
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWavy graphene sheets from electrochemical sewing of corannulene†
Carlo
Bruno
a,
Eleonora
Ussano
a,
Gianni
Barucca
 b,
Davide
Vanossi
c,
Giovanni
Valenti
a,
Edward A.
Jackson
d,
Andrea
Goldoni
e,
Lucio
Litti
f,
Simona
Fermani
a,
Luca
Pasquali
ge,
Moreno
Meneghetti
f,
Claudio
Fontanesi
*g,
Lawrence T.
Scott
*dh,
Francesco
Paolucci
a and
Massimo
Marcaccio
*a
b,
Davide
Vanossi
c,
Giovanni
Valenti
a,
Edward A.
Jackson
d,
Andrea
Goldoni
e,
Lucio
Litti
f,
Simona
Fermani
a,
Luca
Pasquali
ge,
Moreno
Meneghetti
f,
Claudio
Fontanesi
*g,
Lawrence T.
Scott
*dh,
Francesco
Paolucci
a and
Massimo
Marcaccio
*a
aDipartimento di Chimica “G. Ciamician”, Università di Bologna, via Selmi 2, 40126 Bologna, Italy. E-mail: massimo.marcaccio@unibo.it
bDipartimento di Scienze e Ingegneria della Materia, Ambiente ed Urbanistica, Università Politecnica delle Marche, via Brecce Bianche 12, 60131 Ancona, Italy
cDipartimento di Chimica, Università di Modena e Reggio Emilia, via Campi 183, 41100 Modena, Italy
dMerkert Chemistry Center, Boston College, Chestnut Hill, MA 02467-3860, USA. E-mail: lawrence.scott@unr.edu
eSincrotrone Trieste S.C.p.A., s.s. 14 km 163.5 in Area Science Park, 34012 Trieste, Italy
fDipartimento di Chimica, Università di Padova, via Marzolo 1, 35131 Padova, Italy
gDipartimento di Ingegneria ‘E. Ferrari’, Università di Modena e Reggio Emilia, Via Vivarelli 10, 41125 Modena, Italy. E-mail: claudio.fontanesi@unimore.it
hChemistry Department, University of Nevada, Reno, NV 89511, USA
First published on 14th April 2021
Abstract
The presence of non-hexagonal rings in the honeycomb carbon arrangement of graphene produces rippled graphene layers with valuable chemical and physical properties. In principle, a bottom-up approach to introducing distortion from planarity of a graphene sheet can be achieved by careful insertion of curved polyaromatic hydrocarbons during the growth of the lattice. Corannulene, the archetype of such non-planar polyaromatic hydrocarbons, can act as an ideal wrinkling motif in 2D carbon nanostructures. Herein we report an electrochemical bottom-up method to obtain egg-box shaped nanographene structures through a polycondensation of corannulene that produces a new conducting layered material. Characterization of this new polymeric material by electrochemistry, spectroscopy, electron microscopy (SEM and TEM), scanning probe microscopy, and laser desorption-ionization time of flight mass spectrometry provides strong evidence that the anodic polymerization of corannulene, combined with electrochemically induced oxidative cyclodehydrogenations (Scholl reactions), leads to polycorannulene with a wavy graphene-like structure.
Introduction
Graphene, with its flat single layer of carbon atoms arranged in a perfect honeycomb pattern, shows unique electronic, magnetic and optical properties.1 Nevertheless, the presence of some structural defects, which are intrinsically related to the growth process of graphene, significantly modify its physical properties,2,3 as for example the band gap and charge carrier mobility, as well as tunes the graphene behaviour in chemical reactions,4 enhancing such important phenomena as adsorption of atoms or molecules that are crucial in catalysis and for the exploitation of energy storage and conversion devices.5,6 The defects in a graphene sheet are represented by the non-hexagonal rings, mainly pentagons and heptagons, replacing the typical hexagonal rings of the graphene lattice and introduce a surface curvature, thus leading to an undulating or corrugated structure.7,8The insertion of pentagonal rings into the graphene lattice results in a surface with Gaussian curvature. In principle, to have a regularly rippled graphene layer, it is necessary to introduce non-planar aromatic hydrocarbon units in a controlled way during the growth of the graphene sheet, by a bottom-up process consisting of a chemical joining of the various moieties. As a curved aromatic hydrocarbon unit, able to produce non-planarity in graphene, corannulene9 is an ideal building block for the realization of such a structural arrangement.
Corannulene (C20H10, Scheme 1) is the archetype of curved polyaromatic hydrocarbons with a geodesic carbon framework recalling the C60 cage. In fact, the molecular structure of corannulene may be viewed as precisely one-third of a 60-fullerene molecule (i.e., a fullerene cap), with the perimeter valences saturated by hydrogen atoms.10–12 This bowl-shaped polyarene, first synthesized by Barth and Lawton in the late 1960s, became relatively easy to obtain when a flash vacuum pyrolysis synthesis was developed in 1991,13 and it is now accessible in kilogram quantities.14
 | ||
| Scheme 1 Corannulene molecular structure. | ||
The physical chemical properties of corannulene are quite interesting; in particular the electron transfers in reduction have been widely explored,15–18 and a recent paper has experimentally and theoretically demonstrated that three reversible one-electron processes can be electrochemically observed.19,20 The oxidation behaviour of corannulene, by contrast, has been mentioned only rarely in the literature and invariably has been described as a completely irreversible process that causes electrode fouling, thus preventing any further electrochemical investigation. The remarkable luminescence efficiency of corannulene and its derivative species21 makes this class of compounds very attractive candidates for the development of solid-state light emitting devices.22,23 Particularly appealing, the tuning of the electrochemiluminescent colour of corannulene, over a 100 nm wavelength range, was recently published.24
Several examples have been reported concerning the development of bottom-up production of rippled graphene or nanographene25 sheets, mostly based on chemical synthetic procedures that involve the use of curved polynuclear aromatic hydrocarbons (PAH) or their precursors as molecular units to assemble.26,27 In this work we have adopted a different strategy to build up wavy nanographene layers by using corannulene through an anodic electrochemical oligomerization as a sewing process of the curved polyaromatic hydrocarbon.
In this respect, we preliminarily undertook a re-investigation of the anodic electrochemical behaviour of corannulene, paying particular attention to the electrochemical environment. Thus, very low nucleophilicity solvents, such as dichloromethane (DCM) and liquid sulfur dioxide (liq-SO2), have been used, together with electrolytes having low nucleophilicity and high resistance to oxidation. The electrochemical oxidation of corannulene produces a radical carbocation that is extremely reactive towards any nucleophile present in solution.15,19 In our experimental conditions the corannulenium radical cation undergoes a rapid oligomerization process that produces a conducting polymeric film.
Investigation of the chemical and physical–chemical properties of the thin film thus obtained on the electrode surface, through theoretical, structural and electrochemical studies, has revealed that the highly electrophilic corannulenium gives rise to rippled graphene nanostructures. Such oligomeric polycorannulene structures consist, indeed, of wavy graphene strips or ribbons, which grow in two dimensions, terminating eventually with curled edges. These conductive wavy graphene sheets might well find applications in numerous fields, spanning from catalysis to the optoelectronic and energy storage materials sector, and should improve our understanding of how the defects, strains and wrinkles in graphene structures affect the electronic properties.
Results and discussion
Electrochemistry
A prerequisite for the use of corannulene as a building block to synthesize rippled nanographenes is a detailed knowledge of its redox behavior. The first report on the electrochemistry of corannulene was published as soon as it was synthesized for the first time, in the 1960s.15 Recently, it was reinvestigated, mainly looking at the reduction processes, under different experimental conditions and solvents.19 However, the oxidation of corannulene was immediately recognized as a process that is quite tricky to study by voltammetric techniques, and for this reason it has never been explored in depth. In fact, the first oxidation, occurring at about 1.9 V (vs. SCE), is reported to be completely irreversible in acetonitrile, and the products of the follow up reaction bring about the fouling of the electrode surface (vide infra), thus preventing any further investigation.15,24With the preliminary aim to investigate the oxidation behavior of corannulene in aprotic media, solvent as dichloromethane (DCM), electrolyte with very low nucleophilicity and ultra-dry conditions have been chosen, to ensure a high stabilization of the electrogenerated corannulene carbocations and widen the positive potential window. In Fig. 1a the cyclic voltammetric (CV) curve of corannulene in DCM is reported, together with the background curve of the electrolyte solution (tetrabutylammonium hexafluorophosphate, TBAH), for comparison purposes. It can be seen that the oxidation process, with an anodic peak potential (Epa) at 1.86 V, is completely irreversible at a scan rate of 1 V s−1. An increase of the sweep rate does not produce any significant improvement on the reversibility of the electron transfer affected by a chemical process.
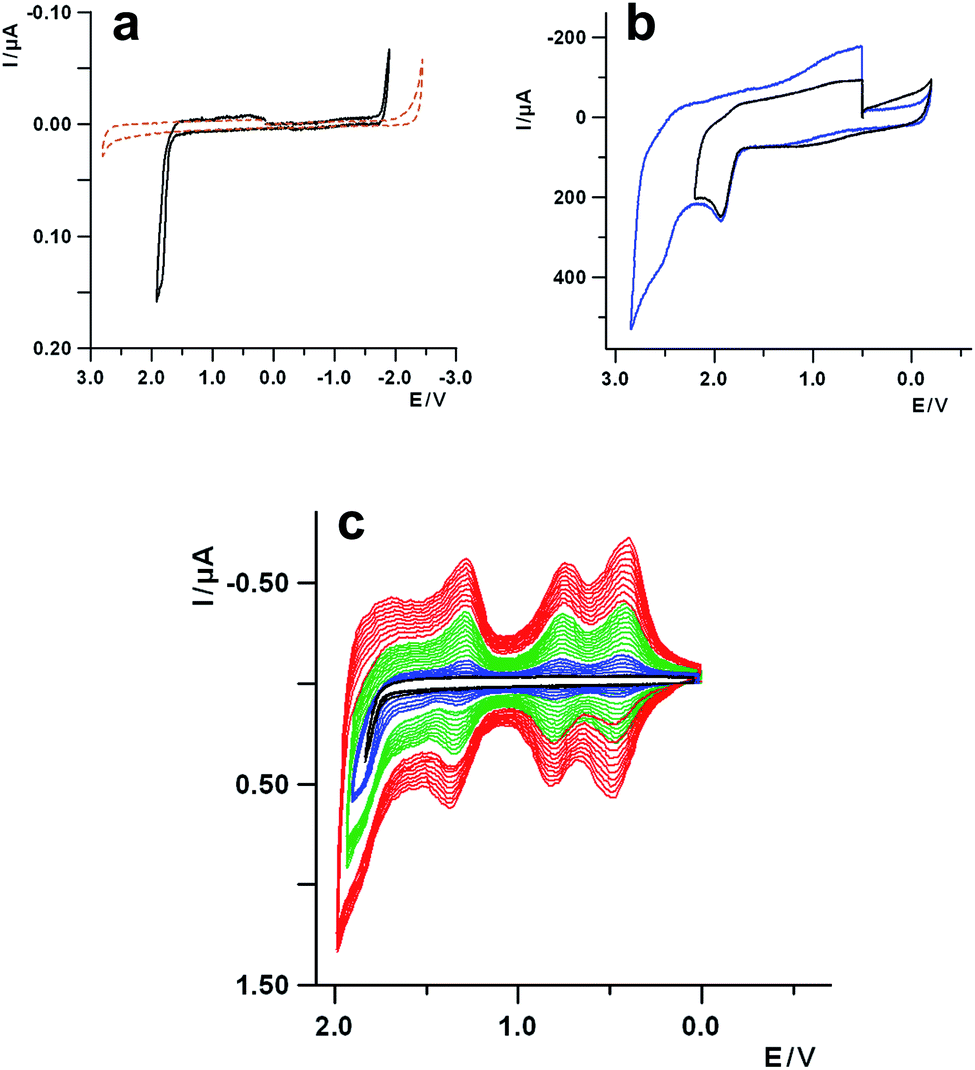 | ||
| Fig. 1 (a) Cyclic voltammetric curves of corannulene 1 mM in a 0.08 M TBAH/DCM solution (black trace) and the background electrolyte solution (brown dashed trace). Working electrode Pt disk 125 μm (diameter); reference electrode SCE; scan rate = 1 V s−1, T = 25 °C. (b) Cyclic voltammetric curves of corannulene 1 mM in a 0.07 M TBAAsF6/liq-SO2 solution. Working electrode Pt disk 0.5 mm (diameter); reference electrode SCE; scan rate = 100 V s−1, T = −65 °C. The Pt electrode surface has been cleaned with alumina slurry and carefully dried before each voltammetric cycle. (c) Multiscan cyclic voltammetric curves of corannulene 1 mM in a 0.08 M TBAH/DCM solution. Working electrode Pt disk 125 μm (diameter); reference electrode SCE; scan rate = 1 V s−1, T = 25 °C. First 3 cycles with a reversing sweep potential Er = 1.80 V (black lines); second subsequent set of 5 cycles with an Er = 1.90 V (blue lines); third subsequent set of 10 cycles with an Er = 1.95 V (green lines); fourth subsequent 10 cycles with Er = 2.00 V (red lines). | ||
To gain further information about the oxidation process, the voltammetric investigation was also carried out with electrolytes of lower nucleophilicity and higher resistance toward oxidation, such as tetrabutylammonium hexafluoroarseniate (TBAAsF6) and tetrabutylammonium tetrakis (perfluoroaryl)borate (TBAFAB), including “unconventional” solvents for electrochemistry, such as liquid sulfur dioxide (liq-SO2), which is suitable for extremely reactive cation species.28 Thus, in Fig. 1b the voltammetric curve of corannulene in liq-SO2 is reported, showing two one-electron oxidations with peak potentials at about 1.9 V for the first process and 2.5 V for the second one, respectively. Both the oxidations are irreversible at 100 V s−1 and low temperature (−65 °C) and they are separated by approximately 0.6 V, a value very close to the energy difference between the first two reductions, as rationalized by DFT calculations, carried out at the B3LYP/cc-pVTZ level of theory, (Fig. S1 and S2 in the ESI†).19 The reversibility of the processes does not improve significantly even when increasing the scan rate up to 200 V s−1 (Fig. S3, ESI†). Thus, concerning only the first process in liq-SO2 the upper limit for the lifetime (t½) of the reactive radical cation can be estimated in the range of μs, on the basis of timescale (τ) of voltammetric experiments (t½ ≪ τ = RT/Fv, with v the scan rate).29
A careful examination of the voltammetric curve of the oxidation of corannulene in DCM, reported in Fig. 1a, highlights some small bumps on the reverse scan, at positive potentials (cathodic part of the curve), in the range comprised between about +1.5 V and 0 V, which might be attributed to some products of the chemical reaction following up the electron transfer. The electrochemical investigation of such voltammetric features can shed some light on the nature of the underlying process. Thus, performing multiple voltammetric cycles which include the first oxidation process, it is possible to observe (Fig. 1c) that, on the subsequent scans after the first one, new voltammetric waves start to grow up at about 0.45 V, 0.80 V and 1.30 V with an overall progressive increase of the current, attributable to the formation of a redox active deposit on the electrode surface as in the case of a conducting polymer.
The successive potential scans provide evidence for a continuous growth in current of the voltammetric pattern developed following the first few cycles (see the green and red multiscan curves in Fig. 1c). The new developing peaks show a Gaussian shape i.e., the typical voltammetric features of species adsorbed onto an electrode, with an anodic-cathodic peak separation in the range 50–70 mV. Such behavior can be observed at different sweep rates, and the voltammetric pattern still remains practically the same even at scan rates as high as 50 V s−1, which is in line with the rather high reactivity of the corannulene carbocation discussed above.
The same electrochemical behavior and developed voltammetric pattern can be observed using different electrolytes (Fig. S4–S7, ESI†) and also on different electrode materials such as platinum, gold and ITO. The thin film deposited on the electrode is brown in colour, mechanically quite stable and it can be easily peeled off (Fig. S8, ESI†).
To investigate further the behaviour of corannulenium, the reactivity of the carbocation has been probed by a different approach, based on the trapping of the electrogenerated highly electrophilic species with a nucleophile such as pyridine (Fig. S9, ESI†). Moreover, the voltammetry of corannulene cation in DCM and in presence of pyridine has also been compared with the results in the nucleophilic solvent acetonitrile (Fig. S10, ESI†), showing that a different film, which is insulating, is formed on the electrode with a kinetic that depends on the strength of the nucleophile.
Nature of the film and spectroscopic characterization
In order to attain some insight into the real structure of the electrochemically deposited film, it was initially characterized by laser desorption-ionization mass spectra (LDITOF-MS). The laser spot impinged directly on the film, and its intensity was carefully adjusted to the lowest possible value that would still allow a good detection of ions, in order to ensure, to the extent possible, that the laser itself did not produce any artifacts due to photoprocesses. A fragmentation pattern between m/z 350 and m/z 2000 is reported in Fig. 2a and S11 (ESI†). In this range, it is possible to observe that the fragmentation features are the same for all the detected oligomeric species and, in particular in Fig. 2a, it is easy to identify the first four oligomers with m/z values of 496, 740, 984 and 1228, which can be safely assigned to the dimer, trimer, tetramer, and pentamer respectively. The spectra analysis indicates that m/z losses of 12, 13 and 24 are predominant and these are consistent with the outgoing of C, CH and C2 units, respectively, during the fragmentation of the various oligomers, together with the dehydrogenation by the peripheral C–H bonds as shown in Fig. S11-1 (ESI†). Moreover, the mass spectra features for the various oligomers are in agreement with the calculated isotopic patterns, although contaminated by different higher oligomeric fragments (see Fig. S11-2 in the ESI†).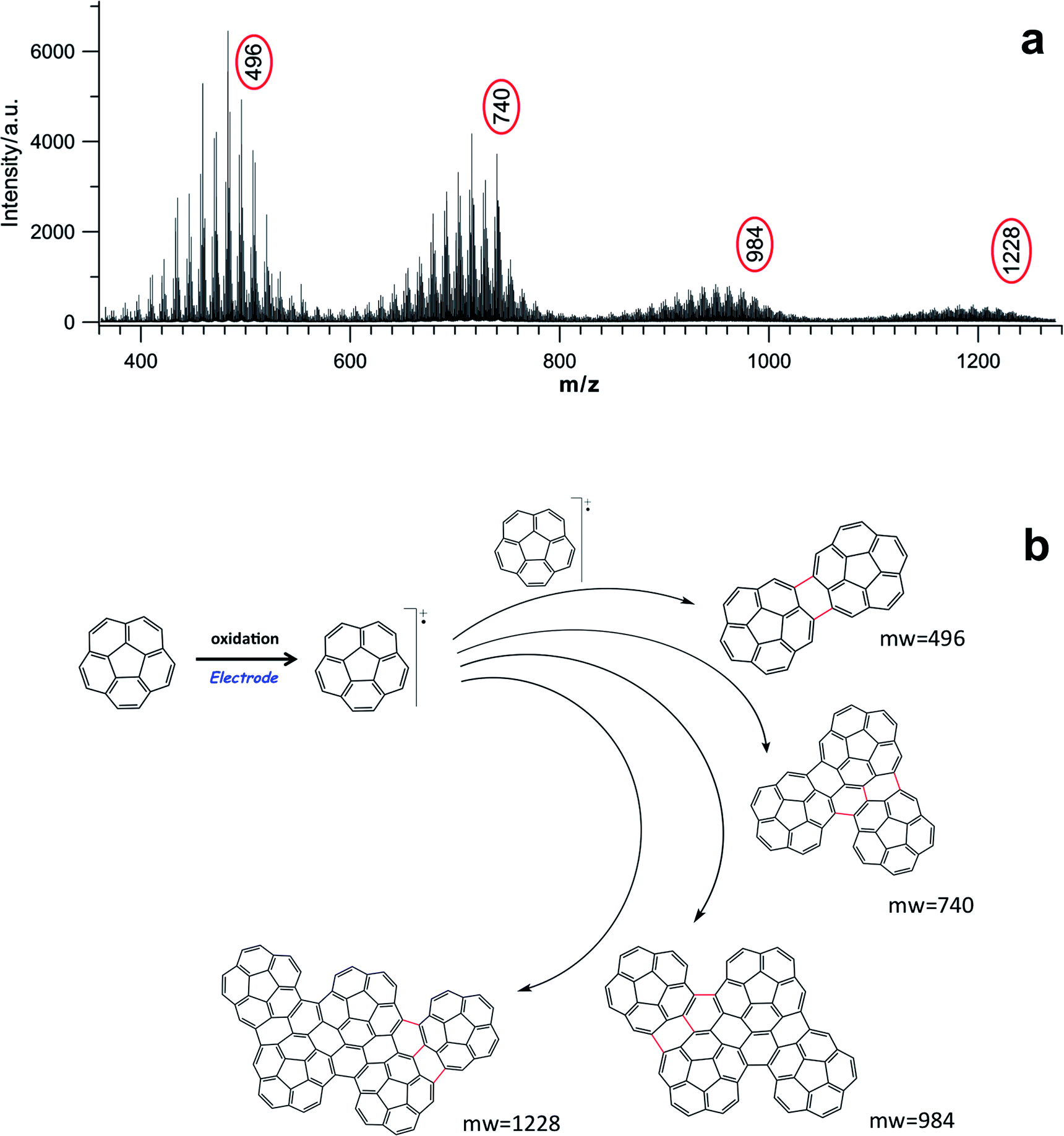 | ||
| Fig. 2 (a) Laser desorption-ionization mass spectrum (LDITOF-MS) recorded in the m/z range between 360 and 1280 of the electrochemically deposited polycorannulene film onto a uflat-ITO substrate from a 1 mM corannulene in 0.08 M TBAH/DCM solution, by repeatedly cycling the potential between 0 V and 2.1 V (vs. SCE). (b) Sketch of the electrochemical process reporting the chemical structures of corannulene oligomers corresponding to the prominent mass peaks in the LDITOF mass spectrum in (a). | ||
Interestingly, for each of these first oligomers there is an m/z loss of 4, 10, 16 and 22 respectively, with respect to the multiples of 250, which is the mass of corannulene. This means that if n is the number of corannulenyl unit, the ions of the first four oligomers have the values (2 × 250)-4, (3 × 250)-10 (4 × 250)-16, and (5 × 250)-22 respectively. Such findings show that there is not a simple straightforward multiple loss of hydrogens accompanying the increase in size of the oligomers, but something more complicated is occurring. In the case of the dimer, for example, 2 hydrogen atoms per corannulene unit are missing, whereas 4 hydrogen atoms are missing per corannulene unit for the tetramer. By extrapolation, the following formula can be written, which predicts the number of hydrogens missing in the oligomer structures:
| Cn×20Hn×10−2(3n−4) with n = 2, 3, 4, 5, etc. | (1) |
Among the possible structures (Fig. S12, ESI†), the most reasonable ones that can rationalize all these facts are sketched in Fig. 2b. Thus, two C–C bonds are formed between the corannulene rims in the dimer, whereas in the case of trimer and tetramer the upcoming corannulene units “condense” around the dimer core, establishing 3 C–C new bonds, most probably due to a cooperative steric constraint.
A plausible mechanism underlying the anodic oligomerization of corannulene can be hypothesized as consisting in a classical polymerization processes of aromatic and heteroaromatic species (e.g., pyrene and thiophene)30,31 to give conducting polymers, followed by an intramolecular oxidative cyclodehydrogenation of an aromatic hydrocarbon, known as a Scholl reaction,32 which is electrochemically triggered, as discussed in the ESI and sketched in Fig. S13.†
As the new formed corannulene oligomers are much more π-conjugated than the parent corannulene they are progressively more easily oxidized (positive doping of film), and this also explains the gradual appearance of a series of new peaks at less positive potentials before the irreversible oxidation of corannulene.
Characterization by XPS and Near edge X-ray absorption at the carbon K-edge (NEXAFS, in s- and p-geometry) measurements, carried out on the film electrochemically deposited on ITO electrode, indicate that the deposit is made, exclusively, of sp2-hybridized carbon atoms, with the binding energy close to that of graphite/graphene (284.5 eV), and the absence of linear dichroic effects, thus suggesting that the polycorannulene units have random average orientation (Fig. S16, ESI†).
The FTIR spectrum recorded on the polymeric film (Fig. S17, ESI†) is in agreement with the IR data reported in the literature for corannulene.33 Concerning the three most prominent bands below 1000 cm−1, those at 735 cm−1 and 693 cm−1 are up-shifted (75 cm−1 and 146 cm−1, respectively) with respect to the out-of-plane vibrations and bending at 660 cm−1 and 547 cm−1 of the pristine corannulene, while that at 837 cm−1 remains unchanged. Such an up-shift of the two bands could be related to the formation of new C–C bonds in the polymeric material.
The Raman spectrum of the polymer, obtained by subtracting the strong fluorescence background and reported in the ESI – Fig. S18† (red lines), is compared to that of corannulene34 (black lines). Comparison of the spectra shows that the polycorannulene seems to have only a reminiscent signature of the two important bands observed for corannulene at 1432 cm−1 and at 1629 cm−1, which have been recently described33 as a mixing among stretching of carbon–carbon bonds and bending of the C–H group. Also, the calculated Raman spectra of corannulene and a tetramer of corannulene (as a model of polycorannulene) are reported and they are in good agreement with the experimental ones.
Finally, the polycorannulene film electrochemically grown has a UV-Vis-NIR absorption peak at about 500 nm and it was also investigated by UV-Vis-NIR spectroelectrochemistry as it was seen to be strongly electrochromic upon positive doping, even during its electrochemical deposition (Fig. S19 and S20, ESI†), despite the very small thickness, thus making the material very attractive for optoelectronic applications. The spectra, recorded at three different positive potentials, show a decrease in the absorption band at about 500 nm of the pristine material and the growth of a strong band at about 630 nm. The absorption spectroscopic behavior thus observed is rather common for conducting π-conjugated polymeric systems, and it is related to the formation of polaronic and bipolaronic states inside the bandgap.31
Morphological and structural characterization
Samples of polycorannulene films on uflat-ITO, with different deposition times, have been prepared and studied by atomic force microscopy, to see the morphology of the film during the early stages of formation of the deposit and its evolution, as well as to check if there is any self-assembling amongst the carbon nanostructures thus obtained. The AFM images in Fig. 3a and b show that there is actually a sort of self-assembling pattern giving rise to nanometric rings with an average diameter ranging between 30 and 40 nm.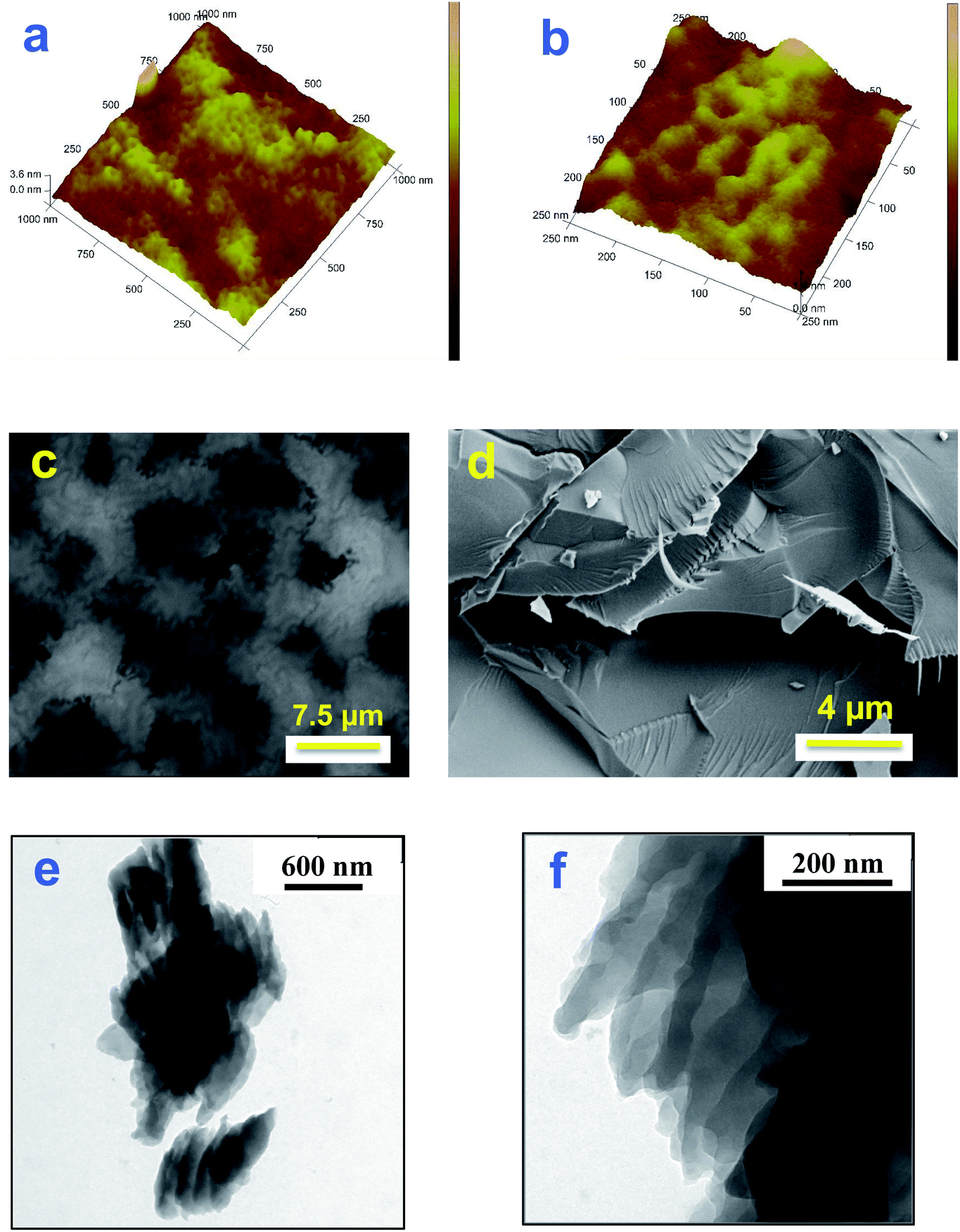 | ||
| Fig. 3 (a) and (b) AFM images of polycorannulene electrochemically deposited on uflat-ITO with 3 voltammetric subsequent cycles at 0.4 V s−1 and T = 25 °C from corannulene 1 mM in a 0.08 M TBAH/DCM solution. (a) 1 × 1 μm2 and (b) 0.25 × 0.25 μm2. (c) and (d) SEM images of polycorannulene electrochemically deposited on uflat-ITO with 40 voltammetric subsequent cycles at 0.4 V s−1 and T = 25 °C from corannulene 1 mM in a 0.08 M TBAH/DCM solution. (e) and (f) TEM images of polycorannulene film by drop-casting on a TEM carbon covered grid an alcohol solution/suspension of the electrochemically synthesized film (on uflat-ITO at T = 25 °C from corannulene 1 mM in a 0.08 M TBAH/DCM solution) obtained scratching the film and suspending the fragments in ethanol. | ||
Investigation by SEM has also been performed, and images of a polycorannulene film, about 300 nm thick, are shown in Fig. 3. In particular, Fig. 3c gives a general view of the sample, revealing that it is composed of a patchwork of carbon structures not completely overlapping. The darker regions are indeed holes or canyons in the film, suggesting a poor electrical connection among the carbon structures. SEM images in Fig. 3d and S21 (ESI†) provide evidence that the polycorannulene film consists of a rather compact lamellar structure.
To further investigate the morphology and the inner structure of the electrochemically deposited film of polycorannulene, TEM analyses were performed on samples prepared in different ways for microscopy observations. Fig. 3e, f and S22 (ESI†) show a selection of TEM bright field images performed on a film detached from the electrode, suspended in ethanol and deposited on a TEM carbon coated grid. The images confirm a layered structure of the film, even at a nanometric scale, and, in some regions, strands of nanometric fibers can also be observed (Fig. S23, ESI†).
The X-ray diffraction characterization carried out on the film of polycorannulene as electrochemically deposited does not evidence any crystalline order (red line in Fig. S24, ESI†). Indeed, no additional peaks besides those of the uflat-ITO substrate (light green line) were observed.
TEM observations were also carried out by dissolving/suspending the electrochemically deposited polycorannulene films both in 1,2,4-trichlorobenzene (TCB) and 1,1,2,2-tetrachloroethane (TCE) solvent, which allowed the observation of some very interesting features. In particular, Fig. 4a shows TEM bright field images of the polycorannulene film dissolved/suspended in TCB. It is noteworthy that amongst the amorphous matrix the presence of some crystalline structures, randomly distributed, can be found. Two of these structures are magnified in the insets of Fig. 4a. They are platelets composed of a few-atomic layers, ranging between two and ten atomic planes, and look very similar to graphene nanoplatelets (see also Fig. S25 and S26, ESI†).35 The inter-layer distance was obtained by Fast Fourier Transform (FFT) of the layered regions evidenced in the insets, providing value of 10.2 ± 0.2 Å, Fig. 4b (vide infra).
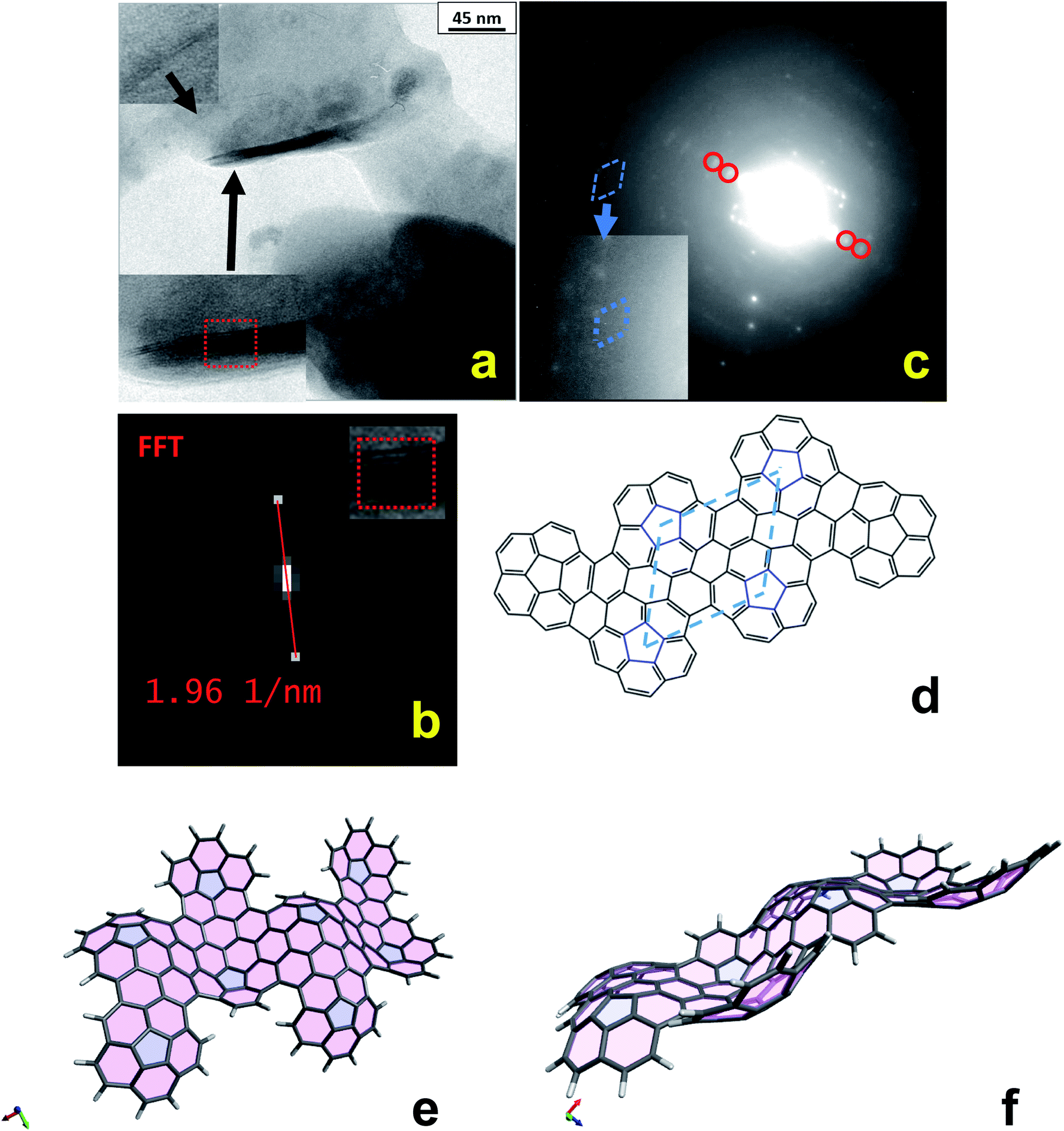 | ||
| Fig. 4 TEM of polycorannulene film by drop-casting on a TEM carbon covered grid a TCB solution/suspension of the electrochemically obtained film (on uflat-ITO at T = 25 °C from corannulene 1 mM in a 0.08 M TBAH/DCM solution). The solution/suspension was obtained by treating the deposited film with TCB solvent (see experimental details). (a) TEM image, together with two zoomed area, to evidence the multilayer graphene-like nanostructures; (b) FFT of the red squared selected area; (c) selected area electron diffraction (SAED) pattern of the sample shown in the panel (a); (d) molecular sketch of a hexa-oligomer structure to show the lattice distances. In the sketched structure, the pentagons are organized, effectively, in a hexagonal lattice, although they are not perfectly equivalent, as it should be in an exact crystalline structure; (e) and (f) tridimensional views of a DFT optimized octa-oligomer structure. | ||
Electron diffraction analyses were performed on the platelets and the selected area electron diffraction (SAED) pattern of the sample imaged in Fig. 4a is shown in Fig. 4c. Although weak diffuse diffraction rings from the amorphous matrix are present, diffraction spots due to the platelets are sufficiently intense, suggesting a large presence of the crystalline phase. The red circles mark diffraction spots corresponding to a lattice distance of 10.1 ± 0.1 Å, in perfect agreement with the interplanar distances observed in the platelets.
Looking at the left of the image, it is possible to observe a symmetrical distribution of diffraction spots, magnified in the inset. Interestingly, such spots are organized in a hexagonal cell with an associated interplanar distance of about 6.5 ± 0.2 Å (blue dashed line cell). Considering those spots originated by a hexagonal lattice and hence having Miller indexes of type (10-10), the corresponding cell parameters can be calculated as a = b = d(10-10)/cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30° = 7.5 ± 0.2 Å. This value is in good agreement with the average distance of about 7.6 Å amongst the pentagons, as measured on the oligomeric structures optimized at the DFT level, and sketched in Fig. 4d for the hexa-oligomer. Such findings, thus represent clear evidence that the electrochemical oxidation of corannulene in DCM produces nanostructures made of graphene-like stacked layers. The lattice cell of these nanostructures can confidently be described as hexagonal with the following parameters: a = b = 7.5 ± 0.2 Å; c = 10.2 ± 0.2 Å. It is important to stress that the other diffraction spots visible in the SAED pattern can be associated with other reflections of the proposed hexagonal cell, suggesting that more platelets are contributing to the diffraction pattern. On the basis of the model proposed to interpret the TEM diffraction data, further considerations can also be made to rationalize the larger value of the c axis with respect to that of graphite and other carbon nanostructures. In fact, due to the bowl geometry of the corannulene units, the resulting electrochemically synthesized nanostructures can be expected to give rise to wavy sheets, as shown in Fig. 4e and f that, when superimposed, produce larger averaged distances among layers. It has been demonstrated that curved aromatic hydrocarbons experience enhanced intermolecular interactions in the concave-convex nested geometries,36 thus supporting that, even for larger interlayer distance between the sheets, the dispersion forces could still be strong enough to help drive the self-assembly of nanostructures.
30° = 7.5 ± 0.2 Å. This value is in good agreement with the average distance of about 7.6 Å amongst the pentagons, as measured on the oligomeric structures optimized at the DFT level, and sketched in Fig. 4d for the hexa-oligomer. Such findings, thus represent clear evidence that the electrochemical oxidation of corannulene in DCM produces nanostructures made of graphene-like stacked layers. The lattice cell of these nanostructures can confidently be described as hexagonal with the following parameters: a = b = 7.5 ± 0.2 Å; c = 10.2 ± 0.2 Å. It is important to stress that the other diffraction spots visible in the SAED pattern can be associated with other reflections of the proposed hexagonal cell, suggesting that more platelets are contributing to the diffraction pattern. On the basis of the model proposed to interpret the TEM diffraction data, further considerations can also be made to rationalize the larger value of the c axis with respect to that of graphite and other carbon nanostructures. In fact, due to the bowl geometry of the corannulene units, the resulting electrochemically synthesized nanostructures can be expected to give rise to wavy sheets, as shown in Fig. 4e and f that, when superimposed, produce larger averaged distances among layers. It has been demonstrated that curved aromatic hydrocarbons experience enhanced intermolecular interactions in the concave-convex nested geometries,36 thus supporting that, even for larger interlayer distance between the sheets, the dispersion forces could still be strong enough to help drive the self-assembly of nanostructures.
Consistent with the TEM results, the X-ray diffraction analysis carried out on a drop-casted film, obtained by treating the electrochemically deposited polycorannulene in 1,2,4-trichlorobenezene solvent (similarly to TEM investigation), allowed the observation of a diffraction peak at a 2θ of about 8.6°, which corresponds to a nanostructured system having an average interlayer separation of 10.3 Å (blue trace in Fig. S24, ESI†). Such an interlayer distance, determined by X-ray, is in very good agreement with the value obtained by TEM analysis.
It is worth noting that the X-ray results and the TEM analysis give evidence for the presence of platelets, as shown in Fig. 4a, only when the samples were obtained by suspending the polycorannulene films in TCB or TCE solvents. These facts indicate an active role played by the solvent both in the reorganization and self-assembly of the nanostructured sheets. In fact, the lack of highly ordered self-assembled polycorannulene formed by direct electrochemical formation on the electrode can almost certainly be attributed to the variability of the oligomer sizes and the molecular structures of the oligomers themselves, as well as to the kinetics of the formation and deposition process. Moreover, solvent molecules act as interlayer penetrating molecules that cooperate in the stabilization of multilayer stacked structures, thus accounting for an unusually large distance of about 10 Å between the layers held together by van der Waals interactions, as also suggested by DFT calculations of two oligomers with two intercalated solvent molecules (Fig. S27, ESI†).
Conclusions
The intriguing oxidation properties of corannulene, the archetype of the curved polynuclear aromatic hydrocarbons, have been thoroughly investigated and the electrophilic radical cation reactivity has been herein reported. The extremely high reactivity of the corannulenium radical leads to the formation of a conducting polymeric film. The nature, morphology and structure of the resulting deposit on the electrode has been studied electrochemically and theoretically by DFT, as well as by mass spectrometry techniques (LDITOF-MS), spectroscopy, and microscopy investigation, SEM, TEM and AFM. The experimental and theoretical findings show that a 2D layered material made of wavy graphene nanostructures are formed, following the electrochemical oxidation of corannulene, when the process is performed under ultra-dry conditions. This new method opens the doors to a feasible and rather easy pathway to perform polymerization involving intramolecular oxidative cyclodehydrogenations (Scholl reactions) of curved polyaromatic hydrocarbons to obtain rippled graphene nanostructures. These can be used in a variety of applications in the field of optoelectronics and as new doped carbon-based materials for heterogeneous catalysis, acting as a scaffold for active nanoparticles (for its topography) as well as an electronically connecting net (e.g., metal or metal-oxide nanoparticles). Also, these bumpy graphene-like sheets could be a useful new material in the field of energy accumulation, considering the highly efficient intercalation-hosting ability demonstrated by the “bowl PAH” corannulene precursor toward lithium ions.Experimental section
Chemicals and electrochemical measurements
Corannulene was synthesized according to the three-step procedure of Scott et al.37 Tetrabutylammonium hexafluorophosphate (TBAH), tetrabutylammonium hexafluoroarsenate (TBAAsF6), tetrabutylammonium tetrakis(perfluoroaryl)borate (TBAFAB) were used as supporting electrolytes and all were electrochemical or analytical grade from Sigma-Aldrich or synthesized and purified as described in the ESI.† Acetonitrile (ACN), and dichloromethane (DCM) were purified and dried as previously reported.38,39 Sulfur dioxide (SO2) was purified and dried just before condensing the vapor into the electrochemical cell.All the conventional solvents were distilled performing the trap-to-trap distillation via a closed system into an electrochemical cell containing the supporting electrolyte and the species under examination, immediately before performing the experiment. Electrochemical experiments were carried out in an airtight single-compartment cell described elsewhere40 using either platinum, indium tin oxide (ITO) on glass (ITO/glass), gold, or glassy carbon (GC) as the working electrode, platinum as the counter electrode and a silver spiral as a quasi-reference electrode. The drift of the quasi-reference electrode was negligible during the time required for an experiment. However, all the E½ values were referenced to the aqueous saturated calomel electrode (SCE).
The cell containing the supporting electrolyte and the electroactive compound was dried under vacuum at 100–120 °C before each experiment. Voltammograms were recorded with an AMEL Mod. 552 potentiostat or a custom made fast potentiostat41 controlled by an AMEL Mod. 568 programmable function generator.
Laser desorption-ionization time of flight mass spectra (LDITOF-MS)
Laser Desorption-Ionization Time of Flight Mass Spectra (LDITOF-MS) were obtained by an Applied Biosystems/MDS SCIEX 4800 MALDI TOF/TOF™ Analyzer, equipped with a Nd:YAG laser at 355 nm wavelength.SEM and TEM microscopy
Scanning Electron Microscopy (SEM) observations were carried out by a Zeiss Supra 40 field emission scanning electron microscope (SEM) equipped with a Bruker Z200 EDX microanalysis.Transmission Electron Microscopy (TEM) analysis was performed by using a Philips CM200 microscope operating at 200 kV and equipped with a LaB6 filament. Samples for TEM observations were prepared by depositing the polycorannulene film on a commercial TEM grid covered with a thin carbon film following the procedures as reported in the ESI.†
All details concerning other characterization methods used in this work are reported in the ESI.†
Computational details
Ab initio and DFT molecular orbital calculations were performed using the Firefly Quantum Chemistry package,42 fully optimizing at the B3LYP/6-31G* level the geometries of the molecules involved in the redox processes and checking the hessian to ensure the nature of stationary point. For the optimized structures, the vertical ionization potential (IPv) and the vertical electron affinity (EAv) were calculated not only at the B3LYP/6-31G* level but also at the B3LYP/cc-pvtz level of theory. Aiming at theoretical evaluation of redox potentials for the various species, the IP and EA values were combined with the solvation free energies using the thermodynamic cycle proposed by Cramer and Truhlar.43Author contributions
C. B., E. U. and M. Marcaccio performed, optimized and analysed the electrochemical, UV-Vis-NIR spectroscopy and AFM experiments; E. A. J. performed the chemical synthesis of corannulene; D. V. and C. F. performed the quantum mechanical calculation; G. B. carried out and analysed the SEM and TEM experiments; G. V. conducted and analysed the LDITOF-MS measurements; A. G. and L. P. performed and analysed the XPS and Near Edge X-ray experiments; L. L. and M. Meneghetti performed and analysed the FTIR and Raman spectroscopy experiments; S.F. performed and analysed the X-ray measurements. C. F., F. P., L. T. S. and M. Marcaccio planned and supervised the research, analysed the data and co-wrote the paper with contributions from other authors. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the University of Bologna, University of Modena and Reggio Emilia, Università Politecnica delle Marche, Università di Padova, United States National Science Foundation (CHE-1149096) and Department of Energy (DE-FG02-93ER14359), the Italian Ministero dell’Istruzione, Università e Ricerca – MIUR and INSTM. The authors would like to thank Dr Diego Pinetti of “Centro Interdipartimentale Grandi Strumenti” (CIGS) of Modena for helping with performing LDITOF mass spectra and prof. Fabio Zeppa for the graphical abstract artwork. The Fondazione CarisBo and ELETTRA (Sincrotrone, Trieste) are also gratefully acknowledged.Notes and references
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- O. V. Yazyev and Y. P. Chen, Nat. Nanotechnol., 2014, 9, 755–767 CrossRef CAS PubMed.
- F. Banhart, J. Kotakoski and A. V. Krasheninnikov, ACS Nano, 2011, 5, 26–41 CrossRef CAS PubMed.
- X. Gao, Y. Wang, X. Liu, T.-L. Chan, S. Irle, Y. Zhao and S. B. Zhang, Phys. Chem. Chem. Phys., 2011, 13, 19449–19453 RSC.
- A. W. Tsen, A. W. Tsen, L. Brown, M. P. Levendorf, F. Ghahari, P. Y. Huang, R. W. Havener, C. S. Ruiz-Vargas, D. A. Muller, P. Kim and J. Park, Science, 2012, 336, 1143–1146 CrossRef CAS PubMed.
- S. Deng and V. Berry, Mater. Today, 2016, 19, 197–212 CrossRef CAS.
- W. Liu, L. Zhao, E. Zurek, J. Xia, Y.-H. Zheng, H.-Q. Lin, J.-Y. Liu and M.-S. Miao, npj Comput. Mater., 2019, 5, 71 CrossRef.
- B. Pacakova, T. Verhagen, M. Bousa, U. Hübner, J. Vejpravova, M. Kalbac and O. Frank, Sci. Rep., 2017, 7, 10003 CrossRef PubMed.
- L. T. Scott, H. E. Bronstein, D. V. Preda, R. B. M. Ansems, M. S. Bratcher and S. Hagen, Pure Appl. Chem., 1999, 71, 209–219 CAS.
- Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS PubMed.
- P. W. Rabideau and A. Sygula, Acc. Chem. Res., 1996, 29, 235–242 CrossRef CAS.
- V. M. Tsefrikas and L. T. Scott, Chem. Rev., 2006, 106, 4868–4884 CrossRef CAS PubMed.
- L. T. Scott, M. M. Hashemi, D. T. Meyer and H. B. Warren, J. Am. Chem. Soc., 1991, 113, 7082–7084 CrossRef CAS.
- A. M. Butterfield, B. Gilomen and J. S. Siegel, Org. Process Res. Dev., 2012, 16, 664–676 CrossRef CAS.
- J. Janata, J. Gendell, C.-Y. Ling, W. Barth, L. Backes, H. B. Mark Jr and R. G. Lawton, J. Am. Chem. Soc., 1967, 89, 3056–3058 CrossRef CAS.
- M. Baumgarten, L. Gherghel, M. Wagner, A. Weitz, M. Rabinovitz, P.-C. Cheng and L. T. Scott, J. Am. Chem. Soc., 1995, 117, 6254–6257 CrossRef CAS.
- T. J. Seiders, K. K. Baldridge, J. S. Siegel and R. Gleiter, Tetrahedron Lett., 2000, 41, 4519–4522 CrossRef CAS.
- A. V. Zabula, A. Filatov, S. N. Spisak, A. Rogachev and M. Petrukhina, Science, 2011, 333, 1008–1011 CrossRef CAS PubMed.
- C. Bruno, R. Benassi, A. Passalacqua, F. Paolucci, C. Fontanesi, M. Marcaccio, E. A. Jackson and L. T. Scott, J. Phys. Chem. B, 2009, 113, 1954–1962 CrossRef CAS PubMed.
- A. Ayalon, A. Sygula, P.-C. Cheng, M. Rabinovitz, P. W. Rabideau and L. T. Scott, Science, 1994, 265, 1065–1067 CrossRef CAS PubMed.
- Y.-T. Wu, D. Bandera, R. Maag, A. Linden, K. K. Baldridge and J. S. Siegel, J. Am. Chem. Soc., 2008, 130, 10729–10739 CrossRef CAS PubMed.
- J. Mack, P. Vogel, D. Jones, N. Kaval and A. Sutton, Org. Biomol. Chem., 2007, 5, 2448–2452 RSC.
- E. Nestoros and M. C. Stuparu, Chem. Commun., 2018, 54, 6503–6519 RSC.
- G. Valenti, C. Bruno, S. Rapino, A. Fiorani, E. A. Jackson, L. T. Scott, F. Paolucci and M. Marcaccio, J. Phys. Chem. C, 2010, 114, 19467–19472 CrossRef CAS.
- M. Treier, C. A. Pignedoli, T. Laino, R. Rieger, K. Müllen, D. Passerone and R. Fasel, Nat. Chem., 2011, 3, 61–67 CrossRef CAS PubMed.
- K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739–744 CrossRef CAS PubMed.
- E. M. Muzammil, D. Halilovic and M. C. Stuparu, Commun. Chem., 2019, 2, 58 CrossRef.
- J. G. Gaudiello, P. R. Sharp and A. J. Bard, J. Am. Chem. Soc., 1982, 104, 6373–6377 CrossRef CAS.
- A. J. Bard, and L. R. Faulkner, Electrochemical Methods. Fundamentals and Applications, II, Wiley, New York, 2001 Search PubMed.
- J. C. Bachman, R. Kavian, D. J. Graham, D. Y. Kim, S. Noda, D. G. Nocera, Y. Shao-Horn and S. W. Lee, Nat. Commun., 2015, 6, 7040 CrossRef PubMed.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS PubMed.
- L. Zhai, R. Shukla, S. H. Wadumethrige and R. Rathore, J. Org. Chem., 2010, 75, 4748–4760 CrossRef CAS PubMed.
- R. Le Parc, P. Hermet, S. Rols, D. Maurin, L. Alvarez, A. Ivanov, J. M. Quimby, C. G. Hanley, L. T. Scott and J.-L. Bantignies, J. Phys. Chem. C, 2012, 116, 25089–25096 CrossRef CAS.
- G. Rouillé, C. Jäger, M. Steglich, F. Huisken, T. Henning, G. Theumer, I. Bauer and H.-J. Knölker, ChemPhysChem, 2008, 9, 2085–2091 CrossRef PubMed.
- U. Coscia, M. Palomba, G. Ambrosone, G. Barucca, M. Cabibbo, P. Mengucci, R. De Asmundis and G. Carotenuto, Nanotechnology, 2017, 28, 194001 CrossRef CAS PubMed.
- M. R. Kennedy, L. A. Burns and C. D. Sherrill, J. Phys. Chem. A, 2012, 116, 11920–11926 CrossRef CAS PubMed.
- L. T. Scott, P.-C. Cheng, M. M. Hashemi, M. S. Bratcher, D. T. Meyer and H. B. Warren, J. Am. Chem. Soc., 1997, 119, 10963–10968 CrossRef CAS.
- A. A. La Pensée, J. Bickley, S. J. Higgins, M. Marcaccio, F. Paolucci, S. Roffia and J. M. Charnock, J. Chem. Soc., Dalton Trans., 2002, 4095–4104 RSC.
- G. Valenti, A. Fiorani, S. Di Motta, G. Bergamini, M. Gingras, P. Ceroni, F. Negri, F. Paolucci and M. Marcaccio, Chem. - Eur. J., 2015, 21, 2936–2947 CrossRef CAS PubMed.
- M. Marcaccio, F. Paolucci, C. Paradisi, M. Carano, S. Roffia, C. Fontanesi, L. J. Yellowlees, S. Serroni, S. Campagna and V. Balzani, J. Electroanal. Chem., 2002, 532, 99–112 CrossRef CAS.
- C. Amatore and C. Lefrou, J. Electroanal. Chem., 1992, 324, 33–58 CrossRef CAS.
- Alex A. Granovsky, Firefly version 7G, accessed November 2020, http://classic.chem.msu.su/gran/firefly/index.html Search PubMed.
- P. Winget, E. J. Weber, C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2000, 2, 1231–1239 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Contains further voltammetric, spectroscopy, quantum chemical calculations and AFM, SEM/TEM microscopy results, electrochemical mechanism discussion and experimental details. See DOI: 10.1039/d1sc00898f |
| This journal is © The Royal Society of Chemistry 2021 |