
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion mediated, tunable isoguanosine self-assemblies: decoding the conformation influence and solvent effects†‡
Mengjia
Liu
a,
Ying
He
a,
Chuan
Shan
 a,
Lukasz
Wojtas
a,
Ion
Ghiviriga
c,
Omar
Fathalla
a,
Yu
Yan
a,
Xiaopeng
Li
ab and
Xiaodong
Shi
*a
a,
Lukasz
Wojtas
a,
Ion
Ghiviriga
c,
Omar
Fathalla
a,
Yu
Yan
a,
Xiaopeng
Li
ab and
Xiaodong
Shi
*a
aDepartment of Chemistry, University of South Florida, 4202 E. Fowler Avenue, Tampa, Florida 33620, USA. E-mail: xmshi@usf.edu
bCollege of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen 518055, People's Republic of China
cDepartment of Chemistry, University of Florida, 125 Buckman Drive, Gainesville, Florida 32611, USA
First published on 26th April 2021
Abstract
Systematic investigations were performed with various substituted groups at C8 purine and ribose. A series of isoG analogs, C8-phenyl substituted isoG were synthesized and applied for Cs+ coordination. The structural proximity between purine and ribose limited pentaplex formation for C8-phenyl substituted isoG derivatives. Based on this observation, deoxy isoG derivative with modification on ribose (tert-butyldimethylsilyl ether) was applied to assemble with the Cs+ cation. Critical solvent (CDCl3 and CD3CN) and anion (BPh4−, BARF−, and PF6−) effects were revealed, leading to the controllable formation of various stable isoG pentaplexes, including singly charged decamer, doubly charged decamer, and 15-mer, etc. Finally, the X-ray crystal structure of [isoG20Cs3]3+(BARF−)3 was successfully obtained, which is the first example of multiple-layer deoxy isoG binding with the Cs+ cation, providing solid evidence of this new isoG ionophore beyond two-layer sandwich self-assembly.
Introduction
The core value of supramolecular chemistry is the efficient construction of highly ordered structures at the molecular level through controllable building block interactions.1 The fundamental scientific tasks in supramolecular chemistry are (A) to develop building blocks that could conduct efficient non-covalent interactions; (B) to understand the driving forces between molecules to reach controllable molecular architecture; and (C) to apply these systems as potential solutions for important scientific problems that are difficult and/or challenging when using alternative covalent approaches.2 To reach these goals, development of basic supramolecular building blocks and understanding the driving forces toward the formation of specific non-covalent interactions are of great importance for tackling challenging scientific questions in alternative covalent approaches.3Isoguanosine (isoG, also known as 2-hydroxy-adenosine) is a structural isomer of guanosine (G).4 IsoG pentaplex is an interesting supramolecular scaffold with intrinsic H-bond donors and acceptors. The 108° H-bond angle allows isoG to form a larger self-assembled core beyond tetrameric structures formed by guanosine (G4).5 In 2000, the Davis group reported the first two X-ray crystal structures that provided the evidence for the formation of the H-bonded isoG5 (isoG-star) self-assembly.6 As shown in Scheme 1, the isoG-star holds an overall planar structure, while the sp3 configuration at the C1′ position gives a twisted conformation between ribose and purine, producing a well-defined bowl-shape assembly, which further led to the formation of a stable sandwiched structure of isoG10.
 | ||
| Scheme 1 IsoG self-assembly with unique C5 symmetry. (A) Formation of isoG-star from isoG self-assembly; (B) tunable deoxy isoG self-assembly. | ||
Unlike the G4-quartet (square planar geometry), the isoG5 gives a star-shaped building block with C5 symmetry and larger pore.7 This unique feature of isoG has enabled it to selectively bind with large size cation Cs+ (radii = 174 pm) in high affinity. The selective binding of Cs+ over other alkali and alkali earth metal cations makes isoG5 a potential host for effective caesium-137 extraction in radioactive waste treatment.8 However, little progress has been made in isoG self-assembly studies since the initial breakthrough.9 This is probably due to the close distance between N9-ribose and N3-purine. Unlike G-quartet, where N3 is not involved in the H-bonding network, the N3 position is crucial in the isoG self-assembly as a key H-bond receptor. In addition, the crystal structure of isoG-star revealed interesting “extra H-bonds” between the ribose O2′ and purine N6-HB (Scheme 1A). The purine–ribose interaction in the isoG pentamer makes it much more challenging for precise conformation control. Thus, new systems that could achieve isoG self-assembly with good stability and structural diversity are highly desirable. Herein, we report a systematic study of the influence of the isoG C8 substitution and ribose modification on the formation of isoG-star. Conditions to achieve controllable isoG5 pentaplex have been investigated, which revealed the critical solvent and anion effects (Scheme 1B).10 The successful preparation of singly charged decamer, doubly charged decamer and pentaplex isoG15 through the anion and ribose conformation control was undoubtedly characterized by 1H NMR, diffusion NMR and ESI-MS. The X-ray crystal structure of [isoG20Cs3]3+(BARF−)3 was successfully obtained, confirming the formation of pentaplex isoG20 beyond the previously reported sandwich structure. These results not only highlighted the critical solvent and anion effects in this supramolecular assembly, but also provided new isoG-based structural motifs for molecular recognition and related potential applications.11
Results and discussion
As reported by Davis, isoG 1 has been used to react with CsBPh4 for the formation of isoG-star (Fig. 1). The relatively rigid ribose modification successfully controlled the formation of a well-defined bowl-shaped structure, which led to the sandwiched isoG10. Notably, as an H-bond receptor, the N3 position is crucial for the isoG-star formation. However, the structural proximity between N3 and ribose limited the choice of modification on ribose or purine. In addition, the crystal structure of isoG-star from isoG 1 revealed interesting “extra H-bonds” between the ribose O2′ and purine N6-HB, which makes one wonder if this purine–ribose interaction is necessary for the stable isoG-star formation. With all these structural analyses, the purine C8 position is the ideal synthetic handle to introduce new functionality without interrupting the needed H-bond in the isoG-star formation. The 8-phenyl modified compound isoG 2 was synthesized (see detailed synthesis in the ESI‡). To evaluate the importance of purine–ribose O2′⋯N6-HB binding, 2-deoxy isoG 3 and 4 were also prepared. All these new isoG derivatives were applied in the self-assembly studies. It is known that, with the large cation radii, Cs+ provides the best match toward the isoG5 pore. As reported previously, treating isoG 1 in CDCl3 solution with CsBPh4 (aqueous solution) gave effective extraction of Cs+ into the organic layer, forming stable [(1)10Cs]+(BPh4−) complexes.12 However, conducting similar experiments with isoG 2 or isoG 3 gave no formation of Cs+ complexes.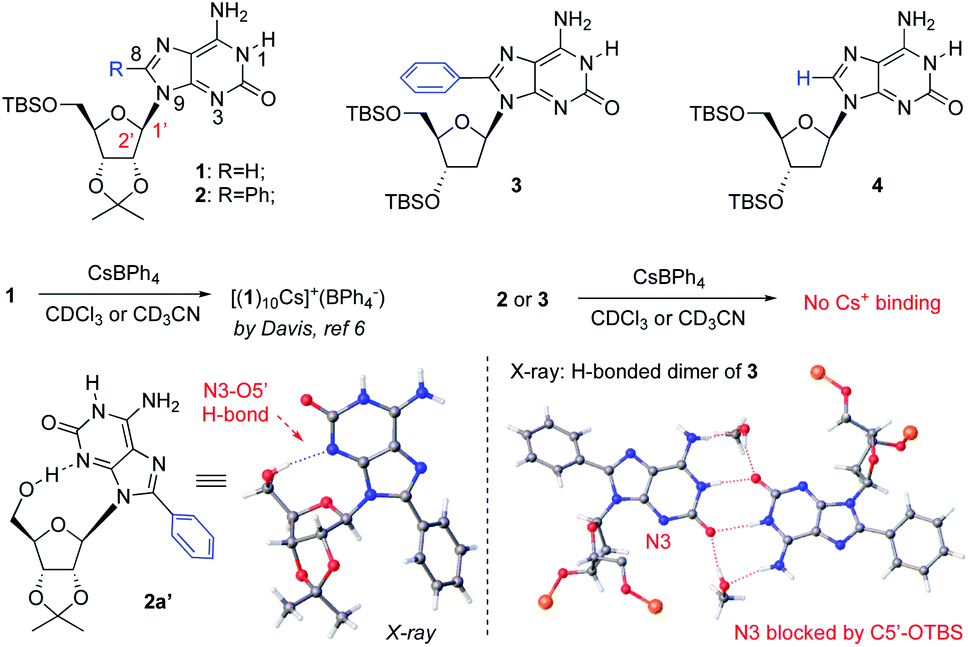 | ||
| Fig. 1 Challenge of isoG-star formation using isoG derivatives. | ||
Fortunately, the X-ray single crystal structures of both compounds were successfully obtained (Fig. 1). As shown in the crystal structures of isoG 2′ (from 5′-TBS-deprotection of 2), the conformation of the free-rotatable C–N bond is influenced by substitution of the purine C8 position. The anti-conformation is energetically favored by avoiding steric repulsion. As a result, O5′ was placed close to N3, blocking critical H-bonding in the isoG5 formation. With the less rigid deoxy-ribose with no O2′, isoG 3 gave a H-bonding dimer since N3 is not accessible for H-bonding, which is critical for the isoG-star formation. These results highlighted the challenge associated with developing new systems for the preparation of isoG-star from small molecular assembly. As revealed by the crystal structures, C8 functionalization prevents N3 from being accessible for H-bonding to form isoG-star.
To examine whether O2′⋯N6-HB H-bonding is necessary for the stable isoG-star formation, deoxy-isoG 4 with no C8 phenyl group and O2′ was applied for self-assembly with Cs+. As shown in Fig. 2E, treating isoG 4 with CsBPh4 (excess amount) led to the formation of multiple sets of new signals in the 1H NMR spectrum which indicated the formation of H-bonded complexes. Interestingly, monitoring the reaction over time suggested the formation of one major set of signals when treating the ligand and the cation for 30 minutes (Fig. 2D). The 1H NMR spectrum integration confirmed that the ratio between isoG 4 and Cs+ is 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. This result is exciting since it suggested the formation of isoG-star could be achieved with deoxy-isoG without the extra ribose–purine H-bond. Various conditions were used to obtain X-ray crystal structures. Fortunately, by switching the anion to BARF−, a stable complex was obtained in a CHCl3–CH3CN solvent mixture with structure confirmed by X-ray crystallography as [(4)20Cs3]3+(BARF−)3 (Fig. 3). Notably, this is not only the first crystal structure of an isoG-assembly from deoxy-ribose but also the first multiple-layer isoG pentaplex beyond the sandwich structure reported so far.
:1. This result is exciting since it suggested the formation of isoG-star could be achieved with deoxy-isoG without the extra ribose–purine H-bond. Various conditions were used to obtain X-ray crystal structures. Fortunately, by switching the anion to BARF−, a stable complex was obtained in a CHCl3–CH3CN solvent mixture with structure confirmed by X-ray crystallography as [(4)20Cs3]3+(BARF−)3 (Fig. 3). Notably, this is not only the first crystal structure of an isoG-assembly from deoxy-ribose but also the first multiple-layer isoG pentaplex beyond the sandwich structure reported so far.
 | ||
| Fig. 2 1H NMR spectra showing formation of isoG monomers and complexes in CDCl3 (A) isoG 1 monomer; (B) [(1)10Cs]+(BPh4−); (C) isoG 4 monomer; (D) treating isoG 4 with CsBPh4 for 30 min; (E) treating isoG 4 with CsBPh4 for 12 h. | ||
 | ||
| Fig. 3 X-ray single crystal structure of the deoxy isoG20, (A) modes of organization in the formation of the complex; (B) side view of [(4)20Cs3]3+(BARF−)3; (C) ion channel in [(4)20Cs3]3+(BARF−)3; (D) outer and inner layer. | ||
As shown in the crystal structure, the isoG20 assembly was formed with the deoxy-ribose ligand. The Cs+ cations bind to isoG5 between layers through ion–dipole interactions, similar to the previously reported isoG10 from isoG 1. The H-bond distance between N1–O2 is 2.71 ± 0.03 Å and N6–N3 is 2.88 ± 0.03 Å, similar to the H-bond distance observed in [(1)10Cs]+. The distances between isoG5 layers in [(4)20Cs3]3+(BARF−)3 are between 3.20 and 3.37 Å, almost identical to the distance observed in the isoG 1 complex, suggesting similar ion–dipole interactions in this deoxy-isoG complexes. The biggest difference between these two types of isoG-assemblies is the ribose conformation. In [(1)10Cs]+(BPh4−), the two isoG5 pentamers stack in a tail-to-tail configuration. With more flexible deoxy ribose, isoG5 from isoG 4 is less sterically hindered, allowing ribose side chain interdigitation between layers. As shown in complex [(1)10Cs]+, the opposite H-bond orientation is crucial for the formation of strong ion–dipole interactions (Cs+ to O2). Similar stacking pattern was observed in [(4)20Cs3]3+(BARF−)3, where the reversed H-bond orientation was observed between two adjacent layers, giving either a head-to-head or tail-to-tail configuration throughout the structures. This result is important since it revealed the possibility to further extend the structure along the c-axis with this new deoxy-ribose isoG for potential applications, such as the Cs+ ion-channel.
With the solid-state structure confirmed by X-ray crystallography, we then focused on the self-assembly in the solution. As discussed above, since this deoxy isoG-star could further extend the assembly along the vertical direction, achieving a controllable pentaplex is critical for potential future applications. To study how deoxy-isoG 4 binds with Cs+ in solution, we first dissolved [(4)20Cs3]3+(BARF−)3 in CDCl3 and monitored by 1H NMR. As shown in Fig. 4A, dissolving [(4)20Cs3]3+(BARF−)3 in CDCl3 gave a mixture of various H-bonded complexes (see full spectra in Fig. S2‡). To identify the structures of these complexes, we conducted a series of titration experiments based on 1H NMR.
 | ||
| Fig. 4
1H NMR spectra of isoG 4 assemblies in CDCl3 (A) dissolving [(4)20Cs3]3+(BARF−)3 crystal in CDCl3; (B) adding excess of Cs+ in solution; (C) adding isoG 4 in solution B; (D) 10:1 ratio of isoG 4:Cs+ in solution; (E) no cation added; (F) model of doubly charged decamer formed; (G) model of singly charged decamer formed. | ||
First, unlike the BPh4− anion (Fig. 2E), treating isoG 4 with excess of CsBARF gives the formation of a complex with 5:1 ratio between isoG 4 and Cs+ (Fig. 4B). Notably, treating isoG 4 with limited amount of Cs+ could form [(4)10Cs]+ (Fig. 4D). Interestingly, titration experiments (addition of isoG 4 into the 5:1 complex) revealed the formation a new complex with three sets of NMR signals in 1:1:1 ratio (complex A, Fig. 4C). To elucidate the composition of the stable 5:1 complex as [(4)5Cs]+ or [(4)10Cs2]2+, diffusion ordered spectroscopy (DOSY) experiments were performed (see the ESI‡), which was firstly reported by Cohen.12 The diffusion coefficients of ribose H1′ were determined as 2.87 ± 0.02 × 10−10 m2 s−1 (5:1 complex) and 3.37 ± 0.01 × 10−10 m2 s−1 (10:1 complex) (Fig. S7 and S8‡). These results strongly suggested the formation of [(4)10Cs2]2+ instead of [(4)5Cs]+. To further confirm this composition analysis, direct comparisons between 5:1 and 10:1 complexes formed from deoxy isoG 4 and isopropylidene isoG 1 were performed. The diffusion coefficients of these four complexes are summarized in Table S1.‡ The diffusion coefficient values of H1′ were observed for the 5:1 complex from isoG1 as 3.11 ± 0.06 × 10−10 m2 s−1 and the 10:1 complex as 3.33 ± 0.09 × 10−10 m2 s−1. These results strongly suggested the formation of [(1)10Cs2]2+ with the BARF− anion in CDCl3 solution, which further confirmed the formation of [(4)10Cs2]2+ as described above.
It has been previously reported that treating isoG 1 with excess CsBPh4 gave [(1)10Cs]+ (Fig. 5B). A similar decamer assembly was formed with the PF6− anion (Fig. 5C). As described above, switching the anion to BARF− gives the formation of a new set of signals (Fig. 5A), suggesting the special anion effect of BARF−. Comparing 1H NMR spectra of [(4)10Cs2]2+(BARF−)2 with free BARF− solution (cryptand complex, Fig. 5G), different chemical shifts were observed, indicating the interaction of the BARF− anion toward the pentaplex in solution (Fig. 5H), which was further confirmed by the diffusion coefficients of BARF− (see Fig. S7 and S8‡).
 | ||
| Fig. 5 Critical anion-effect in the isoG-star coordination with the Cs+ cation (A) [(1)10Cs2]2+(BARF−)2; (B) [(1)10Cs]+(BPh4−); (C) [(1)10Cs]+(PF6−); (D) [(4)10Cs2]2+(BARF−)2; (E) treating 4 with CsBPh4; (F) treating 4 with CsPF6. (G) 1H NMR spectra of (a) [(4)10Cs2]2+(BARF−)2 and (b) free BARF− solution (cryptand complex); (H) model of the doubly charged decamer formed. | ||
With the ability to control the formation of singly charged decamer and doubly charged decamer, we turned our attention to determine the structure of complex A. Considering that the solid-state stable [(4)20Cs3]3+(BARF−)3 was observed from evaporation of CHCl3 and CH3CN mix solvents, we hypothesized that the polarity difference of solvents could be crucial for the formation of complex A. Notably, the Meijer group reported the G-quadruplex self-assembly controlled by the coulombic interaction. The authors suggested that the ion pair separation could be regulated through tuning solvent polarity.13 Inspired by this work, polar solvent (like CH3CN) would cause the dissociation of the ion-pair between isoG-star and the BARF− anion. Therefore, tuning the polarity of solvents might be critical to reach the optimal conditions for the formation of complex A. Solvent titration was performed using a mixture of CDCl3 and CD3CN. The self-assembly process was monitored by 1H NMR (Fig. 6).
 | ||
| Fig. 6
1H NMR spectra of CD3CN titration experiments using [(4)10Cs2]2+(BARF−)2 (A) CDCl3:CD3CN = 9:1; (B) CDCl3:CD3CN = 4:1; (C) CDCl3:CD3CN = 7:3; (D) CDCl3:CD3CN = 2:1; (E) CDCl3:CD3CN = 3:2; (F) CDCl3:CD3CN = 1:1; (G) model of isoG15 formed. | ||
In less polar CDCl3, doubly charged decamer was formed (Fig. 6A). Addition of CD3CN caused increase in polarity with gradual formation of complex A. Finally, in the 1:1 mixture of CDCl3 and CD3CN, complex A was achieved as the dominant structure. 1H NMR spectra integration confirmed the proposed [(4)15Cs2]2+(BARF−)2, consistent with the observed three set of signals (Fig. 6G). In addition, this structure was further confirmed using ESI-MS, giving the doubly charged peak with m/z at 3850.8621 (Fig. S6‡). These experiments confirmed that the more polar solvents which can separate ion pairs favor the formation of larger assemblies. Notably, conducting the similar reaction with isoG 1, gave discrete isoG10 decamer in CD3CN with no formation of multiple-layer pentaplex, which highlighted the versatile coordination ability of deoxy-isoG 4 in the formation of pentaplex and critical solvent effect in the isoG self-assembly process.
Conclusions
In conclusion, influence of the purine C8 position and ribose functionalization in the isoG derivative self-assembly with the Cs+ cation was explored. Although C8 substituents prevent N3 from being accessible for H-bonding formation in the isoG-star, we successfully discovered that 2′-deoxy isoG with no O2′ could serve as a new building block to achieve controllable supramolecular assemblies. This finding suggests that O2′⋯N6-HB H-bonding is not necessary for the stable isoG-star formation. Based on the coordination with the BARF− anion and solvent mixtures, complexes such as isoG10, isoG15, and isoG20 were achieved with structures characterized by 1H NMR, diffusion NMR, ESI-MS and X-ray crystallography. This study not only revealed the important anion and solvent effects in the formation of various isoG self-assemblies, but also paved the crucial foundation for the isoG-star as a new scaffold in molecular architecture for future applications.Author contributions
X. S. conceived the original idea and research direction and supervised the research. X. S. and M. L. designed the study. M. L. carried out the material synthesis and characterization; M. L. and Y. H. analyzed the results and data; C. S. and W. L. performed the X-ray measurements and analyzed the data; Y. Y. and X. L. contributed to the ESI-MS; I. G. performed the DOSY experiments. All authors discussed and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the NSF (CHE-1665122) and NIH (1R01GM120240-01) for financial support. This work was supported in part by the University of South Florida Interdisciplinary NMR Facility and the Chemical Purification, Analysis, and Screening (CPAS) Core Facility.Notes and references
-
(a) L. J. Prins, F. De Jong, P. Timmerman and D. N. Reinhoudt, Nature, 2000, 408, 181–184 CrossRef CAS PubMed
; (b) L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS
-
(a) J. Murray, K. Kim, T. Ogoshi, W. Yao and B. C. Gibb, Chem. Soc. Rev., 2017, 46, 2479–2496 RSC
-
(a) M. García-Arriaga, G. Hobley and J. M. Rivera, J. Am. Chem. Soc., 2008, 130, 10492–10493 CrossRef PubMed
-
(a) F. Fuhrman, G. Fuhrman, R. Nachman and H. Mosher, Science, 1981, 212, 557–558 CrossRef CAS PubMed
-
(a) M. Cai, X. Shi, V. Sidorov, D. Fabris, Y.-f. Lam and J. T. Davis, Tetrahedron, 2002, 58, 661–671 CrossRef CAS
-
(a) X. Shi, J. C. Fettinger, M. Cai and J. T. Davis, Angew. Chem., Int. Ed., 2000, 39, 3124–3127 CrossRef CAS
-
(a) C. Zhong, J. Wang, N. Wu, G. Wu, P. Y. Zavalij and X. Shi, Chem. Commun., 2007, 3148–3150 RSC
-
(a) S. C. Lee, J. D. Lamb, M. Cai and J. T. Davis, J. Inclusion Phenom. Macrocyclic Chem., 2001, 40, 51–57 CrossRef CAS
-
(a) M. Meyer and J. Sühnel, J. Phys. Chem. A, 2003, 107, 1025–1031 CrossRef CAS
-
(a) J. E. Betancourt, M. Martín-Hidalgo, V. Gubala and J. M. Rivera, J. Am. Chem. Soc., 2009, 131, 3186–3188 CrossRef CAS PubMed
-
(a) S. Tirumala and J. T. Davis, J. Am. Chem. Soc., 1997, 119, 2769–2776 CrossRef CAS
- T. Evan-Salem, L. Frish, F. W. van Leeuwen, D. N. Reinhoudt, W. Verboom, M. S. Kaucher, J. T. Davis and Y. Cohen, Chem.–Eur. J., 2007, 13, 1969–1977 CrossRef CAS PubMed
- D. González-Rodríguez, J. L. J. van Dongen, M. Lutz, A. L. Spek, A. P. H. J. Schenning and E. W. Meijer, Nat. Chem., 2009, 1, 151–155 CrossRef PubMed
Footnotes |
| † Dedicated to the 100th anniversary of Chemistry at Nankai University. |
| ‡ Electronic supplementary information (ESI) available: Experimental section, NMR spectra, ESI-MS spectra and crystallographic data. CCDC 2039733, 2039734 and 2073701. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00988e |
| This journal is © The Royal Society of Chemistry 2021 |