
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient electroreduction of CO2 to C2+ products on CeO2 modified CuO†
Xupeng
Yan
ab,
Chunjun
Chen
 *ab,
Yahui
Wu
ab,
Shoujie
Liu
c,
Yizhen
Chen
f,
Rongjuan
Feng
a,
Jing
Zhang
g and
Buxing
Han
*abde
*ab,
Yahui
Wu
ab,
Shoujie
Liu
c,
Yizhen
Chen
f,
Rongjuan
Feng
a,
Jing
Zhang
g and
Buxing
Han
*abde
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid and Interface and Thermodynamics, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: Chenchunjun@iccas.ac.cn; hanbx@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cChemistry and Chemical Engineering of Guangdong Laboratory, Shantou 515063, China
dPhysical Science Laboratory, Huairou National Comprehensive Science Center, Beijing 101400, China
eShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
fHefei National Laboratory for Physical Sciences at the Microscale, Key Laboratory of Strongly-Coupled Quantum Matter Physics of Chinese Academy of Sciences, National Synchrotron Radiation Laboratory, Key Laboratory of Surface and Interface Chemistry and Energy Catalysis of Anhui Higher Education Institutes, Department of Chemical Physics, University of Science and Technology of China, 230026 Hefei, Anhui, People's Republic of China
gInstitute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
First published on 30th March 2021
Abstract
Electrocatalytic reduction of CO2 into multicarbon (C2+) products powered by renewable electricity offers one promising method for CO2 utilization and promotes the storage of renewable energy under an ambient environment. However, there is still a dilemma in the manufacture of valuable C2+ products between balancing selectivity and activity. In this work, cerium oxides were combined with CuO (CeO2/CuO) and showed an outstanding catalytic performance for C2+ products. The faradaic efficiency of the C2+ products could reach 75.2% with a current density of 1.21 A cm−2. In situ experiments and density functional theory (DFT) calculations demonstrated that the interface between CeO2 and Cu and the subsurface Cu2O coexisted in CeO2/CuO during CO2RR and two competing pathways for C–C coupling were promoted separately, of which hydrogenation of *CO to *CHO is energetically favoured. In addition, the introduction of CeO2 also enhanced water activation, which could accelerate the formation rate of *CHO. Thus, the selectivity and activity for C2+ products over CeO2/CuO can be improved simultaneously.
Introduction
Conversion of CO2 into valuable chemicals using electrochemical methods provides a promising way to combat accumulated carbon emissions and also to store renewable energy.1–6 Continuous progress has been made in the field of the electrochemical CO2 reduction reaction (CO2RR), especially for monocarbon products like carbon monoxide (CO) and formate.7–16 However, the manufacture of valuable C2+ products in CO2RR, such as ethylene (C2H4), ethanol (C2H5OH) and n-propanol (n-C3H7OH), still has to balance selectivity and activity,17–25 which obstructs further industrial applications. To achieve a commercial current density (>100 mA cm−2) as well as high selectivity for C2+ products in CO2RR,26–32 highly efficient and robust electrocatalysts are required.Cu-based catalysts are the most promising electrocatalysts for converting CO2 into C2+ products,33–45 owing to their moderate adsorption capacity for the crucial intermediate (*CO). Based on previous reports,1,18,46,47 the selectivity of C2+ products over Cu-based catalysts can be notably improved by the introduction of another component but the understanding of the structure–selectivity relationship remains controversial because the valence state and the microstructure of copper may be influenced simultaneously. What is more, complexity also exists in the production of C2+ products during CO2RR due to the C–C coupling step involved, which not only contains multiple electron-transfer and protonation steps,6 but also exhibits various potential coupling paths on heterogeneous catalysts. As a result, it is necessary to comprehensively reveal the role of another component in the promotion of the selectivity towards C2+ products during CO2RR.
Given the neutral or basic electrolyte used in CO2RR, H2O can serve as the hydrogen source and the activity should be bound up with the activation of H2O in CO2RR.47 According to the Sabatier principle, the energy barrier for the activation of water should be particularly controlled, which could provide enough hydrogen for the hydrogenation of intermediates but not cause excessive production of H2. Considering cerium oxide (CeO2) has a high activity for water activation in CO2 hydrogenation and shows poor activity for the hydrogen evolution reaction (HER),48–50 we can assume that the activity for C2+ products would be improved compared to the CeO2 modified Cu-based catalyst in CO2RR.
Herein, we used CeO2 to modify CuO to obtain CeO2/CuO catalysts, and both a high current density and selectivity towards C2+ products were achieved in CO2RR. A faradaic efficiency (FE) of 75.2% for the C2+ products could be attained on the catalyst with a total current density of 1.21 A cm−2 in a flow-cell system. The experiments and density functional theory (DFT) calculations indicate the energy of generation of *CHO is thermodynamically reduced by the interfacial effect compared to CeO2 modified CuO catalysts and the rapid activation of water around CeO2 accelerates the formation of *CHO kinetically, thus the C–C coupling step is facilitated via the *CHO route, endowing the CeO2/CuO catalyst with an excellent catalytic performance towards C2+ products.
Results and discussion
The Ce(OH)2/Cu(OH)2 catalysts were first prepared by the coprecipitation method, then the CeO2/CuO catalysts were gained by annealing at 600 °C in air. As the amount of Ce in the catalysts increased from 0 to 30%, a set of peaks belonging to the CeO2 phase gradually emerged on the base of the primary CuO phase in the X-ray diffraction patterns (Fig. 1a), indicating the coexistence of CeO2 and CuO in the catalysts, and the CeO2/CuO catalysts were named CCX (X = the molar ratio of Ce and Cu times 100). From scanning electron microscopy (SEM) and transmission electron microscopy (TEM), we can observe that CeO2 nanoparticles below 5 nm were evenly loaded on the surface of CuO (Fig. 1b, c and S2†). Two typical d-spacings of 0.31 nm and 0.23 nm were observed in the image of high-resolution transmission electron microscopy (HR-TEM) for CC20 (Fig. 1d), corresponding to CeO2(111) and CuO(111). According to the distribution of the elements of Cu, Ce and O in the energy dispersive X-ray spectroscopy maps (Fig. 1e), the uniform element dispersion of Cu and Ce over the catalyst confirmed that CeO2 was uniformly dispersed on the CuO.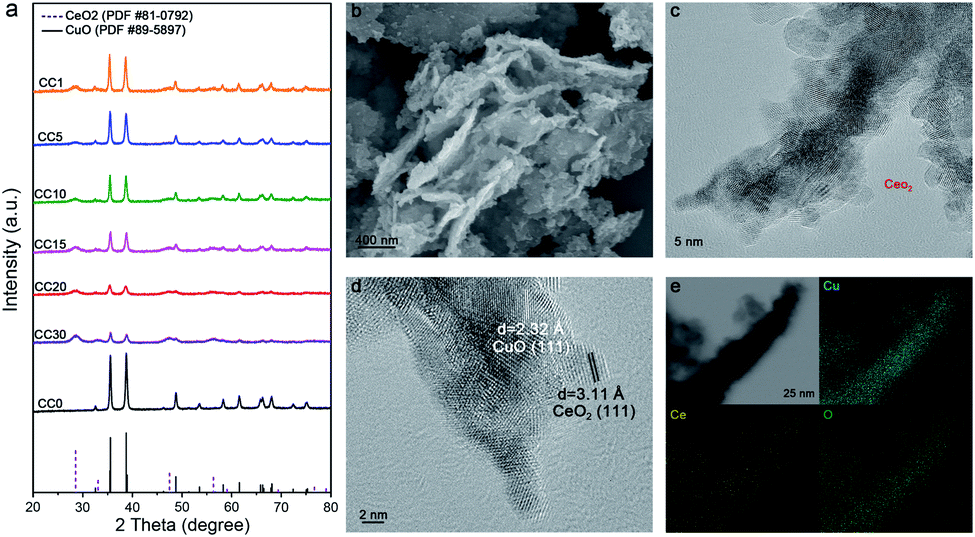 | ||
| Fig. 1 (a) The XRD patterns of the CCX composites with various Ce contents. (b and c) The SEM and TEM images of the CC20 (the red circle represents the CeO2 nanoparticles). (d) The HR-TEM image of the CC20. (e) The energy dispersive X-ray spectroscopy (EDS) maps of CC20. | ||
The electrocatalytic performance of the catalysts was evaluated in the flow cell and 1 M KOH was used as the electrolyte, as reported in our previous work.51 Before the CO2RR, the catalysts were firstly reduced around −0.3 V vs. RHE, which is more negative than the transformation of CuO to Cu (Fig. S3†). The polytetrafluoroethylene (PTFE) membrane (average pore size of 0.22 μm) was used as the gas diffusion electrode, and gaseous and liquid products were analyzed by gas chromatography (GC) and nuclear magnetic resonance (NMR) spectroscopy, respectively (Fig. S4†). The 13C labelled CO2 was used as the source of the reactant gas and the results verified that CO2 was the only carbon source in CO2RR (Fig. S5†).
Based on the performance of the CCX catalysts in CO2RR, a typical volcano plot between FEC2+ and Ce content was observed at −1.02 V (vs. RHE) and CC20 exhibited the best performance at various applied potentials (Fig. 2a and S6–S8†). From the TEM (Fig. S2†), we can observe that the interfaces were produced with the increase of the Ce amount, thus we hypothesized that the selectivity of C2+ was related to the interfaces. However, for the CC30, the selectivity of C2+ products showed a significant decrease because too many Cu sites were covered by the CeO2. Thus, CC20 was chosen for further comparison with CC0. It can be clearly observed that CC20 showed outstanding efficiency for C2+ products in the CO2 reduction (Fig. 2b). The FE of C2+ products for CC20 could reach 75.2% at −1.12 V (vs. RHE), while that over CC0 was only 48.3% at the same condition. Moreover, the evolution of H2 was suppressed over CC20 and the FE of n-propanol was notably improved on CC20 compared to CC0 (Fig. S7–S9†), which might correlate with the escalation of C2 intermediates over CC20 (Fig. S9†). In the meantime, a significant increase was also achieved on the current density on CC20. It is very impressive that the partial current density of C2+ products (jC2+) over CC20 could reach as high as 0.91 A cm−2 at −1.12 V (vs. RHE), which is about 10 times higher than that on CC0 (Fig. 2c). Compared with the state-of-the-art catalysts, the activity and selectivity for C2+ products over CC20 are among the highest values (Fig. 2d and Table S1†). The above results indicate that the introduction of CeO2 could significantly improve both the selectivity and activity for C2+ products. Moreover, the performance of CeO2 was also characterized (Fig. S12†), and only trace CO was detected at −0.87 V and −0.97 V (vs. RHE), while the current density was below 20 mA cm−2 at the applied potentials, indicating that pure CeO2 showed poor activity for CO2RR. Besides, the catalysts were characterized after the reaction and no obvious change was observed in the TEM images and XRD patterns (Fig. S14–S17†). As a result, the proper content of CeO2 would obviously benefit the catalytic performance of the CuO catalyst towards C2+ products in CO2RR.
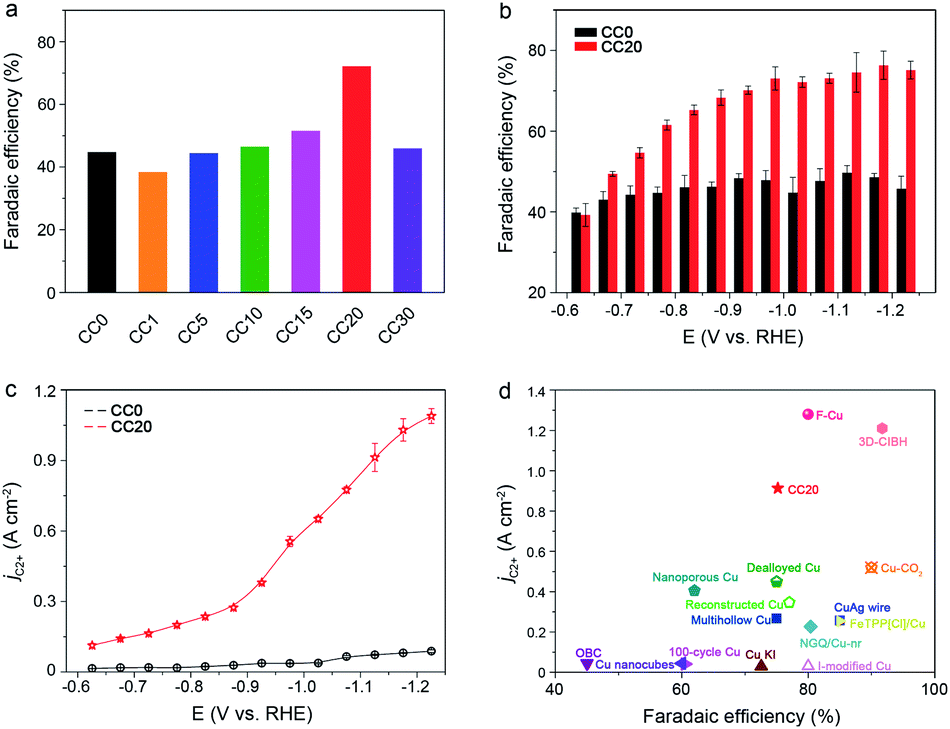 | ||
| Fig. 2 (a and b) The average FEs of C2+ products at various potentials in 1 M KOH over CC0 and CC20, respectively. (c) The partial current density of C2+ products at various potentials in 1 M KOH solution over CC0 and CC20. (d) A comparison of the average FEs and the current density of C2+ products on various reported catalysts and the literature sources are listed in the ESI (Table S1†). | ||
To reveal the reasons for the superior catalytic performance of CC20 in the CO2RR, the electrochemical active surface areas (ECSAs) and electrochemical impedance spectroscopy (EIS) of the catalysts were studied. We can observe that similar ECSAs were obtained over the CCX catalysts with different CeO2 contents (Fig. S18†), indicating the similar surface area of the catalysts at the electrochemical conditions. Moreover, the charge transfer resistance (Rct) for the catalysts was also similar (Fig. S19†), suggesting that the discrepancy of the efficiency for C2+ products did not mainly originate from the slight difference of the ECSAs and electronic conductivity.
The catalytic performance of Cu-based catalysts was closely related to the oxidation state and local structure, which could alter the adsorption of intermediates,30,41,42,52 thus the operando X-ray absorption spectroscopy (XAS) was used to track the evolution of the oxidation state and local structure of Cu and Ce over CC0 and CC20 during CO2RR. At the open circuit potential (OCP), near the Cu K-edge, both the X-ray absorption near edge structure (XANES) and the k3-weighted Fourier-transformed (FT) extended X-ray absorption fine structure (EXAFS) spectra of CC0 and CC20 showed the typical features of CuO (Fig. 3a and b, S20 and S21†), indicating that CuO was dominant in CC0 and CC20 before the reaction. As the potential was applied as −0.62 V (vs. RHE), there was no obvious change in either the XANES or FT-EXAFS spectra. Meanwhile, CC0 and CC20 showed a low FEC2+ and these results could be due to the large proportion of Cu(II). When the applied potential decreased to −0.82 V (vs. RHE), features of Cu with low oxidation states emerged in the XANES spectra and EXAFS analysis also displayed that the Cu first shell coordination switched to the mixture of different Cu species over CC0 and CC20. It can be found that the FEs of C2+ at −0.82 V (vs. RHE) also showed a significant increase compared to that at −0.62 V (vs. RHE), suggesting the potential correlation between the Cu oxidation state and the FEC2+ in CO2RR. Furthermore, according to the XANES spectra, the oxidation state of Cu in the catalysts continued to decrease and the results in the EXAFS data were different from the initial CuO-like state, demonstrating that Cu with a low oxidation state became the main phase. Interestingly, the FE of C2+ in CO2RR still slightly increased from −0.82 V (vs. RHE) to −1.02 V (vs. RHE) on CC0 and CC20, which supported the conclusion that the low-valent Cu species on catalysts were the active phase in CO2RR. Moreover, according to the distribution of various Cu species for CC20 and CC0 (Fig. S22 and S23†), we can observe that the Cu2O species occupied the higher proportion over CC20 than on CC0. These results indicated that the introduction of CeO2 could stabilize the Cu2O, which could be attributed to the interaction between Ce and Cu, and the role of Cu2O in CO2RR will be discussed in the later section. In addition, the operando XANES data at the Ce L3-edge of CC20 showed a negligible change during CO2RR (Fig. 3c), indicating that CeO2 remained stable during CO2RR.
 | ||
| Fig. 3 (a and b) Operando XANES and the corresponding Fourier transforms of k3-weighted EXAFS data at the Cu K-edge at various applied potentials (vs. RHE) over CC20 during CO2RR. (c) Operando XANES at the Ce L3-edge at various applied potentials (vs. RHE) over CC20 during CO2RR. (d) The in situ surface-enhanced Raman spectra for CC0 at various potentials (vs. RHE) during CO2RR. (e) The in situ surface-enhanced Raman spectra for CC20 at various potentials (vs. RHE) during CO2RR. | ||
Generally, the activity and selectivity of C2+ products are closely related to the surface species on the catalysts during the reduction. So, an in situ surface enhanced Raman spectroscopy (SERS) study was carried out to explore the surface species over CC0 and CC20 (Fig. 3d and e, S24†). After the pre-electrolysis at N2 atmosphere, only two weak bands at 524 and 610 cm−1 were observed, which belonged to Cu2O,53 and then disappeared in CO2 electrolysis. Instead, bands at 390 and 536 cm−1 emerged at negative potentials in CO2 electrolysis, which were attributed to the chemisorption of CO2 on the surface Cu.57,58 Furthermore, we can observe that no Cu2O could be found on both CC0 and CC20 during CO2RR from the Raman spectra. Combined with the results of the operando XAFS, we can assume that the Cu2O species exists on the subsurface of the catalysts, due to the Raman spectroscopy being sensitive to the surface species of the catalyst,56,57 which is consistent with previous reports.21,59 In addition, the signals of CeO2 cannot be found on CC20 in the Raman spectroscopy, this may be due to the signals of CeO2 being too weak under the existence of the electrolyte in the in situ experiments.60
As the applied potential negatively moved, both on CC0 and CC20, peaks at 285, 365, 1800–1860 and 2000–2100 cm−1 became cognizable, corresponding to the restricted rotation of adsorbed *CO on Cu, Cu–CO stretching, and bridge and top C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretching, respectively.54–58,61 It is interesting to note that there was a distinct disparity in the performance of the above *CO related peaks over CC0 and CC20. For CC0, at −0.37 V (vs. RHE), *CO related peaks began to be observed and the peak around 1820 cm−1 was weak. On the contrary, those peaks were clearly present over CC20 after −0.17 V (vs. RHE), and the peak between 1800–1860 cm−1 even showed a red shift while the peak at 2000–2100 cm−1 became strong. The difference between those two catalysts supported the conclusion that CO2 could be transformed into CO at lower applied potentials on CC20 than on CC0, indicating the superior activity of CC20 towards CO in CO2RR, which is consistent with the results in the electrochemical tests (Fig. S11†). Moreover, the excellent catalytic capability for the CO product in CO2RR should be favourable for the following steps in CO2RR.
O stretching, respectively.54–58,61 It is interesting to note that there was a distinct disparity in the performance of the above *CO related peaks over CC0 and CC20. For CC0, at −0.37 V (vs. RHE), *CO related peaks began to be observed and the peak around 1820 cm−1 was weak. On the contrary, those peaks were clearly present over CC20 after −0.17 V (vs. RHE), and the peak between 1800–1860 cm−1 even showed a red shift while the peak at 2000–2100 cm−1 became strong. The difference between those two catalysts supported the conclusion that CO2 could be transformed into CO at lower applied potentials on CC20 than on CC0, indicating the superior activity of CC20 towards CO in CO2RR, which is consistent with the results in the electrochemical tests (Fig. S11†). Moreover, the excellent catalytic capability for the CO product in CO2RR should be favourable for the following steps in CO2RR.
DFT calculations were then performed to elucidate the mechanism of the crucial C–C coupling step and to gain insight into the excellent performance of CC20 in CO2RR. According to the above results, the introduction of CeO2 can not only form the interface between CeO2 and Cu, but can also stabilize the subsurface Cu2O. Although both the interface and subsurface Cu2O can promote the CO2RR,62,63 they have been studied separately in previous reports.64,65 Thus, the role of interface and subsurface Cu2O on enhancing the C2+ products should be studied simultaneously and three specific models were used to study the effect of subsurface Cu2O and CeO2 on promoting C–C coupling (Fig. S27†). First, a model with more metallic Cu on the surface and less Cu2O on the subsurface (Cu-M) was built to represent the CC0 (Fig. 4a). Then, a model with less metallic Cu on the surface and more Cu2O on the subsurface (Cu-L) was built to represent the CC20 without CeO2 (Fig. 4b). Last, Cu-L with CeO2 on the surface (CeO2/Cu-L) was built to represent CC20 (Fig. 4c). The Cu(111) and CeO2(111) were chosen as the basic models according to the results of XRD (Fig. S16†), and the ratio of Cu and Cu2O was set according to the results of in situ XAS (Fig. S22 and S23†).
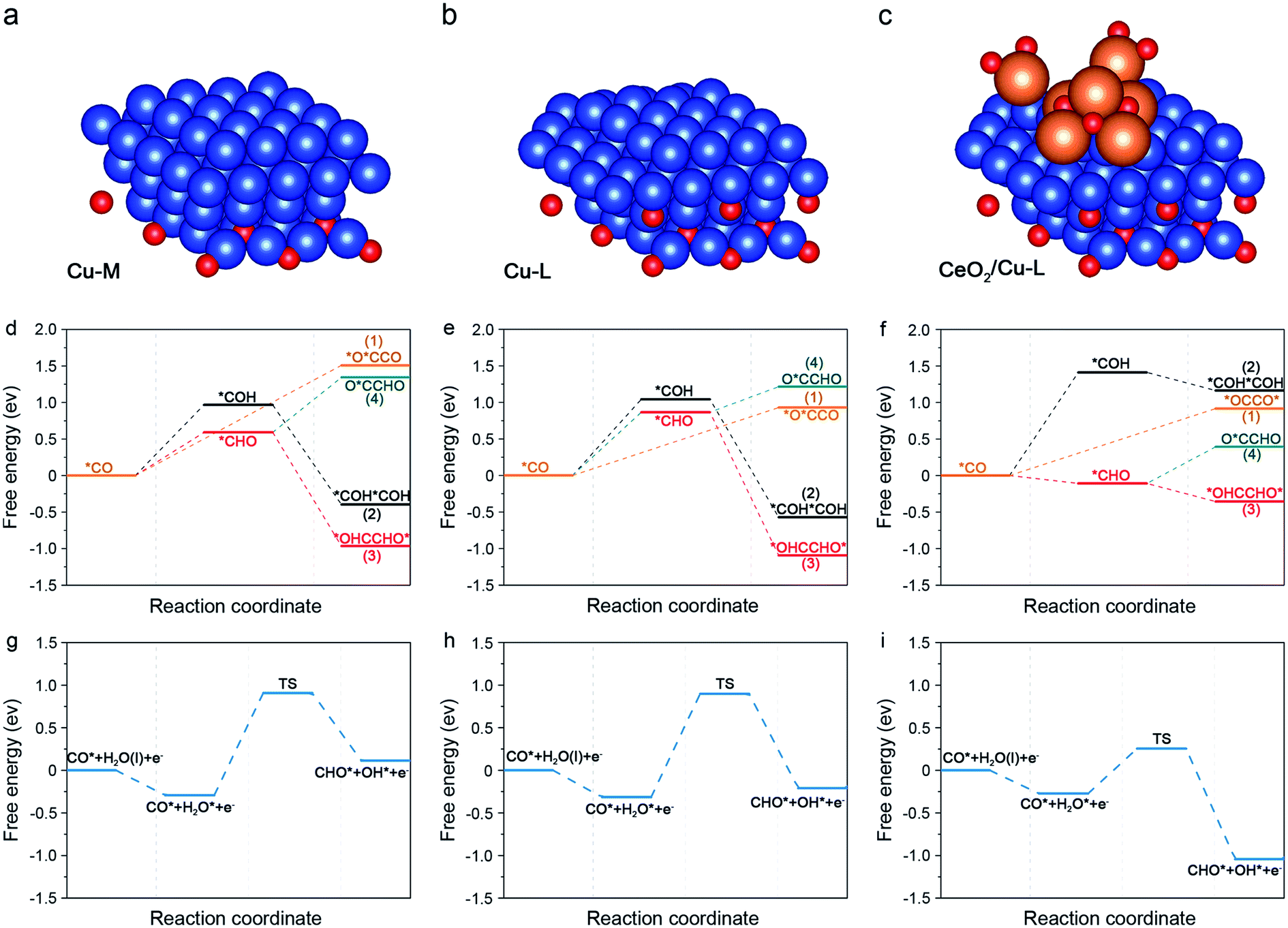 | ||
| Fig. 4 (a–c) The side views of Cu-M, Cu-L and CeO2/Cu-L, in which the blue balls, red balls and orange balls stand for Cu, oxygen, and carbon and hydrogen, respectively. (d–f) The reaction energy diagram for the CO2RR to describe the possible C–C coupling step from *CO on Cu-M, Cu-L and CeO2/Cu-L. (g–i) The reaction energy diagram for *CO hydrogenation to *COH on Cu-M, Cu-L and CeO2/Cu-L, respectively. | ||
Generally, CO2 can be first reduced into CO through the *COOH pathway,28 and the adsorbed CO is regarded as the common intermediate for the C2+ products in CO2RR.66 In this condition, four potential reaction pathways are taken into account in the C–C coupling step and all of them are generated from the vital intermediate *CO (Fig. S28–S33†).
| *CO + *CO → *CO–*CO | (1) |
| *CO + H+ + e− → *COH, *COH + *COH → *COH–*COH | (2) |
| *CO + H+ + e− → *CHO, *CHO + *CHO → *CHO–*CHO | (3) |
| *CO + H+ + e− → *CHO, *CHO + *CO → *CO–*CHO | (4) |
On Cu-M, the energy of 1.51 eV is required for the dimerization of *CO (path 1), higher than that on Cu-L (0.93 eV), indicating that more subsurface Cu2O are beneficial for the C–C coupling through the *CO–*CO route (Fig. 4d and e), which is consistent with previous reports.21,63 Further addition of CeO2 on Cu-L barely alters the energy for the dimerization of *CO (0.92 eV) compared with Cu-L (Fig. 4f). Consequently, we can assume that the energy for dimerization of *CO can be decreased by subsurface Cu2O, however, the energy for the formation of the *O*CCO intermediate was still very high, indicating that C–C coupling through *CO dimerization is difficult.
We notice that both path 2 and path 3 suffer from the endothermic protonation of adsorbed *CO and the subsequent exothermic coupling step in all the models. In terms of the lower energy needed for the generation of *CHO compared to *COH, we can assume that the C–C coupling step would prefer the *CHO route rather than the *COH route. However, the formation of *CHO in each model is different in energy. 0.59 eV is required for the hydrogenation of *CO into *CHO on Cu-M, while a higher energy of 0.87 eV is needed on Cu-L, suggesting that more subsurface Cu2O were not advantageous for the formation of *CHO. This may be due to the fact that the adsorption of *CHO can be affected by the subsurface Cu2O, and the intrinsic reason should be further studied. Surprisingly, the energy for the hydrogenation of *CO into *CHO dramatically declined to −0.11 eV and became exothermic near the interface of CeO2 and Cu-L (Fig. S32 and S33†). The above results convincingly demonstrate that *CHO is easily formed from *CO on CC20 and this should be attributed to the introduction of CeO2 and the formed interface, rather than more subsurface Cu2O. Furthermore, for the following C–C coupling step related to *CHO, the coupling of *CO and *CHO (path 4) is also possible in theory except for the dimerization of *CHO (path 3). Nevertheless, the coupling of *CO and *CHO is endoenergic over all surfaces, suggesting that the exoenergic dimerization of *CHO would be favourable to the coupling process. On the whole, the coupling of *CHO into *OHCCHO* is most favourable in the C–C coupling step among the above possible pathways in the three models and the process even becomes spontaneous in the presence of CeO2. Ma and co-workers also found that the coupling between *CHO showed lower barriers on the Cu(111) surfaces.28 In consequence, the *CHO route (path 3) is favoured on the three models in the C–C coupling step and becomes exothermal on CC20 due to the formed interface, elucidating the high FE for C2+ products on CC20. In addition, we can observe that all the intermediates were mainly adsorbed on the exposed Cu sites, so we can assume that Cu was the active site.
In consideration of the 1 M KOH used in CO2RR, H2O should be considered as the hydrogen donor. As a result, we introduced the water activation process to further study the kinetic process for the formation of *CHO. For Cu-M and Cu-L, water is spontaneously adsorbed on the surface Cu and then the high energy barriers of 1.20 eV and 1.21 eV are needed to form the transient state (TS) for the following formation of *CHO (Fig. 4g, h and S34†), respectively. For the CeO2/Cu-L, H2O would like to be adsorbed around the CeO2 and undergo dissolution to offer active hydrogen. Due to the sufficient active hydrogen, the barrier for TS decreases to only 0.53 eV (Fig. 4i), making the formation of *CHO more kinetically feasible on CC20. In conclusion, the formation of *CHO is faster on CeO2/Cu-L than that on other surfaces without CeO2 due to the rapid water activation around CeO2, which agrees with the high current density for CC20 during CO2RR.
In addition, the DFT calculations were also carried out at the bias of −0.5 V and −1.12 V (Fig. S35 and S36†), respectively, which are the requirement to overcome the C–C coupling step and are consistent with the reaction potential. At the selected potentials, we can observe that hydrogenation of *CO to *CHO and then coupling of *CHO into *CHO–*CHO still remain the favourable path for C–C coupling on each surface during CO2RR. More importantly, both the thermodynamic process and kinetic process for the formation of *CHO on CeO2/Cu-L are more feasible than that on Cu-M or Cu-L. These results elucidate the motivation for the simultaneously enhanced selectivity and activity for C2+ products by the introduction of CeO2.
According to the DFT calculation results, hydrogenation of *CO to *CHO played a crucial role for enhancing the C2+ products, especially incorporated with the activation of H2O. In consequence, the kinetic isotopic effects (KIEs) of H/D over CC0 and CC20 were measured to further ensure the role of water activation in CO2RR (Fig. S37†). As the H2O was replaced by D2O as the solvent in 1 M KOH solution, the formation rate of ethylene significantly decreased on CC0, and the KIE (the ratio of ethylene formation rates in H2O and D2O) was about 2.0, which suggests that dissolution of H2O should be involved in the rate-determining step (RDS) for the ethylene formation. On the contrary, the KIE value on CC20 was nearly 1, suggesting that hydrogen was not related to the rate-determining step over CC20. The above confirmed the results of the DFT calculations that the *CHO route was endothermic on CC0 and exothermic on CC20. In addition, CC20 yielded 312 mA cm−2 at −1.12 V (vs. RHE) for HER under N2 atmosphere, about 2.5 times higher than that on CC0 (Fig. S37†), supporting the argument that CC20 had a superior capability for water activation. Thus, it can be concluded that the existence of CeO2 accelerated the dissolution of H2O to offer enough active hydrogen and thus benefited the generation of *CHO, which enhances the C–C coupling step through the dimerization of *CHO.
Conclusions
In conclusion, the introduction of CeO2 on the surface of CuO significantly enhanced the selectivity and activity towards C2+ products in CO2RR. Experimental and in situ SERS results confirmed the generation of the important intermediate CO was notably enhanced on CC20, which offered abundant precursors for the following steps. More importantly, DFT calculations revealed that the C–C coupling step followed the *CHO route and was facilitated both thermodynamically and kinetically on CC20 by the interfacial effects and the rapid water activation, respectively, findings which were also supported by the KIE experiments. Consequently, the FE of the C2+ products could reach up to 75.2% with the current density of 1.21 A cm−2 at −1.12 V (vs. RHE) in 1 M KOH. We believe that the findings in this work contribute to understanding the role of the introduced component and could help to design efficient catalysts towards C2+ products in CO2RR.Author contributions
X. P. Y., C. J. C. and B. X. H. proposed the project, designed the experiments and wrote the manuscript; X.P. Y. performed the whole experiments; Y. H. W., S. J. L., Y. Z. C., R. J. F. and J. Z. assisted in analyzing the experimental data; B. X. H. supervised the whole project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Key Research and Development Program of China (2017YFA0403102), National Natural Science Foundation of China (21890761 and 21733011), Beijing Municipal Science & Technology Commission (Z191100007219009), and Chinese Academy of Sciences (QYZDY-SSW-SLH013). The operando X-ray adsorption spectroscopy (XAS) measurements were performed using a flow cell at the 1W1B, 1W2B beamline at Beijing Synchrotron Radiation Facility (BSRF), China.Notes and references
- H. X. Wang, Y. K. Tzeng, Y. F. Ji, Y. B. Li, J. Li, X. L. Zheng, A. K. Yang, Y. Y. Liu, Y. J. Gong, L. L. Cai, Y. Z. Li, X. K. Zhang, W. Chen, B. F. Liu, H. Y. Lu, N. A. Melosh, Z. X. Shen, K. R. Chan, T. W. Tan, S. Chu and Y. Cui, Nat. Nanotechnol., 2020, 15, 131–137 CrossRef CAS PubMed.
- T. T. Zheng, K. Jiang and H. T. Wang, Adv. Mater., 2018, 30, e1802066 CrossRef PubMed.
- D. Yang, Q. Zhu and B. Han, Innovation, 2020, 1, 100016 Search PubMed.
- J. Gu, C. S. Hsu, L. C. Bai, H. M. Chen and X. L. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- Z. Liu, Acta Phys.-Chim. Sin., 2020, 36, 1912045 Search PubMed.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- U. O. Nwabara, E. R. Cofell, D. S. Verma, E. Negro and P. J. A. Kenis, ChemSusChem, 2020, 13, 855–875 CrossRef CAS PubMed.
- D. F. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. X. Wang, J. G. Wang and X. H. Bao, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS PubMed.
- L. Dai, Q. Qin, P. Wang, X. J. Zhao, C. Y. Hu, P. X. Liu, R. X. Qin, M. Chen, D. H. Ou, C. F. Xu, S. G. Mo, B. H. Wu, G. Fu, P. Zhang and N. F. Zheng, Sci. Adv., 2017, 3, e1701069 CrossRef PubMed.
- Z. G. Geng, X. D. Kong, W. W. Chen, H. Y. Su, Y. Liu, F. Cai, G. X. Wang and J. Zeng, Angew. Chem., Int. Ed., 2018, 57, 6054–6059 CrossRef CAS PubMed.
- F. Yang, P. Song, X. Z. Liu, B. B. Mei, W. Xing, Z. Jiang, L. Gu and W. L. Xu, Angew. Chem., Int. Ed., 2018, 57, 12303–12307 CrossRef CAS PubMed.
- X. Q. Wang, Z. Chen, X. Y. Zhao, T. Yao, W. X. Chen, R. You, C. M. Zhao, G. Wu, J. Wang, W. X. Huang, J. L. Yang, X. Hong, S. Q. Wei, Y. Wu and Y. D. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed.
- C. W. Lee, N. H. Cho, K. T. Nam, Y. J. Hwang and B. K. Min, Nat. Commun., 2019, 10, 3919 CrossRef PubMed.
- Z. R. Zhang, F. Ahmad, W. H. Zhao, W. S. Yan, W. H. Zhang, H. W. Huang, C. Ma and J. Zeng, Nano Lett., 2019, 19, 4029–4034 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. B. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- S. Gao, Z. T. Sun, W. Liu, X. C. Jiao, X. L. Zu, Q. T. Hu, Y. F. Sun, T. Yao, W. H. Zhang, S. Q. Wei and Y. Xie, Nat. Commun., 2017, 8, 14503 CrossRef CAS PubMed.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed.
- J. J. Fu, W. L. Zhu, Y. Chen, Z. Y. Yin, Y. Y. Li, J. Liu, H. Y. Zhang, J. J. Zhu and S. H. Sun, Angew. Chem., Int. Ed., 2019, 58, 14100–14103 CrossRef CAS PubMed.
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- D. F. Gao, I. Zegkinoglou, N. J. Divins, F. Scholten, I. Sinev, P. Grosse and B. Roldan Cuenya, ACS Nano, 2017, 11, 4825–4831 CrossRef CAS PubMed.
- H. P. Xu, D. Rebollar, H. Y. He, L. N. Chong, Y. Z. Liu, C. Liu, C. J. Sun, T. Li, J. V. Muntean, R. E. Winans, D. J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- Y. F. Li, D. Kim, S. Louisia, C. L. Xie, Q. Kong, S. Yu, T. Lin, S. Aloni, S. C. Fakra and P. D. Yang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 9194–9201 CrossRef CAS PubMed.
- X. L. Wang, J. F. de Araujo, W. Ju, A. Bagger, H. Schmies, S. Kuhl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS PubMed.
- A. Dutta, I. Z. Montiel, R. Erni, K. Kiran, M. Rahaman, J. Drnec and P. Broekmann, Nano Energy, 2020, 68, 104331 CrossRef CAS.
- C. Chen, J. F. K. Kotyk and S. W. Sheehan, Chem, 2018, 4, 2571–2586 CAS.
- F. W. Li, Y. G. C. Li, Z. Y. Wang, J. Li, D. H. Nam, Y. Lum, M. C. Luo, X. Wang, A. Ozden, S. F. Hung, B. Chen, Y. H. Wang, J. Wicks, Y. Xu, Y. L. Li, C. M. Gabardo, C. T. Dinh, Y. Wang, T. T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- W. C. Ma, S. J. Xie, T. T. Liu, Q. Y. Fan, J. Y. Ye, F. F. Sun, Z. Jiang, Q. H. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- J. J. Lv, M. Jouny, W. Luc, W. L. Zhu, J. J. Zhu and F. Jiao, Adv. Mater., 2018, 30, e180311 Search PubMed.
- L. Fan, C. Xia, F. Q. Yang, J. Wang, H. T. Wang and Y. Y. Lu, Sci. Adv., 2020, 6, eaay3111 CrossRef CAS PubMed.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS PubMed.
- D. Higgins, C. Hahn, C. X. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2019, 4, 317–324 CrossRef CAS.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Phys. Chem. B, 2002, 106, 15–17 CrossRef CAS.
- K. J. P. Schouten, E. P. Gallent and M. T. M. Koper, ACS Catal., 2013, 3, 1292–1295 CrossRef CAS.
- X. D. Kong, C. Wang, H. Zheng, Z. G. Geng, J. Bao and J. Zeng, Sci. China: Chem., 2021 DOI:10.1007/s11426-020-9934-0.
- T. T. H. Hoang, S. C. Ma, J. I. Gold, P. J. A. Kenis and A. A. Gewirth, ACS Catal., 2017, 7, 3313–3321 CrossRef CAS.
- C. Choi, T. Cheng, M. F. Espinosa, H. L. Fei, X. F. Duan, W. A. Goddard and Y. Huang, Adv. Mater., 2019, 31, e1805405 CrossRef PubMed.
- M. Zhong, K. Tran, Y. M. Min, C. H. Wang, Z. Y. Wang, C. T. Dinh, P. De Luna, Z. Q. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C. S. Tan, M. Askerka, F. L. Che, M. Liu, A. Seifitokaldani, Y. J. Pang, S. C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- T. T. H. Hoang, S. Verma, S. C. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS PubMed.
- Z. Y. Yin, C. Yu, Z. L. Zhao, X. F. Guo, M. Q. Shen, N. Li, M. Muzzio, J. R. Li, H. Liu, H. H. Lin, J. Yin, G. Lu, D. Su and S. H. Sun, Nano Lett., 2019, 19, 8658–8663 CrossRef CAS PubMed.
- Y. S. Zhou, F. L. Che, M. Liu, C. Q. Zou, Z. Q. Liang, P. De Luna, H. F. Yuan, J. Li, Z. Q. Wang, H. P. Xie, H. M. Li, P. N. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y. W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. Roldan Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- Y. Kim, S. Park, S. J. Shin, W. Choi, B. K. Min, H. Kim, W. Kim and Y. J. Hwang, Energy Environ. Sci., 2020, 13, 4301–4311 RSC.
- J. H. Montoya, A. A. Peterson and J. K. Norskov, ChemCatChem, 2013, 5, 737–742 CrossRef CAS.
- H. Yano, T. Tanaka, M. Nakayama and K. Ogura, J. Electroanal. Chem., 2004, 565, 287–293 CrossRef CAS.
- X. Wang, Z. Y. Wang, F. P. G. de Arquer, C. T. Dinh, A. Ozden, Y. G. C. Li, D. H. Nam, J. Li, Y. S. Liu, J. Wicks, Z. T. Chen, M. F. Chi, B. Chen, Y. Wang, J. Tam, J. Y. Howe, A. Proppe, P. Todorovic, F. W. Li, T. T. Zhuang, C. M. Gabardo, A. R. Kirmani, C. McCallum, S. F. Hung, Y. W. Lum, M. C. Luo, Y. M. Min, A. N. Xu, C. P. O'Brien, B. Stephen, B. Sun, A. H. Ip, L. J. Richter, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Energy, 2020, 5, 478–486 CrossRef CAS.
- K. Chang, H. C. Zhang, M. J. Cheng and Q. Lu, ACS Catal., 2020, 10, 613–631 CrossRef CAS.
- W. Wang, Y. Zhang, Z. Y. Wang, J. M. Yan, Q. F. Ge and C. J. Liu, Catal. Today, 2016, 259, 402–408 CrossRef CAS.
- A. Aitbekova, L. H. Wu, C. J. Wrasman, A. Boubnov, A. S. Hoffman, E. D. Goodman, S. R. Bare and M. Cargnello, J. Am. Chem. Soc., 2018, 140, 13736–13745 CrossRef CAS PubMed.
- C. J. Chen, X. P. Yan, S. J. Liu, Y. H. Wu, Q. Wan, X. F. Sun, Q. G. Zhu, H. Z. Liu, J. Ma, L. R. Zheng, H. H. Wu and B. X. Han, Angew. Chem., Int. Ed., 2020, 59, 16459–16464 CrossRef CAS PubMed.
- Z. Liu, Acta Phys.-Chim. Sin., 2019, 35, 1307–1308 Search PubMed.
- Y. W. Lum and J. W. Ager, Angew. Chem., Int. Ed., 2018, 57, 551–554 CrossRef CAS PubMed.
- W. Akemann and A. Otto, J. Raman Spectrosc., 1991, 22, 797–803 CrossRef CAS.
- W. Y. Shan, R. Liu, H. C. Zhao, Z. L. He, Y. J. Lai, S. S. Li, G. Z. He and J. F. Liu, ACS Nano, 2020, 14, 11363–11372 CrossRef CAS PubMed.
- C. M. Gunathunge, X. Li, J. Y. Li, R. P. Hicks, V. J. Ovalle and M. M. Waegele, J. Phys. Chem. C, 2017, 121, 12337–12344 CrossRef CAS.
- J. Heyes, M. Dunwell and B. J. Xu, J. Phys. Chem. C, 2016, 120, 17334–17341 CrossRef CAS.
- Y. R. Zhao, X. Z. Chang, A. S. Malkani, X. Yang, L. Thompson, F. Jiao and B. J. Xu, J. Am. Chem. Soc., 2020, 142, 9735–9743 CAS.
- Z. Q. Liang, T. T. Zhuang, A. Seifitokaldani, J. Li, C. W. Huang, C. S. Tan, Y. Li, P. De Luna, C. T. Dinh, Y. F. Hu, Q. F. Xiao, P. L. Hsieh, Y. H. Wang, F. W. Li, R. Quintero-Bermudez, Y. S. Zhou, P. N. Chen, Y. J. Pang, S. C. Lo, L. J. Chen, H. R. Tan, Z. Xu, S. L. Zhao, D. Sinton and E. H. Sargent, Nat. Commun., 2018, 9, 3828 CrossRef PubMed.
- M. C. Luo, Z. Y. Wang, Y. G. C. Li, J. Li, F. W. Li, Y. W. Lum, D. H. Nam, B. Chen, J. Wicks, A. N. Xu, T. T. Zhuang, W. R. Leow, X. Wang, C. T. Dinh, Y. Wang, Y. H. Wang, D. Sinton and E. H. Sargent, Nat. Commun., 2019, 10, 5814 CrossRef CAS PubMed.
- A. Vasileff, Y. P. Zhu, X. Zhi, Y. Q. Zhao, L. Ge, H. M. Chen, Y. Zheng and S. Z. Qiao, Angew. Chem., Int. Ed., 2020, 59, 19649–19653 CrossRef CAS PubMed.
- D. F. Gao, I. T. McCrum, S. Deo, Y. W. Choi, F. Scholten, W. M. Wan, J. G. G. Chen, M. J. Janik and B. Roldan Cuenya, ACS Catal., 2018, 8, 10012–10020 CrossRef CAS.
- M. Favaro, H. Xiao, T. Cheng, W. A. Goddard, J. Yano and E. J. Crumlin, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6706–6711 CAS.
- S. B. Varandili, J. F. Huang, E. Oveisi, G. L. De Gregorio, M. Mensi, M. Strach, J. Vavra, C. Gadiyar, A. Bhowmik and R. Buonsanti, ACS Catal., 2019, 9, 5035–5046 CrossRef CAS.
- S. L. Chu, X. P. Yan, C. Choi, S. Hong, A. W. Robertson, J. Masa, B. X. Han, Y. S. Jung and Z. Y. Sun, Green Chem., 2020, 22, 6540–6546 RSC.
- L. Wang, D. C. Higgins, Y. F. Ji, C. G. Morales-Guio, K. Chan, C. Hahn and T. F. Jaramillo, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12572–12575 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01117k |
| This journal is © The Royal Society of Chemistry 2021 |