (PM) that got trapped within its porous network. Due to its close proximity to a DPF, the SCR catalyst is exposed periodically to heating treatments at varying temperatures in the presence of steam. In addition to the water obtained as a byproduct from the reduction reaction, diesel engine emissions may contain up to 10 vol% of H2O,6 whereas crude methanol used in the MTH reaction generally carried out at 400–450 °C contains around 17 wt% of H2O and this concentration increases during the reaction, where one water molecule is generated by each molecule of methanol converted.7
Moreover, before reactions, a pre-treatment of the catalyst at high temperatures is required for the regeneration of their catalytic properties. For instance, in the FCC process, the fast catalyst deactivation requires a continuous regeneration step carried out in the presence of steam at a temperature that can reach up to 760 °C.8,9 These processes make the catalyst vulnerable to structural degradation and hence possessing high hydrothermal stability is a must for its longer catalytic lifetime.
Hydrothermal treatments lead to chemical as well as structural changes which sometimes are irreversible depending on the stability of the zeolitic framework.10 Exposing the zeolite to those conditions causes the breaking of Si–O(H)–Al bonds by hydrolysis and extracting Al atoms (dealumination) from the structure, resulting in the formation of extra-framework aluminum species (EFAL) and decrease of the acidity of the catalysts.
Under severe dealumination conditions, excessive extraction of Al causes the removal of some Si atoms together with the Al atoms from the framework forming mesopores and structural defects that decrease the framework stability, resulting in a total or partial amorphization of the structure.11–14
Based on the knowledge gained from experimental results, several approaches have been developed to overcome the lack of hydrothermal stability of zeolitic materials.
1.1. Synthesis of high-silica zeolites
Zeolite synthesis requires the use of cations that compensate for the negative framework charge induced by the introduction of trivalent elements and at the same time fit the inner zeolite channels and cages. The use of bulky Organic Structure-Directing Agents (OSDAs) that can occupy large volume thus introducing relatively low charge density has allowed preparing zeolites with high adsorption capacities and high Si/Al ratios.15–171.2. Synthesis of a less defective framework
It is not surprising to find that high hydrophobic materials are more stable under steaming conditions. Fluoride-media synthesis has been proven as a method to reduce the formation of structural defects in the synthesis of pure and high-silica zeolites improving the structural stability.18,191.3. Mild dealumination treatments
Controlled mild hydrothermal treatment of zeolite Y, with a high Al content, results in a partial extraction of framework Al, forming EFAL species that remain in solids filling the pores, reducing the number of acid sites, and improving the hydrothermal stability and catalytic activity of the zeolite, known as ultrastable Y (USY).20–22 These EFAL species acting as Lewis acid sites, which have been located in close proximity to the Brønsted acid sites, enhance the stability of the framework.23,241.4. Healing of framework defects
The post-treatment of zeolites with silylating agents allows reducing the number of structural defects and internal and external silanol groups, increasing the hydrophobicity of the catalysts.25–271.5. Incorporation of extra-framework cations
The introduction of extra-framework cations reduces the acidity of the catalysts and at the same time stabilizes the Al in framework positions. The presence of extra-framework rare earth cations has shown a stabilizing effect under harsh conditions by preventing the dealumination of zeolite Y,20,28,29 whereas the interaction between extra-framework phosphate species and framework Al, mainly studied in the H-ZSM-5 zeolite, inhibits the dealumination under steaming conditions.30–32The aim of this review is to consolidate newly developed strategies to improve zeolite stability.
After a brief introduction of the proposed dealumination mechanisms and theoretical studies about the process (Section 2), we will focus on the recent studies on the enhancement of zeolite framework stability. Section 3 covers the effect of the catalyst chemical composition and aluminum distribution on the framework stability and how to control them. Section 4 is focused on the presence of structural defects and the methods to lower their presence in the framework by direct and post-synthesis treatments. Section 5 summarizes the effect and strategies of the incorporation of extra-framework species, viz., phosphorus, rare earth, alkaline and Cu2+ cations. In this review, we will focus on the improvement of aluminosilicate and silicate framework stability from the viewpoint of structural properties. The discussion about the stability of the catalysts in reactions is beyond the scope of this review, although some examples were included in the essay.
2. Zeolite dealumination mechanism
Several factors promote the dealumination of zeolites reducing the hydrothermal stability of the framework. Water accessibility has been proven as one of the main factors determining the reactivity of the Al sites.33 Water adsorption is promoted by the presence of hydrophilic centers, such as silanol groups or framework defects and cations balancing the negative framework charge induced by the introduction of trivalent elements.34Sano et al.35,36 showed linear correlation between adsorbed water molecules and the content of framework Al in the H-ZSM-5 zeolite, and a dealumination rate dependence on the content of Al under hydrothermal treatments at 600–800 °C.
Agostini et al.37 employed in situ time-resolved XRPD and TG, in situ Al K-edge XAS, and ex situ27Al magic angle spinning (MAS) NMR to determine the dealumination mechanism of zeolite Y during steaming at 600 °C. Their results indicate that the formation of extra-framework Al species mainly occurs during the cooling stage (177–227 °C), when water molecules are capable of diffusing through the zeolite framework.
Muller et al.38 studied the dealumination of zeolites *BEA, MOR, MFI and FER by thermal treatment, leaching with oxalic acid and treatment with SiCl4 using 1H, 29Si and 27Al MAS NMR. The zeolite structure, Si/Al ratio and crystal size were found as the main factors determining the dealumination behavior. Furthermore, the presence of defect sites formed during crystallization or calcination results in a more flexible framework that shows less stability against dealumination.
Almutairi et al.39 showed the difference in the stability of the zeolite H-ZSM-5 when it was treated at 400–600 °C for 6 h under mild (8.2 × 10−5 mol min−1) and severe (3.3 × 10−4 mol min−1) steaming conditions. While the removal of framework Al was not observed under mild conditions, an increase in the steam flow resulted in severe dealumination, decrease of Brønsted acid sites, and formation of mesoporosity.
In another study, Iyoki et al.12 found the formation of an amorphous layer on the surface of zeolite Y during hydrothermal treatments which covers the zeolite particles preventing the adsorption of Ar and even the particle core does not suffer from a severe degradation and dealumination.
A controlled dealumination has been used to enhance zeolite stability and obtain high-silica materials from zeolites that can only be obtained with a high Al content.40–42 But an excessive extraction of Al can lead to an undesired effect during the reaction and regeneration of the catalysts, underlining the importance of understanding the dealumination mechanism and avoiding it when it is undesired. The dealumination process has been studied by different characterization techniques, such as solid-state 27Al MAS NMR and Fourier transform infrared spectroscopy (FT-IR), which are powerful and widely used techniques employed in the study of Al atoms.30,39,43
Ristanovic et al.44 studied the differences between mild (500 °C) and severe (700 °C) steaming treatments on ZSM-5 zeolites by measuring the local turnover rates of the oligomerization of furfuryl alcohol using the concept of nanometer accuracy by stochastic chemical reactions (NASCA) microscopy. Mild steamed zeolites showed a highly heterogeneous distribution of acid sites caused by the formation of extra-framework aluminum species and mesoporous defects in the surface region, while the severe treatment caused a significant loss of Brønsted acid sites.
Perea et al.45 employed atom probe tomography to determine the Al–Al bond length in the ZSM-5 zeolite, observing a non-random distribution of the Al atoms before and after steaming treatment, concluding that the porous network acts as a highway for Al transport.
Although the dealumination mechanism has not been fully understood, the experimental results indicate that a severe treatment at high temperatures in the presence of steam leads to the extraction of the Al atoms and the formation of silanol defects.46Fig. 1 shows the proposed four step dealumination mechanism.46–48
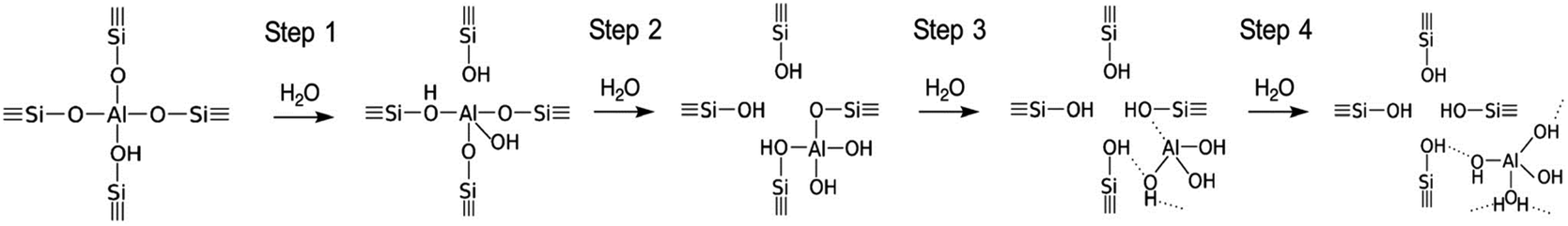 | ||
| Fig. 1 Suggested schema of Al extraction from the zeolitic structure.47,48 Reproduced from ref. 48 with permission from the Royal Society of Chemistry. | ||
Computational simulation has been proven as a very important tool that combined with the experimental results allowed rationalizing the reaction mechanism. Recently, several theoretical studies have been carried out to elucidate the initial steps of the dealumination mechanism.
Silaghi et al.49 reported a dealumination mechanism where one water molecule is adsorbed on the Al atom which adopts a pentahedral coordination leading to bond breaking and resulting in the formation of tetrahedral Al partially bonded to the structure. This initiation mechanism agrees with experimental results; besides, it was confirmed in four different zeolitic frameworks, CHA, MOR, FAU and MFI.
Stanciakova et al.33 proposed four mechanisms involving two water molecules in zeolite MFI as shown in Fig. 2, where the water–water interaction affects the reaction thermodynamics. Prior to any step, the water molecules should be rearranged from stable adsorption modes to active fewer stable modes; therefore, the facility of this rearrangement could have an important influence on the dealumination process. Mechanism I is analogous to that reported by Silaghi et al.49 for one water molecule, while mechanism II is a modified version following a more energetically favorable pathway. In mechanism III the second water molecule interacts with the zeolitic framework before the Al–O(H)–Si bond breaking, whereas mechanism IV is thermodynamically preferred resulting in P2 species that could be reactants in mechanisms I and II. The authors concluded that the water accessibility to the Al sites and treatment temperature are the main factors determining the reactivity of the acid sites.
 | ||
| Fig. 2 Proposed initiation dealumination mechanisms involving two water molecules. Reprinted with permission from ref. 33. Copyright (2019) American Chemical Society. | ||
The above proposed mechanisms were only focused on the first hydrolysis step studying the system from a static approach. Nielsen et al.48 studied the dealumination process in a CHA-type framework by using DFT-based molecular dynamics (DFT-MD) simulations to solve the problem under realistic conditions (atmospheric steam pressure at 450 °C). The free energy barriers of Al–O hydrolysis were calculated for the mechanism where a single molecule was present in every step of the dealumination process, as shown in Fig. 1, and for the system containing four water molecules in the unit cell, as shown in Fig. 3. The authors concluded that water molecules cooperate in the dealumination process by stabilizing the transition state of each step.
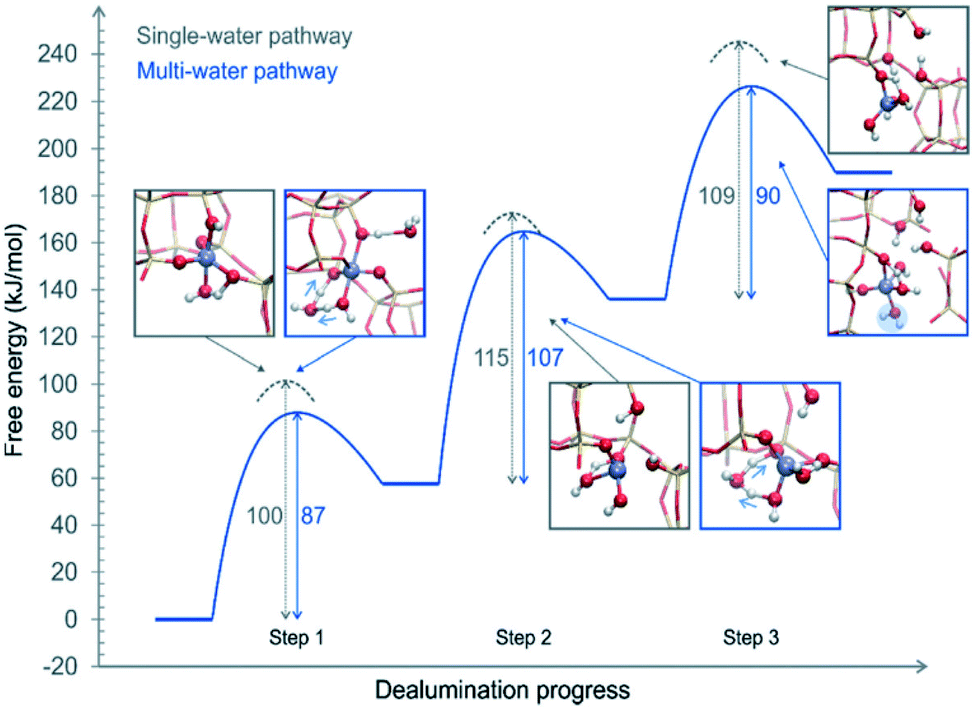 | ||
| Fig. 3 Free energy diagram of a complete dealumination reaction in a CHA-type framework as obtained from DFT-MD umbrella simulations. Reproduced from ref. 48 with permission from the Royal Society of Chemistry. | ||
3. Improvement of thermal/hydrothermal stability by controlling chemical composition and aluminum distribution
High silica zeolites (i.e. Si/Al > 100) are favored in many applications as they show high hydrothermal stability with the exception of ion-exchange processes where a low Si/Al ratio is preferred. Aluminum gets removed from its framework position, which is termed dealumination and occurs due to high temperature treatments either with or without steam and is found to be the major cause of the structural instability of various zeolites.13,143.1. Control of chemical composition by synthesis and post-modification strategies
In order to retain structural integrity and improve the hydrothermal stability of zeolites, researchers focused mainly on the following techniques, i.e. direct synthesis of high-silica zeolites using OSDAs, synthesis using fluoride media, dry gel conversion technique15,18,50–52 and post-synthetic modifications using mild dealumination techniques such as SiCl4 treatment, mild acid treatment, steaming, or combination thereof.13,14,53–55A large number of AlO4/2− ions are required to stabilize the aluminosilicate precursor and lead to the formation of aluminum rich zeolites. Due to the larger content of Al, more negative charges on the framework can be created, which in turn act as an ion-exchange site, an active site for many applications. However, the presence of more aluminum leads to easy dealumination and causes structural collapse, and hence, high-silica zeolites are preferred for applications involving high temperatures in the presence of water vapor.
Researchers found that quaternary ammonium cations due to their bulky nature occupy more space and there is no need for AlO4/2− ions for charge compensation and results in obtaining high-silica zeolites. A major breakthrough has recently been made to synthesize high silica zeolites, viz., *BEA, MTW, MSE, TON, and so on, without the use of OSDAs but with the addition of zeolite seeds.13,40,56–59
Although there are many articles and reviews dealing with the synthesis of high-silica zeolites,15,50,51,60–64 recent study on their hydrothermal stability is scarce; however, to get an overall view of high-silica zeolites including trends in the zeolite synthesis, researchers are encouraged to read those reviews and articles.
In the zeolite synthesis under fluoride media, where the supersaturation of crystallizing species is relatively lower, the formation of metastable phases is largely reduced. It offers the advantages of narrow crystal size distribution, fewer defects and ordered crystallization. Fluoride sources such as HF, NH4F, NaF and KF were used, and F− ions get incorporated into the zeolite framework by replacing OH− ions. This enhances the hydrophobicity of the material, which certainly contributed to the increase in the hydrothermal stability of the obtained zeolites.65–68
The synthesis of hydrothermally stable, all silica zeolites such as BEA* and CHA became possible by the introduction of fluoride ions.18 Kato et al. studied the synthesis of mordenite in fluoride media and explained its stability based on the ordered distribution of Al ascribed to the presence of F− ions in the reactant.69 The effect of zeolite synthesis under fluoride media in limiting defect formation is discussed in detail in Section 4.2.
Lercher and co-workers found that the extra-framework Al species in ion-exchange positions, that were formed as a result of the steaming treatment, played a crucial role in enhancing the hydrothermal stability of H-*BEA zeolites via controlled dealumination and lattice stabilization.55
Post-synthetic dealumination using a mild acid on the as-synthesized zeolite *BEA (with an occluded OSDA) was reported to enhance its hydrothermal stability, resulting in a Si/Al > 1000 and the formed silanol defects were found to be cured by calcination.72 Weckhuysen and co-workers suggested a post-synthetic pressure-controlled dealumination treatment to produce hydrothermally stable, reactive zeolite catalysts.33 A versatile dealumination strategy was also proposed by Shi et al. to stabilize Al-rich zeolites through interaction between the zeolite framework Al and aluminum sulfate.73 Although high silica zeolites are hydrothermally more stable than aluminum rich zeolites, which are more susceptible to dealumination, the lack of exchangeable cation sites makes the former unsuitable for numerous applications. Under such circumstances, it becomes necessary to select zeolites with an optimum Si/Al ratio to maintain a balance between activity and hydrothermal stability.74–76
In this context, attempts were even made to incorporate Al into high silica zeolites under fluoride media using zeolite crystals as an aluminum source.19 The effects of hydrothermal treatment on metal exchanged zeolites were studied by several authors.77–79 With an increase in hydrothermal aging temperature, the structure of zeolites with a higher Cu/Al ratio could easily deteriorate.76,80 As reported, there exists a threshold for the Cu/Al ratio (i.e. 0.35), above which the effect of aging will lead to severe structural disintegration.
Such an effect is not only limited to the Cu/Al ratio as the CuO cluster formation beyond a certain threshold may even lead to hydrothermal aging81,82 and the removal of CuO clusters by washing with a dilute acid followed by ammonium nitrate aqueous solution seemed to improve the hydrothermal stability of these zeolites.83 Recently, the coating of pure silica on the surface of the zeolite was found to not only increase the overall Si/Al ratio but also act as a shield to protect the inner aluminosilicates from dealumination thus endowing it with high hydrothermal stability.84
3.2. Effect of Al distribution
The contents and locations of Al in the zeolite frameworks are the key factors determining the physicochemical properties of zeolites.85–90 The location of Al in the framework plays a vital role in determining the stabilization of exchanged cations, especially di or trivalent ions, and in turn the activity or stability of the catalyst.91–94 Al distribution mainly depends on the Si/Al ratio,95,96 and it controls not only activity but also hydrothermal stability. Yokoi et al.88 reported that the Al atoms located at the intersection of the ZSM-5 zeolite exhibited a shorter catalytic lifetime by investigating the correlation between the location of Al in the framework and catalytic activity/stability. They used high resolution 27Al MAS NMR and 27Al MQMAS NMR techniques to examine the location of Al on the ZSM-5 zeolite synthesized using various OSDAs viz., tetrapropylammonium cations, dipropylamine, hexamethylenimine or cyclohexylamine with or without Na+ cations.Holzinger et al.43 reported a similar observation by investigating the aluminum distribution of four different commercial H-ZSM-5 samples (Samples A, B, C and D of Si/Al ratios of 15, 40, 50 and 140, respectively) before and after steam treatment using the ultrahigh field 27Al MAS NMR technique. In agreement with theoretical studies,90,97 they have arrived at the conclusion, using the normalized data obtained from 27Al NMR for sample B after steaming treatment at 500 °C for about 48 h, that Al in the T sites of the straight and the sinusoidal channels is most stable (T16, T8, T20, and T23; lost only 44–66% of their initial intensity), while framework sites facing the intersections are most prone to dealumination, where more than 77% of Al has been removed (cf.Fig. 4 and 5).
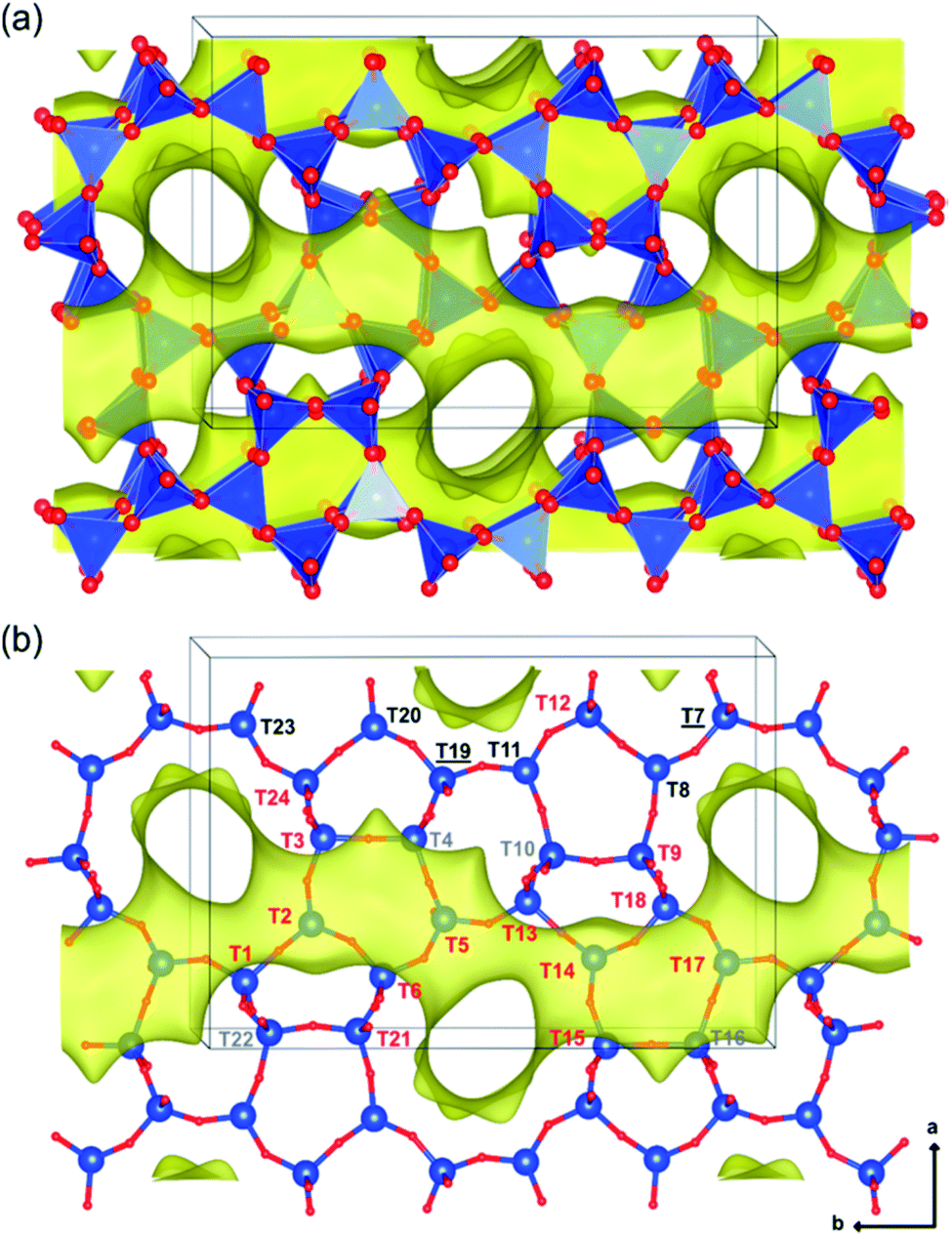 | ||
| Fig. 4 Illustrations of the monoclinic MFI structure, viewed perpendicularly to the straight channels, as tetrahedral units (a) and as bonded atoms (b). The 24 different tetrahedral framework sites are indicated in part b and are denoted in red (intersections), gray (sinusoidal channel only), black (straight channel only), and black underlined (shared by both straight and sinusoidal channels but not located at the intersection). Reprinted with permission from ref. 43. Copyright (2018) American Chemical Society. | ||
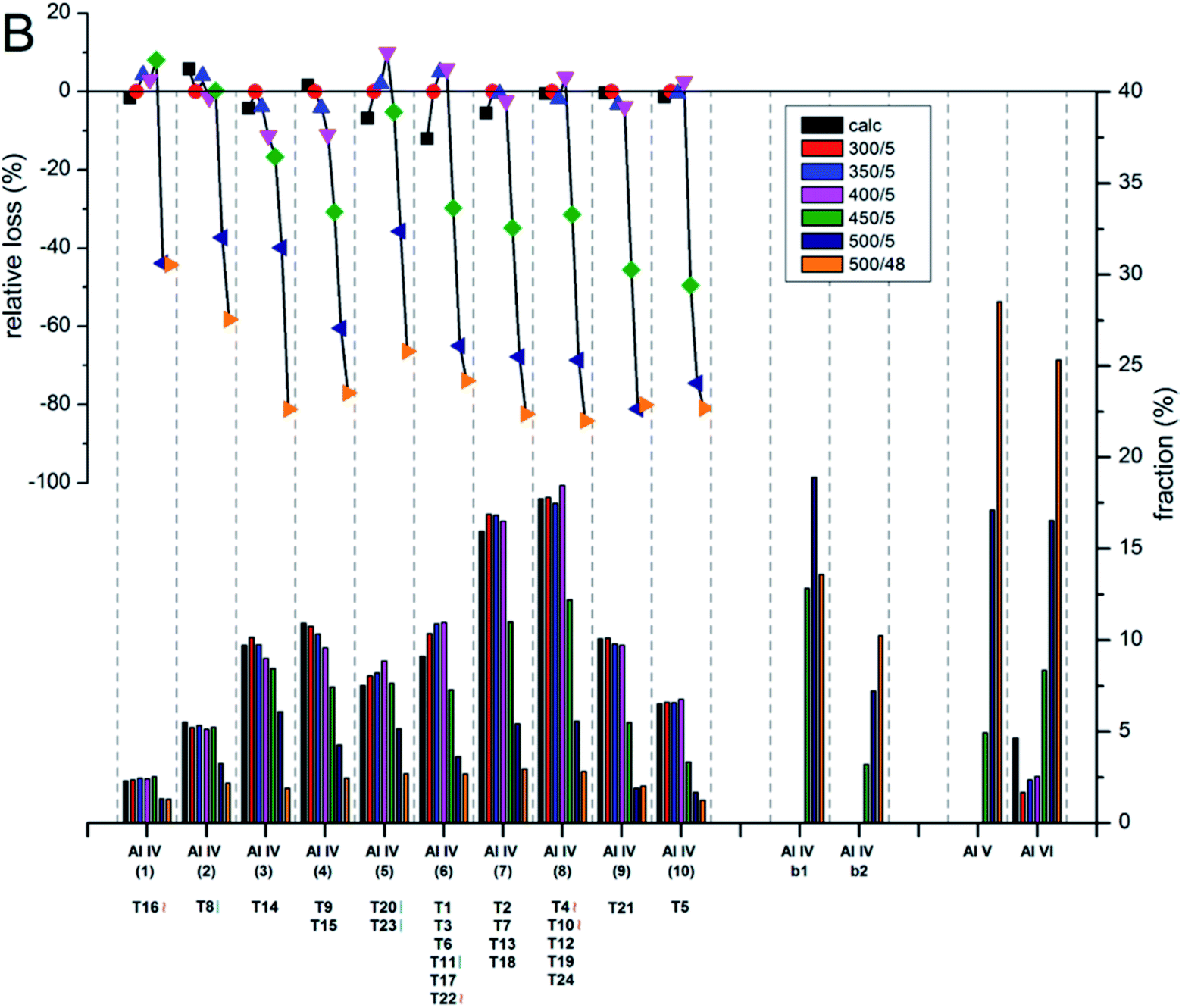 | ||
| Fig. 5 Evolution of the intensities for the different Al sites after the steam treatment of sample B (Si/Al = 40) determined and quantified by 27Al MAS NMR at 22.3 T. The lower bars show the fraction of Al intensity for the individual sites, whereas the symbols in the upper part indicate the relative loss of Al intensity upon steam treatment at different temperatures. Reprinted with permission from ref. 43. Copyright (2018) American Chemical Society. | ||
In addition, Yokoi et al. found a correlation between the relative crystallinity after steam treatment carried out at 800 °C in 40 vol% steam and the Q4 (nAl) and Q3 (nAl) proportions in the parent CHA samples and concluded that Q4 (2Al) is more prone to dealumination compared to Q4 (1Al).98 Davis and co-workers also reported that compared to isolated Al species, the presence of higher proportions of pairing Al species in CHA-type zeolites led to the production of heavier coke in the MTO reaction.99
The location and distribution of framework Al atoms dramatically affect the catalytic activity and selectivity in protonic as well as transition metal ion-containing zeolite catalysts.100–103 The regulation of the Al location or the acid distribution in the framework has proven beneficial for improving the hydrothermal stability of zeolites, thereby making them an industrially useful catalyst.
Zeolite researchers have tackled controlling Al distribution especially on MFI zeolites and partially on *BEA, CHA and AEI; however, the effect of the distribution of Al atoms in the zeolite framework on the hydrothermal stability of most of the zeolites has not been completely understood and it is still a challenging issue to be resolved.
4. Improvement of thermal/hydrothermal stability by decreasing structural defects
4.1. Structure of defects
The term “defect” in a zeolite framework predominantly refers to silanol (SiOH) groups and depending on their location they may be classified as framework or surface defects. Relative to the chemical proportions of the initial multicomponent reactant mixture and its treatment conditions they appear in all shapes and sizes which can be quantified by a combination of characterization techniques such as physisorption, X-ray diffraction (XRD), NMR spectroscopy, vibrational spectroscopy, electronic structure mapping and numerical solution calculations.The influence of synthesis parameters and post-synthesis treatments on the formation and annealing of various kinds of defects in high-silica MFI zeolites has been established systematically using high-resolution solid-state 29Si NMR.104 While the cause of their formation during synthesis and healing after post-synthesis treatments is yet to be fully understood, different models were proposed for their types using various zeolite frameworks (MFI, MTW, AFI and -SVR) by Brunklaus et al.105 with multiple quantum NMR spectroscopy.
The authors observed connectivity defects among MFI, MTW and AFI while -SVR contained ordered vacancy defects. It was suggested that the presence of merged six rings preferentially formed connectivity defects in three zeolites while the fourth lacking it contained vacancy defects. Furthermore, they implied that connectivity defects are preferred over vacancies because the former healed by condensation upon calcination while the latter was not.
Recently, Grosskreuz et al.106 gave an overview of previously established defect models by investigating the alteration of different framework defects in MFI zeolites. The authors suggested that defects for MFI-type zeolites fall within one of the six types shown in Fig. 6. It was established that upon calcination at high temperatures most of the defects get cured, whereas an imperfect condensation leads to empty T-sites in the case of silanol nests. Avoiding defects altogether or healing them if present is crucial and failing to do so would compromise the framework stability and performance of zeolites. There are several studies in the open literature that report different techniques to address the issues related to defect formation in zeolites which will be discussed in the sections to follow.
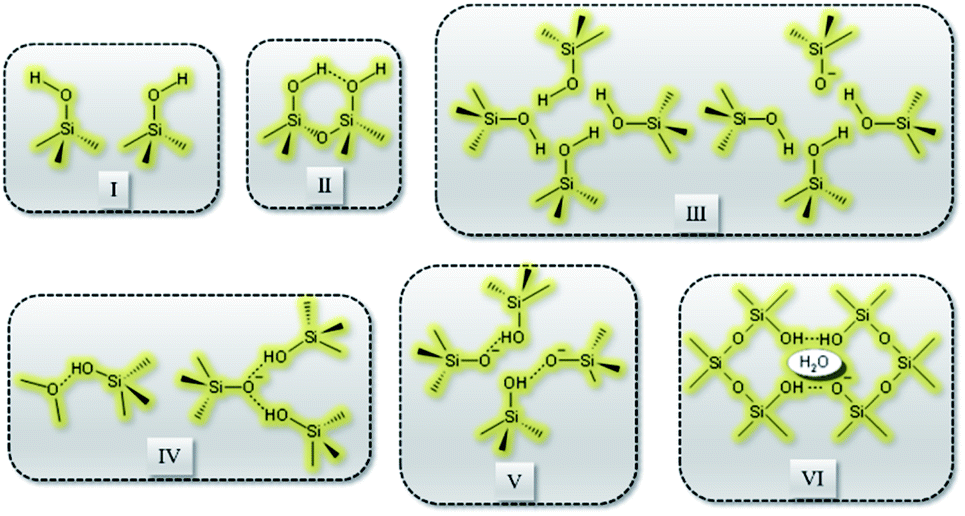 | ||
| Fig. 6 Types of defects: (I) isolated, (II) paired silanol, (III) silanol nests, (IV) combined silanol groups, (V) associated pairs and (VI) clustered pairs with stabilizing H2O. | ||
4.2. Synthesis of zeolites in fluoride media
Conducting the synthesis in fluoride media and post-synthesis silylation are well-known techniques used to decrease the defects in the zeolite structure. The practice of using fluoride ions as a mineralizing agent dates back to 1978 when Flanigen and Patton successfully attempted zeolite preparation in near neutral synthesis media.107 Preparing zeolites in the fluoride medium was found effective in mitigating defects when Chézeau et al. reported the synthesis of a highly siliceous MFI-type zeolite using fluoride anions as the mineralizer.108 Though this technique is instrumental in producing pure or high-silica defect-free zeolites, employing them for preparing aluminosilicate zeolites remained challenging until Moteki and Lobo reported the synthesis of ITW-, CHA-, *BEA- and STT-type zeolites in fluoride media using a FAU-type zeolite as an effective aluminum (Al) source to control the defects as well as the Si/Al ratio.19Recently, fluoride media were used to prepare ZSM-5 model crystals with fewer defects and a near perfect structure–diffusion–activity relationship was established using the same in MTO catalysis. Using this model, it was shown that if the defects were fewer a molecule within the microporous network attains diffusional supermobility or levitation and passes through at least 5 to 6 channel intersections before reaching the nearest acid site thus enhancing the catalytic activity and selectivity.109
Previously, the synthesis of zeolites and zeo-types employing fluoride media was achieved in the pH range 5–9. Preparing zeolites at pH below 5 required seed assistance until the pioneering work of Shi and coworkers opened new possibilities by producing zeolites above the isoelectric point of gel particles in the pH range 2.3–5.110 An in situ IR spectroscopy study on ZSM-5 nanocrystals conducted by Grahn and coworkers showed that lowering the density of internal defects using the fluoride route enhanced their lifetime to 6 times longer than that of the crystals prepared in traditional hydroxyl medium in the methanol to hydrocarbons (MTH) reaction. The abundance of internal defect sites present in the sample was found to accelerate coke deposition in its channel system causing hindered diffusion and percolation effects leading to faster deactivation than the fluoride based one.111
The reports on fluoride-media syntheses aiding the enhancement of catalytic activity, stability and selectivity of zeolites by lowering the defect sites are vast; however, studies exploring their influence on hydrothermal stability are lacking. Conclusions from a recent study investigating such an influence by comparing the hydrothermal stability of ZSM-5 crystals prepared from fluoride and hydroxide media reported by the same group might explain such lacking.
The steam treatment of the ZSM-5 crystals was performed at 1000 °C for 32 h and that prepared from fluoride media exhibited relatively higher framework stability (established from the relative crystallinity) than its hydroxide counterpart. However, it's intriguing to note that the dealumination occurred with comparable rapidity in both cases, necessitating the need for further investigations to understand the role of fluoride anions in stabilizing the framework structure.112
4.3. Treatment of zeolites with a silylating agent (hydrophobization)
Similar to fluoride media synthesis (discussed in Section 4.2), post-synthetic treatment with silylating agents is a well-established method to overcome the influence of defects on the framework stability of zeolites, and even a few studies have identified the latter to be better. One such study by Prodinger et al. states that the role of fluoride anions as a hydrothermal stability enhancer (via defect minimization) was effective only when the concentration of acid sites is lower i.e. on high silica zeolites (Si/Al > 100) and somehow failed to prevent hydrolytic degeneration in zeolites (Si/Al < 50) with a higher concentration of acid sites. Under such circumstances, a selective hydrophobization restricted to the external surface by post-synthetic silylation using long-chain alkyl chlorosilanes is shown to extend the lifetime of defect free Al rich zeolites.113,114Following that study, Vu et al. reported the stability enhancing effect of mono-, di- and tri-chlorosilanes post-synthetically on zeolite Y by systematically varying their alkyl chain lengths for the first time in acidic aqueous media under aqueous-phase processing conditions (i.e. pH ≈ 2, 200 °C and autogenous pressure). Among the silylating agents studied, methyltrichlorosilane was found to improve the hydrothermal stability of zeolite Y significantly.
Unfortunately, a general flaw (i.e. the narrowing effect of the silylating agents on zeolite pores causing their micropore volume to shrink) encountered with all the post-synthetic silylations reported so far was observed in this study as well.115
Although most of the methods discussed so far in this review were able to suppress structural defects and enhance the hydrothermal stability of zeolites to withstand steaming treatment up to a maximum temperature of 850 °C, pushing the limit beyond 1100 °C remained challenging until the following two methods (i.e. synthesis in the presence of alcohol116 and self-defect healing117) were established.
Yoshioka et al. recently unfolded a rapid synthesis technique for preparing a ZSM-5 zeolite in just 2 h by using 1-butanol as an organic additive instead of the usually employed tetrapropylammonium (TPA) cation as the OSDA. Since alcohols are essentially electroneutral and withstand very high synthesis temperatures they are known to generate far fewer defects than TPA cations. Notably, ZSM-5 synthesized in alcoholic media has their Al distributed in a unique fashion leading to a superior catalytic lifetime as well. Compared to TPA, the ZSM-5 synthesized from 1-butanol retained not only 74% of its crystallinity after undergoing a hydrothermal stability test at 1100 °C but also possessed Al atoms in their framework indicating that the synthesis of zeolites in alcohol medium has resulted in widening of the synthesis temperature window and suppression of silanol defects and dealumination.116
4.4. Self-defect healing
In a recent finding, Wakihara and co-workers were able to stabilize the framework of industrially important zeolites (*BEA, MFI and MOR) with three different topologies to withstand extremely harsh steaming conditions exceeding 1100 °C.117 Using a simple and general cooperative liquid-mediated method that involves mutual assistance of pore filling hydroxide and fluoride species, the authors post-synthetically have achieved silicate migration, inducing defect-healing in these zeolites. Unlike many post-synthetic silylation processes,113–115 this new process does not require additional silylating agents, which tends to reduce micropore volume to a significant extent via pore blockage. Fig. 7 shows the highlight of this technique to heal defects to an extreme extent by comparing the relative crystallinities and micropore volumes of the high-silica ZSM-5 zeolite (SiO2/Al2O3 = 1500) both before and after the cooperative liquid-mediated treatment.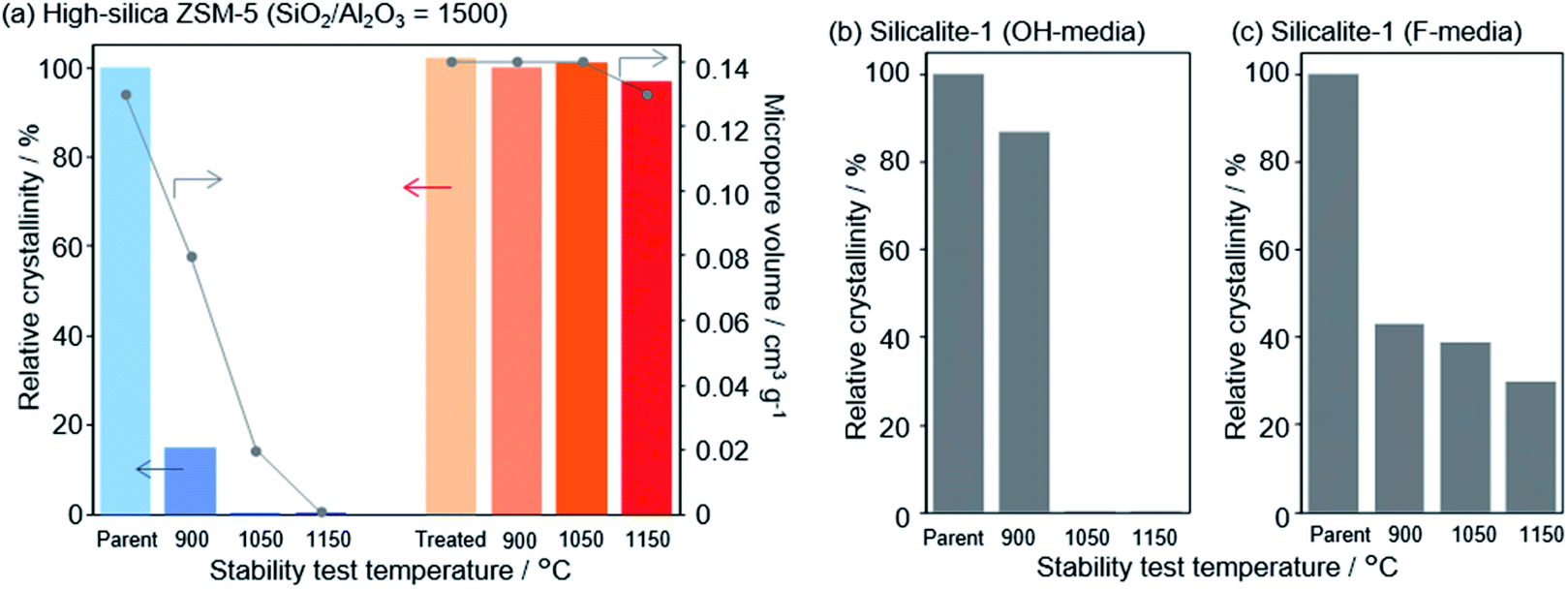 | ||
| Fig. 7 Effect of stabilization treatment on crystallinity and micropore volume. Changes in relative crystallinities and micropore volume of (a) parent and treated high-silica ZSM-5 (SiO2/Al2O3 = 1500) after the stability test and changes in the relative crystallinity of pure-silica ZSM-5 (silicalite-1) synthesized in (b) hydroxide and (c) fluoride media. Reprinted with permission from ref. 117. Copyright (2020) American Chemical Society. | ||
At 900 °C the structure of the untreated ZSM-5 degrades completely while that of the treated ZSM-5 remained intact even a treatment temperature of 1150 °C. Even the pure silica ZSM-5 (Silicalite-1) prepared from hydroxide and fluoride media lost its structural integrity at 900 °C. Also, a self-defect healing mechanism involving the migration of silicate species for this method is proposed in Fig. 8. This new post-synthetic method is found beneficial for improving the hydrothermal stability of different high-silica zeolites falling within a certain range of SiO2/Al2O3. However, the context of improving the hydrothermal stability of low-silica zeolites using this method might require furthermore modifications to address the dominance of dealumination under such high-temperature steaming conditions.117
 | ||
| Fig. 8 Proposed mechanism for self-defect healing. Reprinted with permission from ref. 117. Copyright (2020) American Chemical Society. | ||
5. Improvement of hydrothermal stability by the introduction of extra-framework species
5.1. Incorporation of extra-framework phosphorus species
The improvement of the hydrothermal stability of zeolites by the introduction of extra-framework phosphorus species is well-known, having been studied in many zeolites, especially in MFI and FAU, due to their important industrial applications in catalytic cracking or in the methanol to hydrocarbon reactions.30,118,119The incorporation of extra-framework phosphorus species not only enhances the stability of the zeolitic framework under high temperatures in the presence of steam, but also reduces the number of acid sites, and their strength, because the interaction of phosphates with strong Brønsted acid sites is preferred over weak acid sites.31,32,120 This effect on the acidity has an important influence on the catalytic performance allowing the control of product selectivity.121
The presence of phosphorus also avoids the collapse of the structure, showing low variation of the micropore volume before and after steaming treatment of P-MFI.30
In Section 2, it was shown that after the treatment of the zeolites under high temperature in the presence of steam, a dealumination process occurs. Zeolites modified with P suffered from less dealumination, preserving more Al in framework positions. Nevertheless, it has been reported that there is an optimum P content at which the minimum number of acid sites is lost keeping the maximum stability for the framework aluminum and catalytic performance.30,122
Gao et al.125 compared a P-ZSM-5 zeolite prepared by the impregnation method with a series of samples with different P contents obtained by using the hydrothermal dispersion method, where a mixture of H-ZSM-5 and (NH4)2HPO4 was heated at 140 °C and 0.3 MPa for 2 h. The hydrothermal dispersion enhances the P diffusion through the zeolitic channels, improving the catalytic performance in the butene cracking of the phosphated zeolites. Other reported methods are sample reflux with an aqueous solution of H3PO4 or in an n-octane solution of trimethylphosphite,126 and the gas-phase deposition using as precursor trimethylphosphite127 or trimethylphosphine,128 but they are less used.
Most of the incorporated phosphate species by using the above treatments are monomeric, di- or small chain polyphosphates that after the required calcination step evolve into large polyphosphates which have no interaction with the zeolite framework.
The incorporation of well-dispersed P-species during the zeolite synthesis has been recently explored by using a phosphorus-containing OSDA. This method allowed obtaining new zeolitic structures129–132 and introducing phosphorus into small pore materials that cannot be achieved by standard post-synthesis treatments because of the limited diffusion of phosphates through 8R windows. Small pore zeolites AEI,133–135 CHA,136 AFX,137 LEV,122 GME138 and AFX/CHA139 have been prepared by using a dual-template method where the typical OSDA employed in the crystallization of the zeolite was mixed with tetraethylphosphonium or tetramethylphosphonium hydroxide (Table 1). The presence of tetraalkylammonium (N-OSDA) and tetraalkylphosphonium (P-OSDA) can be confirmed by 13C CP/MAS NMR spectroscopy, and in the case of the AFX zeolite the characteristic shifts of both organic molecules can be distinguished in the 13C CP/MAS NMR spectrum (Fig. 9 left). After calcination, the N-OSDA is decomposed to NOx, CO2 and H2O molecules that can diffuse through the zeolite channels while the P-OSDA decomposes/oxidizes generating PxOy species that remain in the pores and cavities of the material.






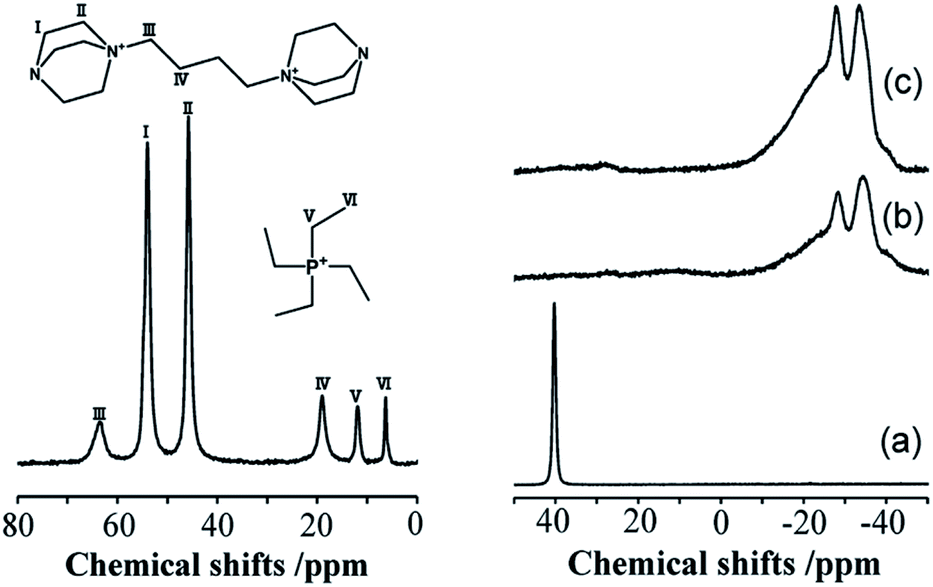 | ||
| Fig. 9 (Left) 13C CP/MAS NMR spectrum of the as-synthesized P-modified AFX and (right) 31P MAS NMR spectra of P-modified AFX zeolites: (a) as-synthesized (run 10), (b) calcined (run 6), and (c) calcined (run 10). Reprinted from ref. 137, with permission from Elsevier. | ||
Besides, 31P MAS NMR spectroscopy is one of the most used techniques to study the phosphorus species located inside zeolite channels and their evolution during heating treatments. On the right side of Fig. 9, the resonance centered at 40 ppm corresponding to tetraethylphosphonium molecules located inside zeolite AFX cavities disappeared upon sample calcination. At the same time, a complex mixture of resonances between −10 and −40 ppm appeared, corresponding to different P-species interacting with the zeolitic framework.
To achieve a further understanding of these complex spectra, Zhao et al.140 calculated the theoretical 31P NMR chemical shifts of extra-framework phosphorus species for the P-CHA system. The authors studied several proposed models of phosphate species interacting with Brønsted acid sites, finding some possible models that could explain the experimental 31P and 27Al MAS NMR spectra. These findings improve the assignment of the 31P NMR resonances in these types of complex systems where broad signals are frequently observed due to the overlap of resonances corresponding to different species and chemical environments.
Danisi et al.141 reported the significant influence of the P content in ZSM-5; at a low phosphorus content, P/Al < 1, a partial dealumination and a decrease of the unit cell volume of 0.25% were observed compared to the parent H-ZSM-5. Meanwhile a high P content, P/Al > 1, leads to more dealumination resulting in the relaxation of the framework and transformation from orthorhombic to monoclinic. This effect agrees with previous studies where it was reported that the orthorhombic/monoclinic phase transformation strongly depends on the chemical composition of the framework and temperature, among other factors.142
The presence of phosphorus species modifies the chemical environment of the Al atoms, as can be observed by 27Al MAS NMR spectroscopy (Fig. 10). Meanwhile the intensity of the characteristic resonance corresponding to tetrahedral framework Al, centered at 55 ppm, decreases with the P content and at the same time the resonances related to distorted tetrahedral and octahedral coordinated Al, appearing between 0 and −10 ppm, increase.143 This P–O–Al interaction is partially reversible, being possible to revert most of the Al to their original tetrahedral coordination by removing the phosphorus species by washing with hot water. However, if a calcination step is performed, the P–O–Al interaction becomes permanent that causes difficulty in the removal of the extra-framework P, which results in the formation of local silicoaluminophosphate interfaces based on the obtained results from 27Al and 31P MAS NMR and K-edge XANES.143
 | ||
| Fig. 10 (a and b) 27Al MAS NMR spectra and (c and d) FT-IR spectra, highlighting the OH-stretch region, and (e and f) 31P MAS NMR spectra of the following samples: H-ZSM-5 impregnated with an aqueous H3PO4 solution (blue) and after washing with hot water (gray), H-ZSM-5 impregnated with an aqueous (NH4)2HPO4 solution (dark green), and after washing with hot water (light green). Reproduced from ref. 143 with permission from the PCCP Owner Societies. | ||
The effect of phosphorus incorporation has been widely studied for the medium pore size MFI zeolite; furthermore, other phosphated 10R structures, such as IMF,144 MWW,121,124 MEL145 and ITH146 have shown enhanced stability against dealumination under steaming conditions.
On the other hand, the stabilization effect of the phosphorus in large pore zeolites *BEA and MOR is still not clear and it seems that thermal treatment after phosphorus incorporation leads to the partial dealumination and formation of extra-framework aluminophosphates.147,148
The dual-template method developed by Sano et al. introduced phosphorus species into small pore zeolites CHA,136 AEI,135 LEV122 and AFX,137 controlling the phosphorus content by adjusting the synthesis conditions and opening the study of the influence of the presence of extra-framework phosphorus species in 8R aluminosilicates. In general, it was observed that the presence of phosphorus avoids the structural collapse of these zeolites after thermal treatments at high temperatures. Zeolites CHA and AEI with a P/Al ratio of 0.51 and 0.69, respectively, presented very high stability, with a relative crystallinity higher than 80% after thermal treatment at 1050 °C (Fig. 11).122,135,136 Meanwhile zeolites LEV (P/Al = 0.23) and AFX (P/Al = 0.20) showed a relative crystallinity close to 60% after thermal treatment at 1000 °C and 900 °C, respectively.122,137 The hydrothermal stability of Cu-containing AEI, CHA and LEV having different P contents was evaluated by a NH3-SCR reaction.122 NO conversion achieved for CHA, AEI and LEV catalysts in the absence of P decreases with the time of hydrothermal treatment at 900 °C in a stream of 10 vol% H2O and 90 vol% air. Meanwhile the presence of P enhances the stability of the zeolites, keeping the NO conversion at high levels, especially in the case of the CHA zeolite, where a NO conversion higher than 80% is observed after 8 hours of hydrothermal treatment, as shown in Fig. 11. The presence of P species not only preserved the zeolite against dealumination but also suppressed the Cu aggregation into CuOx clusters after the hydrothermal treatment.149
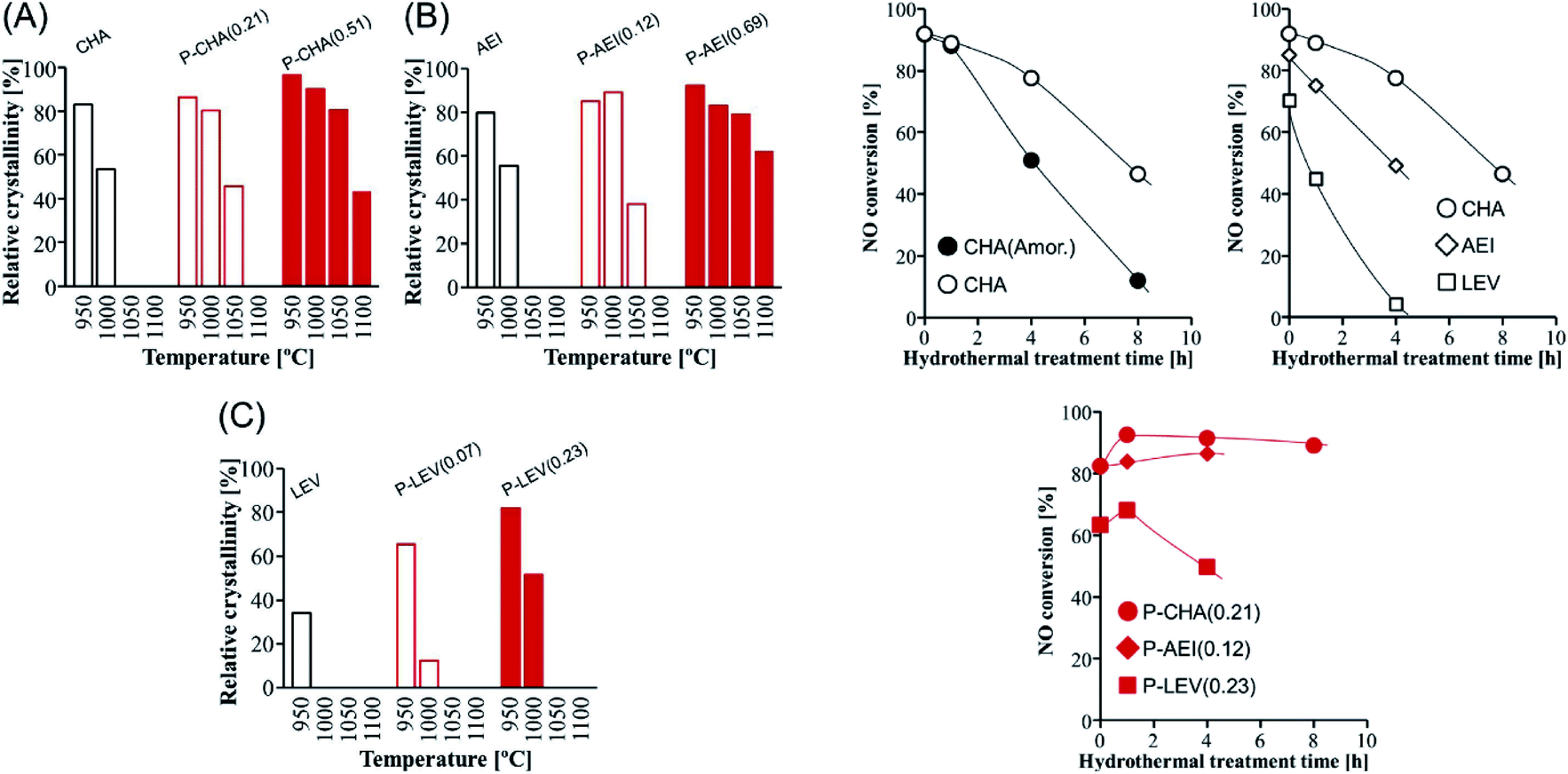 | ||
| Fig. 11 (Left) Relative crystallinity of (A) CHA, (B) AEI, and (C) LEV with and without phosphorus modification after calcination at various temperatures for 1 h. (Right) NO conversion at 200 °C vs. the hydrothermal treatment time over Cu-loaded small-pore zeolite catalysts with and without phosphorus modification. Catalyst = 0.8 g (1.5 mL), Cu content = 1.5 wt%, and flow rate = 1.5 mL min−1 (200 ppm NO, 200 ppm NH3, 10% O2, 3% H2O, and balanced N2). Reprinted from ref. 122, with permission from Elsevier. | ||
Recently, Simancas et al.150 reported the direct synthesis of 8- and 10-ring channel aluminosilicate IFW by using a P-containing OSDA. The presence of extra-framework phosphorus species not only stabilizes the Al in framework positions under steaming conditions at 700 °C but also enhances the catalyst performance in the methanol to olefins reaction.
5.2. Incorporation of extra-framework metal cations
Isolated Cu2+ cations can be found in two locations in the Cu-SSZ-13 zeolite as shown in Fig. 13. Cu2+ balancing 2Al (Cu2+–Z) is located in the d6r and [Cu(OH)]+ interacting with 1Al ([Cu(OH)]+–Z) is located in the CHA cages.81,153 By using DFT simulations, Song et al.153 described the higher hydrothermal stability of Cu2+–2Z than [Cu(OH)]+–Z in the Cu-SSZ-13 zeolite under SCR reaction conditions, which agrees with the experimental results. [Cu(OH)]+–Z species were proposed to be responsible for the Cu agglomeration by hydrolysis and migration through the channels forming small clusters that remain in the pores of the zeolite, confirming the absence of those species on the external surface of the solids by XPS and STEM mapping analysis.
 | ||
| Fig. 13 Schema of Cu2+–2Z and [Cu(OH)]+–Z species and Brønsted acid sites in Cu-SSZ-13. Reprinted with permission from ref. 153. Copyright (2017) American Chemical Society. | ||
Several zeolite topologies have been tested in NH3–SCR reactions but small pore zeolites such as AEI,154 AFX,155 CHA,156 ERI,155 KFI,157 LTA158 and RTH159 showed superior hydrothermal stability. The stabilization of the Cu2+ cation in the double-6-ring (d6r) framework unit in CHA156 and AEI154 zeolites has been shown to be an effective way to enhance the stability properties of the zeolites under steaming conditions and mitigate the dealumination process.
Zeolite LTA showed a similar behavior when the catalyst was fully copper-exchanged. Ryu et al.160 claimed that Cu2+ cations are exclusively located in the single-6-ring (s6r) units, eliminating the requirement of the d6r units in the framework of the catalysts useful for NH3–SCR reactions with good stability properties.
In the case of the Cu–RTH zeolite, the relatively high framework stability after hydrothermal aging at 750 °C was explained by the limited migration of Cu to other locations, keeping good NH3–SCR performance.159 Cu cations, which are commonly incorporated by post-synthesis ion exchange treatments, can also be incorporated by direct synthesis. Cu-SSZ-13 zeolites have been prepared by using a copper–amine complex (Cu–tetraethylenepentamine) as the SDA and Cu source,161,162 which upon calcination treatment decomposes forming CO2, NOx and H2O that diffuse through the zeolitic channels, whereas Cu2+ cations remain inside the cavities of the zeolite.162
Cu2+ cations protect framework Al against dealumination under steaming conditions; however, the hydrothermal stability of the catalyst decreases at high Cu loadings due to the trend of [Cu(OH)]+–Z species to aggregate forming CuOx clusters under harsh conditions.76,161,163 That behavior highlights the importance of the optimization not only of the Cu content but also the cation location in the extra-framework sites and the interaction with isolated or pair Al sites.
The stabilization effect of the introduction of rare earth cations into small pore zeolites has been less explored probably because of the limited diffusion of the relatively large hydrated rare earth ions through the 8R windows. The incorporation of Ce, La, Sm, Y and Yb into the Cu-SSZ-13 zeolites has been reported to enhance the framework stability after hydrothermal aging at 800 °C.161,163,167,168Table 2 shows some recent examples of extra-framework cations used to improve zeolite stability under hydrothermal conditions. Wang et al.163 reported the incorporation of Ce3+ cations to stabilize the Al in framework positions in the zeolite Cu-SSZ-13 after hydrothermal treatment at 800 °C for 20 h; moreover, the H2–TPR results suggested that the Cu2+ cations in the aged Ce-containing samples are mainly located in the 6R, suggesting that the presence of Ce prevents the migration of Cu2+ during the steaming treatment.
| Zeolite | Cation | Treatment | Comments | Ref. |
|---|---|---|---|---|
| USY | La, Ce, Pr, Nd | 100% steam at 800 °C for 2–18 h | Crystallinity retention after steaming follows the order CeY < LaY < PrY < NdY | 165 |
| Cu-SSZ-13 (CHA) | Ce | 10 vol% water vapor in Ar at 720 °C for 10 h (aging) and 800 °C for 20 h (deep aging) | Ce0.93Cu5.65-SSZ-13 prepared for 2 hours of exchange showed the best hydrothermal stability and catalytic performance | 163 |
| Cu-SSZ-13 (CHA) | Ce | 10 vol% water vapor in air at 700, 750, 800 and 850 °C for 5 h | A small amount of Ce (0.2–0.4 wt%) improved the hydrothermal stability of Cu-SSZ-13 (Si/Al = 6.5 and Cu content of 3.4–4.1 wt%) at the same time keeping high activity in the NH3–SCR reaction | 167 |
| Cu-SSZ-13 (CHA) | Ce, La, Sm, Y, Yb | 10 vol% water vapor in air at 800 °C for 16 h | Cu2.8Y1.3-SSZ-13 is the optimum catalyst composition for NH3–SCR activity and hydrothermal stability | 168 |
| Cu-SSZ-13 (CHA) | Ce, Sm | 10 vol% water vapor in air at 800 °C for 8 h | Enhancement of stability by introducing a small amount of Ce or Sm (0.3–0.7 wt%) into Cu-SSZ-13 with a Cu/Al ratio from 0.10 to 0.16 | 161 |
| Cu-SSZ-13 (CHA) | Li, Na, K, Cs, Mg, Ca | 10 vol% water vapor in air at 750 °C for 16 h | Li, Na, K, Cs, Mg, and Ca inhibit Cu-SSZ-13 dealumination | 169 |
| Cu-SSZ-13 (CHA) | Na | 10 vol% water vapor in air at 750 °C or 800 °C for 5 h | Cu2.7Na1.7-SSZ-13 is the optimum catalyst composition for NH3–SCR activity and hydrothermal stability | 170 |
| Cu-SSZ-13 (CHA) | Na | 10 vol% water vapor in air at 750 °C for 16 h | Cu3.9Na0.8-SSZ-13 is the optimum catalyst composition for NH3–SCR activity and hydrothermal stability | 171 |
Zhao et al.168 studied the effect of incorporation of several rare earth cations (Ce, La, Sm, Y, and Yb), finding that Y3+ cations led an enhancement of the hydrothermal stability and catalytic performance of Cu–CHA in the NH3–SCR reaction. However, an excess of Y3+ causes difficulties in the incorporation and diffusion of Cu2+ cations and reduces the activity of the catalyst at low temperatures in the SCR reaction. The preferential location of Y3+ in the 8R was confirmed by two-dimensional 23Na MQ MAS NMR measurements of Na-CHA and Na,Y-CHA, which showed that the signal corresponding to Na located in the 8R decreases for the sample Na,Y-CHA.
The optimum content of rare earth cations is an important parameter. Usui et al.167 studied the effect of different Ce loadings in the Cu-SSZ-13 zeolite introduced by standard wet ion exchange using a cerium acetate aqueous solution and by solid-state ion exchange by mixing cerium acetate monohydrated with Cu-SSZ-13 and subsequently calcinating. The location of Cu and Ce in the structure was studied by several characterization techniques, concluding that the Cu2+ cations are found in the chabazite cage next to the six-membered rings, while the Ce3+ cations are located in the center of the CHA cage.
The enhancement of the Cu-SSZ-13 hydrothermal stability at 850 °C in the presence of a small Ce content (0.2–0.4 wt%) was explained by two possible effects: (1) the cerium cations fill the framework defects protecting the structure and (2) the superior framework stability of the presence of Ce cations on ion-exchange sites instead of protons.
Wang et al.161 also studied the effect of low loadings of Ce and Sm (0.3–0.7 wt%) in Cu-SSZ-13; in this case, the zeolite was prepared by direct synthesis using the complex Cu-tetraethylenepentamine as the structure-directing agent (SDA) and Cu source simultaneously, while Ce and Sm were incorporated by standard ion exchange. The samples modified with Ce and Sm presented a higher fraction of Cu2+–2Z species that are more stable than [Cu(OH)]+–Z (see Section 5.2.1) under steaming conditions, suggesting that the presence of a small amount of Ce and Sm plays an important role in the stability of Cu2+ cations while the contribution to the Al stabilization is limited.
Despite the increasing number of studies, the explanation for the stability enhancement is still not clear. While some authors concluded that the presence of RE mainly stabilizes the zeolite framework167 and limits the dealumination process,168 some others claim that the presence of RE inhibits the agglomeration of Cu2+ into CuOx clusters.161 According to a previous study, the formation of CuOx clusters is the main contribution of the structural collapse of the catalysts during hydrothermal aging.153
Alkali and alkaline earth metals have also been used to improve the hydrothermal stability of zeolites. Gao et al.169 observed that the presence of co-cations, Li+, Na+, K+, Cs+, Mg2+ and Ca2+, prevents the hydrolysis of Si–(OH)–Al bonds of the zeolite Cu-SSZ-13, reducing the dealumination process of the zeolite after treatment in 10% water vapor in an air flow at 750 °C for 16 h. Zhao et al.170 also reported the enhancement of framework stability against the dealumination of Na-containing Cu-SSZ-13 under steaming conditions. However, a further increase of the cation content resulted in the collapse of the structure due to the agglomeration of the active Cu species into CuOx clusters.170–172
6. Summary and outlook
Zeolites have been and are expected to be used as catalysts and adsorbents in various processes. In this review, we introduced the recent research topics focusing on the framework stability improvement of zeolites. The precise control of zeolites at the atomic scale such as structural defects and Al location has a significant effect on stability, while the incorporation of extra-framework species such as phosphorus oxides and metal cations is also crucial.The Al distribution in the framework and the location of the extra-framework phosphorus and Cu cation species showed different stabilization energy, highlighting the importance of the detailed knowledge of the framework to understand and develop new and optimal treatments to enhance the stability. Theoretical studies are potent allies to study those effects and help to explain the experimental results.
The optimization of the improvement of hydrothermal stability strategies described in the review together with the extension of the application to a majority of zeolitic frameworks should open up new applications of zeolites.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the New Energy and Industrial Technology Development Organization (NEDO) Moonshot Project and the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant-in-Aid for Transformative Research Areas(A) JP20A206/20H05880.Notes and references
- http://www.iza-structure.org/databases/, accessed September 26, 2020.
- C. P. Nicholas, Zeolites Ind. Sep. Catal., 2010, 355–402 CrossRef CAS.
- B. Yilmaz and U. Müller, Topics in Catalysis, 2009, vol. 52, pp. 888–895 Search PubMed.
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. F. Peden and J. Szanyi, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC.
- F. Gao and J. Szanyi, Appl. Catal., A, 2018, 560, 185–194 CrossRef CAS.
- İ. A. Reşitoğlu, K. Altinişik and A. Keskin, Clean Technol. Environ. Policy, 2015, 17, 15–27 CrossRef.
- F. J. Keil, Microporous Mesoporous Mater., 1999, 29, 49–66 CrossRef CAS.
- E. T. C. Vogt and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7342–7370 RSC.
- J. Biswas and I. E. Maxwell, Appl. Catal., 1990, 63, 197–258 CrossRef CAS.
- G. Cruciani, J. Phys. Chem. Solids, 2006, 67, 1973–1994 CrossRef CAS.
- A. H. Janssen, A. J. Koster and K. P. de Jong, Angew. Chem., Int. Ed., 2001, 40, 1102–1104 CrossRef CAS.
- K. Iyoki, Y. Yamaguchi, A. Endo, Y. Yonezawa, T. Umeda, H. Yamada, Y. Yanaba, T. Yoshikawa, K. Ohara, K. Yoshida, Y. Sasaki, T. Okubo and T. Wakihara, Microporous Mesoporous Mater., 2018, 268, 77–83 CrossRef CAS.
- Y. Kubota, K. Itabashi, S. Inagaki, Y. Nishita, R. Komatsu, Y. Tsuboi, S. Shinoda and T. Okubo, Chem. Mater., 2014, 26, 1250–1259 CrossRef CAS.
- Y. Wang, R. Otomo, T. Tatsumi and T. Yokoi, Microporous Mesoporous Mater., 2016, 220, 275–281 CrossRef CAS.
- A. Burton, Catal. Rev.: Sci. Eng., 2018, 60, 132–175 CrossRef CAS.
- S. Zones, US Pat., US4544538A, 1985 Search PubMed.
- M. J. Sabater, S. Valencia, F. Rey, A. Corma, J. Rius, S. Francisco, D. Heer and V. Der Zant, Nature, 2004, 431, 287–290 CrossRef PubMed.
- M. A. Camblor, L. A. Villaescusa and M. J. Díaz-Cabañas, Top. Catal., 1999, 9, 59–76 CrossRef CAS.
- T. Moteki and R. F. Lobo, Chem. Mater., 2016, 28, 638–649 CrossRef CAS.
- C. Martínez, D. Verboekend, J. Pérez-Ramírez and A. Corma, Catal. Sci. Technol., 2013, 3, 972–981 RSC.
- G. T. Kerr, J. Phys. Chem., 1967, 71, 4155–4156 CrossRef CAS.
- T. H. Fleisch, B. L. Meyers, G. J. Ray, J. B. Hall and C. L. Marshall, J. Catal., 1986, 99, 117–125 CrossRef CAS.
- S. Li, A. Zheng, Y. Su, H. Zhang, L. Chen, J. Yang, C. Ye and F. Deng, J. Am. Chem. Soc., 2007, 129, 11161–11171 CrossRef CAS.
- C. Schroeder, M. R. Hansen and H. Koller, Angew. Chem., Int. Ed., 2018, 57, 14281–14285 CrossRef CAS PubMed.
- S. Akiyama, H. Mochizuki, H. Yamazaki, T. Yokoi, T. Tatsumi and J. N. Kondo, Mol. Catal., 2017, 433, 48–54 CrossRef CAS.
- Z. Zhu, Z. Xie, Q. Chen, D. Kong, W. Li, W. Yang and C. Li, Microporous Mesoporous Mater., 2007, 101, 169–175 CrossRef CAS.
- S. Prodinger, M. A. Derewinski, A. Vjunov, S. D. Burton, I. Arslan and J. A. Lercher, J. Am. Chem. Soc., 2016, 138, 4408–4415 CrossRef CAS.
- E. F. Sousa-Aguiar, F. E. Trigueiro and F. M. Z. Zotin, Catal. Today, 2013, 218–219, 115–122 CrossRef CAS.
- V. Blay, B. Louis, R. Miravalles, T. Yokoi, K. A. Peccatiello, M. Clough and B. Yilmaz, ACS Catal., 2017, 7, 6542–6566 CrossRef CAS.
- T. Blasco, A. Corma and J. Martínez-Triguero, J. Catal., 2006, 237, 267–277 CrossRef CAS.
- J. A. Lercher and G. Rumplmayr, Appl. Catal., 1986, 25, 215–222 CrossRef CAS.
- Y. Chu, X. Gao, X. Zhang, G. Xu, G. Li and A. Zheng, Phys. Chem. Chem. Phys., 2018, 20, 11702–11712 RSC.
- K. Stanciakova, B. Ensing, F. Göltl, R. E. Bulo and B. M. Weckhuysen, ACS Catal., 2019, 9, 5119–5135 CrossRef CAS.
- C. J. Heard, L. Grajciar, F. Uhlík, M. Shamzhy, M. Opanasenko, J. Čejka and P. Nachtigall, Adv. Mater., 2020, 2003264 CrossRef CAS PubMed.
- T. Sano, N. Yamashita, Y. Iwami, K. Takeda and Y. Kawakami, Zeolites, 1996, 16, 258–264 CrossRef CAS.
- T. Sano, H. Ikeya, T. Kasuno, Z. B. Wang, Y. Kawakami and K. Soga, Zeolites, 1997, 19, 80–86 CrossRef CAS.
- G. Agostini, C. Lamberti, L. Palin, M. Milanesio, N. Danilina, B. Xu, M. Janousch and J. A. van Bokhoven, J. Am. Chem. Soc., 2010, 132, 667–678 CrossRef CAS PubMed.
- M. Müller, G. Harvey and R. Prins, Microporous Mesoporous Mater., 2000, 34, 135–147 CrossRef.
- S. M. T. Almutairi, B. Mezari, E. A. Pidko, P. C. M. M. Magusin and E. J. M. Hensen, J. Catal., 2013, 307, 194–203 CrossRef CAS.
- Y. Ji, J. Birmingham, M. A. Deimund, S. K. Brand and M. E. Davis, Microporous Mesoporous Mater., 2016, 232, 126–137 CrossRef CAS.
- Y. Ji, M. A. Deimund, Y. Bhawe and M. E. Davis, ACS Catal., 2015, 5, 4456–4465 CrossRef CAS.
- R. A. Beyerlein, C. Choi-Feng, J. B. Hall, B. J. Huggins and G. J. Ray, Top. Catal., 1997, 4, 27–42 CrossRef CAS.
- J. Holzinger, P. Beato, L. F. Lundegaard and J. Skibsted, J. Phys. Chem. C, 2018, 122, 15595–15613 CrossRef CAS.
- Z. Ristanović, J. P. Hofmann, G. De Cremer, A. V Kubarev, M. Rohnke, F. Meirer, J. Hofkens, M. B. J. Roeffaers and B. M. Weckhuysen, J. Am. Chem. Soc., 2015, 137, 6559–6568 CrossRef PubMed.
- D. E. Perea, I. Arslan, J. Liu, Z. Ristanović, L. Kovarik, B. W. Arey, J. A. Lercher, S. R. Bare and B. M. Weckhuysen, Nat. Commun., 2015, 6, 7589 CrossRef.
- Z. Yu, A. Zheng, Q. Wang, L. Chen, J. Xu, J.-P. Amoureux and F. Deng, Angew. Chem., Int. Ed., 2010, 49, 8657–8661 CrossRef CAS PubMed.
- S. Malola, S. Svelle, F. L. Bleken and O. Swang, Angew. Chem., Int. Ed., 2012, 51, 652–655 CrossRef CAS PubMed.
- M. Nielsen, A. Hafreager, R. Y. Brogaard, K. De Wispelaere, H. Falsig, P. Beato, V. Van Speybroeck and S. Svelle, Catal. Sci. Technol., 2019, 9, 3721–3725 RSC.
- M.-C. Silaghi, C. Chizallet, E. Petracovschi, T. Kerber, J. Sauer and P. Raybaud, ACS Catal., 2015, 5, 11–15 CrossRef CAS.
- J. Li, A. Corma and J. Yu, Chem. Soc. Rev., 2015, 44, 7112–7127 RSC.
- C. S. Cundy and P. A. Cox, Chem. Rev., 2003, 103, 663–702 CrossRef CAS PubMed.
- H. Al Jabri, K. Miyake, K. Ono, M. Nakai, R. Inoue, Y. Hirota, Y. Uchida, Y. Wang, T. Nishitoba, T. Yokoi, T. Ohnishi, M. Ogura and N. Nishiyama, Microporous Mesoporous Mater., 2020, 297, 109780 CrossRef CAS.
- S. Prodinger and M. A. Derewinski, Pet. Chem., 2020, 60, 420–436 CrossRef CAS.
- L. H. Ong, M. Dömök, R. Olindo, A. C. van Veen and J. a. Lercher, Microporous Mesoporous Mater., 2012, 164, 9–20 CrossRef CAS.
- S. M. Maier, A. Jentys and J. A. Lercher, J. Phys. Chem. C, 2011, 115, 8005–8013 CrossRef CAS.
- K. Iyoki, K. Itabashi and T. Okubo, Microporous Mesoporous Mater., 2014, 189, 22–30 CrossRef CAS.
- Y. Wang, Q. Wu, X. Meng and F. S. Xiao, Engineering, 2017, 3, 567–574 CrossRef CAS.
- B. Xie, J. Song, L. Ren, Y. Ji, J. Li and F.-S. Xiao, Chem. Mater., 2008, 20, 4533–4535 CrossRef CAS.
- Y. Kamimura, W. Chaikittisilp, K. Itabashi, A. Shimojima and T. Okubo, Chem.–Asian J., 2010, 5, 2182–2191 CrossRef CAS PubMed.
- A. W. Burton, S. I. Zones and S. Elomari, Curr. Opin. Colloid Interface Sci., 2005, 211–219 CrossRef CAS.
- S. I. Zones, Y. Nakagawa, L. T. Yuen and T. V. Harris, J. Am. Chem. Soc., 1996, 118, 7558–7567 CrossRef CAS.
- A. Corma and M. E. Davis, ChemPhysChem, 2004, 5, 304–313 CrossRef CAS PubMed.
- A. V. Goretsky, L. W. Beck, S. I. Zones and M. E. Davis, Microporous Mesoporous Mater., 1999, 28, 387–393 CrossRef CAS.
- M. E. Davis and R. F. Lobo, Chem. Mater., 1992, 4(4), 756–768 CrossRef CAS.
- Y. Kalvachev, M. Jaber, V. Mavrodinova, L. Dimitrov, D. Nihtianova and V. Valtchev, Microporous Mesoporous Mater., 2013, 177, 127–134 CrossRef CAS.
- J. Arichi and B. Louis, Cryst. Growth Des., 2008, 8, 3999–4005 CrossRef CAS.
- M. Li, Y. Zhou, C. Ju and Y. Fang, Appl. Catal., A, 2016, 512, 1–8 CrossRef CAS.
- C. A. Fyfe, D. H. Brouwer, A. R. Lewis and J. M. Chézeau, J. Am. Chem. Soc., 2001, 123, 6882–6891 CrossRef CAS.
- M. Kato, K. Itabashi, A. Matsumoto and K. Tsutsumi, J. Phys. Chem. B, 2003, 107, 1788–1797 CrossRef CAS.
- J. M. Silva, M. F. Ribeiro, F. R. Ribeiro, N. S. Gnep, M. Guisnet and E. Benazzi, React. Kinet. Catal. Lett.React. Kinet. Catal. Lett., 1995, 54, 209–215 CrossRef CAS.
- S. Y. Chung, S.-H. Oh, M. H. Kim, I.-S. Nam and Y. G. Kim, Catal. Today, 1999, 54, 521–529 CrossRef CAS.
- E. Bourgeat Lami, F. Fajula, D. Anglerot and T. Des Couriere, Microporous Mater., 1993, 1, 237–245 CrossRef.
- Z. Shi, P. Ji, Z. Zhu, J. G. Jiang, W. Fu, P. Wu, Y. Wang and M. He, ChemCatChem, 2016, 8, 1891–1895 CrossRef CAS.
- H. Jiang, B. Guan, H. Lin and Z. Huang, Fuel, 2019, 255, 115587 CrossRef CAS.
- C. Fan, Z. Chen, L. Pang, S. Ming, X. Zhang, K. B. Albert, P. Liu, H. Chen and T. Li, Appl. Catal., A, 2018, 550, 256–265 CrossRef CAS.
- S. Han, Q. Ye, S. Cheng, T. Kang and H. Dai, Catal. Sci. Technol., 2017, 7, 703–717 RSC.
- S. Shwan, J. Jansson, J. Korsgren, L. Olsson and M. Skoglundh, Catal. Today, 2012, 197, 24–37 CrossRef CAS.
- M. Cheng, B. Jiang, S. Yao, J. Han, S. Zhao, X. Tang, J. Zhang and T. Wang, J. Phys. Chem. C, 2018, 122, 455–464 CrossRef CAS.
- C. H. F. Peden, J. H. Kwak, S. D. Burton, R. G. Tonkyn, D. H. Kim, J. H. Lee, H. W. Jen, G. Cavataio, Y. Cheng and C. K. Lambert, Catal. Today, 2012, 184, 245–251 CrossRef CAS.
- L. Ma, Y. Cheng, G. Cavataio, R. W. McCabe, L. Fu and J. Li, Chem. Eng. J., 2013, 225, 323–330 CrossRef CAS.
- Y. J. Kim, J. K. Lee, K. M. Min, S. B. Hong, I.-S. Nam and B. K. Cho, J. Catal., 2014, 311, 447–457 CrossRef CAS.
- N. Wilken, K. Wijayanti, K. Kamasamudram, N. W. Currier, R. Vedaiyan, A. Yezerets and L. Olsson, Appl. Catal., B, 2012, 111–112, 58–66 CAS.
- Y. Shan, J. Du, Y. Yu, W. Shan, X. Shi and H. He, Appl. Catal., B, 2020, 266, 118655 CrossRef.
- K. Miyake, R. Inoue, T. Miura, M. Nakai, H. Al-Jabri, Y. Hirota, Y. Uchida, S. Tanaka, M. Miyamoto, S. Inagaki, Y. Kubota, C. Y. Kong and N. Nishiyama, Microporous Mesoporous Mater., 2019, 288, 109523 CrossRef CAS.
- Y. Román-Leshkov, M. Moliner and M. E. Davis, J. Phys. Chem. C, 2011, 115, 1096–1102 CrossRef.
- K. Muraoka, W. Chaikittisilp and T. Okubo, J. Am. Chem. Soc., 2016, 138, 6184–6193 CrossRef CAS PubMed.
- S. Wang, Y. He, W. Jiao, J. Wang and W. Fan, Curr. Opin. Chem. Eng., 2019, 23, 146–154 CrossRef.
- T. Yokoi, H. Mochizuki, S. Namba, J. N. Kondo and T. Tatsumi, J. Phys. Chem. C, 2015, 119, 15303–15315 CrossRef CAS.
- A. Vjunov, J. L. Fulton, T. Huthwelker, S. Pin, D. Mei, G. K. Schenter, N. Govind, D. M. Camaioni, J. Z. Hu and J. A. Lercher, J. Am. Chem. Soc., 2014, 136, 8296–8306 CrossRef CAS PubMed.
- A. B. Pinar, R. Verel, J. Pérez-Pariente and J. A. Van Bokhoven, Microporous Mesoporous Mater., 2014, 193, 111–114 CrossRef CAS.
- P. Sazama, E. Tabor, P. Klein, B. Wichterlova, S. Sklenak, L. Mokrzycki, V. Pashkkova, M. Ogura and J. Dedecek, J. Catal., 2016, 333, 102–114 CrossRef CAS.
- J. Dedecek, V. Balgová, V. Pashkova, P. Klein and B. Wichterlová, Chem. Mater., 2012, 24, 3231–3239 CrossRef CAS.
- J. Dědeček, E. Tabor and S. Sklenak, ChemSusChem, 2019, 12, 556–576 CrossRef PubMed.
- V. Pashkova, S. Sklenak, P. Klein, M. Urbanova and J. Dědeček, Chem.–Eur. J., 2016, 22, 3937–3941 CrossRef CAS PubMed.
- B. Lu, T. Kanai, Y. Oumi and T. Sano, J. Porous Mater., 2007, 14, 89–96 CrossRef CAS.
- A. Janda and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 19193–19207 CrossRef CAS PubMed.
- M. C. Silaghi, C. Chizallet, J. Sauer and P. Raybaud, J. Catal., 2016, 339, 242–255 CrossRef CAS.
- T. Nishitoba, N. Yoshida, J. N. Kondo and T. Yokoi, Ind. Eng. Chem. Res., 2018, 57, 3914–3922 CrossRef CAS.
- M. A. Deimund, L. Harrison, J. D. Lunn, Y. Liu, A. Malek, R. Shayib and M. E. Davis, ACS Catal., 2016, 6, 542–550 CrossRef CAS.
- J. Dědeček, Z. Sobalík and B. Wichterlová, Catal. Rev., 2012, 54, 135–223 CrossRef.
- K. A. Tarach, J. Martinez-Triguero, F. Rey and K. Góra-Marek, J. Catal., 2016, 339, 256–269 CrossRef CAS.
- Z. Zhao, Y. Xing, S. Li, X. Meng, F. S. Xiao, R. McGuire, A. N. Parvulescu, U. Müller and W. Zhang, J. Phys. Chem. C, 2018, 122, 9973–9979 CrossRef CAS.
- L. Li, Y. Chen, S. Xu, J. Li, M. Dong, Z. Liu, H. Jiao, J. Wang and W. Fan, J. Catal., 2016, 344, 242–251 CrossRef CAS.
- J. M. Chezeau, L. Delmotte, J. L. Guth and Z. Gabelica, Zeolites, 1991, 11, 598–606 CrossRef CAS.
- G. Brunklaus, H. Koller and S. I. Zones, Angew. Chem., Int. Ed., 2016, 55, 14459–14463 CrossRef CAS PubMed.
- I. Grosskreuz, H. Gies and B. Marler, Microporous Mesoporous Mater., 2020, 291, 109683 CrossRef CAS.
- E. Flanigen and R. Patton, US Pat., US4073865A, 1978 Search PubMed.
- J. M. Chézeau, L. Delmotte, J. L. Guth and M. Soulard, Zeolites, 1989, 9, 78–80 CrossRef.
- P. Losch, A. B. Pinar, M. G. Willinger, K. Soukup, S. Chavan, B. Vincent, P. Pale and B. Louis, J. Catal., 2017, 345, 11–23 CrossRef CAS.
- D. Shi, K. Haw, C. Kouvatas, L. Tang, Y. Zhang, Q. Fang, S. Qiu and V. Valtchev, Angew. Chem., 2020, 132, 19744–19749 CrossRef.
- M. Grahn, A. Faisal, O. G. W. Öhrman, M. Zhou, M. Signorile, V. Crocellà, M. S. Nabavi and J. Hedlund, Catal. Today, 2020, 345, 136–146 CrossRef CAS.
- M. S. Nabavi, M. Zhou, J. Mouzon, M. Grahn and J. Hedlund, Microporous Mesoporous Mater., 2019, 278, 167–174 CrossRef CAS.
- S. Prodinger, H. Shi, S. Eckstein, J. Z. Hu, M. V. Olarte, D. M. Camaioni, M. A. Derewinski and J. A. Lercher, Chem. Mater., 2017, 29, 7255–7262 CrossRef CAS.
- S. Prodinger, H. Shi, H. Wang, M. A. Derewinski and J. A. Lercher, Appl. Catal., B, 2018, 237, 996–1002 CrossRef CAS.
- H. T. Vu, F. M. Harth and N. Wilde, Front. Chem., 2018, 6, 1–9 CrossRef PubMed.
- T. Yoshioka, Z. Liu, K. Iyoki, T. Sano, M. Ando, S. Sukenaga, H. Shibata, T. Okubo and T. Wakihara, Chem. Lett., 2020, 49, 1006–1008 CrossRef CAS.
- K. Iyoki, K. Kikumasa, T. Onishi, Y. Yonezawa, A. Chokkalingam, Y. Yanaba, T. Matsumoto, R. Osuga, S. P. Elangovan, J. N. Kondo, A. Endo, T. Okubo and T. Wakihara, J. Am. Chem. Soc., 2020, 142, 3931–3938 CrossRef CAS PubMed.
- H. E. Van Der Bij, F. Meirer, S. Kalirai, J. Wang and B. M. Weckhuysen, Chem.–Eur. J., 2014, 20, 16922–16932 CrossRef CAS PubMed.
- G. Caeiro, P. Magnoux, J. M. Lopes, F. R. Ribeiro, S. M. C. Menezes, A. F. Costa and H. S. Cerqueira, Appl. Catal., A, 2006, 314, 160–171 CrossRef CAS.
- H. E. van der Bij and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7406–7428 RSC.
- X. Wang, W. Dai, G. Wu, L. Li, N. Guan and M. Hunger, Microporous Mesoporous Mater., 2012, 151, 99–106 CrossRef CAS.
- Y. Kakiuchi, T. Tanigawa, N. Tsunoji, Y. Takamitsu, M. Sadakane and T. Sano, Appl. Catal., A, 2019, 575, 204–213 CrossRef CAS.
- H. E. van der Bij and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2014, 16, 9892–9903 RSC.
- S. Bao, M. Guo, B. Liu, B. Feng, D. Yin, B. Jiang and H. Zhao, React. Kinet., Mech. Catal., 2021, 132, 431–447 CrossRef CAS.
- X. Gao, Z. Tang, H. Zhang, C. Liu, Z. Zhang, G. Lu and D. Ji, Korean J. Chem. Eng., 2010, 27, 812–815 CrossRef CAS.
- A. Jentys, G. Rumplmayr and J. A. Lercher, Appl. Catal., 1989, 53, 299–312 CrossRef CAS.
- Y.-J. Lee, J. M. Kim, J. W. Bae, C.-H. Shin and K.-W. Jun, Fuel, 2009, 88, 1915–1921 CrossRef CAS.
- G. Rumplmayr and J. A. Lercher, Zeolites, 1990, 10, 283–287 CrossRef CAS.
- F. Rey and J. Simancas, in Insights into the Chemistry of Organic Structure-Directing Agents in the Synthesis of Zeolitic Materials, ed. L. Gómez-Hortigüela, 2018, pp. 103–138 Search PubMed.
- J. Simancas, R. Simancas, P. J. Bereciartua, J. L. Jorda, F. Rey, A. Corma, S. Nicolopoulos, P. Pratim Das, M. Gemmi and E. Mugnaioli, J. Am. Chem. Soc., 2016, 138, 10116–10119 CrossRef CAS PubMed.
- Y. Yun, M. Hernández, W. Wan, X. Zou, J. L. Jordá, A. Cantín, F. Rey and A. Corma, Chem. Commun., 2015, 51, 7602–7605 RSC.
- R. Simancas, J. L. Jordá, F. Rey, A. Corma, A. Cantín, I. Peral and C. Popescu, J. Am. Chem. Soc., 2014, 136, 3342–3345 CrossRef CAS PubMed.
- T. Maruo, N. Yamanaka, N. Tsunoji, M. Sadakane and T. Sano, Chem. Lett., 2014, 43, 302–304 CrossRef CAS.
- Y. Kakiuchi, Y. Yamasaki, N. Tsunoji, Y. Takamitsu, M. Sadakane and T. Sano, Chem. Lett., 2016, 45, 122–124 CrossRef CAS.
- T. Sonoda, T. Maruo, Y. Yamasaki, N. Tsunoji, Y. Takamitsu, M. Sadakane and T. Sano, J. Mater. Chem. A, 2015, 3, 857–865 RSC.
- Y. Yamasaki, N. Tsunoji, Y. Takamitsu, M. Sadakane and T. Sano, Microporous Mesoporous Mater., 2016, 223, 129–139 CrossRef CAS.
- E. Mitani, Y. Yamasaki, N. Tsunoji, M. Sadakane and T. Sano, Microporous Mesoporous Mater., 2018, 267, 192–197 CrossRef CAS.
- E. Mitani, N. Tsunoji, M. Sadakane and T. Sano, J. Porous Mater., 2019, 26, 1345–1352 CrossRef CAS.
- Y. Naraki, K. Ariga, K. Nakamura, K. Okushita and T. Sano, Microporous Mesoporous Mater., 2017, 254, 160–169 CrossRef CAS.
- P. Zhao, B. Boekfa, T. Nishitoba, N. Tsunoji, T. Sano, T. Yokoi, M. Ogura and M. Ehara, Microporous Mesoporous Mater., 2020, 294, 109908 CrossRef CAS.
- R. M. Danisi, J. E. Schmidt, A. Lucini Paioni, K. Houben, J. D. Poplawsky, M. Baldus, B. M. Weckhuysen and E. T. C. Vogt, Phys. Chem. Chem. Phys., 2018, 20, 27766–27777 RSC.
- H. van Koningsveld, J. C. Jansen and H. van Bekkum, Zeolites, 1987, 7, 564–568 CrossRef CAS.
- H. E. van der Bij and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2014, 16, 9892–9903 RSC.
- A. Corma, J. Mengual and P. J. Miguel, Appl. Catal., A, 2013, 460–461, 106–115 CrossRef CAS.
- P. Li, W. Zhang, X. Han and X. Bao, Catal. Lett., 2010, 134, 124–130 CrossRef CAS.
- P. Zeng, Y. Liang, S. Ji, B. Shen, H. Liu, B. Wang, H. Zhao and M. Li, J. Energy Chem., 2014, 23, 193–200 CrossRef CAS.
- A. F. Costa, H. S. Cerqueira, J. M. Ferreira, N. M. Ruiz and S. M. Menezes, Appl. Catal., A, 2007, 319, 137–143 CrossRef CAS.
- C. Manrique, A. Guzmán, R. Solano and A. Echavarría, Energy Fuels, 2019, 33, 3483–3491 CrossRef CAS.
- H. Kubota, C. Liu, T. Amada, K. Kon, T. Toyao, Z. Maeno, K. Ueda, A. Satsuma, N. Tsunoji, T. Sano and K. Shimizu, Catal. Today, 2020 DOI:10.1016/j.cattod.2020.07.084.
- R. Simancas, T. Nishitoba, S. Park, J. N. Kondo, F. Rey, H. Gies and T. Yokoi, Microporous Mesoporous Mater., 2021, 317, 111005 CrossRef CAS.
- J. N. Louwen, L. van Eijck, C. Vogt and E. T. C. Vogt, Chem. Mater., 2020, 32, 9390–9403 CrossRef CAS.
- E. Borfecchia, P. Beato, S. Svelle, U. Olsbye, C. Lamberti and S. Bordiga, Chem. Soc. Rev., 2018, 47, 8097–8133 RSC.
- J. Song, Y. Wang, E. D. Walter, N. M. Washton, D. Mei, L. Kovarik, M. H. Engelhard, S. Prodinger, Y. Wang, C. H. F. Peden and F. Gao, ACS Catal., 2017, 7, 8214–8227 CrossRef CAS.
- M. Moliner, C. Franch, E. Palomares, M. Grill and A. Corma, Chem. Commun., 2012, 48, 8264–8266 RSC.
- N. Martín, C. Paris, P. N. R. Vennestrøm, J. R. Thøgersen, M. Moliner and A. Corma, Appl. Catal., B, 2017, 217, 125–136 CrossRef.
- D. W. Fickel and R. F. Lobo, J. Phys. Chem. C, 2010, 114, 1633–1640 CrossRef CAS.
- J. Kim, S. J. Cho and D. H. Kim, ACS Catal., 2017, 7, 6070–6081 CrossRef CAS.
- D. Jo, T. Ryu, G. T. Park, P. S. Kim, C. H. Kim, I.-S. Nam and S. B. Hong, ACS Catal., 2016, 6, 2443–2447 CrossRef CAS.
- Y. Shan, X. Shi, J. Du, Y. Yu and H. He, Catal. Sci. Technol., 2019, 9, 106–115 RSC.
- T. Ryu, N. H. Ahn, S. Seo, J. Cho, H. Kim, D. Jo, G. T. Park, P. S. Kim, C. H. Kim, E. L. Bruce, P. A. Wright, I.-S. Nam and S. B. Hong, Angew. Chem., Int. Ed., 2017, 56, 3256–3260 CrossRef CAS PubMed.
- Y. Wang, X. Shi, Y. Shan, J. Du, K. Liu and H. He, Ind. Eng. Chem. Res., 2020, 59, 6416–6423 CrossRef CAS.
- L. Ren, L. Zhu, C. Yang, Y. Chen, Q. Sun, H. Zhang, C. Li, F. Nawaz, X. Meng and F.-S. Xiao, Chem. Commun., 2011, 47, 9789–9791 RSC.
- J. Wang, Z. Peng, H. Qiao, H. Yu, Y. Hu, L. Chang and W. Bao, Ind. Eng. Chem. Res., 2016, 55, 1174–1182 CrossRef CAS.
- F. E. Trigueiro, D. F. J. Monteiro, F. M. Z. Zotin and E. Falabella Sousa-Aguiar, J. Alloys Compd., 2002, 344, 337–341 CrossRef CAS.
- X. Du, X. Gao, H. Zhang, X. Li and P. Liu, Catal. Commun., 2013, 35, 17–22 CrossRef CAS.
- J. N. Louwen, S. Simko, K. Stanciakova, R. E. Bulo, B. M. Weckhuysen and E. T. C. Vogt, J. Phys. Chem. C, 2020, 124, 4626–4636 CrossRef CAS.
- T. Usui, Z. Liu, S. Ibe, J. Zhu, C. Anand, H. Igarashi, N. Onaya, Y. Sasaki, Y. Shiramata, T. Kusamoto and T. Wakihara, ACS Catal., 2018, 8, 9165–9173 CrossRef CAS.
- Z. Zhao, R. Yu, C. Shi, H. Gies, F.-S. Xiao, D. De Vos, T. Yokoi, X. Bao, U. Kolb, R. McGuire, A.-N. Parvulescu, S. Maurer, U. Müller and W. Zhang, Catal. Sci. Technol., 2019, 9, 241–251 RSC.
- F. Gao, Y. Wang, N. M. Washton, M. Kollár, J. Szanyi and C. H. F. Peden, ACS Catal., 2015, 5, 6780–6791 CrossRef CAS.
- Z. Zhao, R. Yu, R. Zhao, C. Shi, H. Gies, F.-S. Xiao, D. De Vos, T. Yokoi, X. Bao, U. Kolb, M. Feyen, R. McGuire, S. Maurer, A. Moini, U. Müller and W. Zhang, Appl. Catal., B, 2017, 217, 421–428 CrossRef CAS.
- L. Xie, F. Liu, X. Shi, F.-S. Xiao and H. He, Appl. Catal., B, 2015, 179, 206–212 CrossRef CAS.
- Y. Cui, Y. Wang, D. Mei, E. D. Walter, N. M. Washton, J. D. Holladay, Y. Wang, J. Szanyi, C. H. F. Peden and F. Gao, J. Catal., 2019, 378, 363–375 CrossRef CAS.
- W. W. Kaeding and S. A. Butter, J. Catal., 1980, 61, 155–164 CrossRef CAS.
- J. Caro, M. Bülow, M. Derewinski, J. Haber, M. Hunger, J. Kärger, H. Pfeifer, W. Storek and B. Zibrowius, J. Catal., 1990, 124, 367–375 CrossRef CAS.
- J. Zhuang, D. Ma, G. Yang, Z. Yan, X. Liu, X. Liu, X. Han, X. Bao, P. Xie and Z. Liu, J. Catal., 2004, 228, 234–242 CrossRef CAS.
- S. M. Abubakar, D. M. Marcus, J. C. Lee, J. O. Ehresmann, C.-Y. Chen, P. W. Kletnieks, D. R. Guenther, M. J. Hayman, M. Pavlova, J. B. Nicholas and J. F. Haw, Langmuir, 2006, 22, 4846–4852 CrossRef CAS PubMed.
- N. Xue, X. Chen, L. Nie, X. Guo, W. Ding, Y. Chen, M. Gu and Z. Xie, J. Catal., 2007, 248, 20–28 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |