
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree-membered cyclic digermylenes stabilised by an N-heterocyclic carbene†
Zhaowen
Dong
 ab,
Jan Mathis
Winkler
a,
Marc
Schmidtmann
a and
Thomas
Müller
*a
ab,
Jan Mathis
Winkler
a,
Marc
Schmidtmann
a and
Thomas
Müller
*a
aInstitute of Chemistry, Carl von Ossietzky University of Oldenburg, Carl von Ossietzky-Str. 9-11, D-26129 Oldenburg, Federal Republic of Germany. E-mail: thomas.mueller@uni-oldenburg.de
bInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland
First published on 22nd March 2021
Abstract
Treatment of potassium salts of silole dianions with donor stabilised germanium dichlorides gave the anticipated silagermafulvenylidenes R2Si = Ge(Do) (R2Si = 1-silacyclopentadiendiyl, Do = N-heterocyclic carbene (NHC)) only as transient intermediates in a side reaction. They were detected by NMR spectroscopy and, in one case, the formal dimer, 2,4-disila-1λ3,3λ3-digermetane, was isolated. The main products of these reactions are sila-bis-λ3-germiranes, i.e. directly interconnected digermylenes that are part of a three-membered ring. The structural data, supported by the results of density functional calculations confirm the digermylene nature of these products with a long inner cyclic Ge–Ge bond that decreases the inherent high ring strain in silagermiranes.
Introduction
The unusual reactivity of compounds with low-coordinated main group elements has triggered intensive research and a great number of investigations over the last 20 years.1,2 The addition of dihydrogen under ambient conditions to a digermyne3 provided the blueprint for many follow-up reactivity studies on main group compounds including carbene analogues,4–10 heteroalkenes and heteroalkynes,11–14 and frustrated Lewis pairs.15–18 Heavy vinylidenes III, as the formal fusion of carbene analogues I and heteroalkenes II, are, in view of their expected high reactivity, particularly interesting synthetic targets (Fig. 1). Several groups have recently reported the successful synthesis of selected examples of heavy vinylidenes III (Fig. 1) that include donor-stabilised (V–VII)19–22 and not-complexed species (IV).23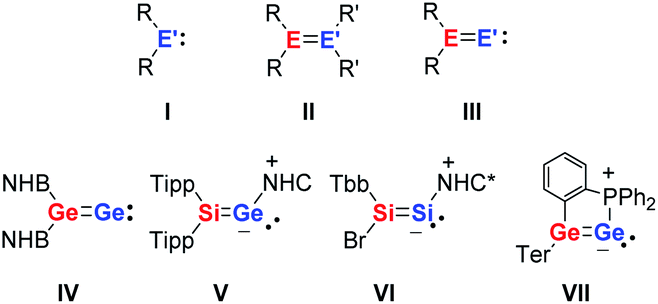 | ||
| Fig. 1 Carbene analogues I, heteroalkenes II, heavy vinylidenes III (E, E′ = Si, Ge) and some examples of isolable sila- and germavinylidenes (IV–VII) (NHB = (HC(Dipp)N)2B, Tipp = 2,4,6-trisopropylphenyl, NHC = (MeC(iPr)N)2C, Tbb = 2,6-((Me3Si)2CH)2-4-tert-butylphenyl, NHC* = (H2C(Dipp)N)2C, Ter = 2,6-bis(2,4,6-trisopropylphenyl)phenyl, and Dipp = 2,6-diisopropylphenyl). | ||
Reduction of germanium(II) halides provides a synthetic route to dicoordinated digermanium compounds that are isomers of digermenylidenes VIII.24–27 These germanium dimers with Ge in the formal oxidation state (+I) exist in two valence isomeric forms, digermynes IX and digermylenes X (Fig. 2).28,29 The digermylene isomer X is preferred when the substituent R is a multidentate ligand.30–39
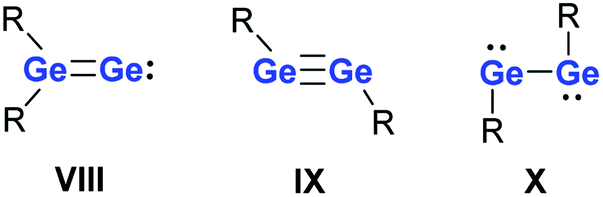 | ||
| Fig. 2 Digermenylidenes VIII, digermynes IX and digermylenes X. | ||
Following previous investigations from our laboratory,40 we probed the potential of salt metathesis of group 14 element dianions XI with group 14 element dihalides XII followed by 1,2-elimination for the synthesis of heavy vinylidenes III (Fig. 3). In contrast to our expectations, two single salt metathesis reactions occurred and the isolated products after subsequent reduction by the excess dianion were cyclic ditetrylenes XIII.
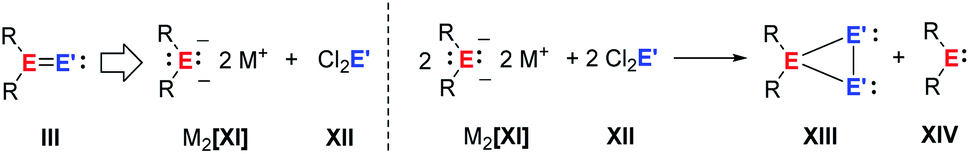 | ||
| Fig. 3 Attempted synthesis of heavy vinylidenes by double salt metathesis and formation of cyclic ditetrylenes after salt metathesis and reduction (E, E′ = Si, Ge). | ||
Results and discussion
The starting materials of choice for us were silole dianions41 and germanium dichloride stabilised by N-heterocyclic carbenes (NHC).42,43 The reaction of dipotassium silacyclopentadiendiides K2[1] with NHC-stabilised dichlorogermylene 2 gave the 3-sila-1λ3,2λ3-digermiranes 5 as main products (Scheme 1). Deep green crystals of siladigermiranes 5 were obtained by recrystallization from diethyl ether at −30 °C in 23% (5a) and 64% (5b) yields. Both spiro-heterocycles were characterised by NMR and UV spectroscopy, MS spectrometry and X-ray diffraction analysis (XRD) of suitable single crystals. 29Si NMR data of siladigermiranes 5 are unremarkable and compare well with those of previously reported siladigermiranes.44–46 The 13C NMR data indicate a silole group with localized C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds (see the ESI material†). The green colour of these compounds results from broad long-wave absorptions at λmax = 610 nm (5a) and λmax = 625 nm (5b). During the reaction of K2[1a] with dichlorogermylene 2, the known NHC-stabilised silylene 6a was detected by NMR spectroscopy as a by-product and was identified by comparison with literature data.47 After isolation of compound 5a, crystallization from diethyl ether allowed a second crop of a few dark red crystals of the tricyclic compound 8 that was identified by XRD analysis (Scheme 1). When the reaction of K2[1a] and dichlorogermylene 2 was followed by NMR spectroscopy, already at −30 °C a mixture of products was detected (see ESI† for details). The 29Si{1H} inverse gated NMR spectra indicated the presence of the NHC-stabilised silylene 6a (δ29Si = −32.9 (SiC4), −11.8), of the bicyclic siladigermirane 5a (δ29Si = −9.8, −7.0 (SiC4)) and of the tricyclic disiladigermetane 8 (δ29Si = +6.0 (SiC4), −7.8). Besides these signals, there are three additional resonances at δ29Si = +157.9 (SiC4), −3.5 and −6.1 (see Fig. S1b† for details). The low field signal is in a typical region for silicon atoms involved in multiple bonds and very close to the 29Si NMR chemical shift reported by the Scheschkewitz group for the NHC-stabilised silagermenylidene 9 (δ29Si(9) = 159).19 At reaction temperatures above −30 °C and prolonged reaction time, the intensity of these signals steadily decreased. Based on the low field 29Si NMR resonance, we identified this intermediate as the NHC stabilised silagermenylidene 7. This assignment is supported by NMR chemical shift calculations, which predict for the tricoordinated silicon atom of silagermenylidene 7δ29Si(calc) = 178, close to the experimental value (see ESI†).
C double bonds (see the ESI material†). The green colour of these compounds results from broad long-wave absorptions at λmax = 610 nm (5a) and λmax = 625 nm (5b). During the reaction of K2[1a] with dichlorogermylene 2, the known NHC-stabilised silylene 6a was detected by NMR spectroscopy as a by-product and was identified by comparison with literature data.47 After isolation of compound 5a, crystallization from diethyl ether allowed a second crop of a few dark red crystals of the tricyclic compound 8 that was identified by XRD analysis (Scheme 1). When the reaction of K2[1a] and dichlorogermylene 2 was followed by NMR spectroscopy, already at −30 °C a mixture of products was detected (see ESI† for details). The 29Si{1H} inverse gated NMR spectra indicated the presence of the NHC-stabilised silylene 6a (δ29Si = −32.9 (SiC4), −11.8), of the bicyclic siladigermirane 5a (δ29Si = −9.8, −7.0 (SiC4)) and of the tricyclic disiladigermetane 8 (δ29Si = +6.0 (SiC4), −7.8). Besides these signals, there are three additional resonances at δ29Si = +157.9 (SiC4), −3.5 and −6.1 (see Fig. S1b† for details). The low field signal is in a typical region for silicon atoms involved in multiple bonds and very close to the 29Si NMR chemical shift reported by the Scheschkewitz group for the NHC-stabilised silagermenylidene 9 (δ29Si(9) = 159).19 At reaction temperatures above −30 °C and prolonged reaction time, the intensity of these signals steadily decreased. Based on the low field 29Si NMR resonance, we identified this intermediate as the NHC stabilised silagermenylidene 7. This assignment is supported by NMR chemical shift calculations, which predict for the tricoordinated silicon atom of silagermenylidene 7δ29Si(calc) = 178, close to the experimental value (see ESI†).
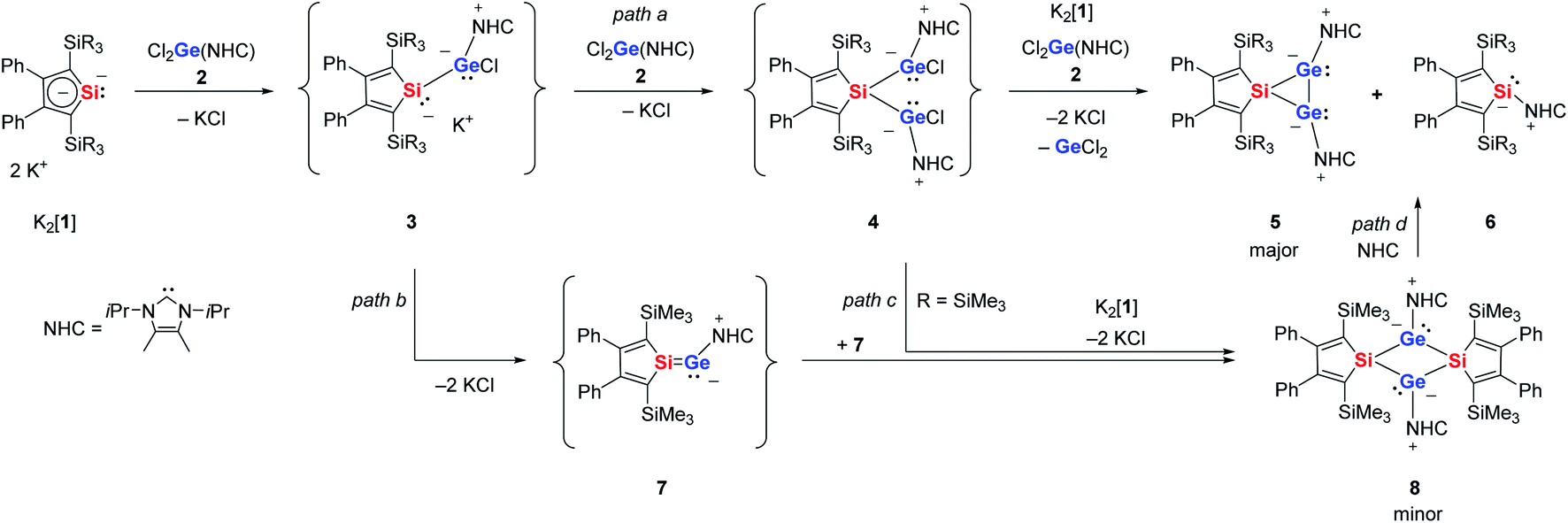 | ||
| Scheme 1 Reaction of dipotassium silole dianions K2[1] with NHC-stabilised dichlorogermylene 2 (a: R = Me; b: R = Et). | ||
Based on these experimental observations, we suggest for this complex reaction the following course (Scheme 1). The main product 5 is formed by two subsequent salt metathesis reactions via the germyl chlorides 3 and 4 (Scheme 1, path a). Such twofold salt metathesis reactions have precedence in stannole dianion chemistry.48 The dichlorides 4 undergo a reductive dehalogenation to form the three-membered ring compounds 5. At this point, the silole dianions 1 act as reducing agents and the NHC-stabilised silylenes 6 are obtained as by-products. The transient silagermenylidene 7 is formed by salt elimination from chloride 3a and subsequently it dimerized to give disiladigermetane 8 (Scheme 1, path b). In view of the very low yield of 8, we suggest that this is only one of several reaction channels for silagermenylidene 7. Likewise, we cannot exclude that compound 8 is formed by double salt metathesis of digermyl chloride 4 with the starting material K2[1] (Scheme 1, path c). Based on the result of DFT calculations, we can discard the possibility of a reversible dimerization of silagermenylidene 7, as the dimerization is highly exothermic and exergonic (ΔE = −266 kJ mol−1, ΔG = −160 kJ mol−1 (T = 298 K), see Fig. S7†). On the other hand, the formation of disilagermirane 5a from disilagermetane 8 by reaction with NHC (Scheme 1, path d) is thermodynamically feasible as it is only slightly endothermic (ΔE = 22 kJ mol−1, ΔG = 3 kJ mol−1 (T = 298 K), see Fig. S7†). We were not able to improve the yields of products 5 by adjusting the stoichiometry of the reaction according to the reaction course shown in Scheme 1. This suggests similar activation barriers for the competing reaction channels. Increasing the steric bulk of the N-heterocyclic carbene resulted in no reaction between the NHC-stabilised germanium dichloride and the dianion even at the temperature of boiling THF.
Deep green single crystals suitable for X-ray diffraction (XRD) analysis were obtained for both siladigermiranes 5 from concentrated Et2O solutions. Their molecular structures in the crystal are very similar and we discuss here only that of siladigermirane 5a in detail (Fig. 4, for details on the crystal structure of siladigermirane 5b, see ESI†). The molecule is almost C2-symmetric and its most prominent feature is the three membered SiGe2-ring with a very long Ge–Ge bond of 259.3 pm, which is longer by 7% than a regular Ge–Ge single bond (242 pm).49 It is however shorter by 28.4 pm than what is found for the Ge–Ge separation in dianion 10 (Ge–Ge = 287.7 pm, Fig. 5)50 and also significantly shorter than the Ge–Ge distance in the neutral germanium(I) dimer 11 (Ge–Ge = 270.9 pm).26 In addition, the Si–Ge bonds in siladigermirane 5a are slightly elongated compared to standard values (mean value in 5a 243.6 pm vs. standard value 237 pm).49 In summary, the metrics of the SiGe2 ring are close to those reported for 3,3-di-tert-butyl-tetramesityl-3-siladigermirane.45 The imidazolium substituents are oriented in a trans-arrangement and the Ge–C bonds (mean value 203.6 pm) are typically longer than the standard value (Ge–C = 196 pm).43,49 The C–Ge–Ge bond angles are small (96.6° and 101.0°) and both rings of the spiro compound enclose a dihedral angle of 68.3°. The coordination environment of the germanium atoms in 5a indicates the presence of a non-bonded electron pair at each of them. This is in agreement with the notion of siladigermiranes 5 as interconnected cyclic digermylenes, which are stabilised by the interaction with two NHCs.
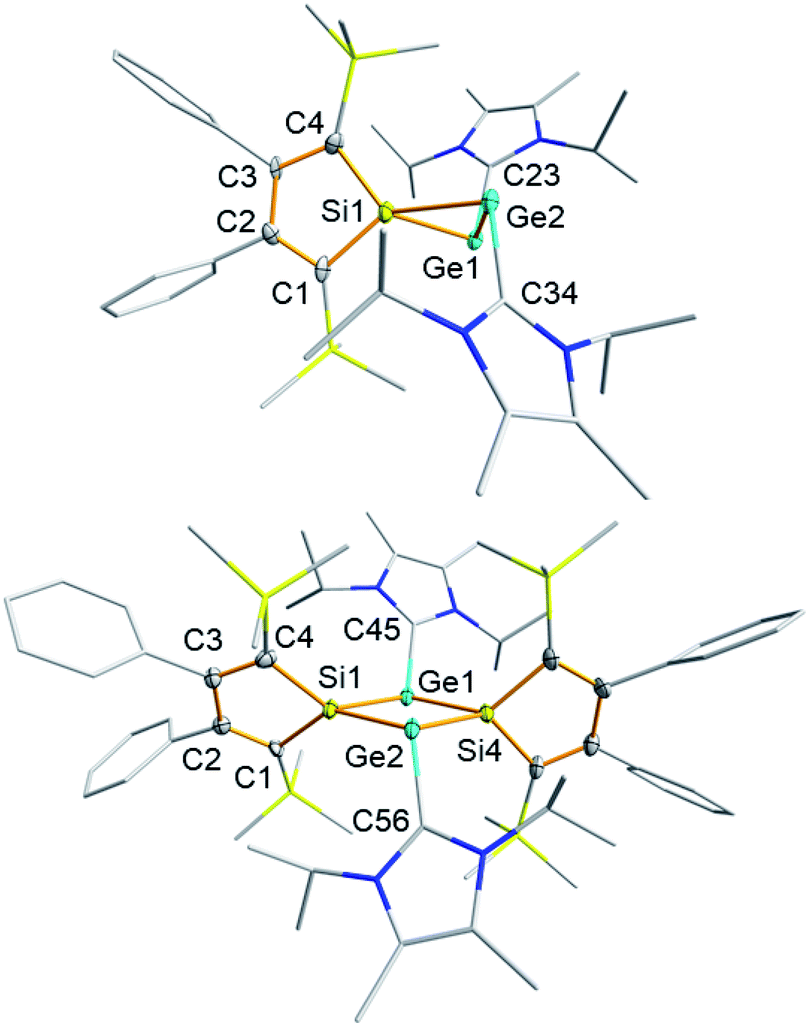 | ||
| Fig. 4 Top: molecular structures of 3-sila-1λ3,2λ3-digermirane 5a in the crystal (thermal ellipsoids at 50% probability, all hydrogen atoms are omitted for clarity. The NHC, Me3Si and Ph substituents are simplified as a wireframe). Selected atomic distances [pm] and angles [°]: Ge1–Ge2 259.3(11), Si1–Ge1 244.6(2), Si1–Ge2 242.5(2), Ge1–C23 203.7(7), Ge2–C34 203.5(7), C23–Ge1–Ge2 96.59(20), C34–Ge2–Ge1 100.98(20), and α(SiC4/SiGe2) 68.3. Bottom: molecular structure of 2,3-disila-1λ3,3λ3-digermetane 8 in the crystal (thermal ellipsoids at 50% probability, all hydrogen atoms are omitted for clarity. NHC substituents are simplified as a wireframe). Selected atomic distances [pm] and angles [°]: Si1–Ge1 246.2(12), Si1–Ge2 245.0(16), Si4–Ge1 244.3(16), Si4–Ge2 243.1(16), Ge1⋯Ge2 339.8(11), Si1⋯Si4 352.1(20), Ge1–C45 200.2(6), Ge2–C56 201.6(5), C45–Ge1–Ge2 122.54(14), C56–Ge2–Ge1 122.74(14), and α(SiC4/Si2Ge2) 58.3. | ||
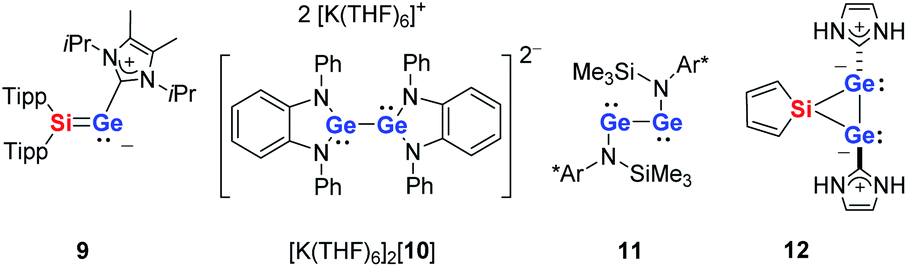 | ||
| Fig. 5 Compounds that are of relevance for the discussion (Tipp: 2,4,6-triisopropylphenyl, Ar*: 2,6-bisdiphenylmethyl-4-methylphenyl). | ||
Dark red single crystals of disiladigermetane 8 suitable for XRD analysis were obtained from the mother liquor of siladigermirane 5a at −30 °C. The dominant feature of the molecular structure of 8 is the almost quadratic (SiGe)2 four-membered ring (Fig. 3). The two silole rings are tilted by 58.3° against this central ring. The Si–Ge bonds are slightly longer (by 3–4%) than the standard value. The trans-cyclic Ge⋯Ge (339.8 pm) and Si⋯Si distances (352.1 pm) are smaller than the sum of the van der Waals radii (ΣvdWr(SiSi): 420 pm, ΣvdWr(GeGe): 422 pm)51 but are longer by 40% (GeGe) and 52% (SiSi) than their respective single bonds.49 This excludes trans-cyclic bonding interaction between these atoms. Similar to the siladigermiranes 5, the NHC-substituents are arranged in a trans fashion at the two Ge-atoms with typically long Ge–C(NHC) bonds. The C–Ge–Ge angles are however less acute (122.5 and 122.7°) and the coordination environment around the two germanium atoms is trigonal pyramidal (sum of the bond angles around germanium, Σα(Ge) = 316.0 and 316.4°).
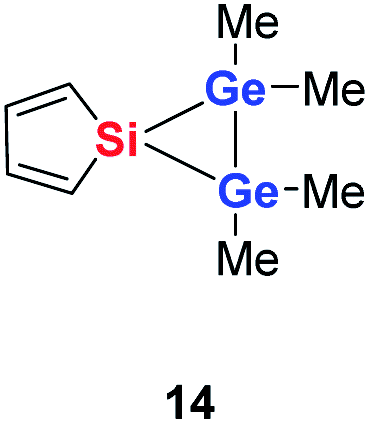
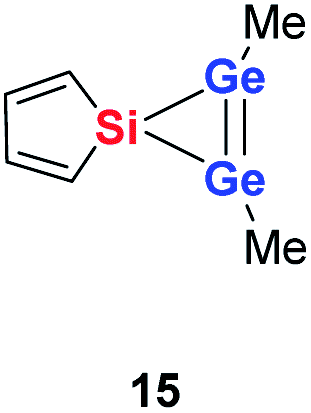
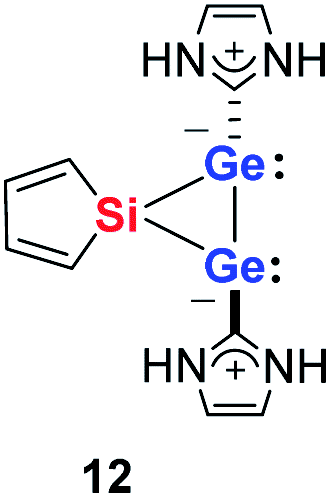
Quantum mechanical calculations at the density functional M06-2X/Def2-TZVP for 5a and for the model siladigermirane 12 were used to investigate the electronic nature of the spiro compounds 5 (see ESI†). The molecular structures optimized for 5a and 12 at this level of theory agree well with the experimental structures that are obtained from XRD analysis. The largest deviation in bond lengths is found for the Ge–C(NHC) linkage showing germanium carbon distances that are elongated by 2% (5a) and by 1% (12) compared to the experimental structure of 5a (see Table S4†). The mean bond dissociation energy (BDE) of the Ge–C(NHC) bond calculated for model compound 12 is substantial (BDE(mean) = 251 kJ mol−1). This indicates the strong stabilization of the siladigermirane by the two NHC substituents at the low coordinated germanium centres. The HOMO of compound 12 is almost exclusively the Ge–Ge σ-bond which is a combination of the atomic Ge(4p) orbitals that are oriented perpendicular to the respective Ge–Si vector. Consequently, the HOMO is slightly bent away from the centre of the three membered ring (Fig. 6). The atomic Ge(4s) orbitals dominate the HOMO−2. The shapes of the HOMO−1 and the LUMO indicate the cross-hyperconjugation between the silole ring and the Ge–Si σ-bonds of the three membered ring (Fig. 6).52,53 A population analysis in the framework of natural bond orbital (NBO) theory54 suggests that the Ge–Ge σ-bond is composed of almost exclusively Ge(4p) orbitals (94% p-orbital contribution, Fig. S9†). The lone pairs at both germanium atoms show large contributions of the 4s(Ge) atomic orbitals (79% s-, 21% p-orbital contribution). The calculated Wiberg bond index (WBI) of the Ge–Ge bond in 12 is very close to that of typical Ge–Ge single bonds (WBI = 0.84 (12) vs. 0.89 (13) and 0.92 (14), Table 1).55 From the wavefunction-based analysis, it is apparent that the long Ge–Ge bond in model compound 12 and likewise in siladigermiranes 5 is the result of the almost pure p-orbital contributions. The complementary analysis of the calculated electron density (quantum theory of atoms in molecules, QTAIM) identifies the Ge–Ge bond in 12 by its properties at the bond critical point (bcp) as being non-polar and covalent (significant electron density ρ, negative Laplacian ∇2ρ, negative total energy density H, see Table 1 and ESI†).56 The electron density at the bcp is however small compared to that calculated for the typical Ge–Ge bond in hexamethyl digermane, 13, and for the Ge–Ge bond in siladigermirane 14. Noticeable is the non-spherical electron distribution at the bcp as expressed by the large ellipticity ε of the electron density at the bcp. It is larger than calculated for the π-bond in siladigermirene 15 and the accumulation of charge extends into an area in the plane of the three-membered ring and directed away from its centre in agreement with the shape of the σ-Ge–Ge bond (Fig. 5). Consistent with the analysis of the experimental structure, the computational results indicate the digermylene nature of compounds 5 and 12 with the two germylene centres interconnected by a Ge–Ge σ-bond as represented by the canonical structure 12A (Fig. 7). Contributions of resonance structures with multiple bonds between the germanium atoms (i.e.12B and 12C) are negligible.
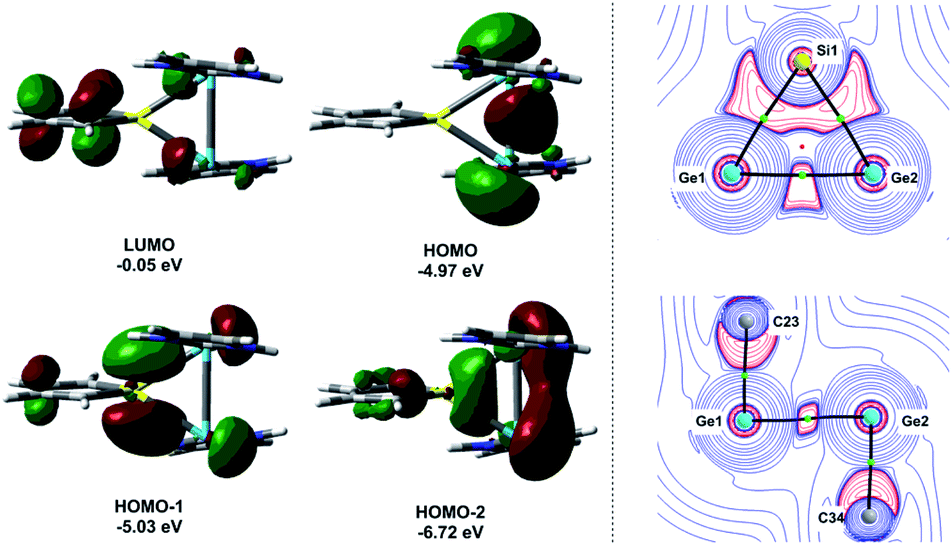 | ||
| Fig. 6 Left: calculated surface diagrams of the frontier molecular orbitals of siladigermirane 12 (isodensity value 0.04, at M06-2X/Def2/TZVP). Right: 2D contour plots of the calculated Laplacian of the electron density, ∇2ρ(r), in the Ge1–Si1–Ge2 plane (top) and in the Ge1–Ge2–C34 plane (bottom) of model compound 12. The molecular graph of model 12 is projected onto the respective contour plot. Solid black lines show the bond paths, which follow the line of maximum electron density between bonded atoms. The corresponding bond critical points (bcp's) are shown as green circles, and ring critical points as red spheres. Red contours indicate regions of local charge accumulation (∇2ρ(r) < 0); blue contours indicate regions of local charge depletion (∇2ρ(r) > 0). | ||
|
|
|
|
|
|
|---|---|---|---|---|
| Ge–Ge [pm] | 245.9 | 245.2 | 224.2 | 261.8 |
| WBI | 0.89 | 0.92 | 1.77 | 0.84 |
| ρ [a.u.] | 0.086 | 0.077 | 0.091 | 0.057 |
| ∇2(ρ) [a.u.] | −0.104 | −0.063 | −0.044 | −0.015 |
| H [a.u.] | −0.041 | −0.036 | −0.050 | −0.020 |
| ε | 0.000 | 0.375 | 0.235 | 0.836 |
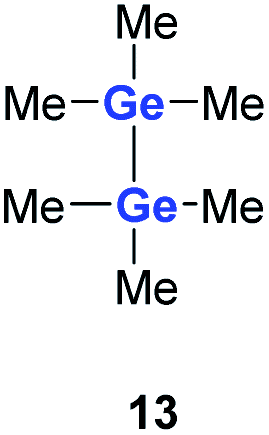
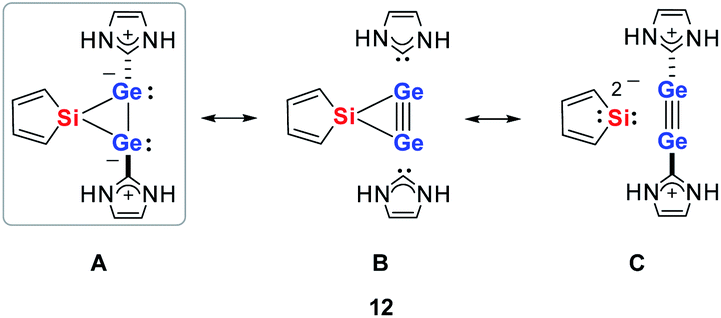 | ||
| Fig. 7 Possible resonance structures of 3-sila-1λ3,2λ3-digermirane 12. | ||
As the ring strain in heavy cyclopropanes (E3H6, E = Si, Ge) is considerably higher than that of cyclopropane (162–165 kJ mol−1vs. 124 kJ mol−1 for C3H6),57,58 we studied the influence of the integration of the Ge2 unit in a three-membered ring using a set of isodesmic reactions. We found that the long Ge–Ge bond reduces the ring strain in siladigermirane 12 by 32 kJ mol−1 compared to a related siladigermirane (see ESI† for details). This factor contributes to the thermodynamic stability of this unusual SiGe2 cycle.
Conclusions
The reaction of K2[1] with NHC stabilised germanium dichloride yields 3-sila-1λ3,2λ3-digermiranes 5 as main products. The targeted silagermylidene 7 was detected only as an intermediate in a side reaction by NMR spectroscopy. Based on the analysis of the structural and computational data the three-membered ring compounds 5 are directly interconnected digermylenes that are stabilised by NHCs.Author contributions
Z. D. was responsible for experimental investigations and formal analysis (lead), computational investigation and visualization, data presentation and writing original draft (equal with T. M.), methodology and conceptualization (supportive). J. M. W. was responsible for experimental investigations (supportive) and validation. M. S. collected the X-ray data and solved the crystal structures. T. M. was in charge for methodology and conceptualization, computational investigations, review and editing of the manuscript (lead), project administration and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Z. D. thanks the state of Lower Saxony for a Georg Lichtenberg Scholarship. This work was supported by the Deutsche Forschungsgemeinschaft (DFG-Mu1440/13-1 and INST 184/108-1 FUGG).References
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- G. H. Spikes, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2005, 127, 12232–12233 CrossRef CAS PubMed.
- G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439–441 CrossRef CAS PubMed.
- D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC.
- M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed.
- Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed.
- T. J. Hadlington, J. A. B. Abdalla, R. Tirfoin, S. Aldridge and C. Jones, Chem. Commun., 2016, 52, 1717–1720 RSC.
- D. Wendel, A. Porzelt, F. A. D. Herz, D. Sarkar, C. Jandl, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 8134–8137 CrossRef CAS PubMed.
- P. P. Power, Acc. Chem. Res., 2011, 44, 627–637 CrossRef CAS PubMed.
- D. Wendel, T. Szilvási, C. Jandl, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 9156–9159 CrossRef CAS PubMed.
- B. Wang, Y. Li, R. Ganguly, H. Hirao and R. Kinjo, Nat. Commun., 2016, 7, 11871 CrossRef CAS PubMed.
- T. Kosai and T. Iwamoto, J. Am. Chem. Soc., 2017, 139, 18146–18149 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- A. Jana, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2013, 52, 12179–12182 CrossRef CAS PubMed.
- A. Jana, M. Majumdar, V. Huch, M. Zimmer and D. Scheschkewitz, Dalton Trans., 2014, 43, 5175–5181 RSC.
- P. Ghana, M. I. Arz, U. Das, G. Schnakenburg and A. C. Filippou, Angew. Chem., Int. Ed., 2015, 54, 9980–9985 CrossRef CAS PubMed.
- K. M. Krebs, D. Hanselmann, H. Schubert, K. Wurst, M. Scheele and L. Wesemann, J. Am. Chem. Soc., 2019, 141, 3424–3429 CrossRef CAS PubMed.
- A. Rit, J. Campos, H. Niu and S. Aldridge, Nat. Chem., 2016, 8, 1022–1026 CrossRef CAS PubMed.
- M. Stender, A. D. Phillips, R. J. Wright and P. P. Power, Angew. Chem., Int. Ed., 2002, 41, 1785–1787 CrossRef CAS.
- Y. Sugiyama, T. Sasamori, Y. Hosoi, Y. Furukawa, N. Takagi, S. Nagase and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 1023–1031 CrossRef CAS PubMed.
- J. Li, C. Schenk, C. Goedecke, G. Frenking and C. Jones, J. Am. Chem. Soc., 2011, 133, 18622–18625 CrossRef CAS PubMed.
- T. J. Hadlington, M. Hermann, J. Li, G. Frenking and C. Jones, Angew. Chem., Int. Ed., 2013, 52, 10199–10203 CrossRef CAS PubMed.
- R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed.
- Y. Peng, R. C. Fischer, W. A. Merrill, J. Fischer, L. Pu, B. D. Ellis, J. C. Fettinger, R. H. Herber and P. P. Power, Chem. Sci., 2010, 1, 461–468 RSC.
- S. Nagendran, S. S. Sen, H. W. Roesky, D. Koley, H. Grubmüller, A. Pal and R. Herbst-Irmer, Organometallics, 2008, 27, 5459–5463 CrossRef CAS.
- W. Wang, S. Inoue, S. Yao and M. Driess, Chem. Commun., 2009, 2661–2663 RSC.
- W.-P. Leung, W.-K. Chiu, K.-H. Chong and T. C. W. Mak, Chem. Commun., 2009, 6822–6824 RSC.
- C. Jones, S. J. Bonyhady, N. Holzmann, G. Frenking and A. Stasch, Inorg. Chem., 2011, 50, 12315–12325 CrossRef CAS PubMed.
- S.-P. Chia, H.-X. Yeong and C.-W. So, Inorg. Chem., 2012, 51, 1002–1010 CrossRef CAS PubMed.
- M. Novák, M. Bouška, L. Dostál, A. Růžička, A. Hoffmann, S. Herres-Pawlis and R. Jambor, Chem.–Eur. J., 2015, 21, 7820–7829 CrossRef PubMed.
- W.-P. Leung, W.-K. Chiu and T. C. W. Mak, Organometallics, 2014, 33, 225–230 CrossRef CAS.
- X. Wang, J. Liu, J. Yu, L. Hou, W. Sun, Y. Wang, S. Chen, A. Li and W. Wang, Inorg. Chem., 2018, 57, 2969–2972 CrossRef CAS PubMed.
- S. Kundu, P. P. Samuel, A. Luebben, D. M. Andrada, G. Frenking, B. Dittrich and H. W. Roesky, Dalton Trans., 2017, 46, 7947–7952 RSC.
- S. P. Green, C. Jones, P. C. Junk, K.-A. Lippert and A. Stasch, Chem. Commun., 2006, 3978–3980 RSC.
- Z. Dong, L. Albers and T. Müller, Acc. Chem. Res., 2020, 53, 532–543 CrossRef CAS PubMed.
- Z. Dong, C. R. W. Reinhold, M. Schmidtmann and T. Müller, Organometallics, 2018, 37, 4736–4743 CrossRef CAS.
- P. A. Rupar, V. N. Staroverov, P. J. Ragogna and K. M. Baines, J. Am. Chem. Soc., 2007, 129, 15138–15139 CrossRef CAS PubMed.
- V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed.
- K. M. Baines and J. A. Cooke, Organometallics, 1991, 10, 3419–3421 CrossRef CAS.
- G. M. Kolleger, W. G. Stibbs, J. J. Vittal and K. M. Baines, Main Group Met. Chem., 1996, 19, 317–330 CAS.
- S. E. Gottschling, A. J. Guivernau Jacobs, M. C. Jennings and K. M. Baines, Silicon Chem., 2002, 1, 1–21 CrossRef CAS.
- D. Tian, X. Li, Y. Liu, Y. Cao, T. Li, H. Hu and C. Cui, Dalton Trans., 2016, 45, 18447–18449 RSC.
- T. Kuwabara and M. Saito, Organometallics, 2015, 34, 4202–4204 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- M. Ma, L. Shen, H. Wang, Y. Zhao, B. Wu and X.-J. Yang, Organometallics, 2020, 39, 1440–1447 CrossRef CAS.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- R. Emanuelsson, A. Wallner, E. A. M. Ng, J. R. Smith, D. Nauroozi, S. Ott and H. Ottosson, Angew. Chem., Int. Ed., 2013, 52, 983–987 CrossRef CAS PubMed.
- A. V. Denisova, J. Tibbelin, R. Emanuelsson and H. Ottosson, Molecules, 2017, 22, 370 CrossRef PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- P. Macchi and A. Sironi, Coord. Chem. Rev., 2003, 238–239, 383–412 CrossRef CAS.
- S. Nagase, Acc. Chem. Res., 1995, 28, 469–476 CrossRef CAS.
- D. A. Horner, R. S. Grev and H. F. Schaefer, J. Am. Chem. Soc., 1992, 114, 2093–2098 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1893832–1893834. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00956g |
| This journal is © The Royal Society of Chemistry 2021 |