
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA donor–acceptor complex enables the synthesis of E-olefins from alcohols, amines and carboxylic acids†
Kun-Quan
Chen
,
Jie
Shen
,
Zhi-Xiang
Wang
 * and
Xiang-Yu
Chen
*
* and
Xiang-Yu
Chen
*
School of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, China. E-mail: zxwang@ucas.ac.cn; chenxiangyu20@ucas.ac.cn
First published on 5th April 2021
Abstract
Olefins are prevalent substrates and functionalities. The synthesis of olefins from readily available starting materials such as alcohols, amines and carboxylic acids is of great significance to address the sustainability concerns in organic synthesis. Metallaphotoredox-catalyzed defunctionalizations were reported to achieve such transformations under mild conditions. However, all these valuable strategies require a transition metal catalyst, a ligand or an expensive photocatalyst, with the challenges of controlling the region- and stereoselectivities remaining. Herein, we present a fundamentally distinct strategy enabled by electron donor–acceptor (EDA) complexes, for the selective synthesis of olefins from these simple and easily available starting materials. The conversions took place via photoactivation of the EDA complexes of the activated substrates with alkali salts, followed by hydrogen atom elimination from in situ generated alkyl radicals. This method is operationally simple and straightforward and free of photocatalysts and transition-metals, and shows high regio- and stereoselectivities.
Introduction
Olefins are the second most common functionalities in natural products and have found a wide range of applications in pharmaceuticals, agrochemicals, materials science and the food industry.1 This has led to the development of quite a number of strategies for effectively generating carbon–carbon double bonds.2 Among them, defunctionalizations have emerged as some of the most valuable transformations for the generation of olefins by using easily available molecules as starting materials.3 In this context, decarboxylation of carboxylic acids at high temperatures4 or using a stoichiometric oxidant,5 enzymes,6 or a high energy lamp7 has been heavily investigated. Recently, the development of metallaphotoredox catalysis8 has enabled decarboxylative olefin synthesis under mild conditions directly9 or indirectly.10 However, all these valuable strategies require a transition metal catalyst, a ligand, or an expensive photocatalyst or lack general, regio- and stereoselective variants (Scheme 1A). In light of the central role played by olefins in organic synthesis and the increasing demand for sustainable chemistry, inventing new synthetic methods starting with readily available materials under transition metal- and photocatalyst-free conditions is highly desirable.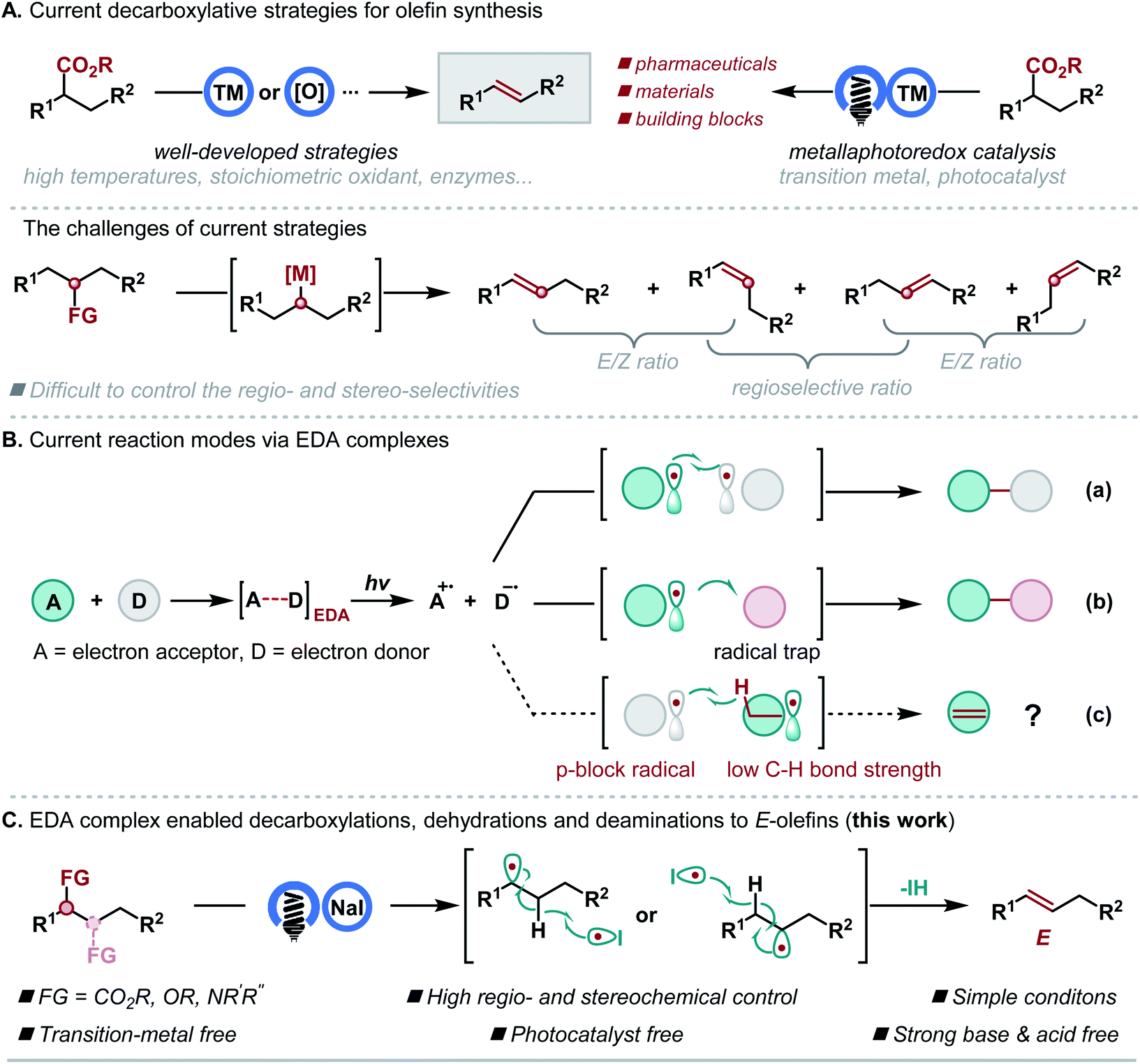 | ||
| Scheme 1 Motivation and synthetic strategy. | ||
In recent years, the photochemistry of electron donor–acceptor (EDA) complexes has emerged as a powerful alternative to metal-based photocatalysts and organic dyes for generating radicals under the excitation of visible light.11 However, limitations exist due to their generation and reaction modes, and their applications in defunctionalative olefin synthesis are unknown. Generally, to date, there are two reaction modes for EDA complexes. One typical mode is the cross-coupling of two substrates (Scheme 1B, mode a). The other mode involves the reaction of an external radical trap, and the generated radical is realized by using a sacrificial or catalytic donor compound (Scheme 1B, mode b). We envision that a new reaction mode would be possible if a p-block radical and an alkyl radical bearing a weak C–H bond could be generated via an EDA complex, and the p-block radical could abstract a C–H bond from the alkyl radical for the olefin formation (Scheme 1B, mode c). If successful, the photoinduced reaction could serve as a general and powerful method in olefin synthesis, using readily available and abundant alcohols, amines and carboxylic acids as starting materials under mild and transition-metal-free conditions. Recently, Shang and Fu realized an EDA complex enabled decarboxylative alkylation, where both PPh3 and NaI were required to form the EDA complex.12 We previously established that the electrostatic effect of N-heterocyclic carbene (NHC) facilitates the formation of an EDA complex to generate alkyl and iodine radicals, and the stabilization effect of NHC on the iodine radical further ensured the following radical–radical couplings.13 Herein, we report an alternative formation of the EDA complex by only using the electrostatic effect of NaI, which differs from previous NaI-promoted photoreactions where PPh3/NHC and the interaction of I-PPh3/NHC are required. In this process, the iodine radical could induce the in situ generated alkyl radical to undergo hydrogen atom elimination, thus forming a carbon–carbon double bond, instead of radical–radical coupling (Scheme 1C).
Results and discussion
Intrigued by this observation, we first examined the decarboxylative elimination of α-amino acids for the synthesis of enamides, which has been widely used for the preparation of lots of biologically and pharmaceutically important compounds.14 Notably, Tunge and co-workers reported an elegant dual photoredox/cobalt catalyst system for their synthesis, where the hydrogen atom transfer was achieved by cobalt catalysis. However, this valuable protocol afforded the desired enamides with moderate E/Z ratios (Scheme 2A).9a To our delight, the reaction of α-amino acid N-hydroxyphthalimide (NHPI) ester proceeded well in the presence of 2.0 equiv. of NaI and acetone as the solvent, affording the desired enamide 1 in 68% yield with high E/Z ratios (Scheme 2B). With this promising result, we further explored the generality of the transformation (Scheme 3). The method showed a broad substrate scope. Besides enamides 2–5, a variety of fine chemicals and natural food flavours – such as anethole 6, isoeugenol 7, isosafrole 8 and polysporin precursor 9 were obtained from the corresponding acids in 65–96% yield with uniformly high E-selectivity. It is worth noting that asymmetric carboxylic acids afforded the internal olefin 10 with excellent regio- and E-selectivities. In addition, both electron-donating (3,5-di-MeO, 4-Me and 4-tBu) and electron-withdrawing (4-Br and 4-Cl) groups on the phenyl ring were tolerated to afford the desired products 11–15 in good yields with high selectivity. The ortho-substituent (2-Cl) was also tolerated in the reaction without apparent change in the yield and selectivity (16). The reaction of 2-(4-methoxyphenyl)hexanoic acid gave the desired product 17 in 77% yield with a 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 E/Z selectivity. The method was also efficient for the synthesis of E-stilbenes (18–20), which widely appear in pharmaceuticals and natural products and play an important role in a large variety of biological activities. Furthermore, the functional groups had no significant effect on the yield and E/Z selectivity. For example, ester, ketone, and alkene substituted carboxylic acids all worked well and gave the desired products with excellent E/Z selectivity (21–23). The reaction scope was further examined by constructing highly substituted terminal olefins, which is another challenge when traditional methods are used. Various 1,1-disubstituted and trisubstituted olefins were obtained in high yields (24–30). Utilizing this simple method, we also synthesized cyclic olefins in good yields (31–35).
:2 E/Z selectivity. The method was also efficient for the synthesis of E-stilbenes (18–20), which widely appear in pharmaceuticals and natural products and play an important role in a large variety of biological activities. Furthermore, the functional groups had no significant effect on the yield and E/Z selectivity. For example, ester, ketone, and alkene substituted carboxylic acids all worked well and gave the desired products with excellent E/Z selectivity (21–23). The reaction scope was further examined by constructing highly substituted terminal olefins, which is another challenge when traditional methods are used. Various 1,1-disubstituted and trisubstituted olefins were obtained in high yields (24–30). Utilizing this simple method, we also synthesized cyclic olefins in good yields (31–35).
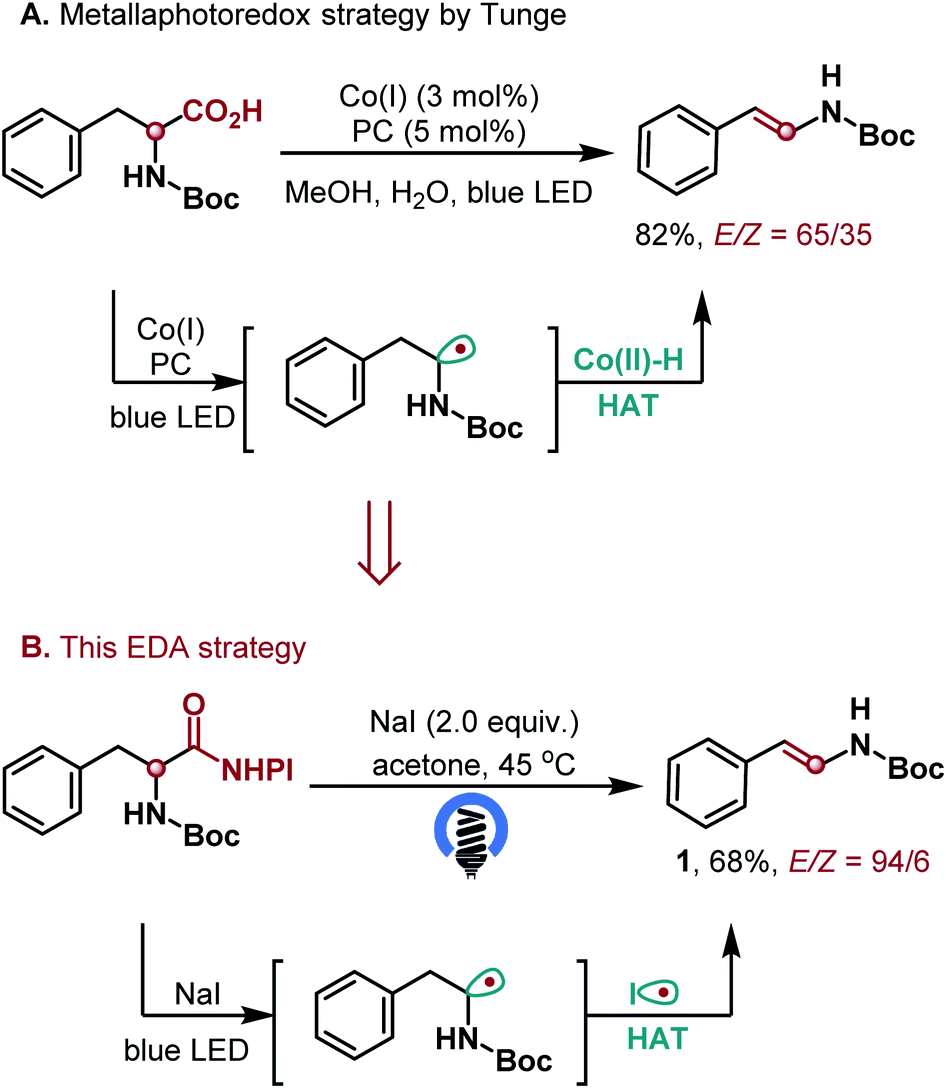 | ||
| Scheme 2 Initial studies. Yield of isolated product 1 after chromatography. | ||
 | ||
| Scheme 3 Substrate scope for decarboxylation. Yield of isolated product given. The E/Z ratios were determined by 1H NMR spectroscopy. DIC = N,N′-diisopropylcarbodiimide and DCM = dichloromethane. | ||
Remarkably, this strategy could serve as a convenient procedure for the late-stage structural modification of natural products and drugs. NHPI esters derived from diverse scaffolds, such as Naproxen 36, Ibuprofen 37, Oxaprozin 38, Ketoprofen 39, Flurbiprofen 40, Carprofen 41, Loxoprofen 42, Zaltoprofen 43 and Bufemid 44, were all converted into the corresponding olefins in good to high yields under mild conditions. Interestingly, the use of commercially available α-hydroxy acids resulted in the desired ketones and aldehydes in high yields (45–47).
On the basis of the photoinduced decarboxylation results, we endeavored to extend this strategy to the deamination of α-amino acids for olefin synthesis. If realized, this protocol would represent a mild and selective olefin synthesis from general amines via EDA photochemistry. Primary amines are also abundant natural feedstocks similar to carboxylic acids and have recently emerged as powerful reagents to generate alkyl radicals.15 However, to our knowledge, the deaminative olefin synthesis remained a daunting synthetic challenge, and there is only one example that exists for this type of transformation, where a high energy UV lamp (800 W) was employed for the deamination of N-phthaloyl amino acids.16 Delightfully, after optimizing the reaction conditions (see Table S2 in the ESI†), we successfully established the photoinduced deamination of α-amino acids by employing Katritzky salts17 as C-centered-radical precursors. A variety of α-amino acids as starting materials worked well, affording the corresponding olefins 21 and 48–52 in good yields and high E/Z selectivities (Scheme 4).
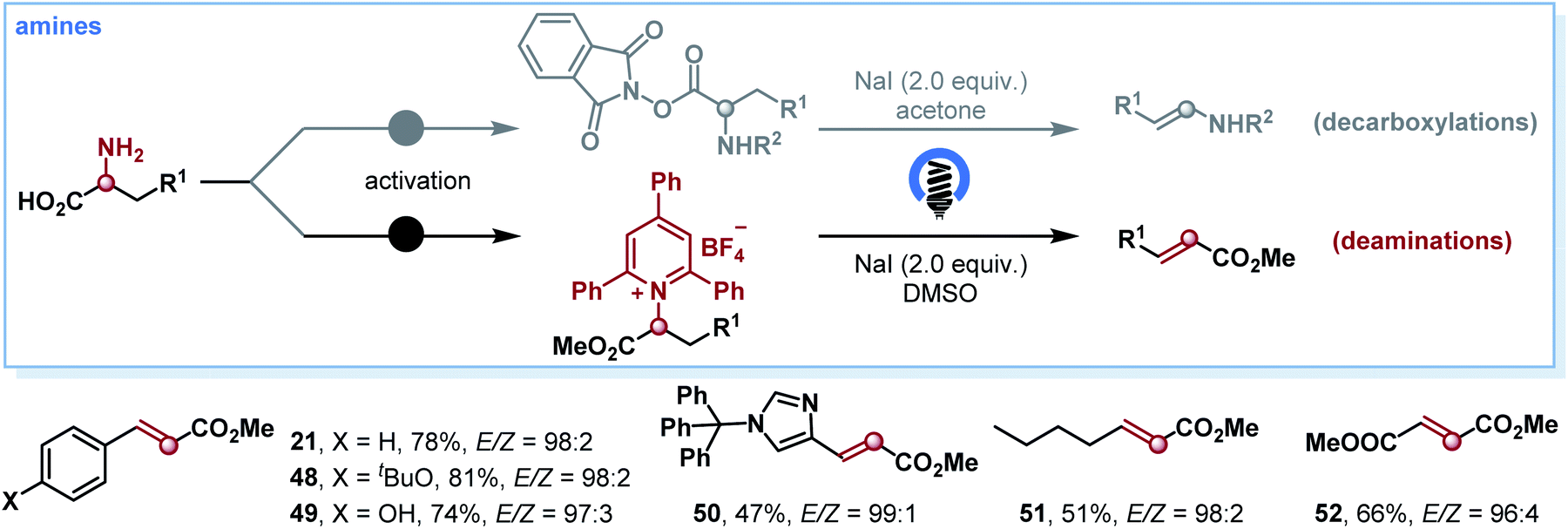 | ||
| Scheme 4 Substrate scope for deamination. Yield of isolated product given. The E/Z ratios were determined by 1H NMR spectroscopy. | ||
In order to examine the generality of the developed new EDA reaction mode, we next turned our attention to the dehydration of alcohols for olefin synthesis. Although known methods for such alcohol transformations have been well studied,18 we wish to provide an alternative protocol for the dehydration and further show the broad applications of the present strategy. Notably, there are only two EDA approaches to generate alkyl radicals from alcohols. One typical entry is the β-scission of an alkoxy radical, which was generated from the EDA complex of N-alkoxyphthalimide and Hantzsch ester (Scheme 5A).19 Another approach involves the formation of the EDA complex with 2-iodophenylthionocarbonates, Et3N and B2cat2 (Scheme 5B).20 We were pleased to find that our newly developed dual alkali cation activation mode could form an EDA complex by using pre-activated alcohols (Scheme 5C). Various alcohols were smoothly converted to the corresponding 1,2-disubstituted olefins 6–11 and 53 in moderate to good yields with excellent E/Z selectivity. With tertiary alcohol, excellent regio- and stereoselectivities were achieved and the corresponding tri-substituted olefins 54 and 55 were obtained in 70% and 81% yield, respectively. 1-(4-Methoxyphenyl)ethan-1-ol was readily accommodated under both reaction conditions (56). The reaction of cyclic alcohol also worked, providing the corresponding product 33 in good yield.
 | ||
| Scheme 5 Substrate scope for dehydration. Yield of isolated product given. The E/Z ratios were determined by 1H NMR spectroscopy. DIAD = diisopropyl azodicarboxylate, DCC = dicyclohexylcarbodiimide, DMAP = 4-dimethylaminopyridine, and PNP = p-nitro-phenyl. aUsing method a. bUsing method b. | ||
To confirm the possible mechanism, we investigated the involvement of the EDA complex. While the NHPI ester, Katritzky salt, alcohol derivatives, NaI or LiBF4 individually showed no significant UV/vis absorption in the visible region of the spectra, an obvious red shift of absorption could be observed in the spectrum of the mixture of NHPI ester and NaI, Katritzky salt and NaI, and the mixture of alcohol derivatives, NaI and LiBF4. These experiments demonstrated the generation of a photoactive EDA complex (Scheme 6A). Although further studies are required to fully understand the detailed mechanism, one possible mechanism is proposed in Scheme 6B. The weak interaction between NHPI ester/Katritzky salts/or alcohol derivatives and NaI allows the formation of an EDA complex. Upon visible-light excitation, the photoactive EDA complex undergoes single-electron transfer, generating the alkyl radical and iodine radical. Subsequently, the iodine radical abstracts a hydrogen atom from the resultant alkyl radical to form the corresponding olefins. Instead of direct hydrogen atom abstraction, an alternative that forms iodoalkanes first, followed by the homolysis of iodoalkanes, is also consistent with our following mechanistic study. According to the mechanism, the E-selectivity of the reaction could be attributed to the antiperiplanar hydrogen atom elimination, which is similar to the classical E2 elimination.
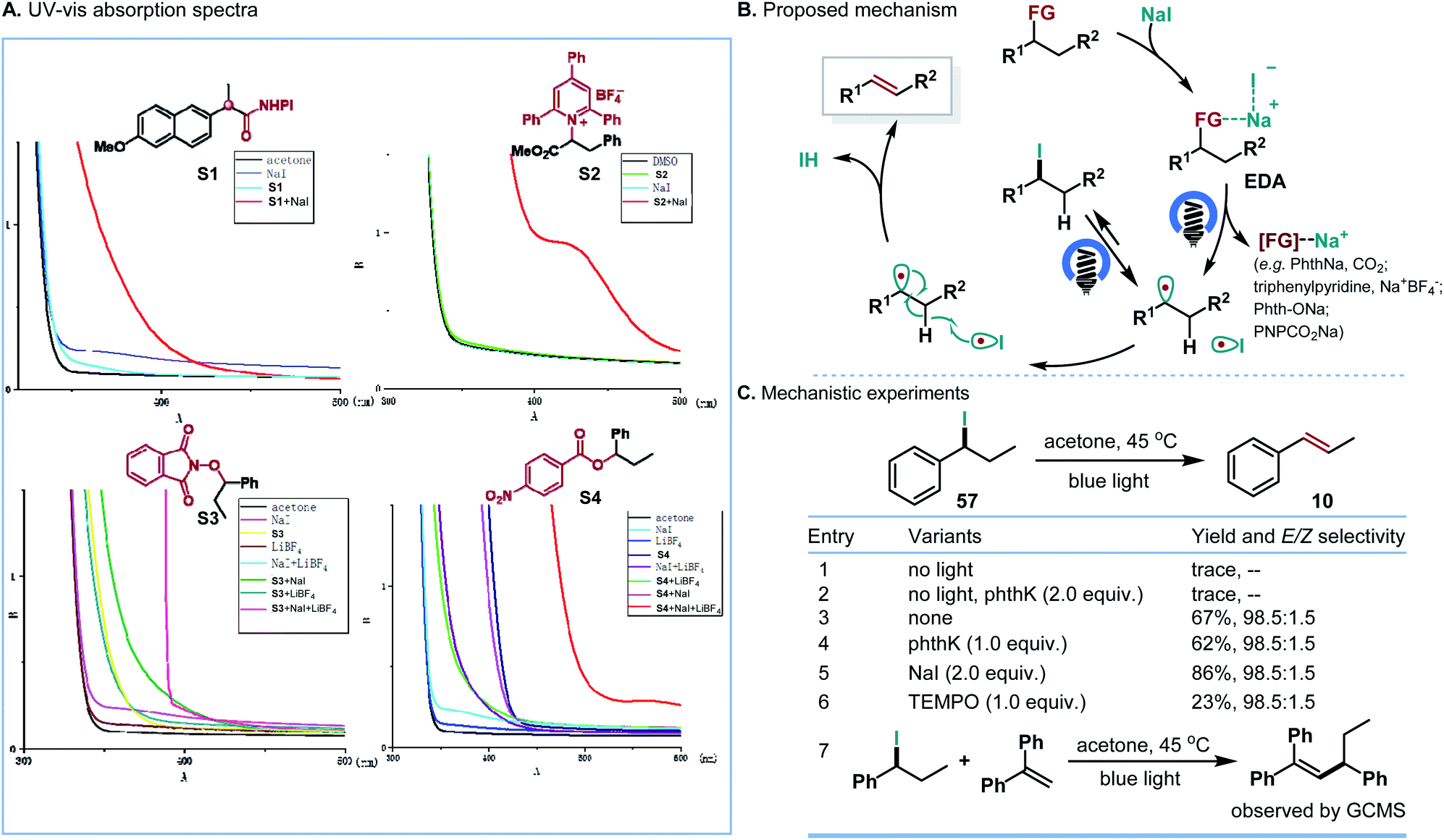 | ||
| Scheme 6 Mechanistic studies. | ||
On the basis of our previous studies which showed that iodoalkanes could be easily generated between NHPI esters and NaI under light irradiation,13 a series of mechanistic experiments were carried out to clarify the possible reaction pathway (Scheme 6C). One possible pathway operating via traditional E2 elimination of iodoalkanes could be ruled out because without irradiation the control experiments (entries 1 and 2) with phthK as the proton extractor only afforded a trace amount of olefins. According to suggestions by Studer and co-workers,21 we envisioned that a light-mediated homolysis of the C–I bond of iodoalkanes could be involved. Supportively, under blue LED irradiation, the corresponding olefins were obtained in good yields and high E/Z selectivities (entries 3–5). The radical inhibition and interception studies (entries 6 and 7) further suggested the involvement of the alkyl radical in the reaction.
Conclusions
In conclusion, we have developed an operationally simple, robust and general EDA strategy to synthesize olefins by using simple and easily available alcohols, amines and carboxylic acids as starting materials. These methods are characterized by high regio- and E/Z selectivities, and cheap alkali metal salts, and do not involve transition metals, photocatalysts, or strong bases and acids, and thus offer a promising solution for various applications including the concise synthesis of industrially relevant olefins and the modifications of commercially available drugs and natural products.Author contributions
K. Q. C. and X. Y. C. conceived and designed the experiments. K. Q. C. and J. S. performed the experiments and analyzed the data. K. Q. C., Z. X. W. and X. Y. C. wrote the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (21773240 and 22001248), Fundamental Research Funds for the Central Universities and the University of the Chinese Academy of Sciences.References
-
(a) G. W. Gribble, J. Nat. Prod., 2000, 63, 735 CrossRef CAS
; (b) P. Ertl and T. Schuhmann, J. Nat. Prod., 2019, 82, 1258 CrossRef CAS PubMed
-
(a) L. F. van Staden, D. Gravestock and D. J. Ager, Chem. Soc. Rev., 2002, 31, 195 RSC
-
(a) S. Kusumoto, T. Tatsuki and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 8458 CrossRef CAS PubMed
-
(a) J. A. Miller, J. A. Nelson and M. P. Byrne, J. Org. Chem., 1993, 58, 18 CrossRef CAS
-
(a) J. D. Bacha and J. K. Kochi, Tetrahedron, 1968, 24, 2215 CrossRef CAS
-
(a) A. Dennig, M. Kuhn, S. Tassoti, A. Thiessenhusen, S. Gilch, T. Bülter, T. Haas, M. Hall and K. Faber, Angew. Chem., Int. Ed., 2015, 54, 8819 CrossRef CAS PubMed
- K. G. M. Kou, J. J. Pflueger, T. Kiho, L. C. Morrill, E. L. Fisher, K. Clagg, T. P. Lebold, J. K. Kisunzu and R. Sarpong, J. Am. Chem. Soc., 2018, 140, 8105 CrossRef CAS PubMed
-
(a) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed
-
(a) K. C. Cartwright and J. A. Tunge, ACS Catal., 2018, 8, 11801 CrossRef CAS
-
(a) A. Tlahuext-Aca, L. Candish, R. A. Garza-Sanchez and F. Glorius, ACS Catal., 2018, 8, 1715 CrossRef CAS
-
(a) C. G. S. Lima, T. de M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389 CrossRef CAS
-
(a) M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429 CrossRef CAS PubMed
-
(a) K.-Q. Chen, Z.-X. Wang and X.-Y. Chen, Org. Lett., 2020, 22, 8059 CrossRef CAS PubMed
- T. Zhu, S. Xie, P. Rojsitthisak and J. Wu, Org. Biomol. Chem., 2020, 18, 1504 RSC
-
(a) F.-S. He, S. Ye and J. Wu, ACS Catal., 2019, 9, 8943 CrossRef CAS
- A. G. Griesbeck, A. Henz, J. Hirt, V. Ptatschek, T. Engel, D. Löffler and F. W. Schneider, Tetrahedron, 1994, 50, 701 CrossRef CAS
-
(a) C. H. Basch, J. Liao, J. Xu, J. J. Piane and M. P. Watson, J. Am. Chem. Soc., 2017, 139, 5313 CrossRef CAS PubMed
- P. Hjerrild, T. Tørring and T. B. Poulsen, Nat. Prod. Rep., 2020, 37, 1043 RSC
- J. Zhang, Y. Li, R. Xu and Y. Chen, Angew. Chem., Int. Ed., 2017, 56, 12619 CrossRef CAS PubMed
- J. Wu, R. M. Bär, L. Guo, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 18830 CrossRef CAS PubMed
- Y. Cheng, C. Mück-Lichtenfeld and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 16832 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01024g |
| This journal is © The Royal Society of Chemistry 2021 |