
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtroposelective synthesis of N-aryl peptoid atropisomers via a palladium(II)-catalyzed asymmetric C–H alkynylation strategy†‡
Yong-Jie
Wu§
a,
Pei-Pei
Xie§
a,
Gang
Zhou
a,
Qi-Jun
Yao
*a,
Xin
Hong
 *a and
Bing-Feng
Shi
*ab
*a and
Bing-Feng
Shi
*ab
aCenter of Chemistry for Frontier Technologies, Department of Chemistry, Zhejiang University, Hangzhou 310027, China. E-mail: 3110000156@zju.edu.cn; hxchem@zju.edu.cn; bfshi@zju.edu.cn
bGreen Catalysis Center, and College of Chemistry, Zhengzhou University, Zhengzhou 450001, China
First published on 9th June 2021
Abstract
The introduction of chirality into peptoids is an important strategy to determine a discrete and robust secondary structure. However, the lack of an efficient strategy for the synthesis of structurally diverse chiral peptoids has hampered the studies. Herein, we report the efficient synthesis of a wide variety of N-aryl peptoid atropisomers in good yields with excellent enantioselectivities (up to 99% yield and 99% ee) by palladium-catalyzed asymmetric C–H alkynylation. The inexpensive and commercially available L-pyroglutamic acid was used as an efficient chiral ligand. The exceptional compatibility of the C–H alkynylation with various peptoid oligomers renders this procedure valuable for peptoid modifications. Computational studies suggested that the amino acid ligand distortion controls the enantioselectivity in the Pd/L-pGlu-catalyzed C–H bond activation step.
Introduction
Peptoids are a new family of non-natural, sequence-specific peptidomimetic oligomers composed of diverse N-substituted glycine units (Fig. 1A).1 Due to the ease of modular synthesis and their enhanced proteolytic stability and increased cell permeability, peptoids hold great potential for therapeutic applications and drug delivery.2a–d Peptoids also have been shown to possess significant promise in materials2e and catalysis.2f Compared to the analogous peptides, one of the major limitations of peptoids is the lack of chirality in the backbone, which inevitably led to the formation of a mixture of right- and left-handed helical secondary structures. Therefore, strategic introduction of chirality into peptoids to determine a discrete and robust conformation has received significant attention. To date, two types of chiral peptoids, including oligomers with N-α-chiral side chains (point chirality) and atropisomeric monomers with bulky ortho-substituted N-aryl side chains (N–C axial chirality), have been reported. The former was synthesized by the assembly of submonomers containing a chiral center, generally derived from a chiral primary amine (R*NH2, Fig. 1B(1)),4 through a classical submonomer synthetic protocol.5 Pioneering studies by Zuckermann and Barron have shown that peptoid backbones may be forced by the presence of N-α-chiral side chains to adopt the conformation close to the helix, despite being devoid of hydrogen bonds, and the helical secondary structures could diversify the functions of peptoids.3,4 The latter was first realized by Kirshenbaum and co-workers in 2011. They reported that peptoids incorporating bulky ortho-substituted N-aryl side chains exhibited confirmationally stable N–C axial chirality, compensating the deficiency of achirality of peptoids (Fig. 1B(2)).6–8 However, the peptoid atropisomers were prepared in racemic form and isolated by preparative chiral HPLC. In sharp contrast to the stereochemistry of natural peptides, the stereochemical features of these atropisomeric N-aryl peptoids could be dynamic at elevated temperature.6a However, due to the lack of efficient methods for the asymmetric synthesis, this novel type of chiral peptoid has not been investigated thoroughly.9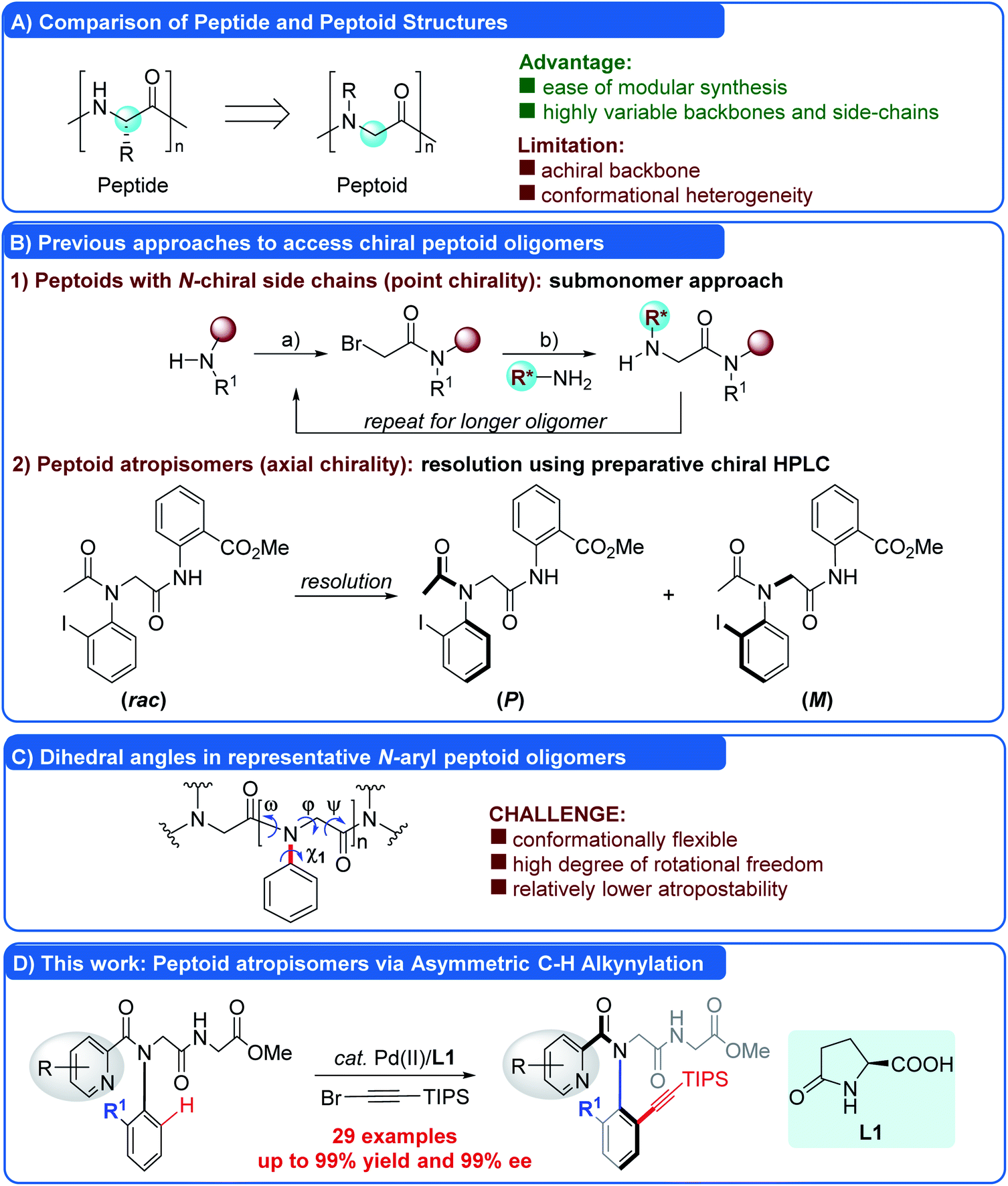 | ||
| Fig. 1 Approaches to access chiral peptoid oligomers and our strategy. | ||
Recently, asymmetric C–H functionalization has emerged as a powerful synthetic protocol in the construction of atropisomers.10 We envisioned that the incorporation of a bulky ortho-substituent via an asymmetric C–H functionalization strategy to constrain the side chain dihedral angle (χ1) (Fig. 1C) would enable the diverse and straightforward access of these N-aryl peptoid atropisomers, a strategy which has not been realized so far. Although this approach sounds promising, the enantioselective C–H functionalization of N-aryl peptoids is not straightforward, due to the following daunting challenges: (1) the peptoid skeleton is generally flexible and multiple rotations around the backbone or side chain (ω, φ, ψ, and χ1 dihedral angles) inherently exist, rendering the conformation and enantiocontrol much more complicated and difficult (Fig. 1C); (2) the asymmetric C–H functionalization11 has to be conducted under mild conditions in order to ensure good enantiocontrol and preserve the enantiopurity because of the relatively low rotation barriers of the resulting atropisomers. Thus, the rational design of the structure of peptoid substrates, such as suitable ortho-substituted N-aryl side chains to constrain the dihedral angle (χ1), would be critical for the success; (3) in order to meet the requirements of high enantioselectivity and atom-economy, the chiral ligand must be readily available and inexpensive. As a part of our longstanding efforts to construct atropisomers via asymmetric C–H activation,9m,12 herein we describe a strategy that surmounts these challenges, enabling the synthesis of N-aryl peptoid atropisomers via Pd(II)-catalyzed C–H alkynylation using readily available L-pGlu-OH as an inexpensive chiral ligand (Fig. 1D). A wide range of N-aryl peptoid atropisomers were obtained in excellent yields and enantioselectivities (up to 99% yield and 99% ee). It offered a streamlined method for the asymmetric synthesis of N-aryl peptoid atropisomers in a highly efficient, practical, atom-economical and isolable manner.
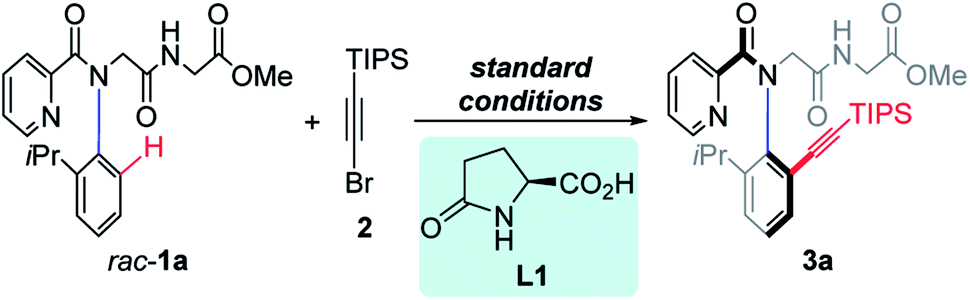
We initiated our research by investigating the reaction of rac-1a with TIPS-protected alkynyl bromide (2). The desired product 3a was obtained in 78% isolated yield with 93% ee when using 10 mol% Pd(MeCN)2Cl2 as the catalyst, 20 mol% L-pGlu-OH (L1) as the chiral ligand and 3.0 equiv. of Ag2CO3 both as the additive and Br-scavenger in HFIP at 60 °C for 8 hours under air (Table 1, entry 1, standard conditions). Then control experiments to study the effect of several additives were performed (entries 2–4). The addition of AcOH or NaHCO3 resulted in reduced yield and ee (entry 2, 76%, 36% ee; entry 3, 60%, 89% ee). Obviously, the acidity of the reaction system was critical to this reaction. The addition of water led to significantly reduced yield with maintained enantioselectivity (entry 4, 37%, 93% ee). A prolonged reaction time resulted in remarkably improved conversion but reduced ee (entry 5, 99%, 71% ee). Reducing the reaction concentration revealed that the reactivity was dramatically decreased without affecting the enantioselectivity (entry 6, 43%, 93% ee). The desired product was obtained in moderate yield and poor enantioselectivity when the reaction temperature was elevated to 80 °C (entry 7, 76%, 46% ee). Only trace racemic product was observed in the absence of L-pGlu-OH (L1) (entry 8, 2%, 0% ee), indicating a dramatic promotion of the ligand. When D-pGlu-OH was used, the other atropisomer was obtained (entry 9, 66%, −93% ee). Notably, both the racemic starting material rac-1a and the enantiopure product 3a exist as a couple of Z/E rotamers as observed in 1H NMR, because of the rotation of the amide bond ω (Fig. 1C), and E-rotamers were formed predominantly.9m,13 The geometry of the E-amide bond also could be observed clearly in the crystal structure of peptoid dimer 3hb.14
|
|
|||
|---|---|---|---|
| Entry | Deviation from standard conditions | Yieldb (%) | eec (%) |
| a Standard conditions: rac-1a (0.1 mmol), 2 (4.0 equiv.), Pd(MeCN)2Cl2 (10 mol%), L1 (0.2 equiv.), Ag2CO3 (3.0 equiv.), HFIP (0.1 mL), 60 °C, 8 h under air. b Determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as the internal standard. c Determined by chiral HPLC. d Isolated yield. | |||
| 1 | None | 78d | 93 |
| 2 | Add AcOH (40 μL) | 76 | 36 |
| 3 | Add NaHCO3 (1.0 equiv.) | 60 | 89 |
| 4 | Add H2O (40 μL) | 37 | 93 |
| 5 | 24 h instead of 8 h | 99 | 71 |
| 6 | HFIP (0.2 M) instead of HFIP (1.0 M) | 43 | 93 |
| 7 | 80 °C instead of 60 °C | 76 | 46 |
| 8 | No L1 | 2 | 0 |
| 9 | D-L1 | 66 | −93 |
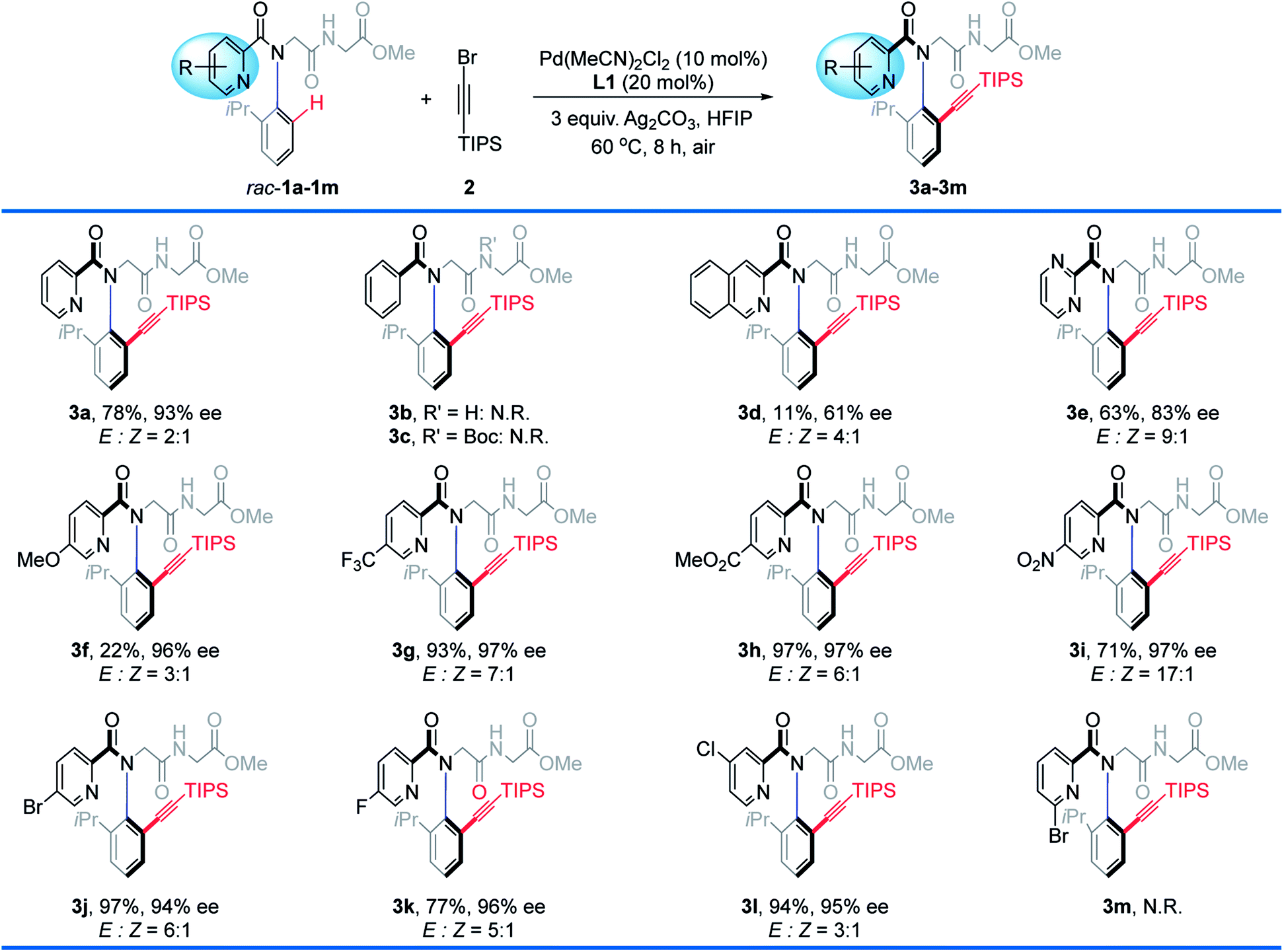
Results and discussion
With the optimal conditions in place, we first tested the scope of picolinamides with different substituents (Table 2). It is worth mentioning that a series of simple peptoid starting materials, such as peptoid monomers and peptoid dimers incorporating various N-aryl side chains can be readily prepared in a sequence-specific form via the Ugi reaction15 in one step (see the ESI‡ for details). The substituents on picolinamides had a dramatic effect on both the reactivity and enantioselectivity. Control experiments using 1b, 1c, and 1m as substrates revealed that: (1) pyridine acts as the coordinating site to facilitate the C–H activation rather than the carbonyl group or peptoid backbone; (2) the coordination ability of palladium is acutely sensitive to steric hindrance in which it lies, as 6′-Br could hamper the coordination (1m). Generally, this protocol is compatible with various electron-withdrawing and -donating substituents on the pyridine ring, albeit those bearing electron-donating substituents gave the alkynylation products in relatively lower yields, compared to the electron-withdrawing counterparts (e.g., 3f, R = 5′-OMe, 22%, 96% ee vs.3g, R = 5′-CF3, 93%, 97% ee; 3h, 5′-CO2Me, 97%, 97% ee; 3i, R = 5′-NO2, 71%, 97% ee; 3j, R = 5′-Br, 97%, 94% ee; 3k, R = 5′-F, 77%, 96% ee).| a Standard conditions. Isolated yields. N.R. = no reaction. |
|---|
|
Then the scope of the peptoid backbone is investigated (Table 3A). We were pleased to find that the deviation on the peptoid backbone was well tolerated, as exemplified by 5a (90%, 93% ee), 5b (99%, 96% ee) and 5c (82%, 96% ee). Peptoid dimers derived from the core structures of natural products were also compatible with this protocol, giving the desired productsin moderate yield with high diastereomeric excess (5d, L-menthol, 51%, 97% de; 5e, dehydroepiandrosterone, 73%, 97% de), irrespective of the existing chiral centers and complexity. Generally, the direct C–H functionalization of longer peptides was expected to be more challenging because the additional amide bonds in longer peptides could deactivate the palladium catalyst.16 To our delight, two representative tripeptides bearing valine (rac-4f), and phenylalanine (rac-4g) residues both reacted smoothly to give alkynylation products in a moderate yield with high diastereomeric excess (5f, 50%, 98% de; 5g, 47%, 97% de).
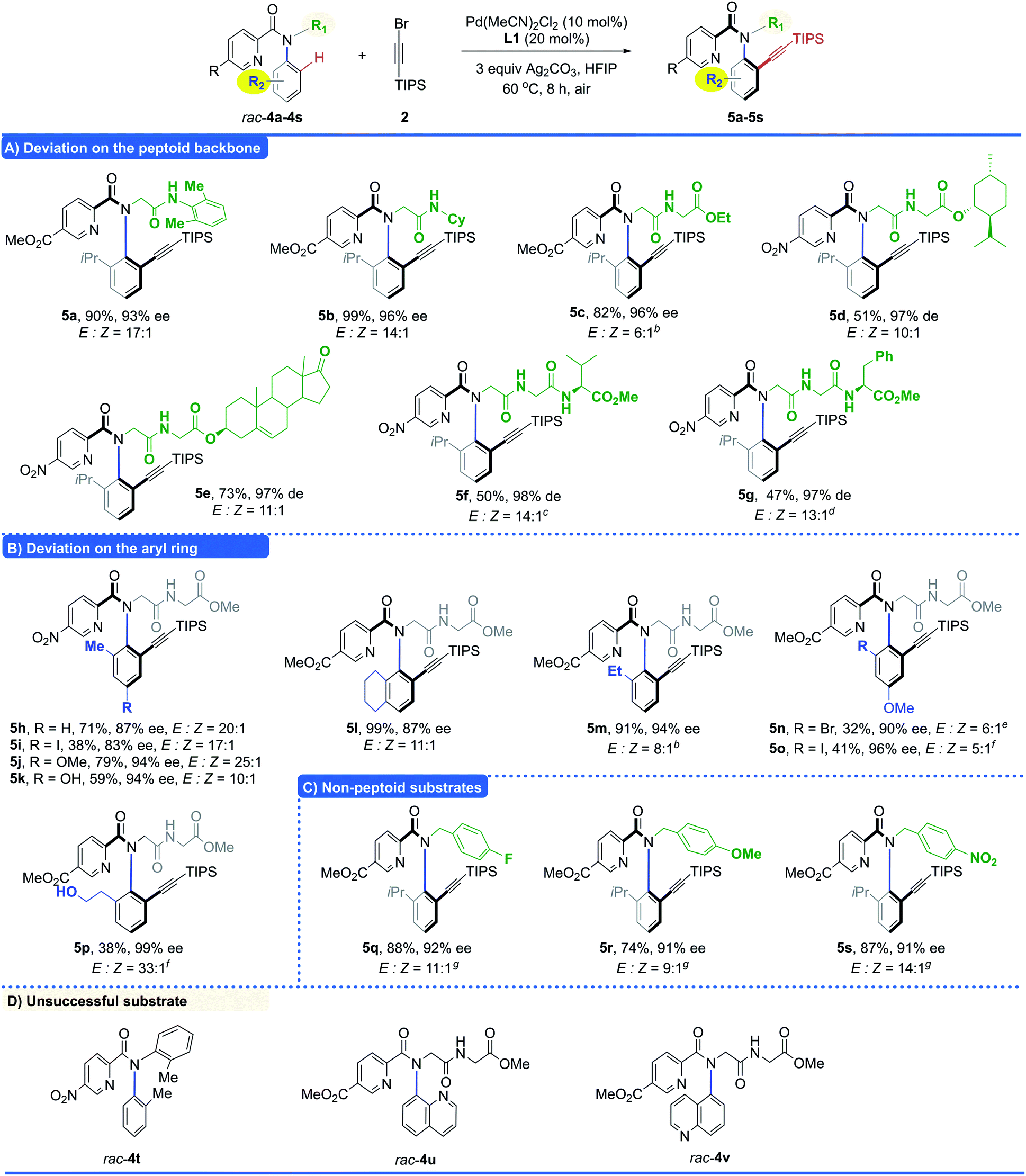
The investigation of the substituents on the aryl ring was then conducted (Table 3B). We found that the electronic effect on the aryl ring is important for both the reactivity and enantioselectivity. As exemplified by 5i–5j, electron-donating substituents led to higher yield and enantioselectivity, compared to their electron-withdrawing counterparts (5i, R = I, 38%, 83% ee; 5j, R = OMe, 79%, 94% ee). Notably, various synthetically useful functional groups, such as hydroxy (rac-4k and rac-4p), bromo (rac-4n) and iodo (rac-4o), were also tolerated. All of these could serve as versatile handles for further transformation, rendering the C–H alkynylation products useful building blocks. To our delight, the scope could also be extended to non-peptoid substrates, providing the desired products in good yields with high enantioselectivities (Table 3C, 5q–5s, 74–88%, 91–92% ee). Unfortunately, substrates bearing an aryl side chain (rac-4t) and quinolinyl units (rac-4u and rac-4v) were not compatible with this protocol (Table 3D). Other alkynyl bromides were also tested, however, no desired alkynylation products were observed under the optimized conditions (Table S10, see the ESI‡ for details).
To further understand the origins of enantioselectivity, density functional theory (DFT) calculations were performed on the key C–H bond activation step based on related mechanistic studies (Fig. 2a).9m,12h,17 We found that the involvement of four HFIP solvents leads to the most favorable C–H bond activation model, compared to other C–H bond activation models with fewer or no HFIP solvent participations (Fig. S1‡). TS6 is 3.4 kcal mol−1 more favorable than TS7, which agrees with the experimental observations that (R)-axial chirality is favored. The DFT-optimized structure of the Pd/L-pGlu-catalyzed C–H bond activation of benzene revealed the intrinsic chiral preference for the L-pGlu amino acid; the arene C–H bond activation prefers to take place in the fourth quadrant (Fig. 2b). This chirality preference matches well with TS6, suggesting that limited structural distortion is required for the formation of (R)-axial chirality during C–H bond activation. The pyramidalization angle of the amino acid nitrogen is similar in TS6 and TS8 (27.6° and 26.3°, respectively). For the formation of (S)-axial chirality, the C–H bond activation transition state TS7 mismatches with the intrinsic chiral preference of TS8, resulting in significant structural distortion in the amino acid and disfavors TS7. The same pyramidalization angle of the amino acid nitrogen is only 8.8° in TS7. Comparing the PdII(L-pGlu) fragment in TS6 and TS7, 4.1 kcal mol−1 energy difference exists between the highlighted fragments (Fig. 2a), which further corroborates the rationale that the amino acid ligand distortion controls the enantioselectivity in the Pd/L-pGlu-catalyzed C–H bond activation step.
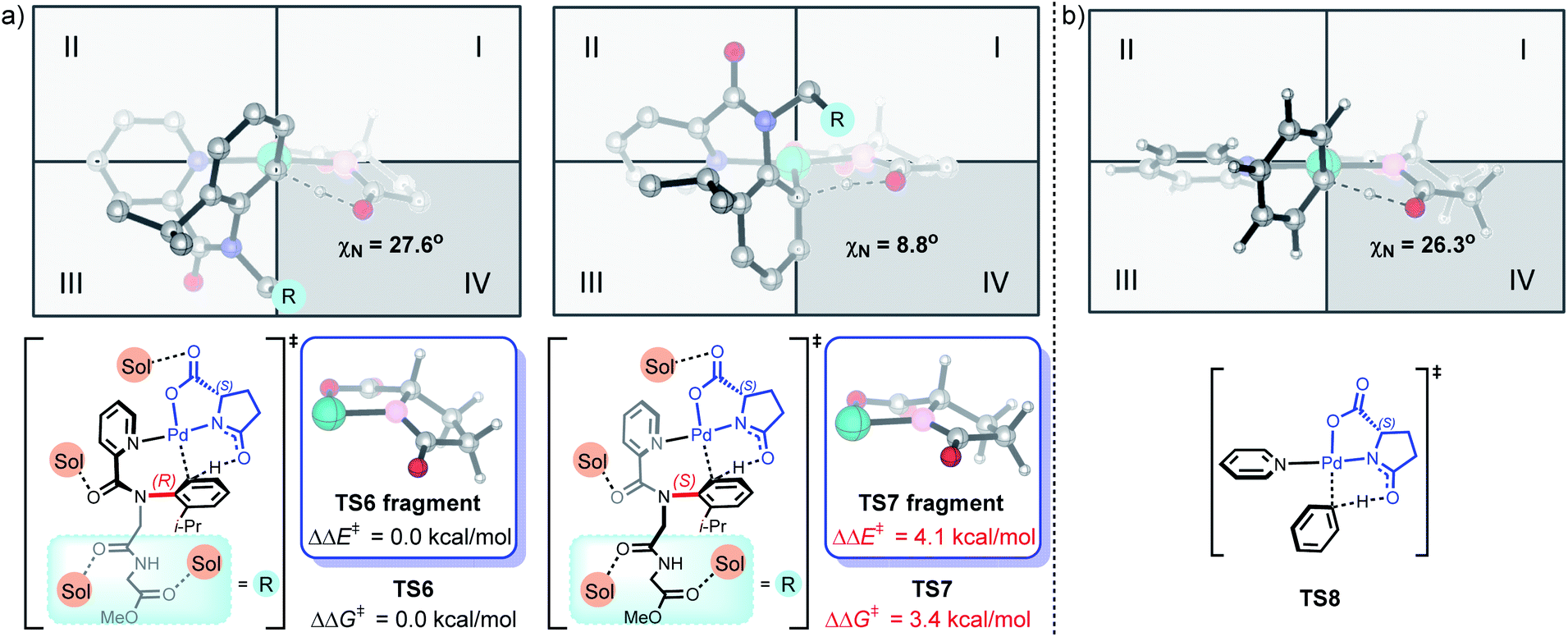 | ||
| Fig. 2 (a) DFT-computed enantioisomeric Pd/L-pGlu-catalyzed C–H bond activation transition states of methyl N-(2-isopropylphenyl)-N-picolinoylglycylglycinate (rac-1a). (b) DFT-optimized structure of the Pd/L-pGlu-catalyzed C–H bond activation transition state of benzene. | ||
To showcase the potential of this C–H alkynylation strategy, a gram-scale synthesis and further diversity transformations were conducted (Fig. 3). A 2.5 mmol-scale reaction of rac-1h with 2 was performed, affording 1.22 g of 3h (80%, 95% ee). When 3h was treated with m-CPBA under mild conditions, the corresponding pyridine N-oxide 3ha was obtained in 59% yield without the erosion of the ee value (95% ee). The TIPS group could be readily removed by treatment with TBAF, giving 3hb in 92% yield with retained chirality (95% ee). The terminal alkyne 3hb could undergo Sonogashira coupling with iodobenzene without significant change in the enantioselectivity (3hc, 91%, 91% ee). Cycloaddition of 3hb with BnN3 gave triazole 3hd (81%, 97% ee). The absolute configuration of the peptoid dimer 3hb was unambiguously confirmed as Ra by X-ray analysis and those of the other products were assigned by analogy.14
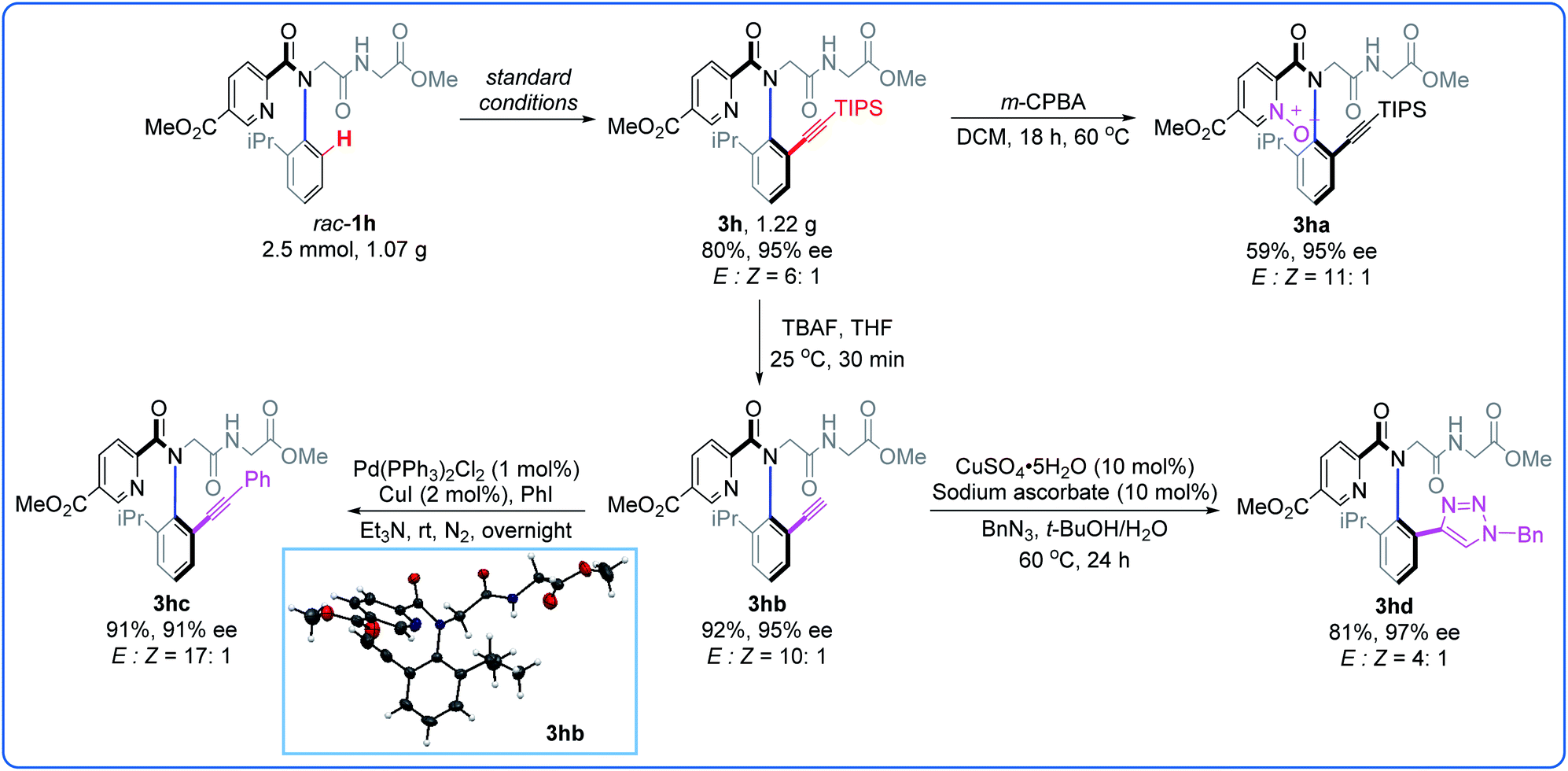 | ||
| Fig. 3 Gram-scale synthesis and transformations. | ||
Conclusions
In summary, we have described a highly practical and efficient strategy for the construction of various isolable N-aryl peptoid atropisomers via Pd(II)-catalyzed enantioselective C–H alkynylation using an inexpensive and commercially available L-pGlu-OH as the chiral ligand. We anticipated that this work will provide a new strategy to define peptoid conformations and promote the development of more chiral peptoid architectures. Further investigations on the applications of these chiral peptoids are underway.Data availability
The electronic supplementary information include experimental detail, computational data, NMR data, X-Ray data and HPLC data.Author contributions
Y.-J. W., Q.-J. Y. and B.-F. S. conceived and designed the project. Y.-J. W. and G. Z. conducted the experiments. X. H. and P.-P. X. performed the theoretical calculations. B.-F. S., X. H., Y.-J. W., and P.-P. X. co-wrote the manuscript. Y.-J. W. and P.-P. X. contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the NSFC (21925109, 21772170 for B.-F. S., 21702182 and 21873081 for X. H.), Out-standing Young Talents of Zhejiang Province High-level Personnel of Special Support (ZJWR0108 for B.-F. S.), Fundamental Research Funds for the Central Universities (2020XZZX002-02 for X. H.), the State Key Laboratory of Clean Energy Utilization (ZJUCEU2020007 for X. H.), and the Center of Chemistry for Frontier Technologies, Department of Chemistry, Zhejiang University, is gratefully acknowledged. Calculations were performed on the high-performance computing system at the Department of Chemistry, Zhejiang University.Notes and references
- (a) R. N. Zuckermann, Pept. Sci., 2011, 96, 545–555 Search PubMed; (b) B. Yoo and K. Kirshenbaum, Curr. Opin. Chem. Biol., 2008, 12, 714–721 Search PubMed; (c) S. B. Y. Shin, B. Yoo, L. J. Todaro and K. Kirshenbaum, J. Am. Chem. Soc., 2007, 129, 3218–3225 Search PubMed.
- (a) N. P. Chongsiriwatana, J. A. Patch, A. M. Czyzewski, M. T. Dohm, A. Ivankin, D. Gidalevitz, R. N. Zuckermann and A. E. Barron, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2794 Search PubMed; (b) R. Zuckermann and T. Kodadek, Curr. Opin. Mol. Ther., 2009, 11, 299–307 Search PubMed; (c) M. M. Reddy, R. Wilson, J. Wilson, S. Connell, A. Gocke, L. Hynan, D. German and T. Kodadek, Cell, 2011, 144, 132–142 Search PubMed; (d) R. J. Simon, R. S. Kania, R. N. Zuckermann, V. D. Huebner, D. A. Jewell, S. Banville, S. Ng, L. Wang, S. Rosenberg and C. K. Marlowe, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9367 Search PubMed; (e) K. T. Nam, S. A. Shelby, P. H. Choi, A. B. Marciel, R. Chen, L. Tan, T. K. Chu, R. A. Mesch, B.-C. Lee, M. D. Connolly, C. Kisielowski and R. N. Zuckermann, Nat. Mater., 2010, 9, 454–460 Search PubMed; (f) G. Maayan, M. D. Ward and K. Kirshenbaum, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13679 Search PubMed.
- K. Kirshenbaum, A. E. Barron, R. A. Goldsmith, P. Armand, E. K. Bradley, K. T. V. Truong, K. A. Dill, F. E. Cohen and R. N. Zuckermann, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4303 Search PubMed.
- (a) P. Armand, K. Kirshenbaum, A. Falicov, R. L. Dunbrack, K. A. Dill, R. N. Zuckermann and F. E. Cohen, Folding Des., 1997, 2, 369–375 Search PubMed; (b) C. W. Wu, T. J. Sanborn, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2001, 123, 2958–2963 Search PubMed; (c) C. W. Wu, K. Kirshenbaum, T. J. Sanborn, J. A. Patch, K. Huang, K. A. Dill, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2003, 125, 13525–13530 Search PubMed.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 Search PubMed.
- (a) B. Paul, G. L. Butterfoss, M. G. Boswell, P. D. Renfrew, F. G. Yeung, N. H. Shah, C. Wolf, R. Bonneau and K. Kirshenbaum, J. Am. Chem. Soc., 2011, 133, 10910–10919 Search PubMed; (b) B. Paul, G. L. Butterfoss, M. G. Boswell, M. L. Huang, R. Bonneau, C. Wolf and K. Kirshenbaum, Org. Lett., 2012, 14, 926–929 Search PubMed.
- (a) D. P. Curran, H. Qi, S. J. Geib and N. C. DeMello, J. Am. Chem. Soc., 1994, 116, 3131–3132 Search PubMed; (b) J. Clayden, Chem. Commun., 2004, 127–135, 10.1039/B307976G.
- R. L. Dunbrack and M. Karplus, J. Mol. Biol., 1993, 230, 543–574 Search PubMed.
- (a) O. Kitagawa, Acc. Chem. Res., 2021, 54, 719–730 Search PubMed; (b) O. Kitagawa, M. Kohriyama and T. Taguchi, J. Org. Chem., 2002, 67, 8682–8684 Search PubMed; (c) J. Terauchi and D. P. Curran, Tetrahedron: Asymmetry, 2003, 14, 587–592 Search PubMed; (d) O. Kitagawa, M. Yoshikawa, H. Tanabe, T. Morita, M. Takahashi, Y. Dobashi and T. Taguchi, J. Am. Chem. Soc., 2006, 128, 12923–12931 Search PubMed; (e) S. Brandes, M. Bella, A. Kjærsgaard and K. A. Jørgensen, Angew. Chem., Int. Ed., 2006, 45, 1147–1151 Search PubMed; (f) K. Tanaka, K. Takeishi and K. Noguchi, J. Am. Chem. Soc., 2006, 128, 4586–4587 Search PubMed; (g) S. Shirakawa, K. Liu and K. Maruoka, J. Am. Chem. Soc., 2012, 134, 916–919 Search PubMed; (h) Y. Liu, X. Feng and H. Du, Org. Biomol. Chem., 2015, 13, 125–132 Search PubMed; (i) M. E. Diener, A. J. Metrano, S. Kusano and S. J. Miller, J. Am. Chem. Soc., 2015, 137, 12369–12377 Search PubMed; (j) S.-L. Li, C. Yang, Q. Wu, H.-L. Zheng, X. Li and J.-P. Cheng, J. Am. Chem. Soc., 2018, 140, 12836–12843 Search PubMed; (k) S. Lu, S. V. H. Ng, K. Lovato, J.-Y. Ong, S. B. Poh, X. Q. Ng, L. Kürti and Y. Zhao, Nat. Commun., 2019, 10, 3061 Search PubMed; (l) D. Li, S. Wang, S. Ge, S. Dong and X. Feng, Org. Lett., 2020, 22, 5331–5336 Search PubMed; (m) Q.-J. Yao, P.-P. Xie, Y.-J. Wu, Y.-L. Feng, M.-Y. Teng, X. Hong and B.-F. Shi, J. Am. Chem. Soc., 2020, 142, 18266–18276 Search PubMed.
- (a) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 Search PubMed; (b) G. Liao, T. Zhou, Q.-J. Yao and B.-F. Shi, Chem. Commun., 2019, 55, 8514–8523 Search PubMed; (c) Q. Wang, Q. Gu and S.-L. You, Acta Chim. Sin., 2019, 77, 690–704 Search PubMed.
- (a) C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173–6214 Search PubMed; (b) C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 Search PubMed; (c) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, eaao4798 Search PubMed; (d) Q. Zhang and B.-F. Shi, Chin. J. Chem., 2019, 37, 647–656 Search PubMed; (e) T. Yoshino, S. Satake and S. Matsunaga, Chem.–Eur. J., 2020, 26, 7346–7357 Search PubMed; (f) G. Liao, T. Zhang, Z.-K. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 19773–19786 Search PubMed; (g) T. K. Achar, S. Maiti, S. Jana and D. Maiti, ACS Catal., 2020, 10, 13748–13793 Search PubMed.
- (a) Q.-J. Yao, S. Zhang, B.-B. Zhan and B.-F. Shi, Angew. Chem., Int. Ed., 2017, 56, 6617–6621 Search PubMed; (b) G. Liao, Q.-J. Yao, Z.-Z. Zhang, Y.-J. Wu, D.-Y. Huang and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 3661–3665 Search PubMed; (c) G. Liao, B. Li, H.-M. Chen, Q.-J. Yao, Y.-N. Xia, J. Luo and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 17151–17155 Search PubMed; (d) S. Zhang, Q.-J. Yao, G. Liao, X. Li, H. Li, H.-M. Chen, X. Hong and B.-F. Shi, ACS Catal., 2019, 9, 1956–1961 Search PubMed; (e) J. Luo, T. Zhang, L. Wang, G. Liao, Q.-J. Yao, Y.-J. Wu, B.-B. Zhan, Y. Lan, X.-F. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2019, 58, 6708–6712 Search PubMed; (f) G. Liao, H.-M. Chen, Y.-N. Xia, B. Li, Q.-J. Yao and B.-F. Shi, Angew. Chem., Int. Ed., 2019, 58, 11464–11468 Search PubMed; (g) B.-B. Zhan, L. Wang, J. Luo, X.-F. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 3568–3572 Search PubMed; (h) L. Jin, Q.-J. Yao, P.-P. Xie, Y. Li, B.-B. Zhan, Y.-Q. Han, X. Hong and B.-F. Shi, Chem, 2020, 6, 497–511 Search PubMed; (i) H. Song, Y. Li, Q.-J. Yao, L. Jin, L. Liu, Y.-H. Liu and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 6576–6580 Search PubMed.
- (a) N. H. Shah, G. L. Butterfoss, K. Nguyen, B. Yoo, R. Bonneau, D. L. Rabenstein and K. Kirshenbaum, J. Am. Chem. Soc., 2008, 130, 16622–16632 Search PubMed; (b) J. R. Stringer, J. A. Crapster, I. A. Guzei and H. E. Blackwell, J. Am. Chem. Soc., 2011, 133, 15559–15567 Search PubMed; (c) P. Armand, K. Kirshenbaum, R. A. Goldsmith, S. Farr-Jones, A. E. Barron, K. T. V. Truong, K. A. Dill, D. F. Mierke, F. E. Cohen, R. N. Zuckermann and E. K. Bradley, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4309 Search PubMed.
- ESI.‡.
- (a) A. Dömling, Chem. Rev., 2006, 106, 17–89 Search PubMed; (b) I. Ugi, Pure Appl. Chem., 2001, 73, 187–191 Search PubMed.
- (a) W. Gong, G. Zhang, T. Liu, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 16940–16946 Search PubMed; (b) T. Liu, J. X. Qiao, M. A. Poss and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 10924–10927 Search PubMed; (c) B.-B. Zhan, M.-X. Jiang and B.-F. Shi, Chem. Commun., 2020, 56, 13950–13958 Search PubMed.
- (a) G.-J. Cheng, P. Chen, T.-Y. Sun, X. Zhang, J.-Q. Yu and Y.-D. Wu, Chem.–Eur. J., 2015, 21, 11180–11188 Search PubMed; (b) X.-M. Zhong, G.-J. Cheng, P. Chen, X. Zhang and Y.-D. Wu, Org. Lett., 2016, 18, 5240–5243 Search PubMed; (c) R. E. Plata, D. E. Hill, B. E. Haines, D. G. Musaev, L. Chu, D. P. Hickey, M. S. Sigman, J.-Q. Yu and D. G. Blackmond, J. Am. Chem. Soc., 2017, 139, 9238–9245 Search PubMed; (d) D. E. Hill, K. L. Bay, Y.-F. Yang, R. E. Plata, R. Takise, K. N. Houk, J.-Q. Yu and D. G. Blackmond, J. Am. Chem. Soc., 2017, 139, 18500–18503 Search PubMed; (e) S. Bag, S. K, A. Mondal, R. Jayarajan, U. Dutta, S. Porey, R. B. Sunoj and D. Maiti, J. Am. Chem. Soc., 2020, 142, 12453–12466 Search PubMed.
Footnotes |
| † Dedicated to the 100th anniversary of Chemistry at Nankai University. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2040388. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01130h |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |