
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEasily accessible non-aromatic heterocycles with handles: 4-bromo-2,3-dihydrofurans from 1,2-dibromohomoallylic alcohols†
Jason
An
,
Jose
Intano
Jr.
,
Alissa
Richard
,
Taehyun
Kim
,
José A.
Gascón
 * and
Amy R.
Howell
*
* and
Amy R.
Howell
*
Department of Chemistry, University of Connecticut, Storrs, CT 06269, USA. E-mail: amy.howell@uconn.edu; jose.gascon@uconn.edu
First published on 29th June 2021
Abstract
The first general preparation of 4-bromo-2,3-dihydrofurans is reported. These non-aromatic heterocycles containing a useful coupling handle are accessed via Cu-catalyzed intramolecular cyclization of 1,2-dibromohomoallylic alcohols, which are themselves available in just two steps from aromatic and aliphatic aldehydes and ketones. Molecular dynamics simulations using the simple substrates and key geometric parameters provide a rationale for the selectivities observed. The synthetic utility of the 4-bromodihydrofurans is also demonstrated.
Introduction
Non-aromatic heterocycles are ubiquitous in nature and widely targeted in synthetic methodology development.1,2 While the ability to diversify aromatic heterocycles has exploded with the development of a range of cross coupling methodologies exploiting aromatic heterocycles with reactive handles,3–8 ready access to non-aromatic heterocycles with such handles is lacking. We have been interested in functionalized non-aromatic heterocycles that can be used as templates for diversification. For example, we have shown that α-methylene-β-lactones 1 undergo cross-metathesis or rhodium catalysed conjugate addition, making them scaffolds with broad utility (Fig. 1A).9–11 It occurred to us that the cyclization of 1,2-dibromohomoallylic alcohols 2 had the potential to provide either 4-bromo-2,3-dihydrofurans 3 or 2-bromomethyleneoxetanes 4 (Fig. 1B). While either outcome would satisfy our interest in diversifiable scaffolds, the utility of this method would be best served by selectivity in the cyclization.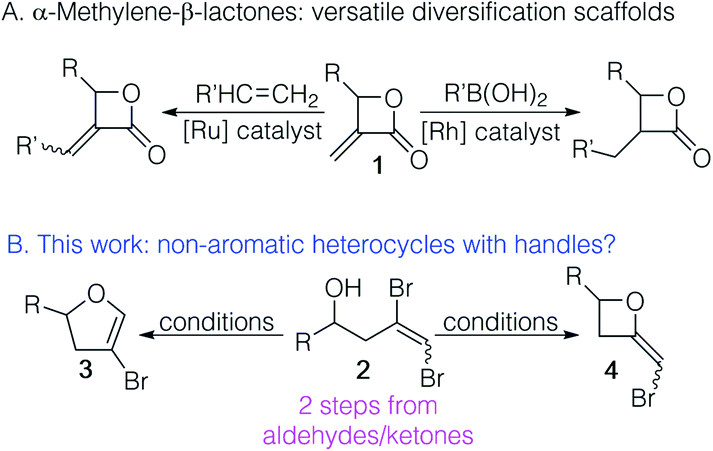 | ||
| Fig. 1 Non-aromatic heterocycles with handles: (A) α-methylene-β-lactones 1 can be used to access diverse β-lactone motifs; (B) 1,2-dibromohomoallyic alcohols could provide non-aromatic heterocycles 3 and/or 4 with a handle for functionalization. | ||
Metal-mediated intramolecular reactions using vinylhalides have been used to prepare oxetanes.12 Intramolecular preparations of dihydrofurans have largely been limited to the cyclization of aryl halides to give dihydrobenzofurans.13,14 These approaches, however, do not inherently provide a handle for diversification subsequent to cyclization. Geminal dibromides have been used extensively in the preparation of benzofurans.15 Although the cyclized products have a handle, the products are almost exclusively aromatic heterocycles; our interest is in non-aromatic heterocycles. To our knowledge, there is no literature precedent using vicinal dihalides (as in 2). We anticipated that the presence of two halogens on the double bond would impact selectivity and yields (due to more competing potential outcomes). Herein we report the results of our exploration of the cyclization of 1,2-dibromohomoallylic alcohols and computational studies to understand the selectivities observed.
Results and discussion
The initially investigated dibromoalkenol 2a was prepared in two steps, propargylation and dibromination (vide infra), from benzaldehyde. It was treated with CuI under typical conditions14 for Ullmann-type cyclizations (Table 1, entry 1) and gave mainly one product in 60% isolated yield. Proton/carbon NMR experiments confirmed that it was dihydrofuran 3a. This result was important because there are no literature reports of a general preparation of 4-bromo-2,3-dihydrofurans. Such systems would be valuable for coupling reactions, leading to functionalized dihydrofurans. Our first goals were to optimize the transformation, then explore its scope. If the 5-endo-cyclization was general, a related goal was to understand the preference for the formation of bromodihydrofurans 3 over bromomethyleneoxetanes 4.|
|
|||
|---|---|---|---|
| Entry | La, base (equiv.) | Solvent | Convb (yield)c |
| a Ligands (L): A = 1,10-phenanthroline; B = L-proline; C = BINOL; D = BINAP. b Conversion was estimated from 1H NMR and is based on relative amounts of starting material and dihydrofuran. c Isolated yield of 3a. d Concentration of 2a = 0.1 M; reaction time = 8 h. e Concentration of 2a = 0.05 M; reaction time = 8 h. f Concentration of 2a = 0.05 M; reaction time = 16 h for entries 3–15. g CuBr (10 mol%) rather than CuI. | |||
| 1d | A, Cs2CO3 (2) | CH3CN | 86 (60) |
| 2e | A, Cs2CO3 (2) | CH3CN | 66 |
| 3f | A, Cs2CO3 (2) | CH3CN | 100 (76) |
| 4f,g | A, Cs2CO3 (2) | CH3CN | 49 |
| 5f | A, Cs2CO3 (1.2) | CH3CN | 100 (80) |
| 6f | A, Cs2CO3 (1.2) | MeNO2 | 0 |
| 7f | A, Cs2CO3 (1.2) | DMF | 29 |
| 8f | A, Cs2CO3 (1.2) | THF | 100 (69) |
| 9f | B, Cs2CO3 (1.2) | CH3CN | 75 |
| 10f | C, Cs2CO3 (1.2) | CH3CN | 12 |
| 11f | D, Cs2CO3 (1.2) | CH3CN | 61 |
| 12f | A, NaOtBu (1.2) | CH3CN | 58 |
| 13f | A, TMSOK (1.2) | CH3CN | 22 |
| 14f | No L, Cs2CO3 (1.2) | CH3CN | 12 |
| 15f | No Cu/L, Cs2CO3 (1.2) | CH3CN | 0 |
Although the initial isolated yield was reasonable (Table 1, entry 1), several byproducts were observed in the crude 1H NMR spectrum. We anticipated that variations described in the literature for intramolecular Ullmann-type coupling would lead to improved yields.14 Decreasing the concentration (entry 2) gave a cleaner reaction, but lower conversion. Doubling the reaction time resulted in complete conversion (entry 3). CuBr promoted clean formation of 3a, but the conversion was significantly lower over the same time period (entry 4). Decreasing the base loading did not substantially change the outcome for 2a (entry 5), but it was found to be critical for ketone-derived substrates (e.g.2j, Table 2), where the higher base loading gave considerable return of initial ketone. Changing the solvent (entries 6–8), the ligand (entries 9–11) and the base (entries 12–13) did not improve the outcome. While a little product was formed without the ligand (entry 14), no conversion occurred without CuI (entry 15). Overall, the best yield for the preparation of 2a resulted from the conditions shown in entry 5 (Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Compound | R | R′ | Yield of 2 | Yield of 3d | Yield of 3c |
| a Procedure A: CuI (10 mol%), 1,10-phenanthroline (20 mol%), Cs2CO3 (1.2 equiv.), CH3CN, 80 °C, 16 h, concentration = 0.05 M. b Procedure B: CuI (10 mol%), 1,10-phenanthroline (20 mol%), Cs2CO3 (1.2 equiv.), 1,4-dioxane, 115 °C, 16 h, concentration = 0.05 M. c Procedure C: CuBr (10 mol%), 1,10-phenanthroline (20 mol%), Cs2CO3 (1.2 equiv.), 1,4-dioxane, 115 °C, 16 h, concentration = 0.05 M. d 2a–n cyclized with procedure A or B contained 1–11% of the corresponding inseparable iodide 6 (see discussion and ESI). Yields are based on just bromide 3. | |||||
| a | Ph | H | 89 | 78a | 88 |
| b | p-Toly | H | 78 | 86a | |
| c | 4-t-BuPh | H | 89 | 74a | 87 |
| d | 4-FPh | H | 86 | 67a | 62 |
| e | 4-CF3Ph | H | 82 | 69a | |
| f | CH3(CH2)8CH2 | H | 85 | 63b | 70 |
| g | Cyclohexyl | H | 80 | 65b | 57 |
| h | PhCH2CH2 | H | 84 | 72b | 74 |
| i | PhCH2OCH2 | H | 25 | 82b | |
| j | Ph | CH3 | 85 | 79a | |
| k | CH3(CH2)7CH2 | CH3 | 86 | 74b | |
| l |
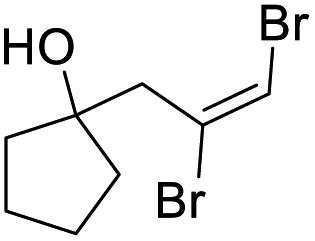
|
85 | 68b | 52 | |
| m |

|
69 | 67b | ||
| n |
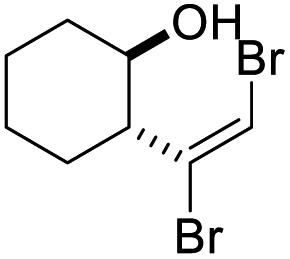
|
61 | 80b | 88 | |
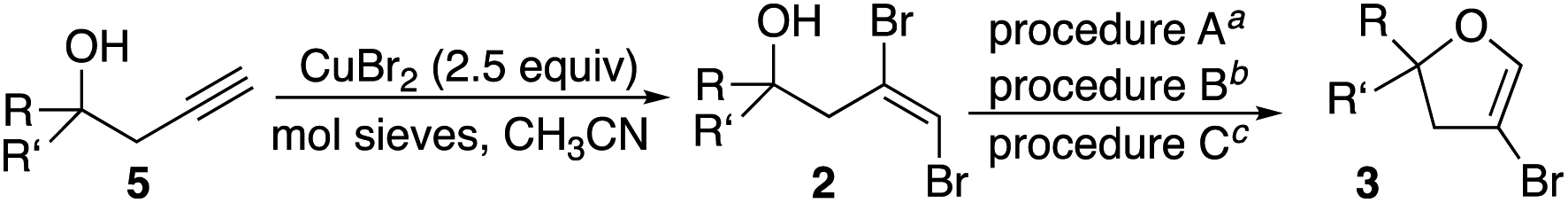
In order to examine the scope of the cyclization, additional dibromoalkenes 2b–n were prepared. This was achieved in two straightforward and efficient steps. Homopropargyl alcohols 5 were readily prepared by zinc-mediated propargylation of the corresponding aldehydes and ketones.16 These were efficiently converted to E-dibromoalkenes 2 by treatment with CuBr2 in the presence of activated molecular sieves.17 Of note, these latter conditions were incompatible with highly electron rich aromatic compounds (e.g., p-MeOPh, thiophene).
The dibromoalkenes 2b–e, derived from aromatic aldehydes, were converted in very good yields to the corresponding 4-bromodihydrofurans 3b–e (Table 2) under the conditions optimized for 2a (procedure A). In contrast, for dibromoalkene 2f, which came from undecanal, procedure A provided only 37% yield of 3f. Although heating in acetonitrile at 80 °C for a longer period improved the conversion, a better outcome was realized by switching to 1,4-dioxane (oil bath temperature 115 °C). Using dioxane, 4-bromodihydrofurans 3f–i were prepared in very good yields (procedure B, Table 2). With the exception of dibromide 2j, which cyclized efficiently under the milder conditions of procedure A (Table 2), dibromoalkenes (2k–m) derived from ketones also cyclized with improved yields when refluxing dioxane was used. Ketone derived 2l and 2m provided interesting spirocyclic bromodihydrofurans 3l and 3m while alcohol 3n, prepared in two steps from cyclohexane oxide, gave fused bromodihyrofuran 3n (see Fig. 2). Overall, a wide range of easily accessible trans-vinyldibromides was converted to 4-bromodihydrofurans in very good to excellent yields.
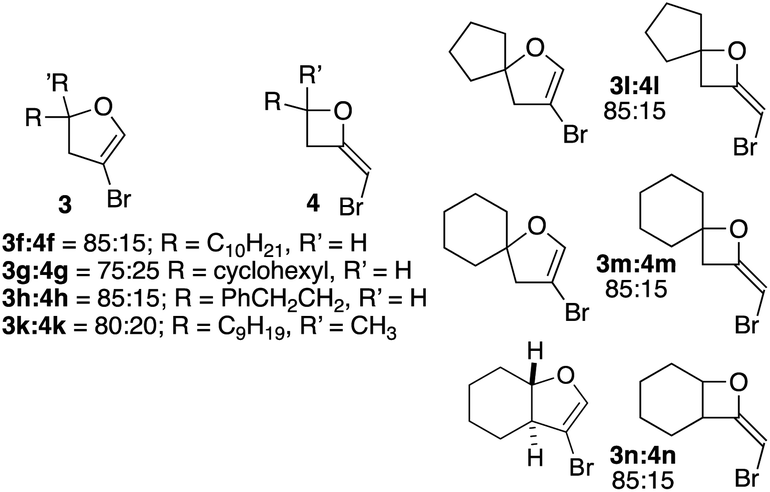 | ||
| Fig. 2 Ratio of 4-bromodihydrofurans 3 to 2-bromomethyleneoxetanes 4 in cyclizations of homopropargylic alcohols derived from aliphatic aldehydes/ketones. | ||
Analysis of the 1H NMRs of spirocyclic furans 3l and 3m led to an interesting observation. For both compounds there was a small doublet just upfield from the doublet for the allylic CH2. Assuming this might be an allylic signal of a related compound, the integration for this unknown product represented ∼5–10% yield in comparison to 3l or 3m. Distinct or shadow peaks in the 13C NMR spectra confirmed the presence of a related product. Attempts to separate this impurity were unsuccessful. Examination of the 1H NMR spectra of other dihydrofurans 3 showed at least hints of upfield allylic proton peaks in all of them. A combination of the similarity of the NMR spectra and a faint pink tinge in NMR solutions that had set for some time led us to suspect that 4-bromo-2,3-dihydrofurans 3l and 3m contained 4-iododihydrofurans 6l and 6m. This was confirmed by GC/MS, which showed that all of the dihydrofurans contained some of the corresponding iodides (from 1–11% – see ESI† for details). The yields of 3 given in Table 2 are for just the bromides. It is noteworthy that the attractiveness of the bromide lies in its utility as a handle for subsequent coupling transformations, and the iodides would be as or more reactive.
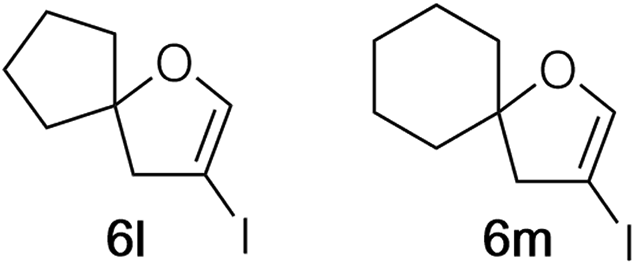
The fact that every 4-bromodihydrofuran prepared contained at least some of the corresponding iodide 6 led us to reconsider CuBr as a catalyst, although we had abandoned it because of the slower conversion to product (Table 1, entry 4). When dibromide 2a was heated in dioxane containing CuBr (10 mol%), 1,10-phenanthroline (20 mol%) and Cs2CO3 (1.2 equiv.) (procedure C, Table 2, last column), 4-bromodihydrofuran 3a was isolated in 88% yield. Gratifyingly, dibromides derived from both aliphatic and aromatic aldehyde precursors could be cyclized with yields similar to those realized with the CuI (Table 2, column 5).
We decided to see if the cyclization strategy could be extended to an internal dibromoalkene and one with substituents on the allylic carbon; so compounds 7 and 10 were prepared (see ESI†). For internal alkene 7, both procedures A and B (Table 2) resulted only in recovery of the starting material. However, when alkene 7 was treated with CuI (20 mol%) in the presence of Cs2CO3 (2 equiv.) without ligand in dioxane (oil bath temperature 115 °C), 1H NMR showed starting material (∼50%) and two products, formed in an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Through NMR experiments we deduced that one of the products was 2-bromomethyleneoxetane 8; the other was the expected bromodihydrofuran 9. In a related outcome, dibromoalkene 10 gave oxetane 11 and dihydrofuran 12, also in an approximately 1:1 ratio. Interestingly, the combined yield of 11/12 was only 24% with some starting material remaining under procedure A (Table 1). Moreover, the crude 1H NMR was unusually messy. In contrast, with procedure C the combined yield of the two products (11:12 ratio of 42:58) was 73%, and the crude 1H NMR showed only the two products. The outcomes with 7 and 10 were significant in that they showed that the preferential, and seemingly exclusive, formation of 5-endo products was not straightforward.
:1 ratio. Through NMR experiments we deduced that one of the products was 2-bromomethyleneoxetane 8; the other was the expected bromodihydrofuran 9. In a related outcome, dibromoalkene 10 gave oxetane 11 and dihydrofuran 12, also in an approximately 1:1 ratio. Interestingly, the combined yield of 11/12 was only 24% with some starting material remaining under procedure A (Table 1). Moreover, the crude 1H NMR was unusually messy. In contrast, with procedure C the combined yield of the two products (11:12 ratio of 42:58) was 73%, and the crude 1H NMR showed only the two products. The outcomes with 7 and 10 were significant in that they showed that the preferential, and seemingly exclusive, formation of 5-endo products was not straightforward.
The fact that 1H NMR chemical shifts for bromomethyleneoxetanes 8 and 11 were generally downfield from those of dihydrofurans 9 and 12 led us to recall earlier observations. Crude 1H NMRs for the cyclizations of aliphatic systems 2f–h and 2k–n had shown varying amounts of a proton signal downfield from that of C2 of the dihydrofurans (see Scheme 1 for numbering). These byproducts were not seen in any fractions from the columns. It occurred to us that this signal could be associated with 2-bromomethyleneoxetanes 4. Our own experience had shown that 2-methyleneoxetanes are generally unstable to normal flash silica gel.18 Running the reactions of 2f–h and 2k–n again, followed by purification on deactivated silica, provided mixtures that contained dihydrofurans 3f–h and 3k–n and the products associated with the signal appearing downfield from the C2 signal of bromodihydrofurans 3. In the case of the cyclization of 2h, running a second column on deactivated silica provided a clean sample of 4h (see Fig. 2 and ESI†). The isolation of 4h and 8 and 11 and comparison of their NMR spectra with those of the mixtures isolated from deactivated silica confirmed that varying amounts of brominated methyleneoxetanes 4f–h and 4k–n were being formed as minor products, along with dihydrofurans 3f–h and 3k–n. It should be stressed that the yields in Table 2 for the formation of 3 accurately reflect those for the dihydrofurans, as bromomethyleneoxetanes 4f–h and 4k–n were destroyed on untreated silica. No bromomethyleneoxetanes were seen in the cyclizations of benzylic alcohols 2a–e and 2j or 2i. The ratios of dihydrofuran to oxetane in the cyclizations of 2f–h and 2k–n (based on crude 1H NMR) are shown in Fig. 2. Taken together, the results with 7 and 10 and with the aliphatic systems 2f–h and 2k–n made us wonder if there is an inherent difference in 4-exo vs. 5-endo-selectivity with these CuI/CuBr promoted reactions.
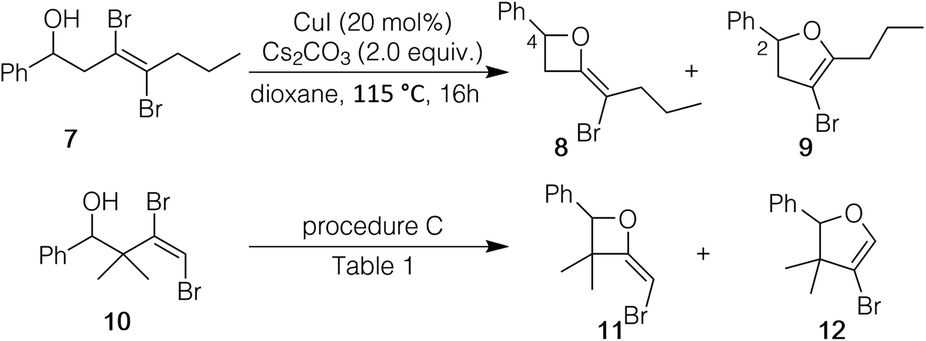 | ||
| Scheme 1 Cyclization of internal dibromoalkene 7 and of a dibromoalkene (10) with allylic substitution. | ||
Fang and Li had conducted selectivity studies in their report on the preparation of 2-methyleneoxetanes by Cu-mediated cyclization of vinyl bromides.12 In their study, alcohols were prepared that had two bromoalkene moieties that allowed for competing product formation via 4-exo- or 5-exo-modes (see 13, Scheme 2) or 4-exo- or 6-exo-modes (see 15). Only the 4-exo-products 14 and 16 were isolated. Dienol 15, which allowed for 4-exo- or 6-endo-product formation, also gave exclusively 4-exo-product (18). However, a 4-exo- vs. 5-endo-competition experiment was not reported. We prepared 19 (see ESI†), and with procedure A (Table 2), complete consumption of the starting material was observed with conversion to both 4-exo-18 and 5-endo-19 (in a 1:1 ratio based on crude 1H NMR). Thus, there is no inherent preference for the formation of 5-endo- over a 4-exo-product in a direct competition. Why, then, are we observing either exclusive or major formation of 5-endo products with 1,2-dibromoalkenes 2 (but not with 7 and 10)?
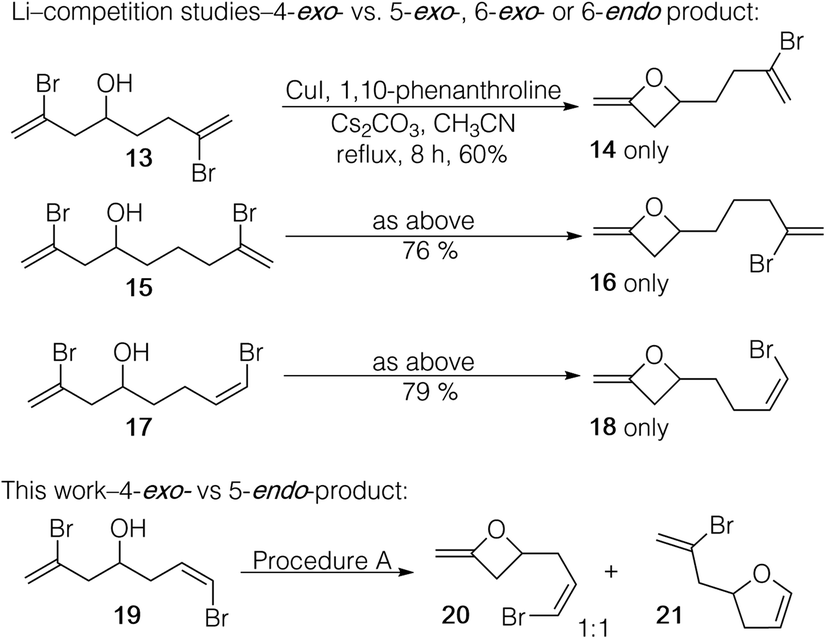 | ||
| Scheme 2 Competition studies. | ||
It was tempting to view the preference for the formation of 5-endo-product as being a result of an electronic effect associated with vicinal 1,2-dibromoalkenes in comparison to the monobromo alkenes shown in Scheme 2. We investigated whether there could be any difference in the electron distribution around the proximal Br (BrP) atom (proximal to O) and the distal Br (BrD) atom (distal to O) (see Fig. 4 for labeling) that could affect selectivity. Fig. 3 shows the electrostatic maps of 2a at its global energy minimum conformation, derived via a combination of force field conformational search and Density Functional Theory (DFT). The electrostatic potential charges (ESP) on the Br atoms are a very small fraction of the unit electron charge. They are an order of magnitude smaller than the charge on the oxygen atom and very similar to each other. Moreover, at finite temperature, we expect the two Br atoms to be electrostatically indistinguishable, ruling out any electronic origin affecting selectivity.
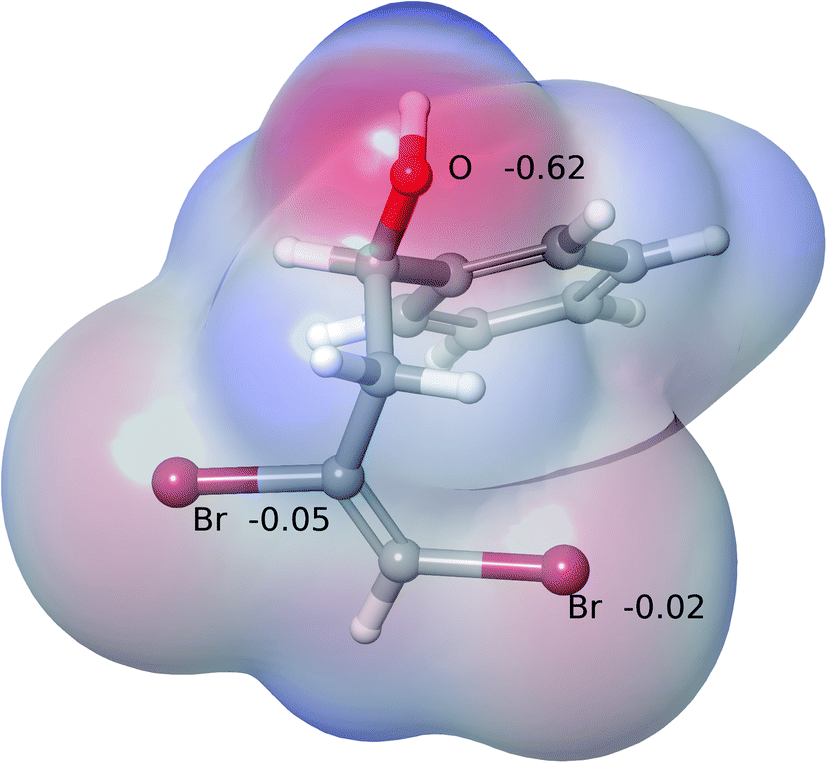 | ||
| Fig. 3 Electrostatic potential map of 2a showing ESP charges on O and Br atoms. | ||
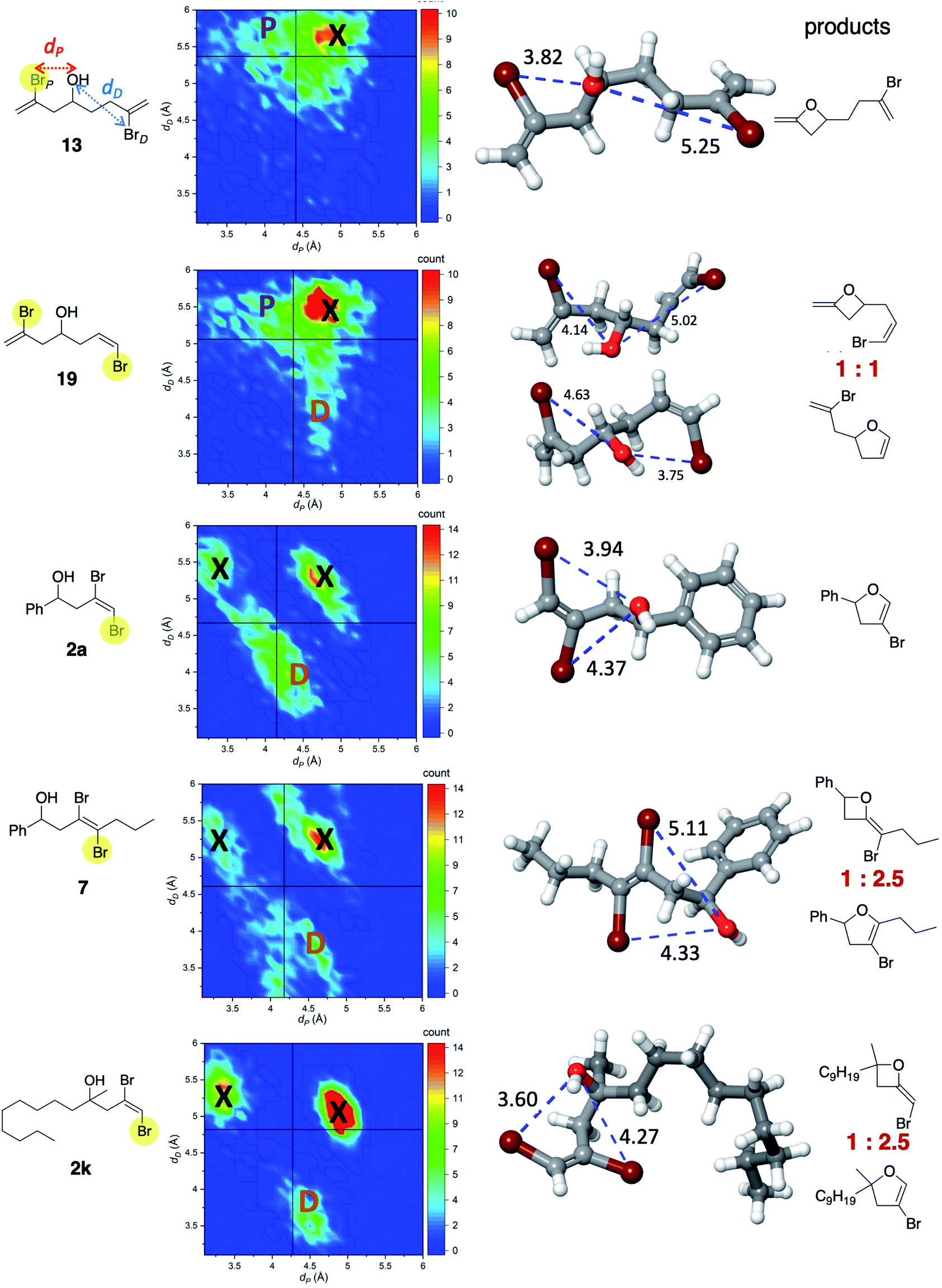 | ||
| Fig. 4 Representative compounds (13, 19, 2a, 7, 2k) for which 4-exo and 5-endo products were experimentally observed. The second column presents a correlation map between dP and dD (annotated on compound 13). The vertical and horizonal black lines correspond to the average value of these two distances. The third column shows typical snapshots of high-density regions in the correlation map, marked up with P (proximal) or D (distal). That is, these snapshots reflect the most abundant configuration that leads to either the oxidative addition of BrP or BrD, and the subsequent formation of either 4-exo or 5-endo, respectively. The ratio x:y (in red) corresponds to the 4-exo:5-endo ratio at 80 °C. Areas marked with X correspond to non-reactive configurations. | ||
Considering Li's work and ours, it has been shown experimentally that Cu, presumably “guided” by its interaction with the alcohol oxygen, promotes the formation of 4-exo and/or 5-endo products. In the 4-exo outcome, the Cu complex undergoes oxidative addition of the proximal C–Br (see Fig. 4), while for the 5-endo product oxidative addition occurs with the distal C–Br. The question is: what controls selectivity? From mechanistic studies on related systems, we assume that Cu (ligated with phenanthroline in most of the cases we consider) first coordinates to oxygen.19,20 Once this occurs, we hypothesize that oxidative addition would, in general, occur with the Br atom (taking into account a dynamical average) that is at an optimal distance to the oxygen atom. If that distance is too large the reaction will not occur. If such distance is too short, this will hinder the activation by Cu. To test this hypothesis and to address the impact of conformation and distance, we selected five representative compounds (13, 19, 2a, 7, 2k) that produce 4- and 5-member rings to various extents. Since these molecules exhibit several rotatable bonds, modeling conformational flexibility via molecular dynamics (MD) is of key importance. Such MD simulations should be at least a few orders of magnitude longer than the slowest vibrational modes, which are typically of the order of 1 ps. Thus, we performed a 10 ns MD simulation at 80 °C for all of these compounds, explicitly solvated by acetonitrile. Since for such time scales a quantum mechanical-based MD would be prohibited, we carried out MD simulations using the OPLS3e force field. This recently developed force field21 represents the largest concerted parameterization effort aimed at organic molecules. In particular, it provides extensive parameterization of valence and torsional terms. MD simulations were carried out with Desmond, within the Schrodinger 2019-4 suite.22 It is also important to note that we made no attempt to model the mechanism of Cu-phenanthroline binding and oxidative addition. Instead, we simply probed whether there could be intrinsic conformations that may alone account for the observed selectivity.
Two key geometric parameters were monitored throughout the MD simulation: dP and dD, as shown in the first structure in Fig. 4. Although there are other internal coordinates that may correlate with oxidative addition selectivity, we found that these two alone presented a simple, qualitative correlation with the outcome of the reactions. The vertical and horizontal lines in the correlation maps correspond to the average of dP and dD, respectively. For instance, for 13, there is a large density of occurrence of short distances in dP (marked with the letter P) and long distances in dD. Those conformations would allow for ready oxidative addition of C–BrP, consistent with the experimental observation of oxetane formation. Notice that high density of distances also occurs in the upper right quadrant (marked as X). However, these points correspond to non-reactive conformations as both dP and dD are clearly too large. A similar map can be observed for 19, except that now there are noticeable patches of short dD distances and long dP distances (marked as D). Remarkably, this is consistent with the observed 1:1 ratio in the outcome of the reaction. For 2a, the highest density of reactive conformations occurs for short dD distances and relatively long dP distances. As a consequence, C–BrP becomes, on average, less accessible to the Cu-complex, and oxidative addition involving C–BrD becomes more probable, consistent with the formation of dihydrofuran 3a. Areas with too short distances are also non-reactive. The distribution of distances for 7 and 2k are similar to 2a in that the larger occurrence corresponds to C–BrD selectivity, also consistent with major formation of dihydrofuran. Interestingly, the maps for 7 and 2k are similar to each other in that the proportion of non-reactive conformations (X) is larger than the reactive conformations. This is consistent with the low conversions we observed for each of these in acetonitrile at 80 °C.
Using MD simulations on just the substrates is attractive in its simplicity. In these cases, where electronic effects can be ruled out and where the two products are likely to result from the same basic pathways, the outcome of the molecular mechanics simulations provides a useful framework for thinking about altering the product distributions.
While the simulations did not include anything related to the catalyst system, it is clear that Cu, its counterion, and the ligand could all influence conformation populations of dP and dD. Although it is not completely straightforward to predict how changes in the catalyst system will affect the balance of products, the understanding that conformation populations play a critical role gave us a clue. For example, 2a under the conditions of procedure A gives solely 3a. The result was the same using CuBr, rather than CuI (procedure C). However, in the absence of a ligand (all other conditions same as procedure C), CuBr provided 3a:4a in a 70:30 ratio. The reaction was slower (3 d), but conversion to the two products was clean. Clearly, the presence or absence of a ligand would impact conformational populations. While the MD simulations with just the starting alcohols did not in themselves reveal how altering the catalyst/ligand would shift the outcome, they did importantly reveal factors to explore.
To illustrate the utility of the 4-bromodihydrofuran products, 3a was coupled with phenyl boronic acid and 3b with phenyl acetylene under standard conditions for the Suzuki–Miyaura and Sonogashira reactions, respectively. Coupled products 22 (61%) and 23 (74%) were isolated in good yields. We also prepared 4-bromodihydrofuran 3o, an intermediate from the patent literature used to prepare pesticides via subsequent coupling reactions (Scheme 3).23 Our three step process from commercial ketone provided 3o in 31% overall yield. The catalysts and ligand used in the sequence are inexpensive and relatively innocuous. The five step route to such trifluoromethylaryl ketone derived 4-bromo-2,3-dihydrofurans reported in the patent literature used a relatively expensive Rh catalyst and ligand and required the use of an autoclave, Br2 and CO. The straightforward preparation of 3o illustrates the utility of this three step preparation of 4-bromodihydrofurans.
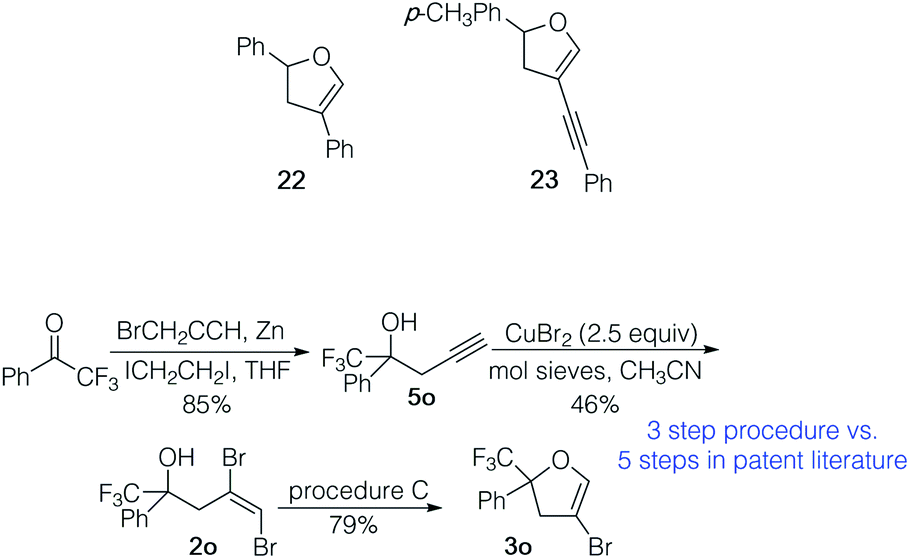 | ||
| Scheme 3 Preparation of patented pesticide precursor 3o. | ||
Conclusions
In conclusion, we have developed a straightforward, efficient three step approach to 4-bromo-2,3-dihydrofurans from aldehydes and ketones. These dihydrofurans are usefully functionalized for subsequent coupling reactions and potential reduction of the alkene to provide tetrahydrofurans. The key transformation involved the possibility of a 4-exo or 5-endo Cu(I) catalyzed O-vinylation of a dibromoalkene in which the latter cyclization predominated. On the other hand, direct competition with divinylalcohol 19, containing appropriately positioned vinylbromide moieties, demonstrated that the 4-exo and 5-endo modes were essentially equally likely to occur. An electronic explanation for the preference for 5-endo products was ruled out via a combination of force field conformational search and DFT. On the other hand, MD simulations, using just the alcohols and based on two key geometric parameters, dP and dD, provided a qualitative rationalization for product distributions between oxetane and dihydrofuran with key representative compounds. These experimental and computational studies have provided important clues for understanding and influencing these useful Ullmann-type cyclizations.Author contributions
J. A., J. I. and T. K. contributed to the experimental work; J. A. G. and A. R. contributed to the computational work. J. A., J. A. G. and A. R. H. contributed to ideation and writing of the paper.Conflicts of interest
There are no conflicts to declare.References
- J. Cornil, L. Gonnard, C. Bensoussan, A. Serra-Muns, C. Gnamm, C. Commandeur, M. Commandeur, S. Reymond, A. Guérinot and J. Cossy, Acc. Chem. Res., 2015, 48, 761–773 CrossRef CAS PubMed.
- Q. Lu, D. S. Harmalkar, Y. Choi and K. Lee, Molecules, 2019, 24, 3778pp CrossRef PubMed.
- A. S. Dudnik and V. Gevorgyan, Angew. Chem., Int. Ed., 2010, 49, 2096–2098 CrossRef CAS PubMed.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- M. K. Lakshman and P. K. Vuram, Chem. Sci., 2017, 8, 5845–5888 RSC.
- K. Morimoto, Chem. Pharm. Bull., 2019, 67, 1259–1270 CrossRef CAS.
- I. Kanwal, A. Mujahid, N. Rasool, K. Rizwan, A. Malik, G. Ahmad, S. A. Shah, U. Rashid and N. M. Nasir, Catalysts, 2020, 10 DOI:10.3390/catal10040443.
- A. Taheri Kal Koshvandi, M. M. Heravi and T. Momeni, Appl. Organomet. Chem., 2018, 32, e4210 CrossRef.
- C. A. Malapit, I. K. Luvaga, D. R. Caldwell, N. K. Schipper and A. R. Howell, Org. Lett., 2017, 19, 4460–4463 CrossRef CAS PubMed.
- K. Camara, S. S. Kamat, C. C. Lasota, B. F. Cravatt and A. R. Howell, Bioorg. Med. Chem. Lett., 2015, 25, 317–321 CrossRef CAS.
- R. Raju and A. R. Howell, Org. Lett., 2006, 8, 2139–2141 CrossRef CAS PubMed.
- Y. Fang and C. Li, J. Am. Chem. Soc., 2007, 129, 8092–8093 CrossRef CAS PubMed.
- S. Bhunia, G. G. Pawar, S. V. Kumar, Y. Jiang and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 16136–16179 CrossRef CAS.
- C. Sambiagio, S. R. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC.
- G. Chelucci, Chem. Rev., 2012, 112, 1344–1462 CrossRef CAS PubMed.
- A. S. Y. Lee, S. F. Chu, Y. T. Chang and S. H. Wang, Tetrahedron Lett., 2004, 45, 1551–1553 CrossRef CAS.
- J. Xiang, R. Yuan, R. Wang, N. Yi, L. Lu, H. Zou and W. He, J. Org. Chem., 2014, 79, 11378–11382 CrossRef CAS PubMed.
- L. M. Dollinger and A. R. Howell, J. Org. Chem., 1996, 61, 7248–7249 CrossRef CAS PubMed.
- G. O. Jones, P. Liu, K. N. Houk and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 6205–6213 CrossRef CAS PubMed.
- H.-Z. Yu, Y.-Y. Jiang, Y. Fu and L. Liu, J. Am. Chem. Soc., 2010, 132, 18078–18091 CrossRef CAS PubMed.
- K. Roos, C. Wu, W. Damm, M. Reboul, J. M. Stevenson, C. Lu, M. K. Dahlgren, S. Mondal, W. Chen, L. Wang, R. Abel, R. A. Friesner and E. D. Harder, J. Chem. Theory Comput., 2019, 15, 1863–1874 CrossRef CAS PubMed.
- A. D. Bochevarov, E. Harder, T. F. Hughes, J. R. Greenwood, D. A. Braden, D. M. Philipp, D. Rinaldo, M. D. Halls, J. Zhang and R. A. Friesner, Int. J. Quantum Chem., 2013, 113, 2110–2142 CrossRef CAS.
- P. Bindschädler, G. K. Datta and W. Von Deyn, World Pat., WO2016102490A1, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic data for all new compounds, computational methods (PDF). See DOI: 10.1039/d1sc01013a |
| This journal is © The Royal Society of Chemistry 2021 |