
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrategic molecular design of closo-ortho-carboranyl luminophores to manifest thermally activated delayed fluorescence†
Dong Kyun
You‡
,
Hyunhee
So‡
,
Chan Hee
Ryu
,
Mingi
Kim
and
Kang Mun
Lee
 *
*
Department of Chemistry, Institute for Molecular Science and Fusion Technology, Kangwon National University, Chuncheon, Gangwon 24341, Republic of Korea. E-mail: kangmunlee@kangwon.ac.kr
First published on 11th May 2021
Abstract
In this paper, we propose a strategic molecular design of closo-o-carborane-based donor–acceptor dyad system that exhibits thermally activated delayed fluorescence (TADF) in the solution state at ambient temperature. Planar 9,9-dimethyl-9H-fluorene-based compounds with closo- and nido-o-carborane cages appended at the C2-, C3-, and C4-positions of each fluorene moiety (closo-type: 2FC, 3FC, 4FC, and 4FCH, and nido-type: nido-4FC = [nido-form of 4FC]·[NBu4]) were prepared and characterized. The solid-state molecular structure of 4FC exhibited a significantly distorted fluorene plane, which suggests the existence of severe intramolecular steric hindrance. In photoluminescence measurements, 4FC exhibits a noticeable intramolecular charge transition (ICT)-based emission in all states (solution at 298 K and 77 K, and solid states); however, emissions by other closo-compounds were observed in only the rigid state (solution at 77 K and film). Furthermore, nido-4FC did not exhibit emissive traces in any state. These observations verify that all radiative decay processes correspond to ICT transitions triggered by closo-o-carborane, which acts as an electron acceptor. Relative energy barriers calculated by TD-DFT as dihedral angles around o-carborane cages change in closo-compounds, which indicates that the structural formation of 4FC is nearly fixed around its S0-optimized structure. This differs from that for other closo-compounds, wherein the free rotation of their o-carborane cages occurs easily at ambient temperature. Such rigidity in the structural geometry of 4FC results in ICT-based emission in solution at 298 K and enhancement of quantum efficiency and radiative decay constants compared to those for other closo-compounds. Furthermore, 4FC displays short-lived (∼0.5 ns) and long-lived (∼30 ns) PL decay components in solution at 298 K and in the film state, respectively, which can be attributed to prompt fluorescence and TADF, respectively. The calculated energy difference (ΔEST) between the first excited singlet and triplet states of the closo-compounds demonstrate that the TADF characteristic of 4FC originates from a significantly small ΔEST maintained by the rigid structural fixation around its S0-optimized structure. Furthermore, the strategic molecular design of the o-carborane-appended π-conjugated (D–A) system, which forms a rigid geometry due to severe intramolecular steric hindrance, can enhance the radiative efficiency for ICT-based emission and trigger the TADF nature.
Introduction
Recently, the icosahedral closo-ortho-carborane (C2B10H12) cluster has gained popularity as an attractive functional component1–4 because it can be considered as a stereoscopic analogue of a benzene ring owing to its high chemical stability and unique electronic characteristic. Furthermore, these properties lead to its extensive use as a three-dimensional unit for novel materials in the field of boron neutron capture therapy (BNCT),2,5–7 building block-ligands of 60° vertex positioning,8–14 and promising optoelectronic devices.15–17 In particular, its strong electron withdrawing property, which originates from the high polarizability of its σ-aromaticity18–22via the high Lewis acidity of ten boron atoms in a cluster, provides the necessary impetus to reveal its intriguing photophysical properties when assembled with π-aromatic conjugated moieties.3,4,15,23–59 These features of the o-carborane induce intramolecular charge transfer (ICT) transitions to form a donor (D)–acceptor (A) dyad system with aromatic groups, wherein o-carborane dominates orbital occupation in the excited state and plays the role of an electron acceptor.23–45 Furthermore, the o-carborane-appended D–A dyad compound exhibits specific emissive behavior during electronic relaxation based on ICT transition. Recent reports demonstrated that the ICT-induced emission is significantly affected by structural variations and molecular geometry. Fox et al. showed that the D–A dyad, C-diazaboryl-o-carborane, exhibits two different excited states (locally excited (LE) and ICT transition states), depending on the rotational motion of diazaboryl derivatives;26 this phenomenon is called the twisted intramolecular charge-transfer (TICT) effect.46–56 The occupation of the lowest unoccupied molecular orbital (LUMO) levels for an o-carborane cage is dramatically altered from 36% to 98% in accordance with the motion. This indicates that the electronic separation between the donor and acceptor moieties is governed by structural motion. Furthermore, our group reported that the structure of the molecular scaffold (i.e., the planarity of the π-aromatic groups directly appended to the o-carborane cages) played a dominant role in efficiently isolating the highest occupied molecular orbital (HOMO; occupied on the π-aromatic group) and LUMO (occupied on o-carborane) levels in the excited states; this helped further enhance the radiative decay of the ICT transition.57–59These results are the distinct inspiration to verify that the o-carborane-appended π-aromatic luminophore exhibits thermally activated delayed fluorescence (TADF).60–65 This is because most closo-o-carboranyl-based luminophores possess minimum orbital overlap between HOMO and LUMO levels in the excited states, which is essential for inducing the narrow energy difference between the first-exited singlet (S1) and triplet (T1) states and for manifesting TADF.66–69 Indeed, carbazole-based o-carborane D–A-type dyad derivatives17 and Ag(I)-cyclized o-carborane complexes70 simultaneously possess aggregation-induced emission (AIE) and strong TADF capabilities with high photoluminescence quantum efficiencies in the solid state. However, the TADF characteristics of ICT-based emissions can be observed only in rigid (solid or at low temperature) states because the structural fluctuations of closo-o-carborane (such as C–C bond elongation and the rotational motion of the cages) severely interrupts the radiative decay process based on the ICT transition in the solution state at ambient temperature.23–26,46,48,50,71–73 All previous reports suggest that the strategic molecular design for the D–A-type o-carboranyl appended π-conjugation system that retains its structural rigidity sufficiently under ambient conditions leads to efficient TADF nature in solution at room temperature. Thus far, to the best of our knowledge, there are no reported studies on closo-o-carboranyl luminophores that exhibit a moderate TADF phenomenon in the solution state.
The effects of structural features and molecular geometry on the photophysical properties of o-carboranyl D–A conjugated systems have been previously reported. These studies provide strong evidence that emissive characteristics can be controlled via strategic molecular design. Specifically, a unique structural formation of appended π-aromatic groups on the o-carborane shows promise. By extension, previous findings demonstrated the promise of these conjugate systems as novel candidates for single-molecule visual sensing materials with a prompt emissive response.
To further examine this intrinsic concept to determine the relationship between the structural rigidity of closo-o-carboranyl luminophores and TADF, we strategically designed and synthesized planar fluorene-based o-carborane compounds (where biphenyl rings are anchored by a C(CH3)2 group) appended at the C2-(2FC), C3-(3FC), and C4-(4FC) positions of each fluorene moiety. Furthermore, a nido-o-carborane-type compound (nido-4FC) and its derivative (4FCH) that possesses a higher fluctuating geometry than that of 4FC were prepared for comparing structural features and photophysical properties. Interestingly, the most rigid compound (4FC) exhibited moderate quantum efficiency and TADF nature in the solution state because of the small energy gap between the S1 and T1 states. This provides an insight into the strategic directivity of using closo-o-carboranyl conjugates as TADF materials. The detailed synthetic procedures, characterization, and optical properties (with theoretical calculations) have been described in this paper.
Experimental
General considerations
All operations were performed in an inert nitrogen atmosphere using standard Schlenk and glove box techniques. Anhydrous solvents (trimethylamine (NEt3), toluene, tetrahydrofuran (THF), and dichloromethane (DCM); Sigma-Aldrich) were dried by passing them through an activated alumina column and stored over activated molecular sieves (5 Å). Spectrophotometric-grade solvents (methanol, ethyl acetate, and n-hexane) were used as received from Alfa Aesar. Commercial reagents purchased from Sigma-Aldrich (9,9-dimethyl-9H-fluorene, 2-bromo-9,9-dimethyl-9H-fluorene, 3-bromo-9,9-dimethyl-9H-fluorene, 4-bromo-9,9-dimethyl-9H-fluorene, phenylacetylene, ethynyltrimethylsilane, n-tetrabutylammonium fluoride, bis(triphenylphosphine)palladium(II) dichloride (Pd(PPh3)2Cl2), copper(I) iodide (CuI), potassium carbonate (K2CO3), magnesium sulfate (MgSO4), diethyl sulfide (Et2S), and poly(methyl methacrylate) (PMMA)) and Alfa Aesar (decaborane (B10H14)) were used without any further purification. Dichloromethane-d2 (CD2Cl2) purchased from Cambridge Isotope Laboratories was dried over activated molecular sieves (5 Å). All NMR spectra were recorded using a Bruker Avance 400 spectrometer (400.13 MHz for 1H and 1H{11B}, 100.62 MHz for 13C, and 128.38 MHz for 11B{1H}) at ambient temperature. Chemical shifts are expressed in ppm and referenced against external Me4Si (1H and 13C) or BF3·Et2O (11B). Elemental analyses were performed on an EA3000 instrument (EuroVector) at the Central Laboratory of Kangwon National University, respectively.General synthetic routes for acetylene derivatives
All acetylene derivatives (2FA, 3FA, 4FA, and 4FASi) were synthesized by following the scheme with an adequate amount of starting materials. Toluene (10 mL) and NEt3 (10 mL) were added via a cannula to the mixture of bromo-fluorene precursors, CuI, and Pd(PPh3)2Cl2 at 298 K. After stirring for 15 min, phenylacetylene (for 2FA, 3FA, and 4FA) or ethynyltrimethylsilane (for 4FASi) of 2 equivalents per bromo-precursor was added to the resulting dark brown slurry. Then, the reaction mixture was refluxed for 24 h. Volatiles were removed with a rotary evaporator affording a dark gray residue. After washing with MeOH followed by n-hexane, the crude product was purified by column chromatography on silica gel ((eluent: DCM/n-hexane (1/20, v/v)) to afford acetylene derivatives.Data for 9,9-dimethyl-2-(phenylethynyl)-9H-fluorene, 2FA
2-Bromo-9,9-dimethyl-9H-fluorene (1.1 g, 4.0 mmol), CuI (76 mg), Pd(PPh3)2Cl2 (280 mg), and phenylacetylene (0.88 mL, 8.0 mmol) afforded 2FA (0.80 g, 2.7 mmol) as a white solid that was purified by column chromatography on silica gel. Yield = 68%. 1H NMR (CD2Cl2): δ 7.75 (m, 1H), 7.73 (d, J = 8.1 Hz, 1H), 7.64 (s, 1H), 7.57 (m, 1H), 7.56 (d, J = 2.4 Hz, 1H), 7.55 (dd, J = 11.8, 4.0 Hz, 1H), 7.47 (m, 1H), 7.37 (m, 5H), 1.51 (s, 6H, –CH3). 13C NMR (CD2Cl2): δ 154.42, 154.20, 139.93, 138.85, 131.89, 131.02, 128.85, 128.65, 128.17, 127.53, 126.35, 123.79, 123.11, 122.04, 120.67, 120.37, 90.53 (acetylene-C), 89.68 (acetylene-C), 47.25 (–C(CH3)2), 27.22 (–CH3). Anal. calcd for C23H18: C, 93.84; H, 6.16. Found: C, 93.55; H, 5.80.Data for 9,9-dimethyl-3-(phenylethynyl)-9H-fluorene, 3FA
3-Bromo-9,9-dimethyl-9H-fluorene (0.52 g, 1.9 mmol), CuI (36 mg), Pd(PPh3)2Cl2 (130 mg), and phenylacetylene (0.42 mL, 3.8 mmol) afforded 3FA (0.40 g, 1.4 mmol) as a pale yellow oil that was purified by column chromatography on silica gel. Yield = 72%. 1H NMR (CD2Cl2): δ 7.97 (s, 1H), 7.80 (d, J = 8.2 Hz, 1H), 7.63 (d, J = 8.1 Hz, 2H), 7.56 (d, J = 7.9 Hz, 1H), 7.49 (t, J = 8.4 Hz, 2H), 7.41 (m, 5H), 1.53 (s, 6H, –CH3). 13C NMR (CD2Cl2): δ 154.41, 154.15, 139.94, 138.73, 131.97, 131.12, 128.88, 128.69, 128.20, 127.55, 123.81, 123.43, 123.19, 123.12, 122.18, 120.59, 90.25 (acetylene-C), 89.14 (acetylene-C), 47.36 (–C(CH3)2), 27.18 (–CH3). Anal. calcd for C23H18: C, 93.84; H, 6.16. Found: C, 93.49; H, 5.90.Data for 9,9-dimethyl-3-(phenylethynyl)-9H-fluorene, 4FA
4-Bromo-9,9-dimethyl-9H-fluorene (0.55 g, 2.0 mmol), CuI (38 mg), Pd(PPh3)2Cl2 (140 mg), and phenylacetylene (0.44 mL, 4.0 mmol) afforded 4FA (0.38 g, 1.3 mmol) as a pale yellow oil that was purified by column chromatography on silica gel. Yield = 65%. 1H NMR (CD2Cl2): δ 8.67 (d, J = 8.8 Hz, 1H), 7.74 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.0 Hz, 1H), 7.53 (d, J = 7.8 Hz, 1H), 7.47 (m, 6H), 7.36 (t, J = 7.6 Hz, 1H), 1.54 (s, 6H, –CH3). 13C NMR (CD2Cl2): δ 154.69, 154.43, 139.47, 139.13, 131.92, 131.72, 128.99, 128.94, 128.17, 127.42, 127.23, 123.77, 123.18, 123.15, 122.79, 116.77, 93.86 (acetylene-C), 88.95 (acetylene-C), 46.69 (–C(CH3)2), 27.29 (–CH3). Anal. calcd for C23H18: C, 93.84; H, 6.16. Found: C, 93.51; H, 5.92.Data for ((9,9-dimethyl-9H-fluoren-4-yl)ethynyl)trimethylsilane, 4FASi
4-Bromo-9,9-dimethyl-9H-fluorene (0.56 g, 1.9 mmol), CuI (37 mg), Pd(PPh3)2Cl2 (135 mg), and ethynyltrimethylsilane (0.53 mL, 3.8 mmol) afforded 4FA (0.19 g, 0.66 mmol) as a white solid that was purified by column chromatography on silica gel. Yield = 34%. 1H NMR (CD2Cl2): δ 8.59 (t, J = 4.8 Hz, 1H), 7.49 (d, J = 8.2 Hz, 1H), 7.46 (dd, J = 7.8, 1.8 Hz, 2H), 7.39 (t, J = 7.8 Hz, 2H), 7.29 (t, J = 8.2 Hz, 1H), 1.48 (s, 6H, –CH3), 0.40 (s, 9H, –Si(CH3)3). 13C NMR (CD2Cl2): δ 154.64, 154.44, 139.68, 139.03, 131.95, 128.20, 127.18, 127.11, 123.38, 123.30, 122.72, 116.71, 104.56 (acetylene-C), 99.52 (acetylene-C), 46.58 (–C(CH3)2), 27.29 (–CH3), 0.06 (–Si(CH3)3). Anal. calcd for C20H22Si: C, 82.70; H, 7.63. Found: C, 82.60; H, 7.55.Synthesis of 4-ethynyl-9,9-dimethyl-9H-fluorene, 4FAH
K2CO3 (0.18 g, 1.3 mmol) was dissolved in methanol (7 mL) and added to a solution of 4FASi (0.19 g, 0.66 mmol) in DCM (3 mL). After stirring for 2 h at 25 °C, the resulting mixture was treated with DCM (30 mL), and the organic layer was separated. The aqueous layer was further extracted with DCM (20 × 2 mL). The combined organic extracts were dried over MgSO4, filtered, and evaporated to dryness to afford a white residue. The crude product was purified by washing with methanol (10 mL) to yield 4FAH as a purple oil, 0.13 g (0.58 mmol). Yield = 88%. 1H NMR (CD2Cl2): δ 8.58 (d, J = 8.0 Hz, m, 1H), 7.50 (dd, J = 4.7, 2.3 Hz, 3H), 7.41 (t, J = 4.8 Hz, 2H), 7.31 (t, J = 7.6 Hz, 1H), 3.59 (s, 1H, –CCH), 1.50 (s, 6H, –CH3). 13C NMR (CD2Cl2): δ 154.69, 154.39, 139.92, 138.85, 132.48, 128.29, 127.35, 127.14, 123.67, 123.21, 122.77, 115.60, 83.01 (acetylene-C), 82.15 (acetylene-C), 46.60 (–C(CH3)2), 27.27 (–CH3). Anal. calcd for C17H14: C, 93.54; H, 6.46. Found: C, 93.24; H, 6.20.General synthetic routes for o-carborane compounds (2FC, 3FC, 4FC, and 4FCH)
An excess amount of Et2S (3.0 equivalent of an acetylene precursor) was added to a toluene solution (12 mL) of decaborane (B10H14, 1.3 equivalent per acetylene derivative) and acetylene precursors (2FA, 3FA, 4FA, and 4FAH) at 298 K. After heating to reflux, the reaction mixture was further stirred for 24 h. The solvent was removed under vacuum. The crude product was purified by column chromatography on basic aluminum oxide (eluent: DCM/n-hexane (1/20, v/v)) and by washing with n-hexane (20 mL) to afford fluorene-based o-carborane compounds as a white solid.Data for 2FC
Decaborane (0.16 g, 1.3 mmol), Et2S (0.28 mL, 11 mmol), and 2FA (0.29 g, 1.0 mmol) in toluene afforded 2FC as a white solid. Recrystallization from a mixture of DCM/n-hexane afforded 0.17 g (0.40 mmol) of 2FC. Yield = 40%. 1H{11B} NMR (CD2Cl2): δ 7.66 (s, 1H), 7.53 (s, 2H), 7.48 (d, J = 8.1 Hz, 2H), 7.39 (s, 2H), 7.33 (s, 2H), 7.19 (d, J = 7.9 Hz, 1H), 7.12 (t, J = 8.2 Hz, 2H), 3.37 (br s, 2H, CB-BH), 2.65 (br s, 2H, CB-BH), 2.57 (br s, 4H, CB-BH), 2.38 (br s, 2H, CB-BH), 1.29 (s, 6H, –CH3). 13C NMR (CD2Cl2): δ 154.61, 153.83, 141.52, 137.87, 131.10, 131.00, 130.71, 130.61, 129.56, 128.64, 127.55, 125.01, 123.08, 120.96, 119.96, 86.65 (CB–C), 86.04 (CB–C), 47.18 (–C(CH3)2), 26.86 (–CH3). 11B{1H} NMR (CD2Cl2): δ −2.63 (br s, 2B), −9.18 (br s, 3B), −10.61 (br s, 5B). Anal. calcd for C23H28B10: C, 66.96; H, 6.84. Found: C, 66.88; H, 6.70.Data for 3FC
Decaborane (0.22 g, 1.8 mmol), Et2S (0.39 mL, 3.6 mmol), and 3FA (0.40 g, 1.4 mmol) in toluene afforded 3FC as a white solid. Recrystallization from a mixture of DCM/n-hexane afforded 0.21 g (0.52 mmol) of 3FC. Yield = 38%. 1H{11B} NMR (CD2Cl2): δ 7.79 (s, 1H), 7.68 (d, J = 6.0 Hz, 1H), 7.51 (d, J = 7.8 Hz, 2H), 7.41 (d, J = 7.2 Hz, 2H), 7.34 (t, J = 3.6 Hz, 2H), 7.21 (d, J = 8.2 Hz, 2H), 7.14 (t, J = 7.3 Hz, 2H), 3.40 (br s, 2H, CB-BH), 2.57 (br s, 6H, CB-BH), 2.37 (br s, 2H, CB-BH), 1.36 (s, 6H, –CH3). 13C NMR (CD2Cl2): δ 156.16, 154.08, 139.87, 138.15, 131.08, 130.98, 130.63, 130.17, 129.88, 128.64, 128.41, 127.51, 123.10, 122.72, 122.67, 120.47, 86.39 (CB–C), 85.99 (CB–C), 47.04 (–C(CH3)2), 26.87 (–CH3). 11B{1H} NMR (CD2Cl2): δ −2.66 (br s, 2B), −9.11 (br s, 3B), −10.56 (br s, 3B), −11.07 (br s, 2B). Anal. calcd for C23H28B10: C, 66.96; H, 6.84. Found: C, 66.80; H, 6.69.Data for 4FC
Decaborane (0.21 g, 1.7 mmol), Et2S (0.37 mL, 3.4 mmol), and 4FA (0.38 g, 1.3 mmol) in toluene afforded 4FC as a white solid. Recrystallization from a mixture of EA/n-hexane afforded 0.06 g (0.14 mmol) of 4FC. Yield = 11%. 1H{11B} NMR (CD2Cl2): δ 8.67 (d, J = 8.0 Hz, 1H), 7.78 (d, J = 8.1 Hz, 1H), 7.43 (d, J = 6.4 Hz, 1H), 7.38 (t, 7.4 Hz, 2H), 7.28 (d, J = 7.3 Hz, 1H), 7.21 (d, J = 7.8 Hz, 2H), 7.11 (q, J = 8.2 Hz, 2H), 6.96 (t, J = 7.8 Hz, 2H), 3.66 (br s, 2H, CB-BH), 2.98 (br s, 2H, CB-BH), 2.58 (br s, 2H, CB-BH), 2.48 (br s, 2H, CB-BH), 1.25 (s, 6H, –CH3). 13C NMR (CD2Cl2): δ 157.22, 154.92, 140.38, 137.66, 135.28, 131.12, 130.68, 130.36, 129.04, 128.30, 128.23, 126.84, 126.58, 126.17, 125.03, 122.72, 90.39 (CB–C), 89.85 (CB–C), 46.00 (–C(CH3)2), 27.67 (–CH3). 11B{1H} NMR (CD2Cl2): δ −2.61 (br s, 1B), −3.65 (br s, 1B), −9.97 (br s, 3B), −11.26 (br s, 5B). Anal. calcd for C23H28B10: C, 66.96; H, 6.84. Found: C, 66.75; H, 6.68.Data for 4FCH
Decaborane (92 mg, 0.75 mmol), Et2S (0.19 mL, 1.7 mmol), and 4FAH (0.13 g, 0.58 mmol) in toluene afforded 4FCH as a white solid. Recrystallization from a mixture of acetone/MeOH afforded 19 mg (55 μmol) of 4FCH. Yield = 10%. 1H{11B} NMR (CD2Cl2): δ 8.21 (d, J = 7.5 Hz, 1H), 7.72 (d, J = 8.0 Hz, 1H), 7.52 (d, J = 7.3 Hz, 2H), 7.36 (m, 2H), 7.32 (t, J = 8.2 Hz, 1H), 4.98 (s, 1H, CB-CH), 2.86 (br s, 2H, CB-BH), 2.74 (br s, 2H, CB-BH), 2.34 (br s, 6H, CB-BH), 1.44 (s, 6H, –CH3). 13C NMR (CD2Cl2): δ 157.60, 155.29, 137.07, 135.80, 131.02, 130.28, 128.51, 127.66, 127.21, 125.71, 124.71, 123.70, 78.44 (CB–C), 60.41 (CB–C), 46.22 (–C(CH3)2), 28.02 (–CH3). 11B{1H} NMR (CD2Cl2): δ −4.23 (br s, 3B), −9.89 (br s, 3B), −11.15 (br s, 2B), −14.31 (br s, 2B). Anal. Calcd for C17H24B10: C, 60.68; H, 7.19. Found: C, 60.55; H, 7.10.Synthesis of nido-4FC, (nido-from of 4FC)·(NBu4)
4FC (18 mg, 43 μmol) was dissolved in 0.17 mL of a 1.0 M THF solution of n-tetrabutylammonium fluoride (TBAF) at 25 °C. The reaction mixture was heated to reflux (60 °C) and stirred for 24 h. After cooling to 25 °C, the resulting mixture was treated with 10 mL of distilled water and 10 mL of DCM, and the organic portion was then separated. The aqueous layer was further extracted with DCM (10 mL). The combined organic portions were dried over MgSO4, filtered, and concentrated to dryness, thereby affording a pale-yellow residue. The crude product upon washing with methanol (15 mL) afforded nido-4FC as a white solid (15 mg, Yield = 54%). 1H{11B} NMR (CD2Cl2): δ 8.37 (d, J = 7.7 Hz, 1H), 7.50 (dd, J = 5.6, 2.5 Hz, 1H), 7.42 (t, J = 8.8 Hz, 1H), 7.25 (s, 2H), 7.06 (d, J = 6.3 Hz, 4H), 6.62 (t, J = 7.1 Hz, 1H), 6.52 (t, J = 7.5 Hz, 2H), 3.03 (m, 8H, n-butyl-CH2), 2.80 (br s, 1H, CB-BH), 2.63 (br s, 1H, CB-BH), 2.18 (br s, 1H, CB-BH), 1.94 (br s, 1H, CB-BH), 1.62 (br s, 5H, CB-BH), 1.56 (m, 8H, n-butyl-CH2), 1.37 (m, 8H, n-butyl-CH2), 1.33 (s, 6H, –CH3), 0.98 (t, J = 9.8 Hz, 12H, n-butyl-CH3), −2.18 (s, 1H, B–H–B). 13C NMR (CD2Cl2): δ 154.00, 153.80, 140.25, 140.11, 139.58, 137.29, 131.47, 131.03, 126.74, 126.34, 126.08, 126.04, 126.00, 125.15, 121.68, 120.28, 59.31 (n-butyl-CH2), 45.70 (–C(CH3)2), 28.46 (–CH3), 26.79, 24.23 (n-buthyl-CH2), 20.02 (n-butyl-CH2), 13.73 (n-butyl-CH3). 11B{1H} NMR (CD2Cl2): δ −8.35 (br s, 1B), −11.82 (br s, 1B), −12.98 (br s, 1B), −14.76 (br s, 1B), −16.82 (br s, 1B), −19.07 (br s, 1B), −20.38 (br s, 1B), −32.58 (br s, 1B), −35.38 (br s, 1B). Anal. calcd for C39H64B9N: C, 72.71; H, 10.01; N, 2.17. Found: C, 72.50; H, 9.99; N, 2.12.X-ray crystallography
Single X-ray quality 2FC, 3FC, 4FC, and 4FCH crystals were grown for X-ray diffractometry from DCM/n-hexane (for 2FC and 3FC), EA/n-hexane (for 4FC), and acetone/MeOH (for 4FCH) mixtures. Each single crystal was coated with Paratone oil and mounted on a glass capillary. Crystallographic measurements were performed using a Bruker D8QUEST CCD area detector diffractometer with graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). The structures of the o-carboranyl compounds were assessed using direct methods, and all nonhydrogen atoms were subjected to anisotropic refinement with the full-matrix least-squares method on F2 using a SHELXTL/PC software package. X-ray crystallographic data are available in the CIF format (CCDC 2057685 for 2FC, 2057686 for 3FC, 2057688 for 4FC, and 2057687 for 4FCH, respectively), and they are provided free of charge by the CCDC.† Hydrogen atoms were placed at their geometrically calculated positions and refined using a riding model on the corresponding carbon atoms with isotropic thermal parameters. Detailed crystallographic data and selected bond lengths and angles are provided in Tables S1 and S2 in the ESI.†UV-vis absorption and photoluminescence (PL) measurements
UV-vis absorption and PL spectra of all o-carboranyl compounds were acquired using V-530 (Jasco) and Fluoromax-4P (HORIBA) spectrophotometers, respectively. UV-vis absorption and PL measurements for the solutions were performed at 298 K in oxygen-free and anhydrous THF with a 1 cm quartz cuvette (3.0 × 10−5 M). Furthermore, PL measurements were performed under various conditions, including different oxygen-free solvents (toluene, DCM, and THF/distilled water mixtures; 3.0 × 10−5 M) in the THF solution (3.0 × 10−5 M) at 77 K, and as a film at 298 K. Thin films of the o-carborane compounds in PMMA were obtained by spin coating a THF solution (1 mL) of PMMA (50 mg) containing each o-carborane compound (5 wt% versus PMMA) on a 10 mm × 10 mm quartz plate (thickness = 1 mm). The absolute photoluminescence quantum yields (PLQYs) in the THF solution (3.0 × 10−5 M) and in the film were obtained at 298 K using a Fluoromax-4P spectrophotometer (HORIBA) equipped with a 3.2-inch integrating sphere (FM-sphere, HORIBA). Fluorescence decay lifetimes and transient PL spectra were measured using a time-correlated single-photon counting spectrometer (FLS920-EDINBURGH instrument at the Central Laboratory of Kangwon National University) equipped with an EPL-375 ps pulsed semiconductor diode laser excitation source and a microchannel plate photomultiplier tube (MCP-PMT, 200–850 nm) detector.Computational calculation studies
The geometries of the o-carboranyl compounds 2FC, 3FC, and 4FC in their ground (S0) and first-excited (S1) states in THF were optimized at the PBE0/6-31G(d,p)74 level of theory. Vertical excitation energies in the optimized S0 and S1 state geometries were calculated using the TD-DFT method75 at the same level of theory. The solvent effects were evaluated using a self-consistent reaction field method based on the integral equation formalism of the polarizable continuum model with THF as the solvent.76 Most stable geometries were determined by constructing one-dimensional potential energy surfaces as a function of each dihedral angle (Ψ) by rotating the o-carborane cage between approximately −180° and 180° at intervals of 30° to yield 18 initial conformations for each compound. Conformations that exhibited physically impossible atomic overlaps were excluded from further geometric optimizations. The dihedral angle was fixed while other geometric variables were fully relaxed for the geometry optimization and energy calculations of the resulting initial conformations using the Gaussian 16 software program.77 The contribution (%) of a group in a molecule for each molecular orbital was calculated using the GaussSum 3.0 software program.78Results and discussion
Synthesis and characterization
Synthetic protocols for 2FC, 3FC, and 4FC with o-carborane cages appended at the C2-, C3-, and C4-positions of each fluorene moiety, respectively, are provided in Scheme 1. To investigate the structural and electronic effects of o-carborane on the photophysical characteristics, corresponding derivatives for 4FC, 4FCH (structure without the terminal phenyl ring from 4FC), and nido-4FC (= [nido-form of 4FC]·[NBu4]) were synthesized (Scheme 1).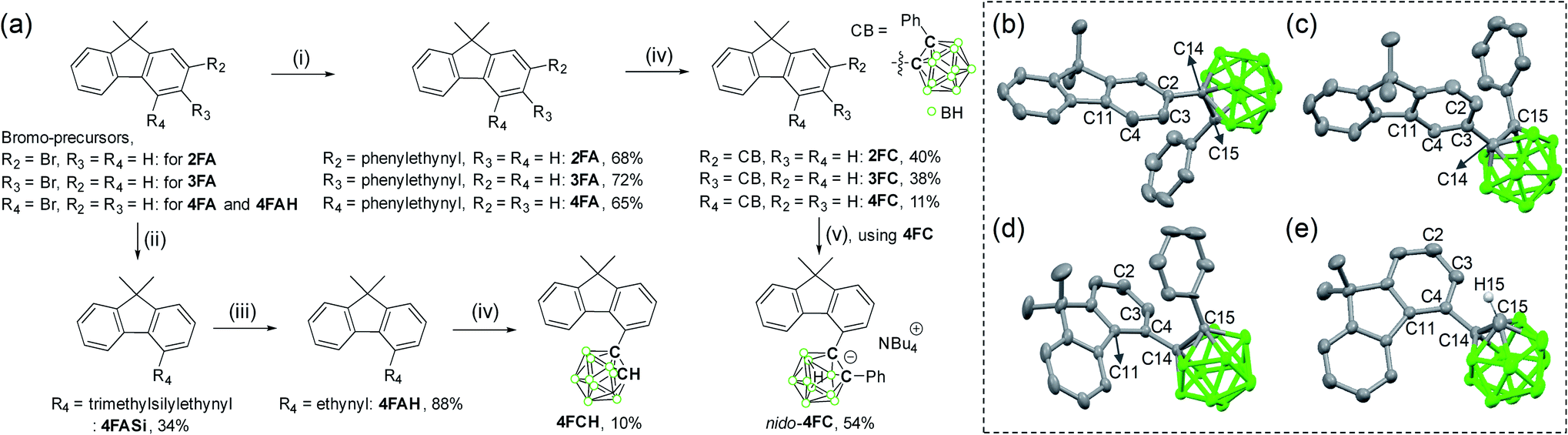 | ||
| Scheme 1 Synthetic routes for the fluorene based o-carboranyl compounds. Reagents and conditions: (i) phenylacetylene, CuI, Pd(PPh3)2Cl2, NEt3/toluene, 110 °C, 24 h. (ii) Ethynyltrimethylsilane, CuI, Pd(PPh3)2Cl2, NEt3/toluene, 110 °C, 24 h. (iii) K2CO3, MeOH/DCM, 25 °C, 2 h. (iv) B10H14, Et2S, toluene, 120 °C, 24 h. (v) TBAF, THF, 60 °C, 24 h. The figures in the box show the molecular structures of (b) 2FC, (c) 3FC, (d) 4FC, and (e) 4FCH (50% thermal ellipsoids, solvent molecules in unit cell and H atoms were omitted for clarity). | ||
Sonogashira couplings between phenylacetylene and either 2-, 3-, or 4-bromo-9,9-dimethyl-9H-fluorene afforded 2FA, 3FA, and 4FA, respectively, in high yields (68, 72, and 65%, respectively). The acetylene precursor 4FAH was prepared by the Sonogashira reaction of 4-bromo-9,9-dimethyl-9H-fluorene with ethynyltrimethylsilane, followed by treatment with a mild base (K2CO3) to deprotect the trimethylsilyl group. The cage-forming reaction79–81 of decaborane (B10H14) with each acetylene precursor (2FA, 3FA, 4FA, and 4FAH) afforded o-carborane compounds 2FC, 3FC, 4FC, and 4FCH, respectively, in relatively low yields (10–40%, Scheme 1). In particular, the cage-forming reaction was difficult for 4FC and 4FCH because of the steric hindrance of the bulky fluorene moiety that leads to significantly low yields (∼10%). Subsequent treatments using 4FC with excess amounts of a weak base like n-tetrabutylammonium fluoride (NBu4F, TBAF) led to the conversion of the closo-carborane species to the nido-species, i.e., nido-4FC (= [nido-form of 4FC]·[NBu4]) with a moderate yield (54%).
All acetylene precursors and fluorene-based o-carboranyl compounds were characterized using multinuclear nuclear magnetic resonance (NMR) spectroscopy (Fig. S1–S15 in the ESI†) and elemental analyses. The 1H{11B decoupled} spectra of the closo-o-carboranyl compounds exhibited expected resonances that can be assigned to the fluorene group, dimethyl groups, terminal aryl rings (for 2FC, 3FC, and 4FC), and the CH of o-carborane (for 4FCH). The broad 1H{11B} peaks present in the upfield region (−2.18 ppm) of the nido-4FC spectrum are characteristic of the B–H–B bridge protons of nido-o-carboranyl cages. Furthermore, observations of three or four broad peaks between −2 and −15 ppm in the 11B{1H} NMR spectra of 2FC, 3FC, 4FC, and 4FCH confirm the presence of a closo-o-carborane cage. Moreover, the 11B{1H} NMR spectra of nido-4FC and nido-CB2 showed nine broad peaks between −8 and −36 ppm; these signals were significantly shifted to the upfield region because of the anionic character of the nido-o-carborane structure, which clearly indicates the presence of the nido-o-carborane cage.
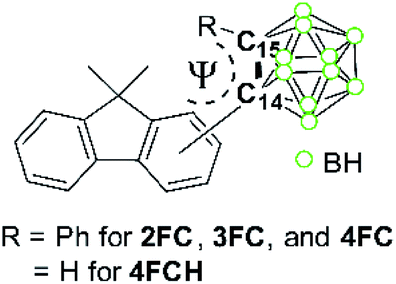
The X-ray diffraction analysis results for 2FC, 3FC, and 4FC demonstrate their solid-state molecular structures (Scheme 1b–d, respectively). Detailed parameters, including the selected bond length and angles, are listed in Tables S1 and S2.† The fluorene group appended to the o-carborane cage in 4FC was significantly distorted, wherein the angle between the two phenyl rings was 10.8° (Fig. 1) and the fluorene plane of both 2FC and 3FC exhibited perfect planarity. These structural features were attributed to the severe steric hindrance around the carborane cage in 4FC, which implies that structural variation centered at the cage was considerably restricted.
 | ||
| Fig. 1 Molecular structure of 4FC exhibiting a distorted fluorene plane formation. | ||
Photophysical analysis for o-carboranyl compounds with theoretical calculations
To investigate the photophysical properties of the fluorene-based o-carboranyl compounds, ultraviolet-visible (UV-vis) absorption and photoluminescence (PL) spectroscopy were performed under various conditions (Fig. 2 for 2FC, 3FC, 4FC, and nido-4FC, Fig. S16† for 4FCH, and Table 1). The major lowest-absorption bands centered at λabs = 302–318 nm for the closo-o-carboranyl compounds in THF were attributed to vibronic structures undergoing spin-allowed π–π* transitions on the fluorene moiety. 9,9-Dimethyl-9H-fluorene exhibited similar strong absorption peaks in this region (Fig. S17,† λabs = 302 nm (ε = 6700 M−1 cm−1) and 291 nm (4440 M−1 cm−1)). Specific features of the low-energy absorption for these closo-compounds include substantially tailing absorption bands at 350 nm and broad absorption features attributed to typical ICT transitions in the o-carborane cages. These ICT characteristics for closo-compounds were verified by the fact that low-energy absorption bands were not observed in the spectrum of 9,9-dimethyl-9H-fluorene (Fig. S17†). Furthermore, nido-4FC did not exhibit low-absorption spectra in the region 310–350 nm (Fig. 2c, black dash-line), which strongly indicates that the anionic feature of nido-o-carborane can inhibit ICT transition, wherein o-carborane acts as an electronic acceptor. The UV-vis spectral properties are highly consistent with the theoretical calculations (vide infra). | ||
| Fig. 2 UV-vis absorption (left side) and PL spectra (right side) for (a) 2FC (λex = 307 nm), (b) 3FC (λex = 300 nm), (c) 4FC and nido-4FC (λex = 318 nm). Black line: absorption spectra in THF (3.0 × 10−5 M); blue line: PL spectra in THF (3.0 × 10−5 M) at 298 K; green line: PL spectra in THF (3.0 × 10−5 M) at 77 K; and orange line: PL spectra in film (5 wt% doped with PMMA) at 298 K. Inset figures show the emission color in each state under irradiation by a hand-held UV lamp (λex = 265 nm). | ||
| λabsa/nm (ε × 10−3 M−1 cm−1) | λex/nm | λem/nm | Φ em | τ obs/ns | k r /× 107 s−1 | k nr /× 108 s−1 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tolb | THFb | DCMb | 77 Ka | Filmc | f w = 90%b,d | THFb | Filmc | f w = 90%b,d | THFb | Filmc | THF | Film | THF | Film | |||
| a 3.0 × 10−5 M in oxygen-free and anhydrous THF. b c = 3.0 × 10−5 M in oxygen-free and anhydrous solvent, observed at 298 K. c Measured in the film state (5 wt% doped in PMMA). d Water fraction in a THF–water mixture. e Absolute PL quantum yield. f k r = Φem/τobs. g k nr = kr(1/Φem − 1). h Not observed because of weak emission. | |||||||||||||||||
| 2FC | 307 (21.2), 283 (23.5) | 307 | —h | —h | —h | 486 | 498 | 503 | <0.01 | 0.29 | 0.26 | —h | 4.6 | — | 6.3 | — | 1.5 |
| 3FC | 302 (6.1), 268 (16.4) | 302 | —h | —h | —h | 498 | 500 | 515 | <0.01 | 0.24 | 0.19 | —h | 4.1 | — | 5.8 | — | 1.8 |
| 4FC | 318 (12.1), 269 (17.1) | 318 | 553 | 577 | 596 | 528 | 523 | 536 | 0.05 | 0.42 | 0.36 | 2.1 | 3.9 | 2.4 | 10.8 | 4.6 | 1.6 |
| nido-4FC | 271 (16.1) | 300 | —h | —h | —h | —h | —h | — | — | — | — | — | — | — | — | — | — |
| 4FCH | 309 (8.1), 271 (10.9) | 309 | 356, 546 | 356, 579 | 356, 586 | 340, 527 | — | — | 0.01 (CT) | 0.04 (CT) | — | -h | 7.6 | — | 0.53 | — | 1.3 |
The origin of electronic transitions in 2FC, 3FC, and 4FC were determined using time-dependent density functional theory (TD-DFT) calculations.75 Ground (S0) and first-excited (S1) states of the compounds were optimized at the PBE0/6-31G(d) level of theory (Fig. 3). Each calculated structure of closo-compounds is based on its X-ray crystal structure (Scheme 1b–d). An integral equation formalism of the polarizable continuum model (IEFPCM) is employed to consider the THF solvent effect.76 S0 state calculations indicate that the lowest-energy electronic transitions for these compounds are associated with transitions from the HOMO to the LUMO (Fig. 3). The HOMO of each compound is predominantly localized on the fluorene moiety (>96% in all closo-compounds, Tables S4, S6, and S8†); however, the LUMOs are substantially distributed over the o-carborane units (>30% in all compounds). These calculation results for S0 states suggest that the low-energy electronic transitions of o-carboranyl-fluorene compounds originate from both local π–π* transitions on the fluorene group and substantial ICT transitions involving o-carborane cages.
 | ||
| Fig. 3 Frontier molecular orbitals for 2FC, 3FC, and 4FC in their ground state (S0) and first excited singlet state (S1) with relative energies obtained from DFT calculations (isovalue 0.04). Transition energy (in nm) was calculated using the TD-PBE0 method with 6-31G(d) basis sets. | ||
The emissive features of o-carboranyl compounds were examined by PL measurements (Fig. 2 and Table 1). While the PL spectra of 4FC in THF at 298 K exhibited significant emission in the region centered at λem = 577 nm (orange emission, inset of Fig. 2c), 2FC and 3FC hardly showed any emissive pattern. Because the PL spectra for 4FC was red-shifted upon increasing solvent polarities (positive solvatochromic effect, λem = 553 nm in toluene to 596 nm in dichloromethane, Δλem = 43 nm, Table 1 and Fig. S18a†), it strongly indicated that the emissive band is attributed to the ICT characteristic. Furthermore, computational results for the S1 states of o-carboranyl compounds supported this observation. Emissions for compounds are dominated by HOMO → LUMO transitions (Fig. 3). The HOMOs are significantly localized on the fluorene moiety (>93% in all compounds, Tables S4, S6, and S8†), whereas the LUMOs are centered on the o-carborane units (>83%). This suggests that emissions originate from ICT processes between the o-carborane and fluorene groups.
Interestingly, these three o-carboranyl compounds demonstrated intense emission at λem = 486–528 nm (Fig. 2 and Table 1) in the rigid state (THF at 77 K and film (5 wt% doped in PMMA)), which can be attributed to ICT-based emission. These features result from the restricted elongation of the C–C bond of the o-carborane triggered by excitation, which prevents radiative decay based on ICT transitions.62–64 The optimized structures of closo-compounds based on their S0 and S1 states demonstrate C–C bond fluctuations (Table 2). The bond lengths of o-carborane cages in the S1 state were 2.39–2.48 Å; these were considerably larger than those in the S0 states (1.67–1.83 Å) and the experimentally measured values (∼1.73 Å) for compounds based on their X-ray structures. Consequently, all the compounds revealed that absolute quantum yields (aem = 29% for 2FC, 24% for 3FC, and 42% for 4FC, Table 1) in the film were dramatically enhanced compared with those (<1% for 2FC and 3FC, and 5% for 4FC) in THF at 298 K. In particular, the additional reason for the enhanced emission of three compounds in the film (solid) state was investigated based on further PL measurements in a THF–water mixture (3.0 × 10−5 M, Fig. S19†). The low-energy emission around 500–550 nm was drastically enhanced by an increased water fraction (fw). Consequently, each maximized aggregation state in THF/water (fw = 90%) for the compounds was associated with intense greenish emission patterns (λem = 503–536 nm) and high Φem values (26%, 19%, and 36% for 2FC, 3FC, and 4FC, respectively; Table 1) similar to those observed in the film state. These observations are characteristic of aggregation induced emission (AIE) phenomena.
|
|
||||||
|---|---|---|---|---|---|---|
| C–C bond/Å | Ψ/° | |||||
| Exp.a | Calc.b | Exp.a | Calc.b | |||
| S 0 | S 1 | S 0 | S 1 | |||
| a Experimental values calculated from their X-ray crystal structures. b Calculated values from their ground state (S0) and first excited singlet state (S1) optimized structures. | ||||||
| 2FC | 1.75 | 1.78 | 2.46 | 87.1 | 87.9 | 0.0 |
| 3FC | 1.73 | 1.77 | 2.44 | 82.9 | 88.3 | 1.8 |
| 4FC | 1.76 | 1.83 | 2.48 | 89.3 | 87.9 | 0.1 |
| 4FCH | 1.72 | 1.67 | 2.39 | 82.7 | 87.0 | 1.6 |
Nido-4FC did not present any emissive traces in THF at 298 K and 77 K (Fig. 2c and Table 1); totally differed from the features of closo-compounds, which strongly suggests that the ICT process can be obstructed by the anionic characteristic of nido-o-carborane. This observation paradoxically reveals that all emissive characteristics of closo-compounds are significantly correlated with the ICT transition involving closo-o-carborane.
Interestingly, 4FCH exhibited a significantly weak dual-emissive trace centered at eem = 356 and 579 nm in the THF at 298 K; however, the emissive pattern was distinctly observed in the rigid state (THF at 77 K and film state, Fig. S16† and Table 1). Based on the PL spectra of 9,9-dimethyl-9H-fluorene in THF at 298 K that exhibits an emission centered at eem = 341 nm (Fig. S17†) and a red-shifted emission in the region of 500–600 nm (positive solvatochromic effect, λem = 546 nm in toluene to 586 nm in dichloromethane, Δλem = 40 nm; Table 1 and Fig. S18b†), high- and low-energy regions predominantly originate from π–π* locally excited (LE) transitions on the fluorene group and ICT transitions between the o-carborane cage and fluorene moiety, respectively. This leads to LE- and ICT-based emissions. Furthermore, high-energy emissions of 4FCH remain unchanged in solvents with different polarities, which strongly indicates LE-characteristics. Consequently, the revelation of LE-based emission leads to a drastic decrease in the efficiency of ICT-based emissions (Φem = 1% in THF at 298 K and 4% in film state, Table 1) because excitons on 4FCH are separated into both LE and ICT transitions.
Such different optical behaviors between 4FC and other closo-compounds were exhibited moderately by only 4FC during the ICT-based emission (Φem = 5%) in THF at 298 K. This suggests that the ICT-based radiative-decay process based on o-carboranyl compounds is severely affected by structural motion. As shown in the structural feature of 4FC (Scheme 1d), the fluorene plane is distorted significantly by intramolecular steric hindrance, and the rotation of the o-carborane cage in 4FC is definitely restricted. However, the o-carboranes of other closo-compounds can be freely rotated. Indeed, the intensity of the Raman spectra of 4FC was much weaker than those of 2FC and 3FC (Fig. S20†), indicating that the structural motion of 4FC is effectively restricted compared to those for 2FC and 3FC. Furthermore, such a restriction of rotational motion for 4FC differs from that for 2FC, 3FC, and 4FCH, and it can be revealed by the energy calculation results of their S0 states upon changing the dihedral angles associated with their o-carboranyl cages (Fig. 5, vide infra). Consequently, the structural fluctuations around the o-carborane cage can decrease the efficiency of ICT-based radiative decay for 2FC and 3FC, and it can further reinforce LE-based emission instead of the ICT process.
Comparative analysis of the ICT-based radiative decay of o-carboranyl compounds in the solid state
To analyze and compare the efficiency of the ICT-based emission from each closo-o-carboranyl compound in the film (solid), radiative (kr) and nonradiative (knr) decay constants were estimated using Φem and emission decay lifetimes (τobs) (Table 1). The τobs values for all closo-compounds determined at 298 K were found to be similar on the nanosecond time scale (1.3–4.3 ns, Fig. 4 for 4FC; Fig. S21† for 2FC, 3FC, and 4FCH; and Table 1), which demonstrated their fluorescent nature. The kr value calculated using the Φem and τobs of 4FC in the film was estimated to be 1.1 × 108 s−1 (Table 1). This was approximately two times more than those for 2FC (6.3 × 107 s−1) and 3FC (5.8 × 107 s−1), and 20 times larger than that of 4FCH (5.3 × 106 s−1). Furthermore, the knr (1.6 × 108 s−1) of 4FC was similar to those of other compounds (1.5 × 108 s−1 for 2FC, 1.8 × 108 s−1 for 3FC, and 1.3 × 108 s−1 for 4FCH). Quantitative results for the ICT-based emission strongly suggest that the structural formation around o-carborane cages is determined by structural rigidity and has a significant relevance to the efficiency of radiative decay based on ICT transitions. Furthermore, this can lead to the enhancement of ICT-based emissions.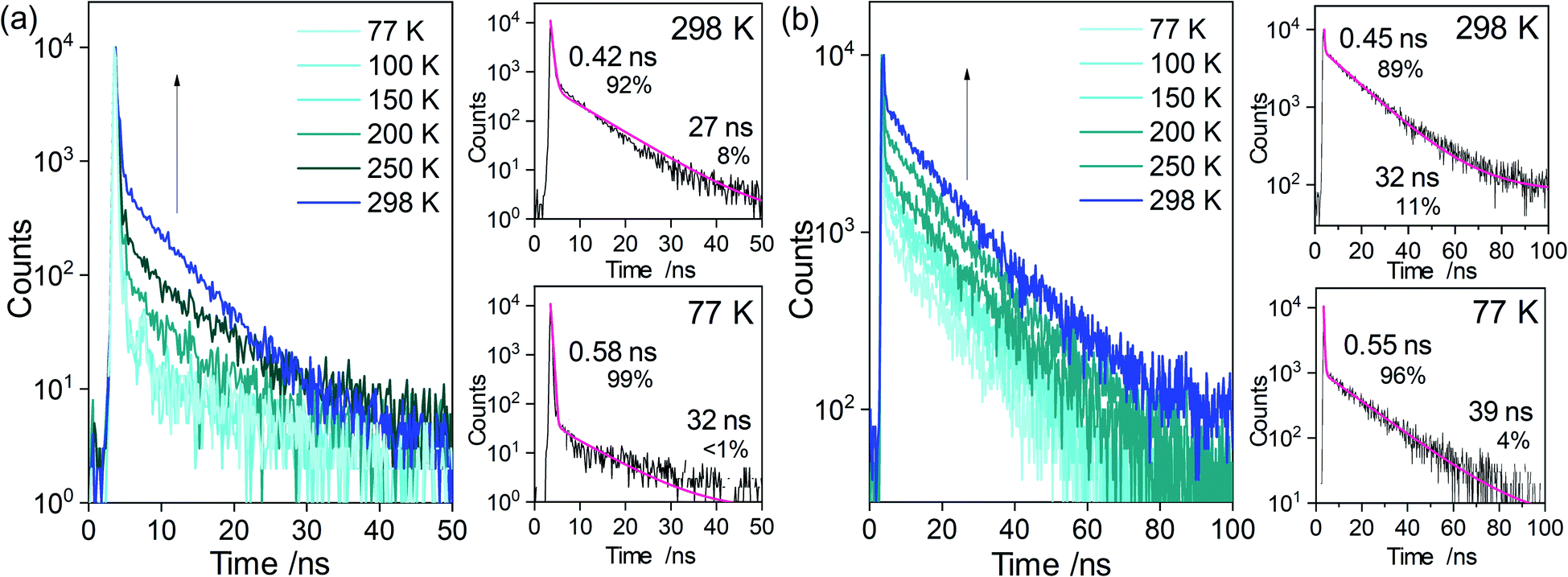 | ||
| Fig. 4 Transient PL decay profiles for 4FC (a) in oxygen-free and anhydrous THF (3.0 × 10−5 M) detected at 528 nm and (b) in the film state (5 wt% doped in PMMA) detected at 523 nm based on changes in temperature. Each red line represents its double exponential fitting curve. | ||
The calculated relative energy of each closo-o-carboranyl compound in its ground state (S0, black solid-dot line in Fig. 5) demonstrated the structural environment around the o-carborane cage. These results were calculated as functions of dihedral angles (Ψ = from −180° to 180° range in 30° steps) between the fluorene plane and the C–C bond of o-carborane (Table 2) using the PBE0 functional and 6-31G(d) basis set.
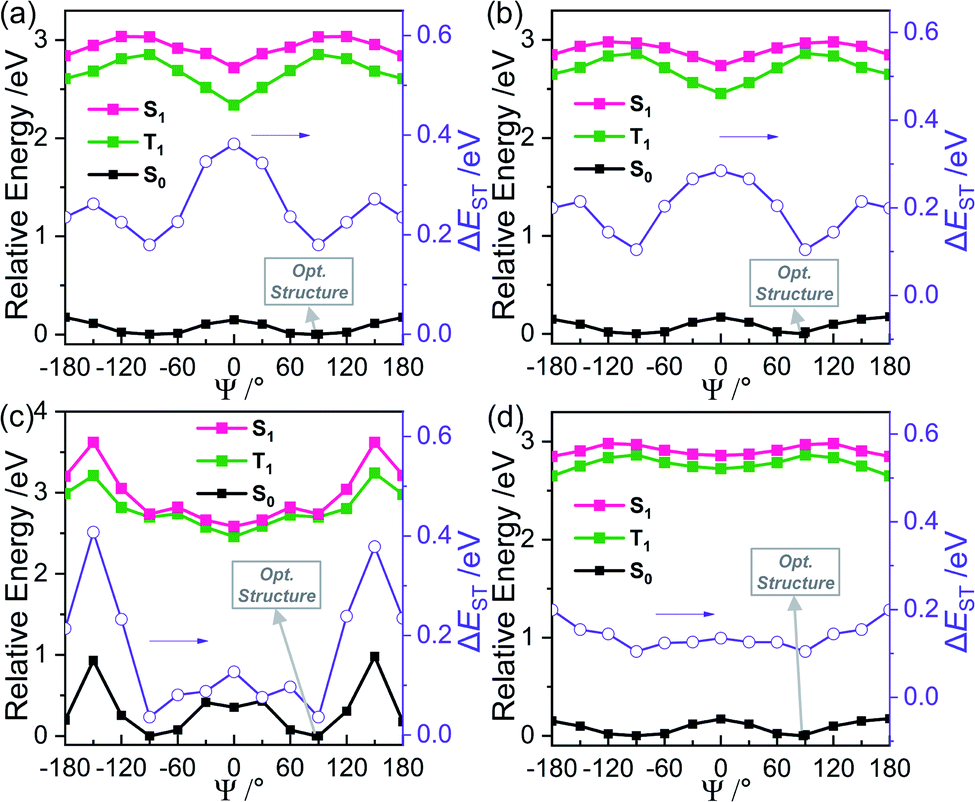 | ||
| Fig. 5 Relative energy diagrams of S0, S1, and T1 states for (a) 2FC, (b) 3FC, (c) 4FC, and (d) 4FCH on changing the dihedral angle (Ψ). Each purple line indicates the energy gap between the relative energy for the S1 and T1 states. | ||
The relative energy of each compound was presented against the thermodynamic energy of each S0-optimized structure in THF (energy for each S0-optimized state = 0 eV). The Ψ values of the S0-optimized structure for the closo-compounds were estimated to be ∼88°, which closely reflected the experimental values for compounds, obtained by X-ray diffraction analyses (82.7–87.1°, Table 2). Meanwhile, values for the S0-optimized state indicated that the C–C bond of the o-carborane became parallel to the fluorene plane (Ψ = 0.0–1.8°). These results strongly suggest that the rotation motion of o-carborane cages for all closo-compounds can occur during the radiative excitation and relaxation processes. Furthermore, the relative energy barrier on the S0-states for 2FC, 3FC, and 4FCH did not exceed 0.18 eV throughout the entire Ψ range (Fig. 5), while the lowest energy points of 4FC were observed around Ψ = 90 and −90°; moreover, the energy maximum point was ∼0.95 eV (Ψ = 120°). Considering the thermal energy value at 298 K = 0.59 kcal mol−1 (∼0.025 eV mol−1)—calculated as kBT (kB = Boltzmann constant, 1.98 cal K−1 mol−1)—the features indicate that the structural formation of 4FC was fixed at Ψ ≈ 90° in all states, whereas o-carborane cages in 2FC, 3FC, and 4FCH can be freely rotated in the solution state at ambient temperature and can further exist in various structural forms in the solid state. Consequently, calculation and experimental results confirmed that the structural rigidity of 4FC led to efficient radiative decay based on ICT transitions associated with o-carborane.
TADF characteristics revealed by the rigid structural formation
When investigating the radiative decay lifetime of 4FC, a decay feature was observed, wherein exponential components are separated as prompt and delayed decay profiles. In particular, transient PL decay for the ICT-based emission of 4FC in THF at 298 K revealed that PL decay curves are best fitted with short-lived (τobs = 0.42 ns, Fig. 4a) and comparatively long-lived (27 ns) decay components, respectively. Furthermore, PL spectral profiles of 4FC in aerated THF at 298 K exhibited slightly lower intensities (Fig. S22†) compared to that in oxygen-free THF (the PL in Fig. 2c); this indicates that the oxygen-sensitive radiative decay component related to the triplet (T1) excited state is slightly involved with the ICT-based emission of 4FC. These results suggest that long-lived emissions can be assigned to the delayed fluorescence that arises from the S1 state through upconversion from the T1 state; i.e., from the reverse intersystem crossing.Furthermore, the intensity of the delayed component (∼30 ns) gradually decreases as the temperature is decreased from 298 K to 77 K (8% at 298 K to <1% at 77 K, Fig. 4a, inset); this confirms that the delayed emission can be assigned to TADF. In addition, the delayed fluorescence was found to be more intensely observed in the film (solid) state than in solution. Indeed, transient PL decays of 4FC in the film demonstrated a greater portion (4–11%, Fig. 4b) of the delayed component (32–39 ns) than that of the prompt component. Temperature-dependent PL decay profiles in the film state verified the TADF characteristic of the delayed emission. Furthermore, the PL decay profiles of the other closo-o-carboranyl compounds 2FC, 3FC, and 4FCH remained unchanged with temperature (Fig. S20†), which indicates their normal fluorescent nature. These results imply that fluorene-based o-carboranyl compounds exhibit the TADF property, which is affected by the rigidity of their structural formation in accordance with the substitution position of the o-carborane cage in the fluorene plane. The TD-DFT calculation results for the S1 states revealed that the orbital overlap-integral (IH/L) between HOMO and LUMO levels for 4FC was considerably lower (3.3%) than those for 2FC (8.9%) and 3FC (8.4%), which implies a discrete D–A structural feature. To verify the relationship between the structural fluctuation around the o-carborane cages and the TADF phenomenon, the energy gap (ΔEST) between the S1 and T1 states was estimated based on changes in the Ψ values for all closo-compounds (Fig. 5). The ΔEST value for 4FC at Ψ = 90° is nearly same as that (87.9°, Table 2) of the S0-optimized structure; it is significantly small (0.032 eV). The result and the fact that 4FC is fixed at around Ψ = 90° because of the high-energy barrier suggests that 4FC can maintain a small ΔEST in all states, which can sufficiently evoke an efficient TADF. The experimental ΔEST value for 4FC, which is calculated using the gap of onset wavelengths for prompt and delayed PL (phosphorescent nature), was significantly similar (0.027 eV, Fig. S23†) to that value. To validate these small ΔEST values for 4FC in both THF and the film state, ΔEST values were again calculated from the Arrhenius plots using the activation energy of the reverse intersystem crossing (RISC) (Fig. S24†). Indeed, temperature dependent rate constants of the RISC (kRISC) were determined from the prompt and delayed fluorescence components and using previously reported equations.82,83 Moreover, kRISC in THF and the film at 298 K could be estimated as 4.0 and 3.5 × 107 s−1, respectively. The ΔEST values evaluated from the slope of temperature dependence of the kRISC plot (Arrhenius plot) was determined to be 8.9 meV in THF and 13.5 meV in the film. Thus, the narrow ΔEST value and relatively large kRISC in both solution and the film support the efficient RISC process and TADF feature of 4FC.
In contrast, ΔEST values for the other closo-compounds around each S0-optimized structure (Ψ ≈ 90°) were found to be greater than 0.10 eV; furthermore, those of 2FC and 3FC were found to be greater than 0.28 eV at Ψ = 0°. Based on the structural features of the compounds wherein the free rotation of o-carborane cages is comparatively easy to achieve owing to the low-energy barrier, these results indicate why closo-compounds besides 4FC exhibit normal fluorescent characteristics in the film state.
Consequently, the strategic molecular design of o-carborane-appended fluorene compounds can lead to a rigid-structural formation, which enhances radiative efficiency for ICT transitions corresponding to o-carboranes and reveals their intriguing TADF nature.
Electrochemical and thermal stabilities of the o-carboranyl compounds
Electrochemical and thermal stabilities of organometallic materials are considerably important for wide practical applications. To obtain the stabilities for the closo-o-carboranyl compounds, 2FC–4FC, cyclic voltammetry (CV) measurements (Fig. 6a), thermogravimetric analysis (TGA, Fig. 6b), and differential scanning calorimetry (DSC, Fig. 6b) were performed. The HOMO and LUMO energy levels for the compounds obtained using the measured oxidation (Vox,onset, 1.39 V for 2FC, 1.37 V for 3FC, and 1.42 V for 4FC → HOMO: −6.19 eV for 2FC, −6.17 eV for 3FC, and −6.22 eV for 4FC) and reduction onset potentials (Vred, onset, −1.18 V for 2FC, −1.23 V for 3FC, and −1.27 V for 4FC → LUMO: −3.62 eV for 2FC, −3.57 eV for 3FC, and −3.53 eV for 4FC) were significantly similar to the HOMO levels on S0-optimized structures and the LUMO levels on S1-optimized structures obtained by TD-DFT calculations, respectively (Fig. 3, see the TD-DFT results). Furthermore, all the compounds underwent reversible and consistent oxidation and reduction processes during five consecutive cycles (Fig. S25†), implying outstanding electrochemical stability.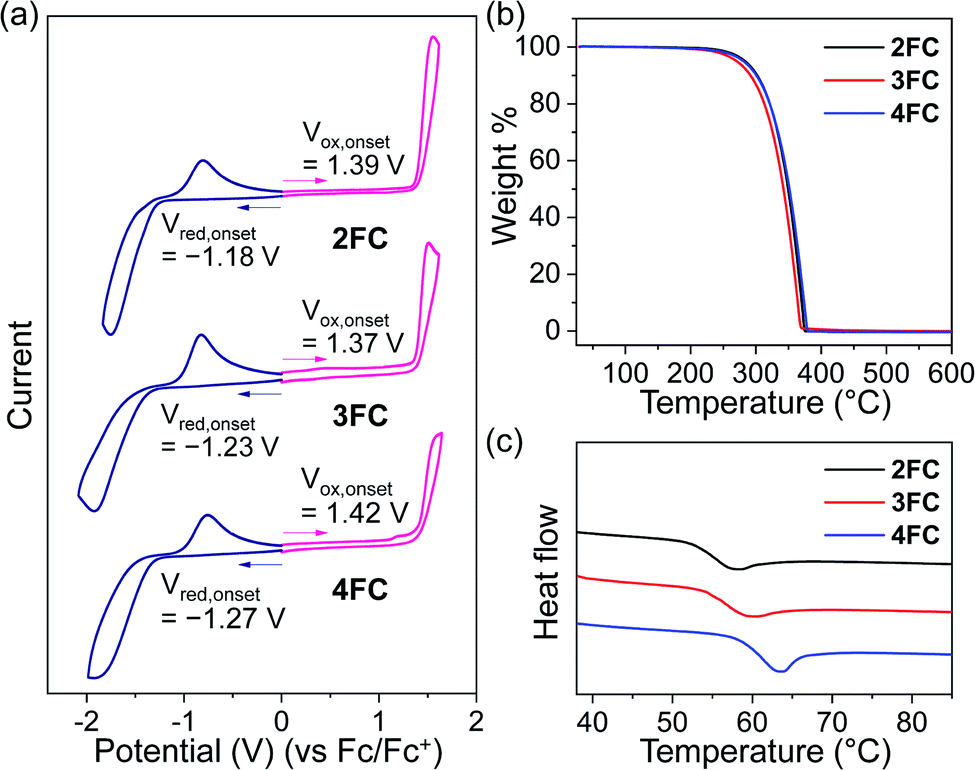 | ||
| Fig. 6 (a) Cyclic voltammograms of 2FC, 3FC, and 4FC showing oxidation (pink solid line) and reduction (navy) potentials (5 × 10−4 M in DCM, scan rate = 100 mV s−1). (b) TGA and (c) DSC curves for 2FC, 3FC, and 4FC. | ||
The thermal stabilities of the compounds were investigated using Td5 (the temperature at 5 wt% degradation) and Tg (the glass transition temperature) values from TGA and DSC (Fig. 6b). 2FC, 3FC, and 4FC exhibited relatively high Td5 and Tg values (284 °C and 58 °C for 2FC, 271 °C and 61 °C for 3FC, and 281 °C and 64 °C for 4FC, respectively). These features could be attributed to the rigid and robust structure of the closo-o-carborane cluster. Consequently, these findings further support the potential of materials that can be applied to the optoelectronic industry.
Conclusions
We synthesized and characterized 2FC, 3FC, 4FC, 4FCH, and nido-4FC. The molecular structures for all closo-compounds were determined by single crystal X-ray diffraction analysis. The crystal structure of 4FC exhibited a significantly distorted fluorene plane, which revealed severe intramolecular steric hindrance. Interestingly, 4FC demonstrated intense ICT-based emission in all states (solution at 298 K and 77 K, film, and aggregated states), while other closo-compounds demonstrated emissions in only the rigid state. In addition, nido-4FC did not exhibit any emissive trace, which indicated that all radiative decay processes definitely corresponded to the ICT transition triggered by the existence of closo-o-carborane. Relative energy barriers and dihedral angles around o-carborane cages changed in closo-compounds, which indicated that o-carborane cages in all the compounds except for 4FC could be freely rotated at ambient temperature, whereas the structural formation of 4FC was fixed around its S0-optimized structure. Furthermore, 4FC demonstrated that separated PL decay components were short-lived (∼0.5 ns) and long-lived (27–39 ns) in the solution at 298 K and in the film state, respectively, which could be attributed to prompt fluorescence and TADF, respectively. Such a characteristic for 4FC was revealed by maintaining the significantly small ΔEST in the structural fixation. Consequently, the strategic molecular design of the o-carborane-appended π-conjugated (D–A) system, which can form rigid structural geometries, can enhance radiative efficiency for ICT-based emission and trigger intriguing photophysical properties such as TADF. To the best of our knowledge, this is the first example of a closo-o-carboranyl D–A type compound demonstrating TADF nature in the solution state at ambient temperature; it demonstrates that the closo-o-carborane can act as a novel functional group of electronic acceptors for TADF.Author contributions
D. K. You and H. So were responsible for experimental investigations and formal analysis (lead), data presentation, computational investigation, collected the X-ray data, solved the crystal structures, and visualization. C. H. Ryu and M. Kim were responsible for experimental investigations (supportive), computational investigation (lead) and visualization. K. M. Lee was in charge for methodology and conceptualization, computational investigations, writing original draft (lead), review and editing of the manuscript (lead), project administration and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant (NRF-2020R1A2C1006400 and NRF-2016M3A7B4909246 for K. M. Lee) funded by the Ministry of Science and ICT and Ministry of Education. We thank Dr Dongwook Kim (Center for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS)) for assistance with X-ray diffraction experiments.Notes and references
- V. I. Bregadze, Chem. Rev., 1992, 92, 209–223 CrossRef CAS
.
- F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS PubMed
- R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. Fabrizi de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef PubMed
- J. Ochi, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2020, 59, 9841–9855 CrossRef CAS PubMed
- M. F. Hawthorne, Angew. Chem., Int. Ed., 1993, 32, 950–984 CrossRef
- A. H. Soloway, W. Tjarks, B. A. Barnum, F. G. Rong, R. F. Barth, I. M. Codogni and J. G. Wilson, Chem. Rev., 1998, 98, 1515–1562 CrossRef CAS PubMed
- M. F. Hawthorne and A. Maderna, Chem. Rev., 1999, 99, 3421–3434 CrossRef CAS PubMed
- H. Jude, H. Disteldorf, S. Fischer, T. Wedge, A. M. Hawkridge, A. M. Arif, M. F. Hawthorne, D. C. Muddiman and P. J. Stang, J. Am. Chem. Soc., 2005, 127, 12131–12139 CrossRef CAS PubMed
- Y. Shirai, J.-F. Morin, T. Sasaki, J. M. Guerrero and J. M. Tour, Chem. Soc. Rev., 2006, 35, 1043–1055 RSC
- B. P. Dash, R. Satapathy, E. R. Gaillard, J. A. Maguire and N. S. Hosmane, J. Am. Chem. Soc., 2010, 132, 6578–6587 CrossRef CAS PubMed
- A. M. Spokoyny, C. W. Machan, D. J. Clingerman, M. S. Rosen, M. J. Wiester, R. D. Kennedy, C. L. Stern, A. A. Sarjeant and C. A. Mirkin, Nat. Chem., 2011, 3, 590–596 CrossRef CAS PubMed
- Z. Qiu, S. Ren and Z. Xie, Acc. Chem. Res., 2011, 44, 299–309 CrossRef CAS PubMed
- Z.-J. Yao and G.-X. Jin, Coord. Chem. Rev., 2013, 257, 2522–2535 CrossRef CAS
- S. P. Fisher, A. W. Tomich, S. O. Lovera, J. F. Kleinsasser, J. Guo, M. J. Asay, H. M. Nelson and V. Lavallo, Chem. Rev., 2019, 119, 8262–8290 CrossRef CAS PubMed
- K.-R. Wee, Y.-J. Cho, S. Jeong, S. Kwon, J.-D. Lee, I.-H. Suh and S. O. Kang, J. Am. Chem. Soc., 2012, 134, 17982–17990 CrossRef CAS PubMed
- J. Guo, D. Liu, J. Zhang, J. Zhang, Q. Miao and Z. Xie, Chem. Commun., 2015, 51, 12004–12007 RSC
- R. Furue, T. Nishimoto, I. S. Park, J. Lee and T. Yasuda, Angew. Chem., Int. Ed., 2016, 55, 7171–7175 CrossRef CAS PubMed
- R. Núñez, C. Viñas, F. Teixidor, R. Sillanpää and R. Kivekäs, J. Organomet. Chem., 1999, 592, 22–28 CrossRef
- F. Teixidor, R. Núñez, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2000, 112, 4460–4462 CrossRef
- R. Núñez, P. Farràs, F. Teixidor, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2006, 45, 1270–1272 CrossRef PubMed
- J. O. Huh, H. Kim, K. M. Lee, Y. S. Lee, Y. Do and M. H. Lee, Chem. Commun., 2010, 46, 1138–1140 RSC
- K. M. Lee, J. O. Huh, T. Kim, Y. Do and M. H. Lee, Dalton Trans., 2011, 40, 11758–11764 RSC
- K. Kokado and Y. Chujo, J. Org. Chem., 2011, 76, 316–319 CrossRef CAS PubMed
- B. P. Dash, R. Satapathy, E. R. Gaillard, K. M. Norton, J. A. Maguire, N. Chug and N. S. Hosmane, Inorg. Chem., 2011, 50, 5485–5493 CrossRef CAS PubMed
- K.-R. Wee, W.-S. Han, D. W. Cho, S. Kwon, C. Pac and S. O. Kang, Angew. Chem., Int. Ed., 2012, 51, 2677–2680 CrossRef CAS PubMed
- L. Weber, J. Kahlert, R. Brockhinke, L. Böhling, A. Brockhinke, H.-G. Stammler, B. Neumann, R. A. Harder and M. A. Fox, Chem. - Eur. J., 2012, 18, 8347–8357 CrossRef CAS PubMed
- A. Ferrer-Ugalde, E. J. uárez-Pérez, F. Teixidor, C. Viñas and R. Núñez, Chem. - Eur. J., 2013, 19, 17021–17030 CrossRef CAS PubMed
- L. Weber, J. Kahlert, R. Brockhinke, L. Böhling, J. Halama, A. Brockhinke, H.-G. Stammler, B. Neumann, C. Nervi, R. A. Harder and M. A. Fox, Dalton Trans., 2013, 42, 10982–10996 RSC
- L. Weber, J. Kahlert, L. Böhling, A. Brockhinke, H.-G. Stammler, B. Neumann, R. A. Harder, P. Low and M. A. Fox, Dalton Trans., 2013, 42, 2266–2281 RSC
- S. Kwon, K.-R. Wee, Y.-J. Cho and S. O. Kang, Chem. - Eur. J., 2014, 20, 5953–5960 CrossRef CAS PubMed
- H. J. Bae, H. Kim, K. M. Lee, T. Kim, Y. S. Lee, Y. Do and M. H. Lee, Dalton Trans., 2014, 43, 4978–4985 RSC
- J. Poater, M. Solà, C. Viñas and F. Teixidor, Angew. Chem., Int. Ed., 2014, 53, 12191–12195 CrossRef CAS PubMed
- H. Naito, Y. Morisaki and Y. Chujo, Angew. Chem., Int. Ed., 2015, 54, 5084–5087 CrossRef CAS PubMed
- B. H. Choi, J. H. Lee, H. Hwang, K. M. Lee and M. H. Park, Organometallics, 2016, 35, 1771–1777 CrossRef CAS
- S. Mukherjee and P. Thilagar, Chem. Commun., 2016, 52, 1070–1093 RSC
- J. Poater, M. Solà, C. Viñas and F. Teixidor, Chem. - Eur. J., 2016, 22, 7437–7443 CrossRef CAS PubMed
- R. Núñez, I. Romero, F. Teixidor and C. Viñas, Chem. Soc. Rev., 2016, 45, 5147–5173 RSC
- K. O. Kirlikovali, J. C. Axtell, A. Gonzalez, A. C. Phung, S. I. Khan and A. M. Spokoyny, Chem. Sci., 2016, 7, 5132–5138 RSC
- H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, J. Mater. Chem. C, 2017, 5, 10047–10054 RSC
- D. Tu, P. Leong, S. Guo, H. Yan, C. Lu and Q. Zhao, Angew. Chem., Int. Ed., 2017, 56, 11370–11374 CrossRef CAS PubMed
- K. Nishino, K. Uemura, K. Tanaka, Y. Morisaki and Y. Chujo, Eur. J. Org. Chem., 2018, 12, 1507–1512 CrossRef
- H. Mori, K. Nishino, K. Wada, Y. Morisaki, K. Tanaka and Y. Chujo, Mater. Chem. Front., 2018, 2, 573–579 RSC
- I. Nar, A. Atsay, A. Altındal and E. Hamuryudan, Inorg. Chem., 2018, 57, 2199–2208 CrossRef CAS PubMed
- H. Jin, H. J. Bae, S. Kim, J. H. Lee, H. Hwang, M. H. Park and K. M. Lee, Dalton Trans., 2019, 48, 1467–1476 RSC
- K. L. Martin, J. N. Smith, E. R. Young and K. R. Carter, Macromolecules, 2019, 52, 7951–7960 CrossRef CAS
- K.-R. Wee, Y.-J. Cho, J. K. Song and S. O. Kang, Angew. Chem., Int. Ed., 2013, 52, 9682–9685 CrossRef CAS PubMed
- A. Ferrer-Ugalde, A. González-Campo, C. Viñas, J. Rodríguez-Romero, R. Santillan, N. Farfán, R. Sillanpää, A. Sousa-Pedrares, R. Núñez and F. Teixidor, Chem. - Eur. J., 2014, 20, 9940–9951 CrossRef CAS PubMed
- K. Nishino, H. Yamamoto, K. Tanaka and Y. Chujo, Org. Lett., 2016, 18, 4064–4067 CrossRef CAS PubMed
- K. Nishino, H. Yamamoto, K. Tanaka and Y. Chujo, Asian J. Org. Chem., 2017, 6, 1818–1822 CrossRef CAS
- H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2017, 56, 254–259 CrossRef CAS PubMed
- X. Wu, J. Guo, Y. Cao, J. Zhao, W. Jia, Y. Chen and D. Jia, Chem. Sci., 2018, 9, 5270–5277 RSC
- J. Li, C. Yang, X. Peng, Y. Chen, Q. Qi, X. Luo, W.-Y. Lai and W. Huang, J. Mater. Chem. C, 2018, 6, 19–28 RSC
- X. Wu, J. Guo, J. Zhao, Y. Che, D. Jia and Y. M. Chen, Dyes Pigm., 2018, 154, 44–51 CrossRef CAS
- A. V. Marsh, N. J. Cheetham, M. Little, M. Dyson, A. J. P. White, P. Beavis, C. N. Warriner, A. C. Swain, P. N. Stavrinou and M. Heeney, Angew. Chem., Int. Ed., 2018, 57, 10640–10645 CrossRef CAS PubMed
- S. Kim, J. H. Lee, H. So, J. Ryu, J. Lee, H. Hwang, Y. Kim, M. H. Park and K. M. Lee, Chem. - Eur. J., 2020, 26, 548–557 CrossRef CAS PubMed
- S. Kim, J. H. Lee, H. So, M. Kim, M. S. Mun, H. Hwang, M. H. Park and K. M. Lee, Inorg. Chem. Front., 2020, 7, 2949–2959 RSC
- N. Shin, S. Yu, J. H. Lee, H. Hwang and K. M. Lee, Organometallics, 2017, 36, 1522–1529 CrossRef CAS
- H. So, J. H. Kim, J. H. Lee, H. Hwang, D. K. An and K. M. Lee, Chem. Commun., 2019, 55, 14518–14521 RSC
- M. Kim, C. H. Ryu, J. H. Hong, J. H. Lee, H. Hwang and K. M. Lee, Inorg. Chem. Front., 2020, 7, 4180–4189 RSC
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS
- H. Kaji, H. Suzuki, T. Fukushima, K. Shizu, K. Suzuki, S. Kubo, T. Komino, H. Oiwa, F. Suzuki, A. Wakamiya, Y. Murata and C. Adachi, Nat. Commun., 2015, 6, 8476 CrossRef CAS PubMed
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330–336 CrossRef CAS PubMed
- M. Z. Shafikov, A. F. Suleymanova, R. Czerwieniec and H. Yersin, Chem. Mater., 2017, 29, 1708–1715 CrossRef CAS
- J. Llop, C. Viñas, F. Teixidor, L. Victori, R. Kivekäs and R. Sillanpää, Organometallics, 2001, 20, 4024–4030 CrossRef CAS
- J. M. Oliva, N. L. Allan, P. v. R. Schleyer, C. Viñas and F. Teixidor, J. Am. Chem. Soc., 2005, 127, 13538–13547 CrossRef CAS PubMed
- A. Ferrer-Ugalde, E. J. Juárez-Pérez, F. Teixidor, C. Viñas, R. Sillanpää, E. Pérez-Inestrosa and R. Núñez, Chem. - Eur. J., 2012, 18, 544–553 CrossRef CAS PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision B.01; Gaussian. Inc., Wallingford, CT, 2016 Search PubMed
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed
-
R. Dennington, T. A. Keith and J. M. Millam, GaussView 6, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed
- M. F. Hawthorne, T. E. Berry and P. A. Wegner, J. Am. Chem. Soc., 1965, 87, 4746–4750 CrossRef CAS PubMed
- T. E. Paxson, K. P. Callahan and M. F. Hawthorne, Inorg. Chem., 1973, 12, 708–709 CrossRef CAS
- W. Jiang, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 1996, 35, 3056–3058 CrossRef CAS
- L. Zhang and K. W. Cheah, Sci. Rep., 2018, 8, 8832 CrossRef PubMed
- K. J. Kim, G. H. Kim, R. Lampande, D. H. Ahn, J. B. Im, J. S. Moon, J. K. Lee, J. Y. Lee, J. Y. Lee and J. H. Kwon, J. Mater. Chem. C, 2018, 6, 1343–1348 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: 1H, 1H{11B}, 13C, and 11B{1H} NMR spectra, X-ray crystallographic data in CIF format, UV-vis absorption and PL spectra for o-carboranyl compounds and 9,9-dimethyl-9H-fluorene, emission decay curves, and computational calculation details. CCDC 2057685 for 2FC, 2057686 for 3FC, 2057688 for 4FC, and 2057687 for 4FCH, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00791b |
| ‡ The first and second authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |