
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed asymmetric reductive cross-coupling of α-chloroesters with (hetero)aryl iodides†
Travis J.
DeLano
a,
Sara E.
Dibrell‡
 a,
Caitlin R.
Lacker‡
a,
Adam R.
Pancoast
b,
Kelsey E.
Poremba
a,
Leah
Cleary
a,
Matthew S.
Sigman
b and
Sarah E.
Reisman
*a
a,
Caitlin R.
Lacker‡
a,
Adam R.
Pancoast
b,
Kelsey E.
Poremba
a,
Leah
Cleary
a,
Matthew S.
Sigman
b and
Sarah E.
Reisman
*a
aThe Warren and Katharine Schlinger Laboratory for Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. E-mail: reisman@caltech.edu
bDepartment of Chemistry, University of Utah, 315 South 1400 East, Salt Lake City, Utah 84112, USA
First published on 26th April 2021
Abstract
An asymmetric reductive cross-coupling of α-chloroesters and (hetero)aryl iodides is reported. This nickel-catalyzed reaction proceeds with a chiral BiOX ligand under mild conditions, affording α-arylesters in good yields and enantioselectivities. The reaction is tolerant of a variety of functional groups, and the resulting products can be converted to pharmaceutically-relevant chiral building blocks. A multivariate linear regression model was developed to quantitatively relate the influence of the α-chloroester substrate and ligand on enantioselectivity.
Introduction
Carboxylic acid derivatives containing α-aryl stereogenic centers are useful synthetic building blocks and are found in a number of biologically active compounds, including non-steroidal anti-inflammatory drugs such as naproxen and ibuprofen (Fig. 1a). Often these compounds are synthesized in enantioenriched form by chiral resolution or through the use of chiral auxiliaries.1 In order to streamline the synthesis of such compounds, there has been significant effort aimed at the development of enantioselective transition metal-catalyzed enolate arylation reactions.2 A challenge of this approach is the need for a strong base, which can give rise to racemization of the newly formed stereocenter under the reaction conditions. To address this challenge, several teams have investigated the cross-coupling of α-halo carbonyl compounds with aryl nucleophiles using chiral Ni, Co, or Fe catalysts,3–5 which can proceed under mild conditions to give products with good levels of enantiomeric excess (Fig. 1b).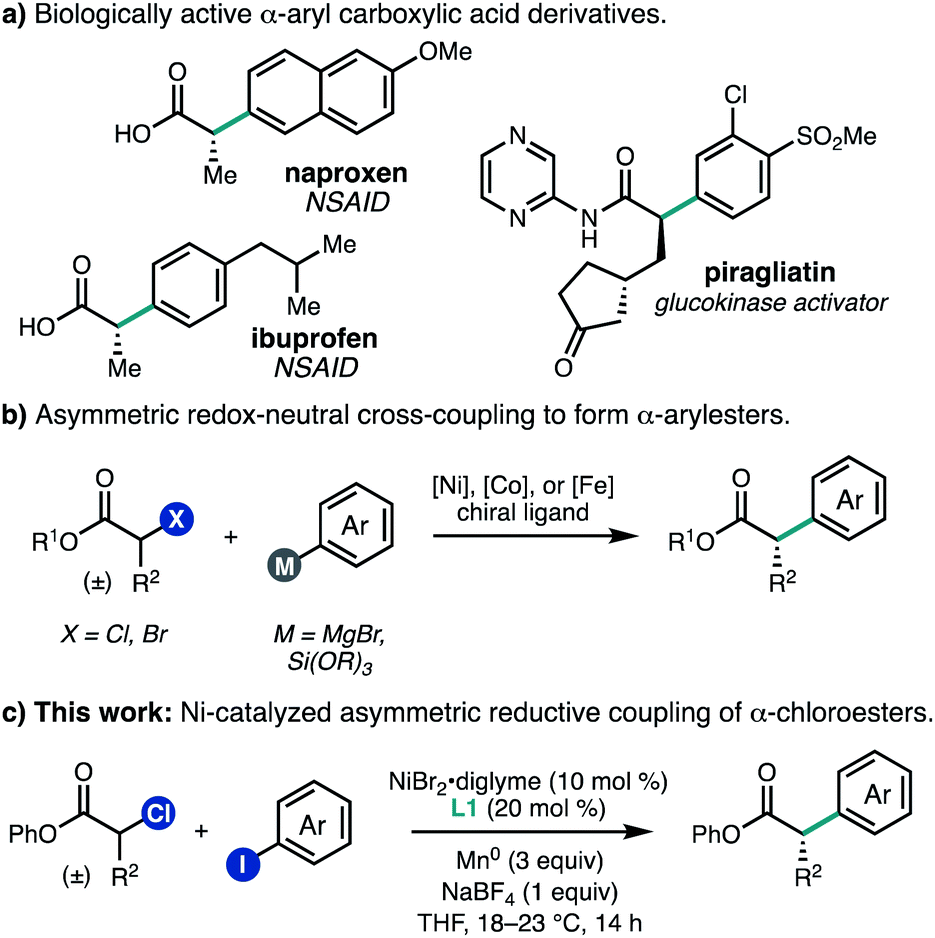 | ||
| Fig. 1 Enantioenriched α-aryl carboxylic acid derivatives. | ||
As an alternative approach to enantioenriched α-aryl carboxylic acids, we envisioned developing a Ni-catalyzed asymmetric reductive cross-coupling of α-chloroesters with (hetero)aryl iodides. Such cross-electrophile couplings have emerged as versatile methods for C(sp2)–C(sp3) bond formation. One advantage over traditional cross-coupling reactions is that no pre-generated organometallic reagents are required, which can improve the functional group tolerance. In this context, our laboratory has developed Ni-catalyzed enantioselective cross-electrophile couplings for a range of electrophile pairs.6 In the racemic sense, early studies by Durandetti and coworkers established that Ni catalyzes the reductive cross-coupling of α-chloroesters and aryl iodides using either Mn0 or electrochemical reduction to turn over the catalyst;7,8 however, the scope of investigations were limited to methyl 2-chloropropanoate and methyl 2-chloroacetate. As we were completing our own investigations,9 Mao, Walsh, and coworkers reported a Ni-catalyzed asymmetric coupling of α-chloroesters and aryl iodides using a metallaphotoredox approach.10 Here, we report the development of a nickel-catalyzed enantioselective reductive cross-coupling between α-chloroesters and a variety of aryl and heteroaryl iodides (Fig. 1c). This system performs particularly well for β-branched substrates, providing access to α-aryl carboxylic acid derivatives that are both difficult to prepare and underrepresented in reported methods. Additionally, multivariate linear regression (MLR) informs how steric matching between the ligand and substrate controls the enantioselectivity observed for the reaction.
Results and discussion
We began our study by investigating the coupling between phenyl 2-chloropropanoate (1a) and pyridyl iodide 2a. An initial evaluation of reaction parameters identified the BiOX family of ligands as most promising for this transformation, using NiBr2·diglyme as the Ni source, THF as the solvent, and Mn0 as the terminal reductant (Table 1, entry 1). The use of NaBF4 (1.0 equiv.) as an additive was found to be critical for the formation of 3a (entry 2).11 BiOX ligands with branched alkyl substituents were found to perform best, with 4-heptylBiOX (L1) giving the highest combination of yield and enantioselectivity (entries 3 and 4). Replacing the BiOX alkyl substituent with a phenyl (L4) resulted in loss of reactivity (entry 5).12 The ligand loading could be lowered with a modest reduction in both yield and ee (entries 6 and 7).|
|
|||
|---|---|---|---|
| Entry | Deviation from standard conditions | Yieldb (%) | eec (%) |
| a Reactions conducted in duplicate on 0.2 mmol scale. b Determined by 1H NMR analysis using 1,1,2,2-tetrachloroethane as an internal standard. c Determined by SFC using a chiral stationary phase. TDAE = tetrakis(dimethylamino)ethylene. | |||
| 1 | None | 92 | 86 |
| 2 | No NaBF4 | 0 | — |
| 3 | L2 instead of L1 | 62 | 76 |
| 4 | L3 instead of L1 | 89 | 70 |
| 5 | L4 instead of L1 | 0 | — |
| 6 | 10 mol% L1 | 81 | 83 |
| 7 | 12 mol% L1 | 83 | 85 |
| 8 | Zn0 instead of Mn0 | 29 | 81 |
| 9 | TDAE instead of Mn0 | 0 | — |
| 10 | DMA instead of THF | 67 | 84 |
| 11 | 1,4-Dioxane instead of THF | 0 | — |
| 12 | Methyl ester | 40 | 84 |
| 13 | t-Butyl ester | 20 | 89 |
| 14 | 1 equiv. 2a | 85 | 84 |
| 15 | 4 instead of 2a | 10 | 84 |
| 16 | 5 instead of 1a | 0 | — |
| 17 | No NiBr2·diglyme | 0 | — |
| 18 | No L1 | 0 | — |
| 19 | No Mn0 | 0 | — |
|
|||
Zn0 proved less effective than Mn0 as a reductant (entry 8) and use of TDAE failed to afford any of the desired product (entry 9). Whereas the reaction performed reasonably well in DMA, no reaction was observed in 1,4-dioxane, a solvent previously applied to other [L1·Ni]-catalyzed asymmetric reductive coupling reactions (entries 10 and 11).6d,e Use of the methyl or tert-butyl esters instead of the phenyl ester gave coupled product in similar enantioselectivity but reduced yields (entries 12 and 13). The amount of 2a could be reduced to 1.0 equiv. with only a slight decrease in the yield of 3a (entry 14). When pyridyl bromide 4 was employed instead of 2a, substantially lower yields of 3a were obtained (entry 15), while use of α-bromoester 5 failed to give any of the desired product and was recovered unreacted (entry 16). Control experiments confirmed that nickel, ligand, and Mn0 are all required for product formation (entries 17–19). We note that these conditions offer some potential advantages over the metallaphotoredox reductive coupling:10 (1) the use of only 1.5 equiv. of aryl halide coupling partner (vs. 3.0 equiv.); (2) the use of an easy-to-functionalize ester derived from inexpensive phenol (vs. 2,2,3-trimethylbutanol); (3) the use of an inexpensive terminal reductant (Mn0vs. Hantzsch ester); and (4) shorter reaction times (14 vs. 48 h).
To investigate the scope of the reaction, a series of aryl iodides were coupled with 1a under standard conditions (Fig. 2). The reaction tolerates both electron-rich (3g and 3k) and electron-poor (3c and 3e) aryl iodides, as well as heteroaryl iodides (3a, 3d, and 3j). Protected heteroatoms (3j and 3k) and an aryl chloride (3i) were tolerated, giving enantioenriched products poised for further elaboration. Whereas para- and meta-tolyl substrates 3g and 3h coupled efficiently, ortho-tolyl iodide resulted in significantly reduced yield and enantioselectivity.13 Analysis of this series of substrates suggests that the reaction is relatively insensitive to the electronic properties of the arene.
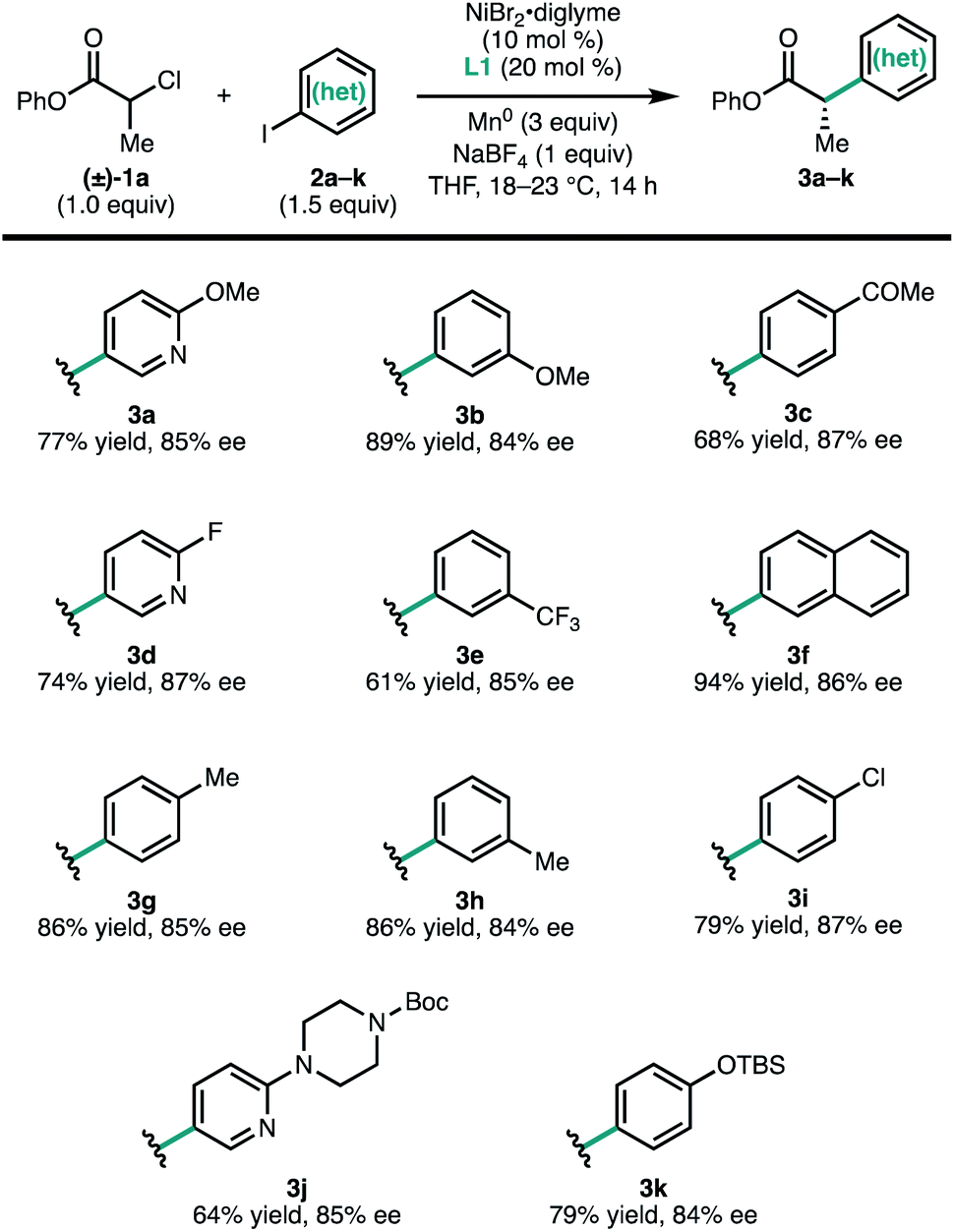 | ||
| Fig. 2 Scope of (hetero)aryl iodides. Reactions conducted on 0.2 mmol scale. Isolated yields are provided; ee was determined by SFC using a chiral stationary phase. | ||
In contrast to the aryl iodides, the enantioselectivity of the reaction was found to be quite sensitive to the structure of the α-chloroesters (Fig. 3). For a series of substrates where the α-substituent is changed from methyl (3a) to ethyl (6a) to iso-propyl (6b), the ee of the product increased from 85% to 88% to 96%, respectively. A similar trend was observed in the formation of 3b, 6c, and 6d. The α-iso-propyl chloroester 1c can be coupled with a variety of aryl iodides to give the corresponding α-arylesters in good yield and uniformly high ee (6b, 6d, 6e, 6k, and 6l). Similar yields and ee were obtained with the α-cyclopentyl and α-cyclohexyl substituents (6g and 6h). Using the α-chloroester derived from L-isoleucine (1f), either the S,S- or R,S-diastereomer (6i and 6j) could be obtained simply by changing the enantiomer of L1 that was used, demonstrating that products with vicinal stereogenic centers can be prepared with catalyst control over the configuration of the α-carbon. We note that, qualitatively, the increase in enantioselectivity moving from α-methyl to α-iso-propyl does not come at the expense of yield, in contrast to related transformations.6d,14 However, this trend did not hold true for the α-tert-butyl-α-chloroester: in this case, the cross-coupled product 6f was not observed when L1 was employed. By using L2, a ligand with a smaller steric profile, 6f could be formed in 16% yield and 89% ee. We note that in the related photoredox coupling reported by Walsh and Mao, the introduction of β-branching did not lead to higher enantioselectivity.10
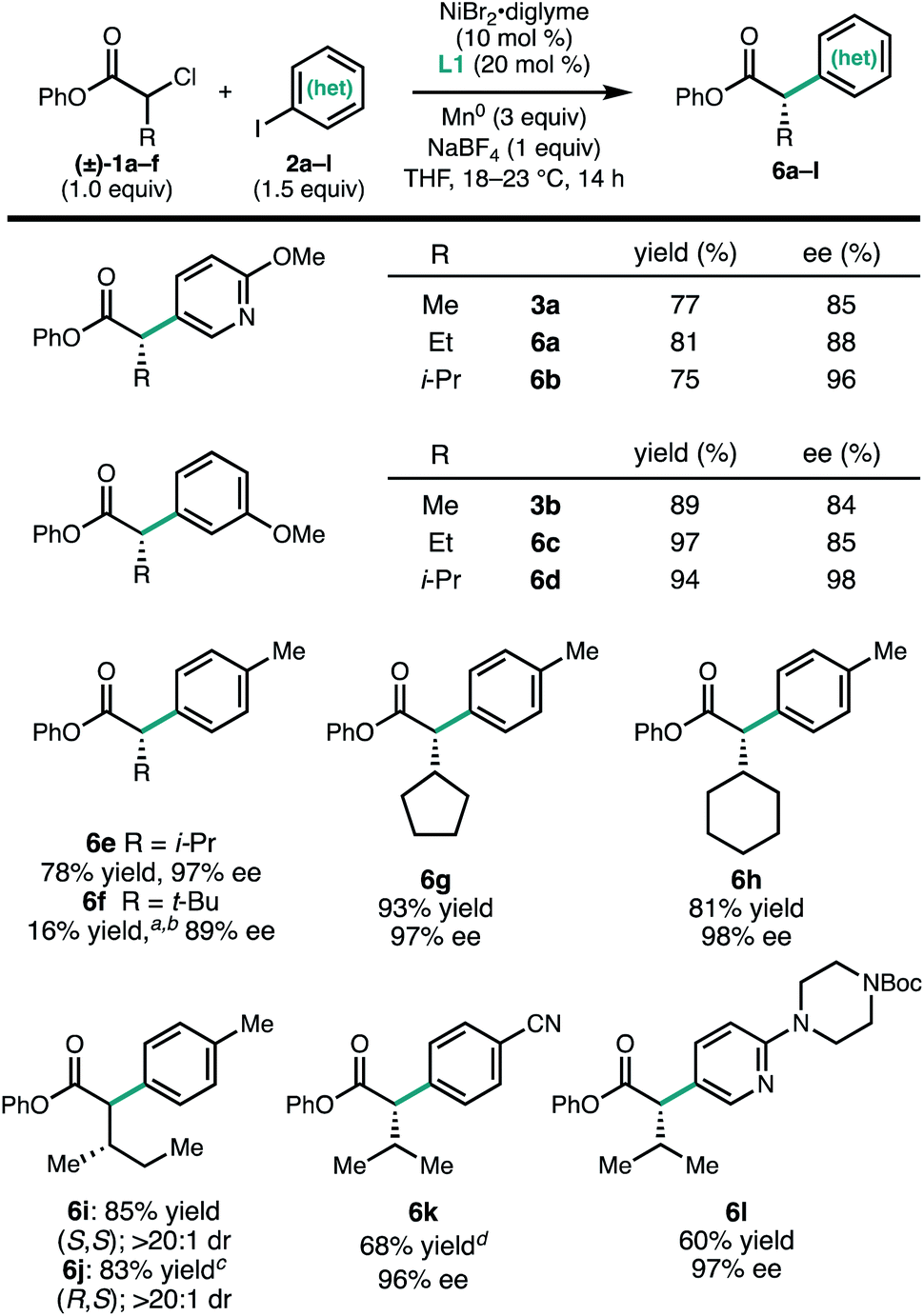 | ||
| Fig. 3 Scope of α-chloroesters. Reactions were conducted on 0.2 mmol scale. Isolated yields are provided; ee was determined by SFC using a chiral stationary phase. aDetermined by 1H NMR. bL2 was used. c(S,S)-L1 was used. dReaction time was 48 h. | ||
Given that the enantioselectivity improves as a function of the size of the α-substituent, we hypothesized that a synergistic interaction between the substrate and the ligand may be at play. In order to quantify this, we used statistical modelling of substrate/ligand features with the observed enantioselectivity by evaluating a matrix of six α-chloroesters with five BiOX ligands.15 Utilizing a workflow previously reported by one of our labs,16,17 conformers with a 2.4 kcal mol−1 energy range were identified via a conformational search using the OPLS3e force field (see ESI for details†).18 Each conformer was then submitted to DFT level geometry optimization, followed by single point energy calculations of the optimized structures at the M06-2x/def2-TZVP level of theory.19,20 Various molecular features, including Boltzmann-weighted descriptors, were acquired from these optimized structures.16 The ensuing library was then split into a training set (20 points) and a test set (5 points) by an automated process21 using a test ratio of 0.20. Using both the experimental ee (expressed as ln(er)) and the computationally derived molecular features, a forward stepwise linear regression algorithm was used to yield a statistical model (Fig. 4a).22
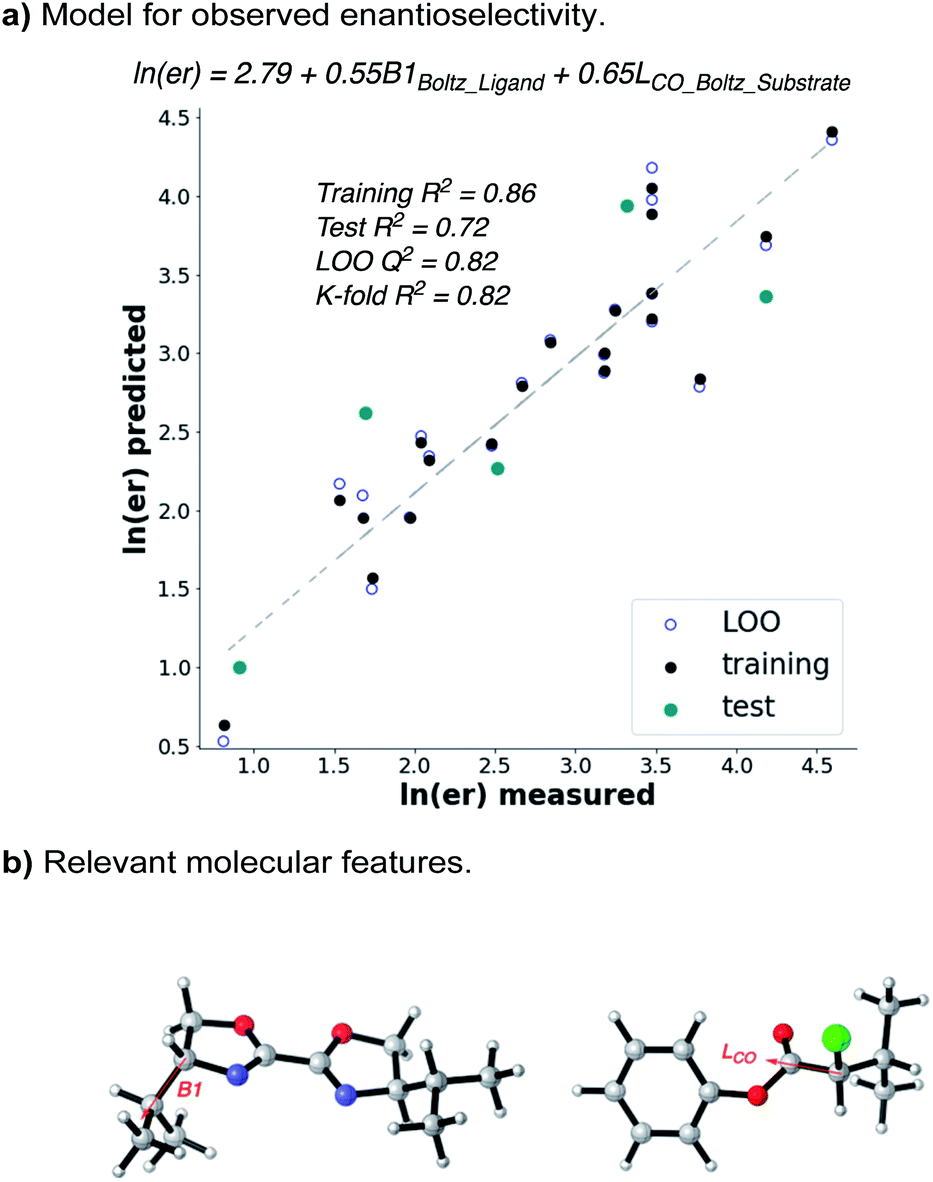 | ||
| Fig. 4 Multivariate linear regression shows that the ee depends on the size of both ligand and α-chloroester. | ||
The resulting statistical model reveals a clear correlation between the observed enantioselectivity and the Boltzmann-weighted minimum width (B1) of the ligand and the Boltzmann-weighted length (L, down the C–CO axis) of the substrate (Fig. 4b). The statistics of the model indicate a high level of accuracy (R2 = 0.86) and the model robustness is also high as indicated by cross-validations (leave-one-out (LOO) Q2 = 0.82 and K-fold = 0.82). The model indicates that steric matching between the catalyst and substrate is responsible for high selectivity, as evidenced by the fact that the ligand with the largest Boltzmann B1 value (L1) and the α-chloroester with the largest Boltzmann L value (6h) give the best selectivity while those with the smallest values give poorer selectivity. This simple model should be highly predictive if either a new catalyst or a new substrate is considered for application of this reaction.
To demonstrate the utility of this method to access pharmaceutically-relevant α-aryl carboxylic acids, we prepared the non-steroidal anti-inflammatory drug (S)-naproxen (9, Scheme 1). Coupling of α-chloroester 1a with napthyl iodide 7 under standard conditions afforded ester 8 in 93% yield and 84% ee on 1.0 mmol scale. Hydrolysis of the phenyl ester gave 9 in 83% ee. The ee of 9 could be further enriched to 92% by recrystallization as its octylammonium salt (76% recovery). This synthesis allowed the unambiguous assignment of the configuration of 8 as S.23
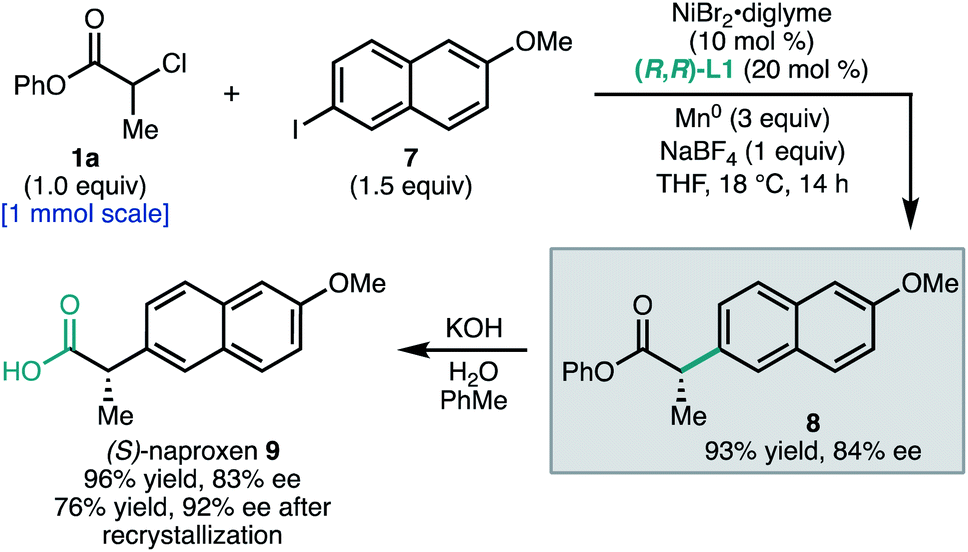 | ||
| Scheme 1 Product elaboration to naproxen. | ||
It is has become accepted that many Ni-catalyzed cross-coupling reactions of alkyl halides involve oxidative addition by a radical mechanism.24 However, α-chloroesters such as 1 could potentially react via an in situ-generated manganese enolate. To investigate this possibility, Mn enolate 10 was prepared and subjected to aryl iodide 2g under the standard reaction conditions; however, no product 3g was observed (Fig. 5a). Control experiments determined that the reductive cross-coupling could still proceed in 73% yield and 81% ee when the byproducts from Mn enolate formation were doped into the standard reaction (LiCl, hexanes, and iPr2NH).25 No consumption of 1a was observed under standard conditions in the absence of Ni and 2g. A stoichiometric experiment was carried out in which pre-complexed [L1·Ni0] was added dropwise to aryl iodide 2g; subsequent addition of chloroester 1a afforded 3g in 72% yield and comparable ee to the catalytic reaction (Fig. 5b).
 | ||
| Fig. 5 Mechanistic experiments. aDetermined by 1H NMR. | ||
Taken together, these experiments do not support the formation of a Mn enolate, but do indicate that product formation does not require reduction of [L1·NiIIArX].26 Similar studies were used by Weix and coworkers to implicate oxidative addition of the alkyl halide by a radical chain mechanism in the coupling between aryl halides and unactivated alkyl halides.27 Consistent with a possible radical-type oxidative addition, use of α-chloro-α-cyclopropylester 11 failed to give the desired product, but instead provided ring opened product 12 in 10% yield.13
Conclusions
In conclusion, we have developed a nickel-catalyzed asymmetric reductive cross-coupling of α-chloroesters and (hetero)aryl iodides. The transformation is enabled by a chiral BiOX ligand previously developed by our group, and forms α-arylated esters in good yields and enantioselectivities under mild conditions. These products are useful chiral building blocks with potential applications in pharmaceuticals, agrochemicals, and materials. The reaction proves especially selective when β-branched substrates are employed; a trend not observed in prior art from Walsh and Mao.10 An MLR model has been developed to quantitatively demonstrate the cooperative influence of the substrate and ligand steric profiles on enantioselectivity.Author contributions
S. E. R., L. C., K. E. P., and T. J. D. conceptualized the project. T. J. D., S. E. D., C. R. L., K. E. P., and L. C. carried out the experimental investigations. A. R. P. carried out the MLR modeling studies. M. S. S. supervised the modeling studies. S. E. R. supervised the experimental investigations. T. J. D., S. E. D., C. R. L., K. E. P., A. R. P., M. S. S. and S. E. R. wrote and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Scott Virgil and the Caltech Center for Catalysis and Chemical Synthesis are gratefully acknowledged for access to analytical equipment. We thank Yoshihiro Ogura for early studies and Raymond Turro for the preparation of L1. M. S. S. thanks the NIH (R35GM136271) for support. Fellowship support was provided by the NSF (T. J. D., S. E. D., C. R. L., K. E. P., Grant No. DGE-1144469). S. E. R. is a Heritage Medical Research Institute Investigator, and acknowledges financial support from the NIH (R35GM118191).Notes and references
- P. J. Harringto and E. Lodewijk, Org. Process Res. Dev., 1997, 1, 72 CrossRef.
- Y.-J. Hao, X.-S. Hu, Y. Zhoo, J. Zhao and J.-S. Yu, ACS Catal., 2020, 10, 955 CrossRef CAS.
- X. Dai, N. A. Strotman and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 3302 CrossRef CAS PubMed.
- J. Mao, F. Liu, M. Wang, L. Wu, B. Zheng, S. Liu, J. Zhong, Q. Bian and P. J. Walsh, J. Am. Chem. Soc., 2014, 136, 17662 CrossRef CAS PubMed.
- M. Jin, L. Adak and M. Nakamura, J. Am. Chem. Soc., 2015, 137, 7128 CrossRef CAS PubMed.
- (a) K. E. Poremba, S. E. Dibrell and S. E. Reisman, ACS Catal., 2020, 10, 8237 CrossRef CAS PubMed; (b) A. H. Cherney, N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 7442 CrossRef CAS PubMed; (c) A. H. Cherney and S. E. Reisman, J. Am. Chem. Soc., 2014, 136, 14365 CrossRef CAS; (d) N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2015, 137, 10480 CrossRef CAS PubMed; (e) K. E. Poremba, N. T. Kadunce, N. Suzuki, A. H. Cherney and S. E. Reisman, J. Am. Chem. Soc., 2017, 139, 5684 CrossRef CAS PubMed; (f) T. J. DeLano and S. E. Reisman, ACS Catal., 2019, 9, 6751 CrossRef CAS PubMed.
- M. Durandetti, C. Gosmini and J. Périchon, Tetrahedron, 2007, 63, 1146 CrossRef CAS.
- (a) M. Durandetti, J.-Y. Nédélec and J. Périchon, J. Org. Chem., 1996, 61, 1748 CrossRef CAS; (b) M. Durandetti and J. Périchon, Synthesis, 2004, 18, 3079 CrossRef.
- Disclosure of this study was delayed by the COVID-19 pandemic. The use of L1 for this transformation was disclosed in 2019 in the following thesis: K. E. Poremba, Development of Nickel-Catalyzed Asymmetric Reductive Cross-Coupling Reactions, PhD Thesis, California Institute of Technology, Pasadena, CA, 2019, https://resolver.caltech.edu/CaltechTHESIS:05222019-135022700.
- H. Guan, Q. Zhang, P. J. Walsh and J. Mao, Angew. Chem., Int. Ed., 2020, 59, 5172 CrossRef CAS.
- G. A. Molander, K. M. Traister and B. T. O'Neill, J. Org. Chem., 2014, 79, 5771 CrossRef CAS.
- Whereas 5-iodo-2-methoxypyridine failed to give 3a when L4 was used, 1-iodo-4-methylbenzene could be coupled in 41% yield and 80% ee with L4 as the ligand.
- See ESI.†.
- J. L. Hofstra, A. H. Cherney, C. M. Ordner and S. E. Reisman, J. Am. Chem. Soc., 2018, 140, 139 CrossRef CAS PubMed.
- A. Verloop, W. Hoogenstraaten and J. Tipker, Development and Application of New Steric Substituent Parameters in Drug Design, Academic Press, New York, 1976, vol. VII Search PubMed.
- M. S. Sigman, K. C. Harper, E. N. Bess and A. Milo, Acc. Chem. Res., 2016, 49, 1292 CrossRef CAS PubMed.
- C. B. Santiago, J.-Y. Guo and M. S. Sigman, Chem. Sci., 2018, 9, 2398 RSC.
- K. Roos, C. Wu, W. Damm, M. Reboul, J. M. Stevenson, C. Lu, M. K. Dahlgren, S. Mondal, W. Chen, L. Wang, R. Abel, R. A. Friesner and E. D. Harder, J. Chem. J. Chem. Theory Comput., 2019, 15, 1863 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- The y equidistant method of splitting the data into training and test sets was chosen based on the observation that this method resulted in the best spread of data for the test set. This method chooses points that evenly span the experimental output of the dataset. However, other methods of splitting the data resulted in models with similar statistics. The reader is directed to Fig. S4 in the ESI† for examples of said models.
- J.-Y. Guo, Y. Minko, C. B. Santiago and M. S. Sigman, ACS Catal., 2017, 7, 4144 CrossRef CAS.
- Assignment of configuration of the products in Fig. 2–4 are made in analogy based on the configuration of product 9.
- J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 2, 1411 RSC.
- The reactivity of the Mn enolate was confirmed in a standard alkylation reaction. See ESI.†.
- Since [L1·NiIIArX] could not be cleanly isolated and independently subjected to 1a, we cannot rule out radical generation by NiI species formed through a comproportionation mechanism.
- S. Biswas and D. J. Weix, J. Am. Chem. Soc., 2013, 135, 16192 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00822f |
| ‡ These authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2021 |