
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCeramic boron carbonitrides for unlocking organic halides with visible light†
Tao
Yuan
a,
Meifang
Zheng
*a,
Markus
Antonietti
 b and
Xinchen
Wang
*a
b and
Xinchen
Wang
*a
aState Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou 350116, China. E-mail: mfzheng@fzu.edu.cn; xcwang@fzu.edu.cn
bMax-Planck Institute of Colloids and Interfaces, Department of Colloid Chemistry, Research Campus Golm, 14424 Potsdam, Germany
First published on 23rd March 2021
Abstract
Photochemistry provides a sustainable pathway for organic transformations by inducing radical intermediates from substrates through electron transfer process. However, progress is limited by heterogeneous photocatalysts that are required to be efficient, stable, and inexpensive for long-term operation with easy recyclability and product separation. Here, we report that boron carbonitride (BCN) ceramics are such a system and can reduce organic halides, including (het)aryl and alkyl halides, with visible light irradiation. Cross-coupling of halides to afford new C–H, C–C, and C–S bonds can proceed at ambient reaction conditions. Hydrogen, (het)aryl, and sulfonyl groups were introduced into the arenes and heteroarenes at the designed positions by means of mesolytic C–X (carbon–halogen) bond cleavage in the absence of any metal-based catalysts or ligands. BCN can be used not only for half reactions, like reduction reactions with a sacrificial agent, but also redox reactions through oxidative and reductive interfacial electron transfer. The BCN photocatalyst shows tolerance to different substituents and conserved activity after five recycles. The apparent metal-free system opens new opportunities for a wide range of organic catalysts using light energy and sustainable materials, which are metal-free, inexpensive and stable.
Introduction
The renaissance of chemistry with radicals in organic synthesis is also related to the revival of photochemistry.1 The growth of the field comes from the direct conversion of solar energy to chemical energy, thus enabling mild conversions, even uphill reactions, as well as efficient access to open shell reactive intermediates.2 Photocatalysts, which can be classified into transition metal-based complexes, organic dyes and semiconductors, have all been widely applied in a range of organic transformations with light irradiation.3 Most previous contributions used homogeneous photosensitizers, for example, Ru(ppy)3Cl2,1fac-Ir(ppy)3,3 eosin Y,4 and acridinium salts.5 Previous reports on homogeneous molecular photosensitizers are inevitably limited by photo-corrosion as well as catalyst deactivation due to the strong interaction with strong nucleophiles/electrophiles and reactive radical intermediates.5–8 Besides photobleaching, there is also a general concern about the sustainability and price of widely-used metal complexes containing precious metals, such as Ru and Ir, as well as related health risks.In contrast to homogeneous catalysts, heterogeneous catalysts feature the advantage of easy recyclability by simple filtration due to their insoluble nature, while still conserving reactivity during prolonged photochemical operation. The electron transfer mechanism of heterogeneous semiconductor photocatalysts is otherwise similar to that of homogeneous photosensitizers, as illustrated in Fig. 1.9 In the case of molecular photosensitizers, one electron transfers to the LUMO (lowest unoccupied molecular orbital) upon light excitation and then transfers to the substrates.10 To close the catalytic cycle, reductive or oxidative quenching is required to bring the photosensitizer back to the ground state (Fig. 1a). For semiconductor materials, electron–hole pairs are produced under light irradiation. Similarly to the frontier orbital theory, the CB (conduction band) can donate electrons (like the LUMO), forming reductive intermediates, and the VB (valence band) can act as an electron acceptor (like the HOMO), forming oxidative intermediates, which together participate in yielding the final products (Fig. 1b).11 When considering a semiconductor for certain transformations thermodynamically, the light absorption and the related magnitude of the bandgap are important, as well as the absolute redox potentials (the positions of the VB and CB) with respect to those of the targeted substrates.12 Electron transfer to and from the substrate must be thermodynamically allowed, otherwise the reaction cannot take place.
 | ||
| Fig. 1 Schematic illustrations of molecular and semiconductor photoredox catalytic electron transfer modes in the activation of substrates and the specific process of photochemical organic transformations. | ||
Semiconductor photocatalysts have been broadly investigated and applied to the water splitting reaction aiming to mitigate the issues of the impending energy shortage and the utilization of light energy.13–16 However, the developed metal-based semiconductors, such as metal oxides, (oxy)nitrides and (oxy)sulfides, are either visible light-irresponsive or photocorrosive, and the involved precious metals compromise the sustainability. Metal-free semiconductors, for example, conjugated microporous polymers (CMPs),14 covalent organic frameworks (COFs),17,18 and graphitic carbon nitrides (g-CN),15,19 are the emerging, superior heterogeneous photocatalysts for light energy conversion and environmental remediation. The ceramic semiconductor boron carbonitride (BCN) has just recently been developed but is already well-known in the area of artificial photosynthesis.20–22 BCN materials are inorganic semiconductor photocatalysts, which in principle feature a lower binding energy of excitons and faster charge migration properties than polymeric photocatalysts.23 Analogous to hexagonal boron nitride (h-BN), BCN exhibits excellent adsorption properties for organic molecules because of the existence of the polar B–N bond and the high specific surface area.24–26 BCN ceramics are visible-light-responsive semiconductors with an adjustable band gap energy and energy levels depending on the amount of carbon incorporated into the boron nitride (BN) frameworks.27–29 The ternary BCN materials allow a tunable band gap energy in a wide range of 0–5.5 eV by varying the concentration of B, C and N, which indeed enables extended use of light irradiation to run the organic reactions.30–32 In addition, the cheap and simple preparation also makes BCN (a few euros per kilogram) a promising alternative as a metal-free heterogeneous photocatalyst for organic photosynthesis.
Despite possessing obvious advantages including easy separation, high photo/thermal/chemical stability, and recyclable features, only a few explorations of organic transformations have been made with BCN. We herein report the application of ceramic BCN as heterogeneous photoredox catalysts for the radical coupling of organic halides using visible light. Over the past decades, significant advances in the use of halides in cross-coupling have been made in organic chemistry, usually under the involvement of transition metal complexes, enabling the conversion of C–X (carbon–halogen) into carbon–carbon, –nitrogen, –sulfur, and –oxygen bonds, such as the Suzuki, Ullman, and Kumada reactions.33–35 The interest in developing mild conditions has propelled new advances in the transformation of halides and has made photocatalytic techniques an attractive alternative.36–38 Reports exemplified fac-Ir(ppy)3 as a photosensitizer, which converted non-activated C–I into C–H bonds via photoinduced electrons or energy transfer processes to substrates under visible light irradiation.39 The MacMillan group reported that combination of iridium complexes and nickel organocatalysts can drive the coupling of C(sp2/sp3)–Br with C(sp2/sp3)–H (Fig. 1c).36,40,41 Despite the widespread success of transition-metal-catalyzed coupling methodologies, “metal residue” is a concern in the synthesis of medicinally important or biologically active compounds, and realizing a metal-free methodology has aroused extensive research interest. To get rid of “metal residues” in the final products, reduction of electron-deficient aryl halides has been achieved with PDI (perylene-diimide),42 Rh-6G,43 phenolate anion,44 phenothiazine45etc. under visible light illumination in homogeneous systems. Here we show that the ceramic BCN semiconductors can efficiently catalyze the general coupling of halides in heterogenous and metal-free systems under mild reaction conditions, with good to excellent selectivity and yields in a gram-scalable manner (Fig. 1d).
Results
Hydrodehalogenations of (het)aryl and alkyl halides
Hydrodehalogenation is a kind of reaction in which a halogen atom is formally substituted by a hydrogen atom.46 It not only serves as a deprotection toolkit in the multistep synthesis of complex natural products, but also applies to the degradation of artificial and environmentally toxic halogen-containing organic pollutants, such as residual pesticides, hard-metabolizable pharmaceuticals, and fire retardants.47 Herein, BCN was chosen as a dehalogenation photocatalyst because of its high surface area, its morphology of porous, two-dimensional nanosheets, and its ability for light absorption.20 Control experiments demonstrated that the presence of light and BCN are crucial for the reaction to occur, as no product 2a was detected in the absence of light or photocatalyst (Table S1,† entries 9–10). Other metal-free semiconductors, such as polymeric carbon nitride (CNU and mpg-CN), have been previously reported for some oxidation reactions19,48 and the C–N cross-coupling of aryl bromides with the Ni complex cocatalyst,49 however, they were not able to photocatalyze the hydrodehalogenation reactions successfully with visible light (Table S1,† entries 12–13).The analyzed cases of BCN photocatalytic dehalogenation are shown in Table 1 (the valence band position of BCN was determined to be +1.58 V vs. SCE, see ESI, Fig. S7†). iPrOH acts as both the hydrogen and electron donor (Eox = +1.27 V vs. SCE, see ESI, Fig. S13†), which was confirmed by the deuteration experiment (Fig. S22†). The aryl radical intermediate could be captured by TEMPO, demonstrating the existence of a single-electron-transfer (SET) process in our metal-free heterogeneous system (Fig. S23†). Aryl bromides with methoxy and methyl substituents at different positions provided the corresponding arenes in good to excellent yields (2a–2f) (Table 1a). A 58% yield of 2g was isolated, revealing methylthio substituent tolerance. 4-Bromobiphenyl was reduced to biphenyl (2h) in 95% yield, and the substituted naphthalenes showed excellent yields (2i–2j) (Table 1a). Besides, ketone, ester, and carboxyl functionalities were maintained with good to excellent yields (2k–2q) (Table 1a). Moreover, heteroarenes such as benzothiophene (2r), carbazole (2s), quinoline (2t), and 1,4-benzodioxan (2u) can be achieved from the reduction of the corresponding bromides with good to excellent yields (Table 1a). In addition, the system is also feasible for dechlorination, which is a thermodynamically challenging process. Pleasingly, hydrodechlorination of 4-chloroanisole occurred smoothly leading to an 87% yield of 2v. An 82% yield of 2w was isolated using 2-bromo-1,4-dimethoxybenzene as the substrate. 2-Hydroxy-benzoicacimethylester (2x), a natural perfume, was obtained in 70% isolated yield under a prolonged irradiation time (Table 1b). Aryl chloride derivatives with electron-withdrawing groups also went through the reduction reaction successfully with excellent yields of 2y (87%), 2z (93%), and 2aa (96%), respectively (Table 1b). 1-Chloro-4-phenylbutane could be reduced to 2bb with a 36% yield of 2bb in this system (Fig. S12†). However, trace amounts of quinoline (<5%, 2cc) were detected after 15 h of illumination (Table 1b).

C(sp2)–H (het)arylations with C(sp2)–Br
The metal-free BCN photocatalyst was efficient for the coupling of C(sp2)–H and C(sp2)–Br as well (Fig. 2a). Intramolecular cyclization of inactive o-brominated alkylaryl ethers proceeded smoothly, and dihydrobenzofuran (3a) and dihydrobenzopyran (3b) were obtained with satisfying isolated yields in 72 h (Fig. 2a). This success of the intramolecular coupling under photoreductive conditions inspired the idea of exploring other cross-coupling reactions.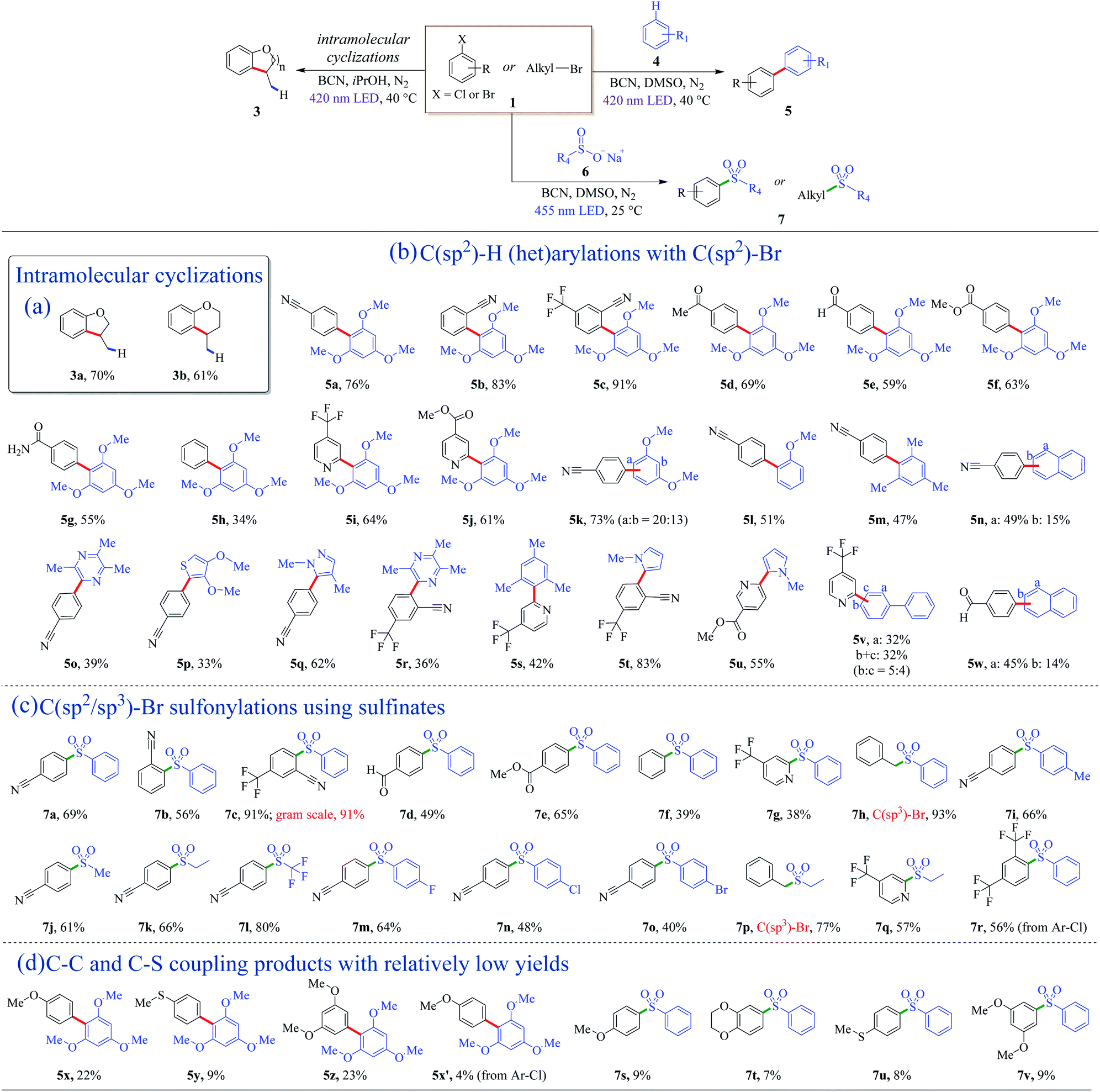 | ||
| Fig. 2 Photoredox coupling of organic halides with (het)arenes and sulfinates to afford new C–C and C–S bonds using the ceramic BCN photocatalyst. Detailed reaction conditions can be found in Methods (see General procedure 2–4 in this article). It should be noted that the yields of 5x–5x′ and 7s–7v were determined by GC-MS. | ||
Biaryl structures are ubiquitous motifs in pharmaceutical molecules, biochemistry, and polymeric materials.50–52 In the previous dehalogenation examples, the photo-generated holes were consumed and removed from the reaction by sacrificial agents. To get full utilization of all charge carriers, arenes were introduced into the system, leading to the formation of heteroarene cation radicals, resulting in the C–C cross-coupling biaryl motifs. To get rid of the hydrogenation products, the aprotic solvent DMSO was used instead of protic solvents. The valence band (VB) of BCN is +1.58 V vs. SCE (Fig. S7†), which is thermodynamically capable of oxidizing 1,3,5-trimethoxybenzene (4a, E4a/4a˙+ = +1.34 V vs. SCE, Table S4†) to generate an arene cation radical. With the optimized reaction condition, a 76% yield of 5a was obtained (Table S2†). Examples of direct C(sp2)–H (het)arylations using C(sp2)–Br as nucleophilic agents are displayed in Fig. 2b. Aryl bromide derivatives with cyano or trifluoromethyl substituents, coupling with 1,3,5-trimethoxybenzene (4a), afforded good to excellent yields (5a–5c). The mild and free-of-base-and-metal system shed light on the maintenance of the aldehyde, ester, and amido groups, and moderate yields of products (5d–5g) were obtained (Fig. 2b). Bromobenzene can couple with C(sp2)–H with BCN as the photocatalyst (5h). Electron-rich aryl halides proceeded with relatively low yields, as shown in Fig. 2d (5x–5x′, Fig. S14–S17†). Except for the arenes, heteroaromatic derivatives are reactive as well, and the corresponding products were achieved in good yields (5i and 5j) (Fig. 2b).
Next, the scope of C(sp2)–H was explored. When 1,3-dimethoxybenzene was used as an aromatic substrate, the coupling products at the a and b-position (5k) were obtained in a 73% yield with 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13 of the mixture (Fig. 2b). Anisole and 1,3,5-trimethylbenzene can also react with 4-bromobenzonitrile (1b), affording biaryl compounds 5l and 5m with moderate yields of 51% and 47%. Moreover, a- and b-position substituted aryl naphthalenes were successfully synthesized with the present metal-free system (5n). Among them, the cyano-containing compound 5n-a is a direct precursor for the one-pot synthesis of 2,4,6-tris(4(naphthalen-1-yl)phenyl)-1,3,5-triazine (TNPT), which is valuable as an excellent hole blocking layer (HBL) material in long lifetime organic light emitting diodes (OLEDs).53 Several heteroarenes went through cross-coupling successfully, and heterocyclic C(sp2)–H arylation products were isolated with general yields of 33–62% (5o–5s). Biologically relevant pyrrole derivatives were also compatible with the reaction conditions, producing the het-aryl and het–het biaryl motifs in generally excellent yields (5t and 5u). These unique structures could serve as the backbone units of drug molecules.54 Polycyclic biaryl structures could be acquired with moderate yields as well (5v and 5w).
:13 of the mixture (Fig. 2b). Anisole and 1,3,5-trimethylbenzene can also react with 4-bromobenzonitrile (1b), affording biaryl compounds 5l and 5m with moderate yields of 51% and 47%. Moreover, a- and b-position substituted aryl naphthalenes were successfully synthesized with the present metal-free system (5n). Among them, the cyano-containing compound 5n-a is a direct precursor for the one-pot synthesis of 2,4,6-tris(4(naphthalen-1-yl)phenyl)-1,3,5-triazine (TNPT), which is valuable as an excellent hole blocking layer (HBL) material in long lifetime organic light emitting diodes (OLEDs).53 Several heteroarenes went through cross-coupling successfully, and heterocyclic C(sp2)–H arylation products were isolated with general yields of 33–62% (5o–5s). Biologically relevant pyrrole derivatives were also compatible with the reaction conditions, producing the het-aryl and het–het biaryl motifs in generally excellent yields (5t and 5u). These unique structures could serve as the backbone units of drug molecules.54 Polycyclic biaryl structures could be acquired with moderate yields as well (5v and 5w).
C(sp2/sp3)–Br sulfonylations using sulfinates
The introduction of sulfonyl groups could enhance the surface hydrophilicity and/or decrease the rates of enzymatic metabolization, improving the pharmacokinetic properties of lead candidates in drug discovery.55 Several reports employed homogeneous nickel/photoredox dual catalysis to introduce sulfonyl groups by cross-coupling of sodium sulfonates with halides.56,57 It is mild and odorous thiols and strong oxidizing reagents can be avoided. Herein, a metal-free photocatalytic system was introduced to install sulfonyl motifs into aryls. Solvents and a series of metal-free catalysts were screened first to optimize the reaction conditions (Table S3†). The reaction is thermodynamically favorable due to the valence band (VB) position of BCN (+1.58 V vs. SCE) outdistancing the oxidation potential of sodium benzenesulfinate (+0.37 V vs. SCE, Table S4†). According to the results, BCN had a product yield of 51% at 24 h under 3 W blue LED irradiation. Polymeric carbon nitrides (CNU, mpg-CN, and CCN) as reference metal-free photocatalysts did not give the desire sulfonyl product (Table S3†). With prolonged illumination time, a satisfactory yield of 7a (78%) was obtained using BCN as the photocatalyst under mild conditions.Examples of C(sp2/sp3)–Br sulfonylations using sulfinates as coupling reagents are depicted in Fig. 2c. Diverse sulfone motifs were obtained via BCN photoredox catalysis. For aryl bromides, the protocol could tolerate a variety of functional groups, including cyano (7a, 7b), trifluoromethyl (7c), aldehyde (7d), and ester (7e) groups (Fig. 2c). Bromobenzene can be the coupling partner with a 39% yield of sulfonyldibenzene (7f), as well as a p-bromopyridine motif (7g). Moreover, the C(sp3)-centered reactant gave the corresponding product with an excellent yield of 93% (7h). Electron-rich aryl bromides resulted in low yields, as shown in Fig. 2d (7s–7v, Fig. S18–S21†). A wide range of structurally diverse sulfonyl reagents were tolerant for this transformation. Sodium sulfinates with phenyl and alkyl groups went through the sulfonyl process smoothly with good yields (7i–7k). Noteworthily, sodium trifluoromethanesulfinate can retain the sulfonyl group, and an 80% yield of sulfonylation product was formed (7l). Besides, haloarenes were compatible with the reaction conditions, leading to the corresponding products (7m–7o) in moderate to good yields. Alkyl and heterocyclic sulfones were also afforded (7p and 7q). In addition, this protocol was applicable to aryl chloride with a 56% yield (7r).
Discussion
For the mechanistic studies, a series of monochromatic light experiments with different wavelengths were carried out. Apparently, the trend of the wavelength-dependent yields of 5a matches well with the optical absorption intensity of BCN, supporting the photo-induced nature of the process (Fig. 3a).55 The variation of the concentration of reactant 1b, C–H arylation product 5a, and hydrodehalogenation by-product during the photoredox reaction is shown in Fig. 3b. The concentration of 1b declined gradually while the arylation product 5a steadily increased. After 24 h irradiation, 1b was completely consumed. The hydrodehalogenation by-product was minor but unavoidable. In addition, the relative reactivities of different para-substituted aryl bromides toward C(sp2)–H arylation were examined (Fig. 3c and Table S5†). A positive linear correlation between the calculated log(kR/kH) values and σp constants was observed from the Hammett plot analysis, disclosing that the reaction proceeds through nucleophilic aromatic substitution.58 Besides, the recovered BCN could be used for the corresponding reactions for five repeats, with slightly decreased yields. This illustrates the photo and chemical stability of the photocatalyst (Fig. 3d), which was further confirmed by structural characterizations before and after the reaction (Fig. S30 and S31†), essentially reflecting no change. Thus, the photochemical stabilities of BCN allows easy scaling to gram quantities, and the gram-scale reaction for the preparation of 7c proceeded smoothly with an isolated yield of 91% (Fig. 3e–g, also see Fig. S9†). | ||
| Fig. 3 (a) Wavelength-dependent yields of C(sp2)–H arylation product (5a), the reaction time was 12 h. (b) Variation of the amount of 5a, the hydrodehalogenation by-product and reactant 1b during the C(sp2)–H arylation reaction. (c) Hammett plots for the arylation of C(sp2)–H with substituted aryl bromides. Hammett plots were obtained from a ratio of the yield with a reaction time of 24 h. (d) Evaluation of catalysts recycling for different reactions (dehalogenation, C–H arylation, and C–Br sulfonylation performed in a sequence yielding 2a, 5a, and 7a, respectively) under standard conditions, respectively. Upscaling experiments, gram-scale preparation of 7c using ceramic BCN as the photocatalyst are displayed in (e–g): (e) the required reagents and reaction flask; (f) the photochemical reaction device under blue LED irradiation; and (g) the recovered BCN and isolated product 7c. | ||
Kinetic isotope effect (KIE) and radical capture experiments were conducted to further characterize the metal-free C(sp2)–H arylation process. As shown in Fig. 4a and S24,† the value of KIE was 1.25 (KH:KD), suggesting that the C–H bonds cleavage of arenes is not the rate-determining step. In addition, the yield of 5a declined sharply when two equivalents of 2,2,6,6-tetramethylpiperidinoxyl (TEMPO) were added as the radical scavenger, and the radical intermediate from 4a was trapped by TEMPO with the molecular weight of the adduct being detected by GC-MS (Fig. S25†), proving the oxidation step of arenes by BCN. Thereafter, EPR study using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as the radical scavenger further proved the formation of a carbon-centered radical of 4a in the mixture, and the signal values (aN = 15.2, aH = 24.9) were nearly identical to the reported values of the carbon-centered radical (Fig. 4b–d).59
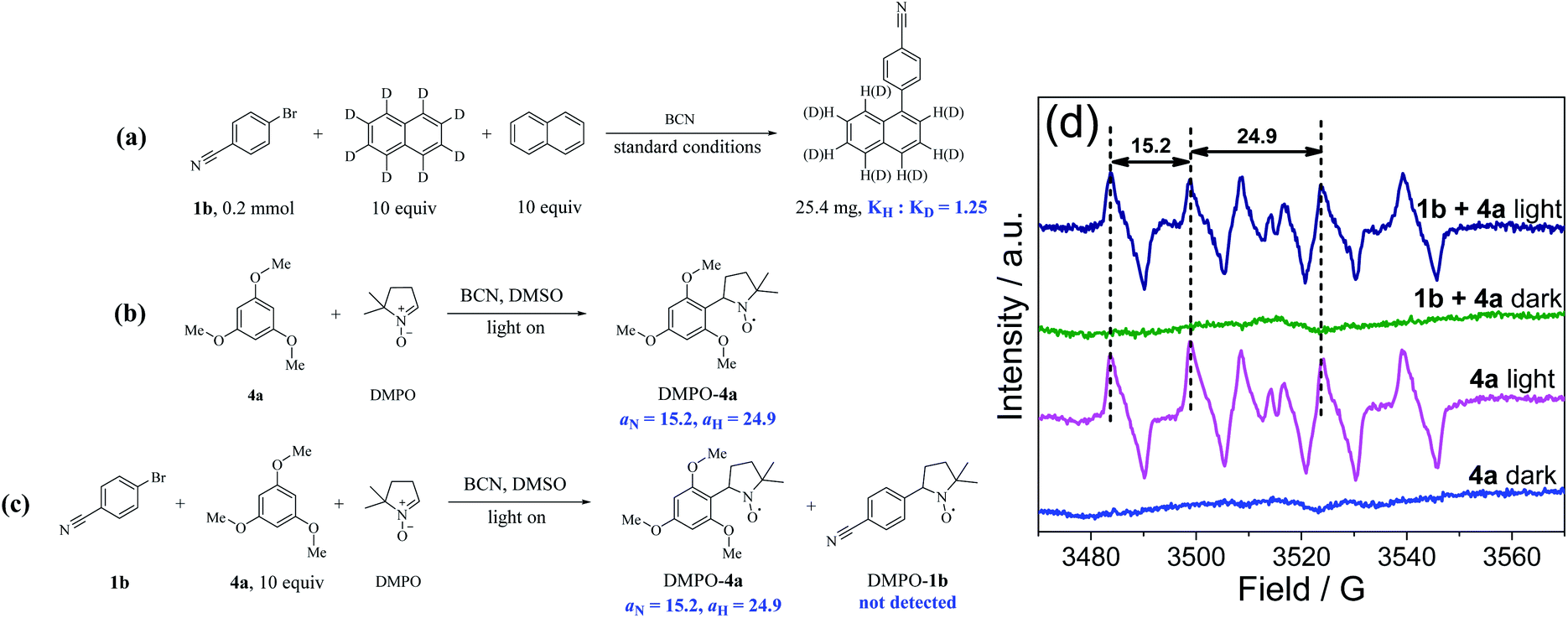 | ||
| Fig. 4 The mechanistic insights of C(sp2)–H arylations with C(sp2)–Br. (a) The intermolecular KIE experiment. EPR measurements of DMSO solution of DMPO and BCN with addition of (b) 4a and (c) 1b/4a mixture upon 3 min visible light irradiation. (d) EPR spectrum of measurement results. | ||
Several control experiments were also carried out for the C–Br sulfonylation reaction. The reaction was restrained with a product yield of 14% under the open air (Fig. S27a†), indicating that the presence of oxygen is detrimental to the reaction. Additionally, the yield declined with the loading amount of TEMPO (the yield declined to minor with 5 equivalents of TEMPO, Fig. S27b†), suggesting a radical pathway. With the addition of 1,1-diphenylethylene, the sulfonylation process was prohibited, meanwhile, the sulfonyl and aryl radicals were captured by 1,1-diphenylethylene, with molecular weights of 258 and 281, respectively (Fig. S27c and S28†). The results indicated the involvement of both photogenerated-hole oxidation and photogenerated-electron reduction.
Based on the above experimental results, we put forward a possible reaction mechanism for C–X couplings, as illustrated in Fig. 5. Upon visible light irradiation, BCN is excited to generate photo-induced electrons at the conduction band (CB, −1.09 V vs. SCE) and holes at the valence band (VB, +1.58 V vs. SCE). Then the photogenerated electron–hole pair separates and migrates to the surface of the photocatalyst. For the hydrogenation or cyclization process, the hole oxidizes sacrificial agent 1A with the release of a proton and electron to afford 1B, while the electron on the CB reduces the C–X substrate (1C or 2C) to form the active intermediate 1D or 2D. Subsequently, mesolytic C–X cleavage generates a halogen anion and (het)aryl radical (1E or 2E). The radical (1E or 2E) then accepts one electron and one proton with the formation of a hydrogenation or cyclization product (Fig. 5a and b). Moreover, the reaction might be further thermodynamically favored through a proton-coupled electron transfer (PCET) process that overcomes the disfavored energetic limitation.60,61 For the construction of C–C and C–S bonds, the sacrificial agent is replaced with the corresponding synthons – (het)arene and sulfinate (3A and 4A). Afterward, the oxidative electron transfer involves these substrates (3A and 4A) with the formation of an arene cation radical (3B) or sulfonyl radical (4B′). Aryl bromide 3C then couples with 3B to form the intermediate 3D followed by the subsequent reduction by the electron on the CB, finally releasing the bromide anion and C–C coupling product (Fig. 5c). Alternative possible mechanisms for C–H (het)arylations are initiated with reductive cleavage of C–Br bond (Fig. S26†). Different from C–C coupling, the C–X substrate (4C) accepts an electron and develops to an activated radical anion (4D) through reductive electron transfer. The coupling of two radicals (4D and 4B′) affords the final C–S coupling product (Fig. 5d).
 | ||
| Fig. 5 The proposed mechanism for (a) hydrodehalogenation, (b) intramolecular cyclization, (c) C–H (het)arylation, and (d) C–Br sulfonylation to form new C–H, C–C, and C–S bonds by BCN under visible light. | ||
Conclusions
The ceramic BCN semiconductor was introduced as a visible-light-responsive photocatalyst for the coupling of halides with various synthons. A wide range of organic halides including aryl-, alkyl-, bromides and chlorides can be effectively reduced to carbon-centered radicals in good to excellent yields and with good substituent tolerance. The good performance is due to the accessible photocatalytic sites of BCN. In this system, hydrogenations take advantage of electron reduction and coupling reactions orchestrate oxidative and reductive electron transfer. All these are achieved free of any transition-metal catalysts or ligands. The protocol provides an ideal alternative for the construction of C–H, C–C, and C–S bonds to classical transition metal catalysts and organic dyes. We believe that the generality of this methodology will be useful also for other organic coupling chemistry. The protocol provides an avenue to organic synthesis in an environment-friendly, atom-economic, and sustainable manner, which may be valuable for large scale synthesis or even industrial production.Methods
Synthesis of ceramic BCN
Typically, urea (1.6 g), boric acid (0.8 g), and glucose (5.6 g) were ground fully with an agate mortar for 20 min. Then, 3 g KCl was poured into the mortar and the mixture was ground again for 10 min. Afterwards, the mixed precursor was put into a horizontal tube furnace. Ammonia was pumped into the tube for 20 min to expel air before heating up. Then the mixture was heated to 1250 °C for 5 h at a heating rate of 5 °C min−1 under a flow of ammonia (200 mL min−1). The obtained product was washed with 200 mL water 5 times and dried overnight at 80 °C in a vacuum oven. The resulting final sample was denoted BCN.General procedure 1
Hydrodehalogenation of (het)aryl and alkyl halides. To a Schlenk tube containing a stirring bar was added BCN (10 mg), iPrOH (6 mL) and halide (Br or Cl, 0.2 mmol), then the tube was degassed in vacuo and refilled with N2 5 times. With the purple LED (420 nm, 15 W) switched on, the reaction mixture was stirred for a certain time (noted in the text) at 40 °C until the reactant was fully consumed which was determined by GC-MS. After the reaction, the mixture was filtered and extracted by CH2Cl2, the solvent was removed under reduced pressure, and the residue was purified by column chromatography on silica gel using petroleum ether/ethyl acetate and pentane/diethyl ether as the eluent to afford the final hydrodehalogenation product.General procedure 2
Intramolecular cyclizations of inactive o-alkenyl bromobenzenes. Typically, to a Schlenk tube containing a stirring bar was added BCN (20 mg), K2CO3 (55 mg, 2 equiv.), iPrOH (6 mL) and reactant o-alkenyl bromobenzene (0.2 mmol), then the tube was degassed in vacuo and refilled with N2 5 times. With the purple LED (420 nm, 15 W) switched on, the reaction mixture was stirred for 72 h at 40 °C, and the reaction progress was monitored by TLC or GC-MS. After the reaction, the mixture was filtered and extracted by CH2Cl2, the solvent was removed under reduced pressure, and the residue was purified by column chromatography on silica gel using pure petroleum ether as the eluent to afford the final intramolecular cyclized product.General procedure 3
C(sp2)–H (het)arylations with C(sp2)–Br. To a Schlenk tube containing a stirring bar was added BCN (20 mg), (het)aryl bromide (0.2 mmol), and (het)arenes (20 equiv.), then DMSO (2 mL) was added to the mixture. After that, the tube was degassed in vacuo and refilled with N2 for 5 times. With the purple LED (420 nm, 15 W) switched on, the reaction mixture was stirred for 24 h at 40 °C, and the reaction progress was monitored by TLC or GC-MS. After the reaction, the mixture was filtered and extracted with ethyl acetate several times, the organic layers were combined and dried over anhydrous Na2SO4, the solvent was removed under reduced pressure, and the residue was purified by column chromatography on silica gel using petroleum ether/ethyl acetate as the eluent to afford the final C–C cross-coupling product.General procedure 4
C(sp2/sp3)–Br sulfonylations using sulfinates. To a Schlenk tube containing a stirring bar was added BCN (10 mg), (het)aryl halide (Br or Cl, 0.2 mmol), and sulfinate (1 mmol, 5 equiv.), then DMSO (2 mL) was added to the mixture. After that, the tube was degassed in vacuo and refilled with N2 5 times. With the blue LED (455 nm, 3 W) switched on, the reaction mixture was stirred for 72 h at 25 °C and the reaction progress was monitored by TLC or GC-MS. After the reactions, the mixture was filtered and extracted with ethyl acetate several times, the organic layers were combined and dried over anhydrous Na2SO4, the solvent was removed under reduced pressure, and the residue was purified by column chromatography on silica gel using petroleum ether/ethyl acetate as the eluent to afford the final C–S cross-coupling product.Author contributions
X. W. and M. Z. conceived and directed the project. M. Z. and T. Y. designed the experiments. T. Y. performed and analyzed the experiments. T. Y., M. Z., M. A. and X. W. prepared the manuscript. All authors contributed to the analysis and interpretation of the data and commented on the final draft of the manuscript.Conflicts of interest
There are no competing interests.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (U1905214, 21961142019, 22071026, 22032002, 21702030, 21761132002, 21861130353, 21902031, and 21425309), the National Key Technologies R & D Program of China (2018YFA0209301), the National Basic Research Program of China (2013CB632405), the Chang Jiang Scholars Program of China (T2016147), and the 111 Project (D16008).Notes and references
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- I. M. Dixon, J. P. Collin, J. P. Sauvage, L. Flamigni, S. Encinas and F. Barigelletti, Chem. Soc. Rev., 2000, 29, 385–391 RSC.
- D. M. Yan, J. R. Chen and W. J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 378–380 CrossRef CAS PubMed.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed.
- J. B. McManus and D. A. Nicewicz, J. Am. Chem. Soc., 2017, 139, 2880–2883 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- J. J. Devery, J. J. Douglas, J. D. Nguyen, K. P. Cole, R. A. Flowers and C. R. J. Stephenson, Chem. Sci., 2015, 6, 537–541 RSC.
- A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765–773 CrossRef CAS PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of molecular photochemistry: an introduction, University Science Books, Sausalito, CA, 2009 Search PubMed.
- W. Che, W. R. Cheng, T. Yao, F. M. Tang, W. Liu, H. Su, Y. Y. Huang, Q. H. Liu, J. K. Liu, F. C. Hu, Z. Y. Pan, Z. H. Sun and S. Q. Wei, J. Am. Chem. Soc., 2017, 139, 3021–3026 CrossRef CAS PubMed.
- K. Hu, A. D. Blair, E. J. Piechota, P. A. Schauer, R. N. Sampaio, F. G. L. Parlane, G. J. Meyer and C. P. Berlinguette, Nat. Chem., 2016, 8, 853–859 CrossRef CAS PubMed.
- C. M. Wolff, P. D. Frischmann, M. Schulze, B. J. Bohn, R. Wein, P. Livadas, M. T. Carlson, F. Jackel, J. Feldmann, F. Wurthner and J. K. Stolarczyk, Nat. Energy, 2018, 3, 862–869 CrossRef CAS.
- W. Huang, J. Byun, I. Rorich, C. Ramanan, P. W. M. Blom, H. Lu, D. Wang, L. C. da Silva, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8316–8320 CrossRef CAS PubMed.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Y. X. Fang, Y. Zheng, T. Fang, Y. Chen, Y. D. Zhu, Q. Liang, H. Sheng, Z. S. Li, C. C. Chen and X. C. Wang, Sci. China: Chem., 2020, 63, 149–181 CrossRef CAS.
- S. Bi, C. Yang, W. Zhang, J. Xu, L. Liu, D. Wu, X. Wang, Y. Han, Q. Liang and F. Zhang, Nat. Commun., 2019, 10, 2467 CrossRef PubMed.
- E. Q. Jin, Z. A. Lan, Q. H. Jiang, K. Y. Geng, G. S. Li, X. C. Wang and D. L. Jiang, Chem, 2019, 5, 1632–1647 CAS.
- I. Ghosh, J. Khamrai, A. Savateev, N. Shlapakov, M. Antonietti and B. König, Science, 2019, 365, 360–366 CrossRef CAS PubMed.
- C. Huang, C. Chen, M. Zhang, L. Lin, X. Ye, S. Lin, M. Antonietti and X. Wang, Nat. Commun., 2015, 6, 7698 CrossRef PubMed.
- M. F. Zheng, J. L. Shi, T. Yuan and X. C. Wang, Angew. Chem., Int. Ed., 2018, 57, 5487–5491 CrossRef CAS PubMed.
- M. F. Zheng, T. Yuan, J. L. Shi, W. C. Cai and X. C. Wang, ACS Catal., 2019, 9, 8068–8072 CrossRef CAS.
- Y. X. Fang and X. C. Wang, Angew. Chem., Int. Ed., 2017, 56, 15506–15518 CrossRef CAS PubMed.
- Z. S. Luo, Y. X. Fang, M. Zhou and X. C. Wang, Angew. Chem., Int. Ed., 2019, 58, 6033–6037 CrossRef CAS PubMed.
- J. Hao, J. M. Wang, S. Qin, D. Liu, Y. W. Li and W. W. Lei, J. Mater. Chem. A, 2018, 6, 8053–8058 RSC.
- Y. Z. Hao, S. Z. Wang, Y. L. Shao, Y. Z. Wu and S. D. Miao, Adv. Energy Mater., 2020, 10, 1902836 CrossRef CAS.
- F. S. Guo, P. J. Yang, Z. M. Pan, X. N. Cao, Z. L. Xie and X. C. Wang, Angew. Chem., Int. Ed., 2017, 56, 8231–8235 CrossRef CAS PubMed.
- J. M. Wang, J. Hao, D. Liu, S. Qin, D. Portehault, Y. W. Li, Y. Chen and W. W. Lei, ACS Energy Lett., 2017, 2, 306–312 CrossRef CAS.
- W. W. Lei, S. Qin, D. Liu, D. Portehault, Z. W. Liu and Y. Chen, Chem. Commun., 2013, 49, 352–354 RSC.
- M. F. Zheng, W. C. Cai, Y. X. Fang and X. C. Wang, Nanoscale, 2020, 12, 3593–3604 RSC.
- J. Shi, T. Yuan, M. Zheng and X. Wang, ACS Catal., 2021, 11, 3040–3047 CrossRef CAS.
- M. Zhou, Z. Chen, P. J. Yang, S. B. Wang, C. J. Huang and X. C. Wang, Appl. Catal., B, 2020, 276, 118916 CrossRef CAS.
- D. Tilly, F. Chevallier, F. Mongin and P. C. Gros, Chem. Rev., 2014, 114, 1207–1257 CrossRef CAS PubMed.
- F. S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC.
- H. Ye, C. Xiao, Q. Q. Zhou, P. G. Wang and W. J. Xiao, J. Org. Chem., 2018, 83, 13325–13334 CrossRef CAS PubMed.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052, DOI:10.1038/s41570-017-0052.
- J.-H. Shon, D. Kim, M. D. Rathnayake, S. Sittel, J. Weaver and T. S. Teets, Chem. Sci., 2021, 12, 4069–4078 RSC.
- C. Kerzig and O. S. Wenger, Chem. Sci., 2019, 10, 11023–11029 RSC.
- J. D. Nguyen, E. M. D'Amato, J. M. Narayanam and C. R. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. C. MacMillan, Nature, 2018, 560, 70–75 CrossRef CAS PubMed.
- C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. C. MacMillan, Nature, 2016, 536, 322–325 CrossRef CAS PubMed.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- L. Marzo, I. Ghosh, F. Esteban and B. König, ACS Catal., 2016, 6, 6780–6784 CrossRef CAS.
- K. J. Liang, Q. Liu, L. Shen, X. P. Li, D. L. Wei, L. Y. Zheng and C. F. Xia, Chem. Sci., 2020, 11, 6996–7002 RSC.
- S. F. Jin, H. T. Dang, G. C. Haug, R. He, V. D. Nguyen, V. T. Nguyen, H. D. Arman, K. S. Schanze and O. V. Larionov, J. Am. Chem. Soc., 2020, 142, 1603–1613 CrossRef CAS PubMed.
- Y. Y. Wang, Q. Zhu, Y. Wei, Y. J. Gong, C. C. Chen, W. J. Song and J. C. Zhao, Appl. Catal., B, 2018, 231, 262–268 CrossRef CAS.
- F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2002, 102, 4009–4092 CrossRef CAS PubMed.
- A. Savateev, I. Ghosh, B. König and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 15936–15947 CrossRef CAS PubMed.
- S. Gisbertz, S. Reischauer and B. Pieber, Nat. Catal., 2020, 3, 611–620 CrossRef CAS.
- L. Huang, L. K. G. Ackerman, K. Kang, A. M. Parsons and D. J. Weix, J. Am. Chem. Soc., 2019, 141, 10978–10983 CrossRef CAS PubMed.
- J. A. Caputo, L. C. Frenette, N. Zhao, K. L. Sowers, T. D. Krauss and D. J. Weix, J. Am. Chem. Soc., 2017, 139, 4250–4253 CrossRef CAS PubMed.
- C. L. Sun, H. Li, D. G. Yu, M. Yu, X. Zhou, X. Y. Lu, K. Huang, S. F. Zheng, B. J. Li and Z. J. Shi, Nat. Chem., 2010, 2, 1044–1049 CrossRef CAS PubMed.
- T. Kim, K. H. Lee and J. Y. Lee, J. Mater. Chem. C, 2018, 6, 8472–8478 RSC.
- M. C. Hilton, X. Zhang, B. T. Boyle, J. V. Alegre-Requena, R. S. Paton and A. McNally, Science, 2018, 362, 799–804 CrossRef CAS PubMed.
- X. Tang, L. Huang, Y. Xu, J. Yang, W. Wu and H. Jiang, Angew. Chem., Int. Ed., 2014, 53, 4205–4208 CrossRef CAS PubMed.
- H. Yue, C. Zhu and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 1371–1375 CrossRef CAS PubMed.
- M. J. Cabrera-Afonso, Z. P. Lu, C. B. Kelly, S. B. Lang, R. Dykstra, O. Gutierrez and G. A. Molander, Chem. Sci., 2018, 9, 3186–3191 RSC.
- Z. B. Dong, G. Manolikakes, L. Shi, P. Knochel and H. Mayr, Chem.–Eur. J., 2010, 16, 248–253 CrossRef CAS PubMed.
- Q. Guo, F. Liang, X. B. Li, Y. J. Gao, M. Y. Huang, Y. Wang, S. G. Xia, X. Y. Gao, Q. C. Gan, Z. S. Lin, C. H. Tung and L. Z. Wu, Chem, 2019, 5, 2605–2616 CAS.
- J. W. Darcy, B. Koronkiewicz, G. A. Parada and J. M. Mayer, Acc. Chem. Res., 2018, 51, 2391–2399 CrossRef CAS PubMed.
- E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01028j |
| This journal is © The Royal Society of Chemistry 2021 |