
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational design of thioamide peptides as selective inhibitors of cysteine protease cathepsin L†
Hoang Anh T.
Phan
 a,
Sam G.
Giannakoulias
a,
Taylor M.
Barrett
a,
Chunxiao
Liu
ab and
E. James
Petersson
*a
a,
Sam G.
Giannakoulias
a,
Taylor M.
Barrett
a,
Chunxiao
Liu
ab and
E. James
Petersson
*a
aDepartment of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA. E-mail: ejpetersson@sas.upenn.edu
bKey Laboratory for Northern Urban Agriculture of Ministry of Agriculture and Rural Affairs, Beijing University of Agriculture, Beijing 102206, P. R. China
First published on 19th July 2021
Abstract
Aberrant levels of cathepsin L (Cts L), a ubiquitously expressed endosomal cysteine protease, have been implicated in many diseases such as cancer and diabetes. Significantly, Cts L has been identified as a potential target for the treatment of COVID-19 due to its recently unveiled critical role in SARS-CoV-2 entry into the host cells. However, there are currently no clinically approved specific inhibitors of Cts L, as it is often challenging to obtain specificity against the many highly homologous cathepsin family cysteine proteases. Peptide-based agents are often promising protease inhibitors as they offer high selectivity and potency, but unfortunately are subject to degradation in vivo. Thioamide substitution, a single-atom O-to-S modification in the peptide backbone, has been shown to improve the proteolytic stability of peptides addressing this issue. Utilizing this approach, we demonstrate herein that good peptidyl substrates can be converted into sub-micromolar inhibitors of Cts L by a single thioamide substitution in the peptide backbone. We have designed and scanned several thioamide stabilized peptide scaffolds, in which one peptide, RS1A, was stabilized against proteolysis by all five cathepsins (Cts L, Cts V, Cts K, Cts S, and Cts B) while inhibiting Cts L with >25-fold specificity against the other cathepsins. We further showed that this stabilized RS1A peptide could inhibit Cts L in human liver carcinoma lysates (IC50 = 19 μM). Our study demonstrates that one can rationally design a stabilized, specific peptidyl protease inhibitor by strategic placement of a thioamide and reaffirms the place of this single-atom modification in the toolbox of peptide-based rational drug design.
Introduction
In recent decades, there has been an increased interest in the development of peptides as therapeutics and imaging agents.1–3 Peptide-based drugs offer advantages such as high selectivity and potency, low tissue accumulation, relatively predictable metabolism, and safety. Additionally, the ease of obtaining high biological and chemical diversity from standard synthetic procedures makes peptide therapeutics attractive.1,4 Peptides thus stand out as promising candidates to fill the gap between the two main drug categories – traditional small-molecule drugs (smaller than 500 Da) and biologics (larger than 5000 Da).4 Most of the peptides that are currently clinically approved or under active development are targeted for metabolic diseases and cancer.2 However, despite the numbers of known targets for peptide therapeutics and existing peptide libraries, peptides still display certain disadvantages that hinder them from more easily becoming effective drugs.1,2 Peptides are subject to rapid proteolysis, oxidation, display short half-lives and fast renal clearance in vivo, as well as low membrane permeability, thereby exhibiting suboptimal pharmacokinetics.1,2 To address the metabolic stability issues of peptides, modifications at protease cleavage sites using techniques such as synthetic substitutions of amino acid sidechains or the peptide backbone have been developed and utilized to increase resistance to proteolysis.3,5–8Backbone thioamidation is a promising tool that has been shown to improve proteolytic stability of both linear and macrocyclic peptides.9–20 Our laboratory previously demonstrated that a thioamide substitution near the scissile bond of glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide (GIP), two therapeutically relevant peptides for diabetes treatment, significantly enhances their proteolytic stability against dipeptidyl peptidase 4.21 Thioamidation of GLP-1 and GIP increased their half-lives up to 750-fold without significantly compromising their cellular activity; the thioamide GLP-1 analogue was also biologically active in rats and exhibited improved potency for glycemic control compared to its native, all-amide GLP-1 counterpart.21 Motivated by these results showing thioamide stabilization effects at P2 and P1 positions (positions numbered from the scissile bond by convention), our laboratory developed a fluorescence sensor design to systematically study the positional effects of thioamide substitution against different cysteine proteases (papain, cathepsins L, V, K, B, and S) and serine proteases (trypsin, chymotrypsin, and kallikrein).22–24 Intriguingly, we found that thioamide positional effects differ not only between serine proteases and cysteine proteases, but also between members of the same protease family despite their high homology (31–59% sequence identity) and mechanistic similarity.22,24 We also successfully utilized data from these systematic studies to design a two-site stabilized thioamide peptide specifically targeting neuropeptide Y1-receptor expressing MCF-7 breast cancer cells.22 With the experimental data from these systematic studies, we recently developed a Rosetta machine learning model that accurately classifies positional effects of thioamides on proteolysis by these cysteine and serine proteases which can be used to rationally design stabilized peptides for therapeutic and imaging applications.23
Given this precedent, in this study, we aim to further utilize the strategic incorporation of thioamides to develop stabilized peptides as protease inhibitors, more specifically, inhibitors of the cysteine protease cathepsin L (Cts L). Among the 500–600 proteases identified in mouse and human, the cathepsin (Cts) family includes proteases that orchestrate numerous critical physiological processes and are involved in many different diseases such as neurological disorders, cardiovascular diseases, arthritis, obesity, and cancer.25,26 Cysteine Cts proteases, which comprise 11 members in humans (Cts B, C, F, H, K, L, O, S, V/L2, X, and W), belong to the papain-like cysteine protease family. They have been shown to be upregulated in many cancer types and play critical roles in cancer progression.27,28 Cts L is an ubiquitously expressed endopeptidase that is uniquely involved in the major histocompatibility complex (MHC) class II processing pathway,29 prohormone or proneuropeptide processing,30–32 and autophagy,33 as well as cardiac homeostasis and signal transduction.34–37 Cts L is highly expressed in tumors associated with breast cancer, colorectal cancer, and pancreatic adenocarcinoma.27,38 Cts L participates in the degradation of epithelial cadherins, transmembrane receptors, and extracellular domains of cell adhesion molecules in cancer cells, thereby disrupting cell adhesion, promoting tumor invasion, and possibly underlying resistance to chemotherapy.27,39,40 Importantly, Ou et al. recently showed that lysosomal activation of SARS-CoV-2's spike (S) glycoproteins by the host cell's Cts L, but not Cts B, is critical for its cellular entry via endocytosis during infection.41 These researchers showed that treatment with Cts L inhibitor SID 26681509 decreased SARS-CoV-2 pseudovirus entry into HEK 293/hACE2 cells by more than 76%, highlighting the role of Cts L in lysosomal priming of the virus upon entry.41 There is also evidence for elevated Cts L circulating level in COVID-19 patients.42 This is significant as Cts L inhibitors have now been identified as promising therapeutic agents to inhibit SARS-CoV-2 for potential treatment of COVID-19.41–44 It has been proposed that a protease inhibitor cocktail composed of a Cts L-specific inhibitor as well as serine protease inhibitors could be a safe and novel treatment for COVID-19 patients.45 Although it is desirable to develop Cts L inhibitors, there are currently no specific inhibitors for Cts L that have advanced to clinical trials as it is challenging to obtain selectivity against closely related Cts family members.46
Many of the known Cts L inhibitors, which are mostly small molecules, resemble its physiological substrate and often have electrophilic “warheads” (e.g. epoxide ring, acyloxymethyl ketone, aziridine, vinylsufonate, nitrile, or thiosemicarbazone) that are strategically placed to trap the catalytic Cys25 residue of Cts L.46 This follows a logic common to the development of covalent protease inhibitors, wherein a good protease substrate is converted into an inhibitor by strategic incorporation of such warheads.46–48 Inspired by this principle, we demonstrate herein that good peptidyl substrates of Cts L, designed by combining knowledge about substrate sequence specificity from previous positional scanning with our protease sensor studies, can be converted into good inhibitors of Cts L by a single thioamide substitution to the peptide backbone. Unlike the warhead strategy, thioamide modification only renders the substrate inert to proteolysis and does not result in covalent inhibition. There are many advantages to our strategy as concerns about the use of covalent enzyme inhibitors linger in spite of several successes with the aforementioned warhead approach.46,49,50 With our thioamidation approach, we hope to potentially overcome challenges with selectivity and off-target effects of Cts inhibitors that are normally encountered with small molecule protease inhibitors.46 An inhibitor with high specificity for a single Cts is a powerful tool compound for studying its role in health and disease and can serve as a therapeutic lead where such specificity is necessary to avoid undesirable side effects. In this study, we examine the stability of several thioamide peptide scaffolds toward Cts proteolysis and identify one peptide that shows resistance to Cts L, Cts V, Cts K, Cts S, and Cts B while inhibiting only Cts L. We also show that this stabilized thioamide peptide can inhibit Cts L in human hepatocellular liver carcinoma (HepG2) whole cell lysate. To our knowledge, this is the highest affinity thioamide-based protease inhibitor to date. Our studies show the potential of utilizing thioamides to stabilize and convert good peptidyl substrates into specific protease inhibitors.
Results and discussion
Designing and examining thioamide peptide inhibitors of cathepsin using a fluorescence protease sensor system
Previously, our laboratory has designed a fluorescent protease sensor system that capitalizes on the fluorophore quenching property of thioamides to monitor real-time protease activity.22,24,51,52 For our first-generation sensors, a thioamide and a fluorophore are placed on opposite sides of the scissile bond, thereby leading to a turn on of fluorescence upon cleavage.51,52 Building upon this design, we generated a series of peptides with 7-methoxycoumarin-4-yl-alanine (Mcm; μ) at both the N-terminus and C-terminus to systematically study thioamide positional effects on proteolysis of cysteine24 and serine22 protease substrates. Once the doubly-labeled peptide is cleaved, there will be a turn on in fluorescence as one of the fluorophores will be separated from the thioamide, regardless of the placement of the thioamide.22,24 This allows real-time monitoring of proteolysis kinetics. From our previous systematic studies with cysteine proteases (Cts V, Cts K, Cts S, Cts B, Cts L, and papain), we learned that thioamide substitution at the P1 position significantly slowed the proteolysis rates of the generic μLLKAAAμ substrate by Cts V, Cts K, Cts S, and Cts L, but not significantly by Cts B and papain.23,24 By convention, amino acids N-terminal to the scissile bond are denoted PX positions (e.g. P1, P2, P3; non-primed positions), while those C-terminal to the scissile bond are considered PX′ positions (e.g. P1′, P2′, P3′; primed positions). Interestingly, we found that the P1 thioamide peptide, μLLKSAAAμ (KSP1), not only showed the highest level of protease resistance to Cts L, but also served as a potent inhibitor of Cts L (KI = 0.87 μM; Fig. S13 and Table S12†). Although this preliminary inhibition data was exciting, the KSP1 peptide would not be very stable in vivo since it could still be efficiently cleaved by other cysteine proteases (papain, Cts B, Cts V, Cts K, and Cts S)23,24 as the sequence of this peptide was designed to be generic. Motivated by these results, we thus envisioned advancing this approach to design and scan for a sequence-optimized, thioamide-containing peptide specific inhibitor to Cts L, yet being stabilized in the presence of other closely related cathepsins without inhibiting them.The rationale of our peptide design entails two main steps: (1) to design all-amide peptides that are good substrates of Cts L, then (2) to turn those substrates into stabilized peptides inhibiting Cts L by strategic placement of a single thioamide. In this study, utilizing the peptide sensor design from our previous studies, our peptide inhibitor candidates contained two coumarins (μ residues) at their termini, allowing for quick initial identification of peptides that showed resistance to proteolysis by cathepsins via steady-state protease assays (Fig. 1).22,24 To design the amino acid sequences for initial scanning, the primed positions of these peptides were kept generic and consistent with our previous studies by retaining alanine at the P1′, P2′, and P3′ positions. For the non-primed positions, sequence design was guided by a comprehensive substrate profiling study using a synthetic library of 160![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 fluorogenic tetrapeptides by Choe et al. (Fig. S1†).53 With the knowledge of different amino acid preferences by different cathepsins, we identified peptide sequences that might be specific to Cts L. At the P1 position, all human cathepsins prefer basic residues, so arginine and lysine were clearly the choice for this position, with arginine being preferred by Cts L.53 P2 is considered the major determinant for substrate specificity of Cts L that differentiates it from Cts K, Cts S, and Cts B, as Cts L has a unique preference for aromatic residues (phenylalanine, tryptophan, tyrosine) at this position.53,54 Similar preference for aromatic residues at P2 position is only observed in Cts V, which is most closely related to Cts L by sequence identity (78% sequence identity).53 As Cts V favors tryptophan and tyrosine over phenylalanine, phenylalanine seemed to be the best choice for P2. For P4, Cts L shows a preference for histidine, prompting us to choose histidine at this position. At P3, however, Cts L has less well defined specificity, but displays some preference for basic residues as well as a few aliphatic amino acids.53 Since we already included a positively charged residue at the P1 position, we decided to incorporate an aliphatic amino acid at P3 to reduce the potential for multiple Cts L cleavage sites. We chose leucine for the P3 position since the data from Choe et al. suggested that Cts L prefers leucine at P3 among the aliphatic amino acids (Fig. S1†).53 Regarding placement of the thioamide, the P1 position was chosen based on our previous systematic studies with the cysteine proteases that showed P1 thioamide peptide KSP1 gave the highest level of protease resistance.23,24 Using this rationale, the first series of peptides synthesized via solid-phase peptide synthesis for initial scanning with steady state protease assays were: μHLFRAAAμ (R3A), μHLFKAAAμ (K3A), and their P1 thioamide analogs (RS3A and KS3A) (Fig. 1A). The thioamide position is denoted as a superscript “S” in the peptide sequences.
000 fluorogenic tetrapeptides by Choe et al. (Fig. S1†).53 With the knowledge of different amino acid preferences by different cathepsins, we identified peptide sequences that might be specific to Cts L. At the P1 position, all human cathepsins prefer basic residues, so arginine and lysine were clearly the choice for this position, with arginine being preferred by Cts L.53 P2 is considered the major determinant for substrate specificity of Cts L that differentiates it from Cts K, Cts S, and Cts B, as Cts L has a unique preference for aromatic residues (phenylalanine, tryptophan, tyrosine) at this position.53,54 Similar preference for aromatic residues at P2 position is only observed in Cts V, which is most closely related to Cts L by sequence identity (78% sequence identity).53 As Cts V favors tryptophan and tyrosine over phenylalanine, phenylalanine seemed to be the best choice for P2. For P4, Cts L shows a preference for histidine, prompting us to choose histidine at this position. At P3, however, Cts L has less well defined specificity, but displays some preference for basic residues as well as a few aliphatic amino acids.53 Since we already included a positively charged residue at the P1 position, we decided to incorporate an aliphatic amino acid at P3 to reduce the potential for multiple Cts L cleavage sites. We chose leucine for the P3 position since the data from Choe et al. suggested that Cts L prefers leucine at P3 among the aliphatic amino acids (Fig. S1†).53 Regarding placement of the thioamide, the P1 position was chosen based on our previous systematic studies with the cysteine proteases that showed P1 thioamide peptide KSP1 gave the highest level of protease resistance.23,24 Using this rationale, the first series of peptides synthesized via solid-phase peptide synthesis for initial scanning with steady state protease assays were: μHLFRAAAμ (R3A), μHLFKAAAμ (K3A), and their P1 thioamide analogs (RS3A and KS3A) (Fig. 1A). The thioamide position is denoted as a superscript “S” in the peptide sequences.
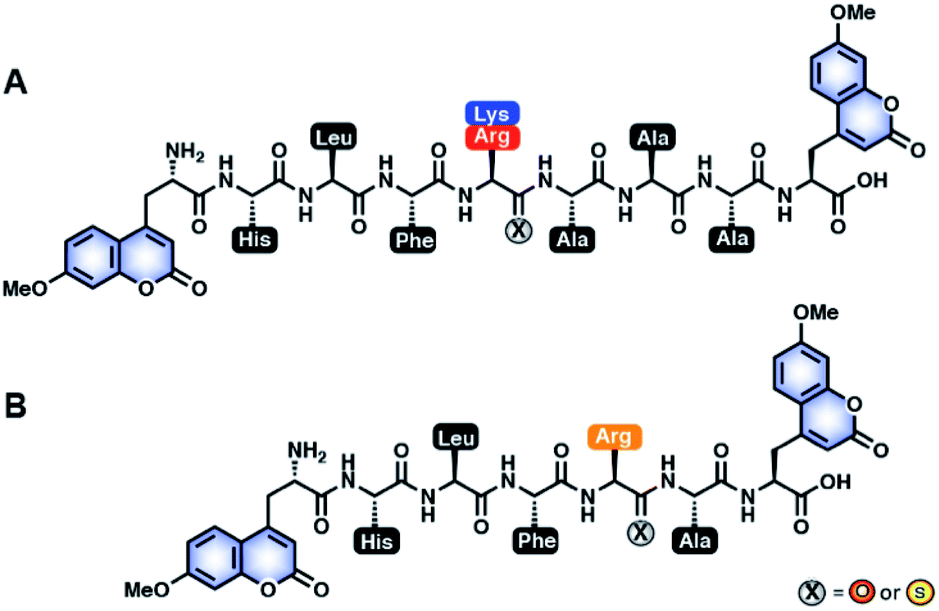 | ||
| Fig. 1 All-amide peptides and thioamide peptides investigated in this study. (A) Sequence-optimized all-amide and thioamide peptides of K3A (μHLFKAAAμ) and R3A (μHLFRAAAμ) for initial steady-state protease scanning with Cts L, Cts V, Cts K, Cts S, and Cts B and inhibition studies with Cts L. The peptides contain 7-methoxycoumarin-4-yl-alanine (MCM; μ) residues at both termini, and either an amide (X = O) or a thioamide (X = S) residue at the denoted P1 position. (B) Truncated all-amide and thioamide R1A peptide (μHLFRAμ). The RS1A peptide, which shows stabilization against all five proteases, was further investigated for specificity and inhibitory effect in HepG2 whole cell lysate. | ||
To validate our design, we needed to first confirm whether the all-amide peptides were good substrates of the proteases before proving that the thioamide substitution could transform them into stabilized peptide inhibitors. For ease of comparison between thioamide positions and protease, raw fluorescence measurements were normalized and are presented in Fig. 2 (primary data are shown in ESI, Fig. S2–S6†). Initial rates of proteolysis were determined for each cleavage reaction (Table 1). High performance liquid chromatography (HPLC) and matrix-assisted laser desorption ionization mass spectrometry (MALDI MS) were used to confirm the cleavage sites in all assays (Tables S6–S11 and Fig. S7–S12;† cleavage sites summarized in Table S5†). In the absence of protease, no significant changes in fluorescence intensities nor degradation of the peptides in the assay buffers were observed. Both of the all-amide peptides K3A and R3A were recognized and efficiently cleaved by all five proteases, confirming that these were indeed good substrates of Cts L (Fig. 2 and Table 1). It is worth noting that the all-amide peptides K3A and R3A were both cleaved at the P1 position by all of the proteases, consistent with the fact that these five cysteine cathepsin proteases have high preferences for recognizing and cleaving their substrates at basic residues (Table S5†). Interestingly, for the all-amide peptides K3A and R3A, cleavage sites other than the expected P1 position was also observed with Cts L, Cts V, and Cts K, while the peptides were cleaved at only the P1 position by Cts S and Cts B (Tables S6, S8 and Fig. S7, S9†). This likely reflects the fact that Cts L, Cts V, and Cts K are most closely related in sequence identity, resulting in similar preferences for substrate specificity. We then performed the assays with the thioamide analogs, where P1 thioamide stabilization was observed with Cts L, Cts V, Cts K, and Cts S (Fig. 2 and Table 1). This supports our choice of thioamide placement at the P1 position and is consistent with our previous findings reported in Liu et al. and Giannakoulias et al., where we observed that P1 thioamides retarded proteolysis by Cts V, Cts K, Cts S, and Cts L, but not Cts B.23,24 Substituting the thioamide at the P1 position thus not only stabilized the P1 position, but also resulted in multiple-site stabilization effects in the cases with Cts L, Cts V, and Cts K (Tables S7, S9 and Fig. S8, S10†). We have previously observed similar multiple-site stabilization with thioamide substrates of serine proteases, which we were able to exploit to stabilize cancer cell imaging peptides at two positions with a single thioamide modification.22 Overall, having a thioamide at the P1 position here rendered the KS3A and RS3A peptides completely resistant to proteolysis by Cts V, Cts K, and Cts S, while significantly slowing the rate of proteolysis by Cts L (Table 1). The only exception to this P1 thioamide effect was with Cts B, where the KS3A and RS3A peptides were cleaved at the penultimate C-terminal alanine residue as indicated by the slashes – μHLFKSAA/Aμ or μHLFRSAA/Aμ (Tables S7, S9 and Fig. S8, S10†). This pattern of cleavage by Cts B aligns well with the fact that Cts B is known to be both an endopeptidase and a carboxydipeptidase (exopeptidase).28 The KS3A and RS3A peptides were also slowly cleaved by Cts L at the same position. We therefore postulated that a truncated version of this peptide, μHLFRSAμ (RS1A; Fig. 1B), would eliminate this cleavage site by Cts L and Cts B and stabilize the peptide. As expected, the all-amide version of this shorter peptide (R1A; μHLFRAμ) was recognized and cleaved by all five cathepsins – Cts L, Cts V, Cts K, Cts S, and Cts B (Fig. 2, S11 and Tables 1, S10) while the corresponding thiopeptide RS1A was left intact (Fig. 2, S12 and Tables 1, S11). Since preceding literature and our inhibition assays with the K3A and R3A peptides suggested that the arginine substrates have higher affinity for Cts L, we proceeded to investigate in depth the RS1A peptide instead of its lysine analog (μHLFKSAμ) as discussed in the next section.53
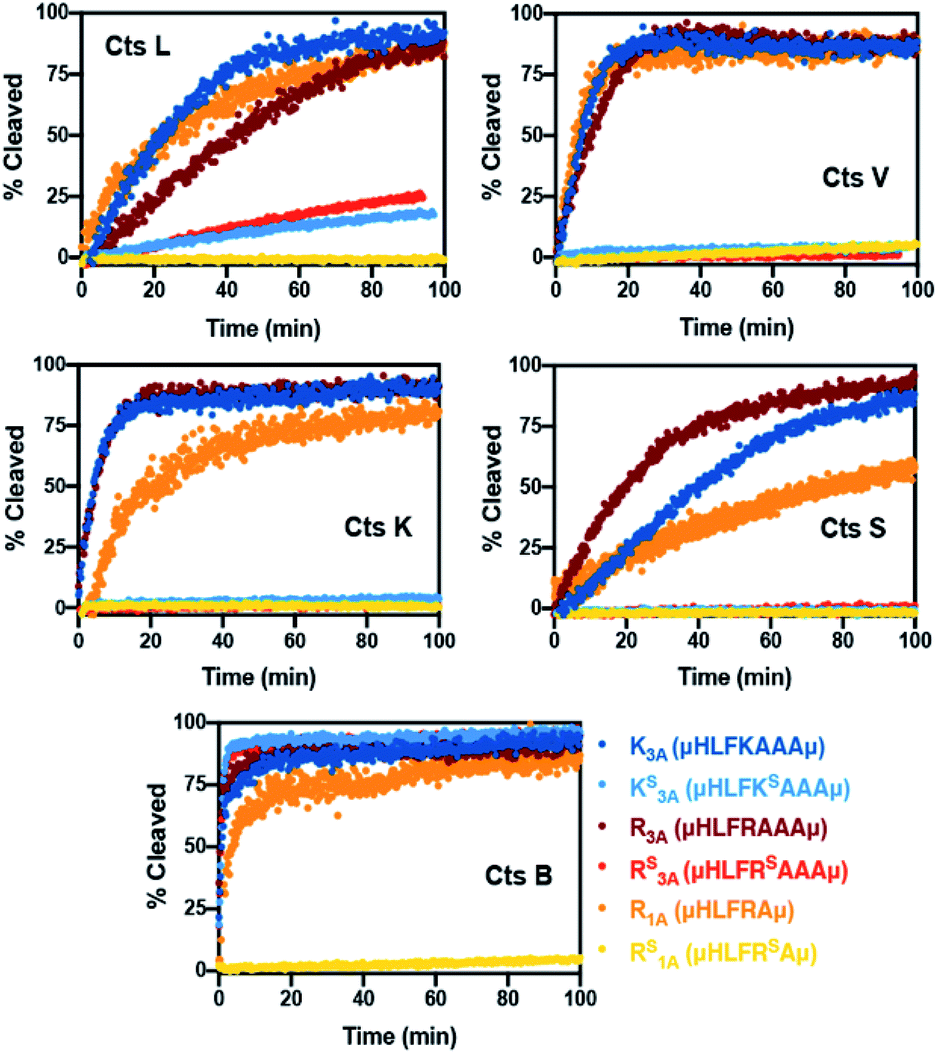 | ||
| Fig. 2 Summary of normalized cleavage data with cysteine cathepsin proteases. Peptides (7.5 μM) were incubated in the absence or presence of Cts B (37.6 nM), Cts K (42.6 nM), Cts L (30.3 nM), Cts S (21.6 nM) or Cts V (20.5 nM) in 100 mM sodium acetate, 100 mM NaCl, 1 mM EDTA, 5 mM DTT, and pH 5.5 at 27 °C. The original fluorescence was originally monitored at 390 nm with an excitation wavelength of 325 nm, then the fluorescence data was converted to percent cleavage rates. All traces are the average of three independent trials. Raw fluorescence data and more details of the assays are described in the ESI.† | ||
| Peptide | Sequence | Cts L | Cts V | Cts K | Cts S | Cts B |
|---|---|---|---|---|---|---|
| a All rates are reported in μM min−1. Rates and standard errors are calculated by fitting to linear regression function in Prism 8. b MALDI MS and HPLC confirmed essentially no cleavage with these peptides. Details are reported in the ESI. | ||||||
| K3A | μHLFKAAAμ | 0.181 ± 0.003 | 0.511 ± 0.025 | 0.689 ± 0.045 | 0.110 ± 0.002 | 1.921 ± 0.206 |
| KS3A | μHLFKSAAAμ | 0.014 ± 0.000 | 0.002 ± 0.000b | 0.002 ± 0.000b | 0.000 ± 0.000b | 3.643 ± 0.153 |
| R3A | μHLFRAAAμ | 0.112 ± 0.003 | 0.363 ± 0.014 | 0.564 ± 0.024 | 0.189 ± 0.004 | 2.747 ± 0.265 |
| RS3A | μHLFRSAAAμ | 0.023 ± 0.000 | 0.002 ± 0.000b | 0.001 ± 0.000b | 0.002 ± 0.000b | 3.968 ± 0.209 |
| R1A | μHLFRAμ | 0.125 ± 0.003 | 0.553 ± 0.021 | 0.402 ± 0.021 | 0.053 ± 0.000 | 1.034 ± 0.074 |
| RS1A | μHLFRSAμ | 0.000 ± 0.000b | 0.005 ± 0.000b | 0.000 ± 0.000b | 0.000 ± 0.000b | 0.003 ± 0.000b |
Investigating inhibitory effects with cathepsin proteases
Inhibition assays with Cts L were performed with all of the peptides from the first series (K3A and R3A peptides and their P1 thiopeptides) as well as the RS1A peptide (Tables 2, S13–S18 and Fig. S14–S19†). For these assays, Z-Phe-Arg-AMC (Z-FR-AMC, where Z is benzyl and AMC is 7-aminomethylcoumarin), which is a commercial fluorogenic substrate of Cts L, was used. As the Z-FR-AMC substrate was cleaved by Cts L, its turn-on fluorescence was monitored at 460 nm with an excitation wavelength of 380 nm, which is different from the wavelength used for monitoring potential cleavages of our peptides with the μ residues (λexcitation = 325 nm; λemission = 390 nm), allowing them to be separately monitored without interference. The all-amide peptides (K3A and R3A) showed some inhibitory effects, which was expected since these substrates compete for the active site of Cts L (Table 2). The corresponding thioamide peptides, KS3A and RS3A, were very good inhibitors of Cts L, with respective KI values of 0.60 ± 0.15 μM and 0.52 ± 0.12 μM (Table 2). The RS1A peptide (μHLFRSAμ), which showed resistance to proteolysis by all five cathepsins, was also a good inhibitor of Cts L with a KI value of 1.11 ± 0.22 μM. Although the RS3A and KS3A exerted slightly better inhibitory effects than the truncated peptide RS1A, they are not as ideal because we established in the steady-state protease assays that they could be cleaved by Cts L and by Cts B at the C-terminus. Lastly, the role of the two coumarins was examined with the coumarin-free peptide HLFRSA (RS1A*). Although this peptide showed resistance to cleavage by Cts L (Fig. S20†), it was a significantly weaker inhibitor of Cts L (KI = 13.23 ± 6.89 μM), indicating an important role for the coumarins in binding. The finding that the thioamide peptides could serve as potent inhibitors of Cts L was exciting because our previous investigations of thioamide-stabilized protease substrates had found them to be only fairly weak inhibitors, implying that the thioamide primarily acted to disrupt binding to the protease.21,22,24 Indeed, earlier investigations of thioamide effects on proteolysis had found similar results for di- and tripeptides.14,15,20,55 Thus, we wished to further investigate the mechanism of inhibition.| Peptide | K I (μM) | α | αK I or K′I (μM) |
|---|---|---|---|
| a Data was obtained by fitting to the mixed inhibition model that allows us to simultaneously determine the KI and the mechanism of inhibition using the output “alpha” (α) in GraphPad Prism 8 software.56,57 Detailed analysis are described in the ESI. | |||
| K3A | 3.05 ± 0.64 | 1.91 ± 0.92 | 5.81 ± 3.06 |
| KS3A | 0.60 ± 0.15 | 2.18 ± 1.35 | 1.30 ± 0.87 |
| R3A | 1.68 ± 0.49 | 1.33 ± 0.78 | 2.23 ± 1.47 |
| RS3A | 0.52 ± 0.12 | 1.83 ± 0.99 | 0.95 ± 0.56 |
| RS1A | 1.11 ± 0.22 | 1.61 ± 0.69 | 1.79 ± 0.85 |
| RS1A* | 13.23 ± 6.89 | 1.71 ± 1.89 | 22.58 ± 27.61 |
From initial evaluation of the kinetic parameters obtained by fitting data to a Michaelis–Menten model (details are shown in the ESI; Tables S13–S18†), there was generally a decrease in Vmax, but either a minor increase or no significant change in KM as the concentration of the inhibitors was increased. This eliminates the possibility of these peptides as acting as purely competitive inhibitors or uncompetitive inhibitors, suggesting that they are likely mixed-type inhibitors of Cts L based on the traditional categorization of inhibitors. This can be easily visualized in Lineweaver–Burke plots (Fig. S14–S19†), confirming the high likelihood of a mixed-type mechanism of inhibition, as shown in Fig. 3 for the RS1A peptide. To further evaluate the mechanism of inhibition and to obtain the KI values, kinetics data were fitted using a non-linear regression analysis with the mixed inhibition model that allows us to determine the KI and the mechanism of inhibition using the output “alpha” (α) using GraphPad Prism software.56 All of the α values are consistently between 1–2, confirming these peptides are likely mixed-type inhibitors, with element of competitive inhibition since a value of α > 1 suggests tighter binding to the free enzyme (Table 2).57 Interestingly, mixed inhibitors of Cts L have been previously shown to be promising antiviral candidates. A study with a high-throughput screening of 5000 molecules discovered a small-molecule inhibitor of Cts L (5705213) with a mixed inhibition mechanism that can inhibit Cts L-mediated cleavage of the viral glycoproteins derived from all four viruses – SARS-CoV, Ebola, Hendra, and Nipah viruses, a process that is essential for entry into host cells.58
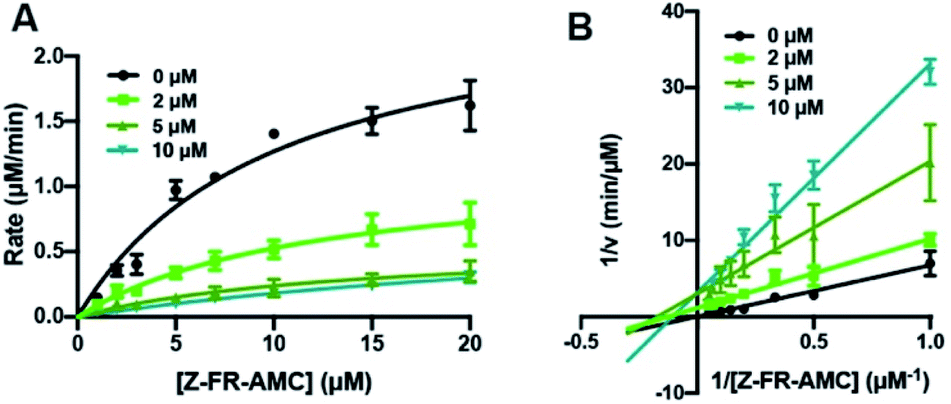 | ||
| Fig. 3 Cts L proteolysis inhibition by RS1A peptide (μHLFRSAμ). (A) Michaelis–Menten analysis and (B) Lineweaver–Burke plot of Cts L activity in the absence of presence of three different concentrations of the RS1A peptide (μHLFRSAμ). Various concentrations of the fluorogenic Z-Phe-Arg-AMC (Z-FR-AMC) were incubated in the presence of 37.93 nM Cts L. Averages of three trials along with the standard deviations are shown. Raw fluorescence traces are reported in Fig. S18.† These suggest that the peptide is likely a mixed-type inhibitor, with element of competitive inhibition since a value of α = 1.61 (α > 1) suggests tighter binding to the free enzyme. | ||
To serve as useful specific inhibitors of Cts L, the thioamide peptides must also be inert to cleavage by other proteases that may be present in vivo while not inhibiting them. Since the sequence-optimized RS1A peptide herein showed resistance to proteolysis by all five proteases, we then assessed the specificity of inhibition by the RS1A peptide by determining whether it could also effectively inhibit Cts V, Cts K, Cts S, and Cts B using assays similar to the Z-FR-AMC used with Cts L. The RS1A peptide exhibited a 26-fold increase in KI and is a weak mixed-inhibitor of Cts V, with a KI of 26.22 ± 8.42 μM (Fig. S21 and Table S19†). No significant differences in the values of kcat and KM were observed for proteolysis of Z-Leu-Arg-AMC (Z-LR-AMC) by Cts K or Cts S in the presence of >30 μM concentrations of RS1A peptide (Fig. S22, S23 and Tables S20, S21†). Similarly, essentially no differences in kcat and KM were observed for Cts B proteolysis of the Z-Arg-Arg-AMC (Z-RR-AMC) substrate in the presence of up to 50 μM RS1A peptide with Cts B (Fig. S24 and Table S22†). In summary, in addition to acting as a potent inhibitor of Cts L (KI = 1.11 ± 0.22 μM), the RS1A peptide is ≥25-fold selective against other Cts family members tested, as it only weakly inhibits the closely related Cts V (78% sequence identity with Cts L) and shows little to no significant inhibitory effects with Cts K, Cts S, and Cts B (58%, 55%, and 26% respective sequence identity with Cts L).
Evaluation of cathepsin L inhibition in HepG2 whole cell lysate
Cts L has been considered an appealing target for cancer treatment because its expression has been linked to tumor progression and metastases of different types of cancers.38,61 In particular, it has been previously shown that increased Cts L expression is associated with worse outcome in hepatocellular carcinoma patients62 and elevated Cts L activity has been found in malignant liver cancer HepG2 cells.63 To further validate our RS1A peptide inhibitor of Cts L, we investigated whether it could effectively inhibit Cts L activity in whole cell lysate from the HepG2 human hepatocellular liver carcinoma cell line. Using a commercially available fluorescence based Cts L activity kit, we incubated different doses of RS1A peptide with HepG2 whole cell lysate. We found that the RS1A peptide could effectively inhibit fluorescent reporter activity in the HepG2 whole cell lysate (IC50 = 19.3 ± 4.5 μM) (Fig. 4). Significantly, MALDI MS and HPLC data showed that this peptide's half-life was 28.6 hours, which was approximately 238 times more stable than its all-amide counterpart, R1A, with a half-life of only 7.2 minutes in HepG2 whole cell lysate (Fig. 4C and S26†). The fact that this thioamide peptide showed great stability in the presence of other proteases and cellular components in the HepG2 whole cell lysate further corroborated the enhanced stability we previously observed in steady-state protease assays with individual cathepsins (Cts L, V, K, S, and B). Excitingly, our preliminary data showed that RS1A peptide could also inhibit Cts L in human MDA-MB-231 breast cancer cells overexpressing Cts L (Fig. S47†).42,64 These findings establish exciting precedent for translating RS1A to in vivo assays to determine the impact of highly specific Cts L inhibition on processes such as cancer cell growth and viral uptake.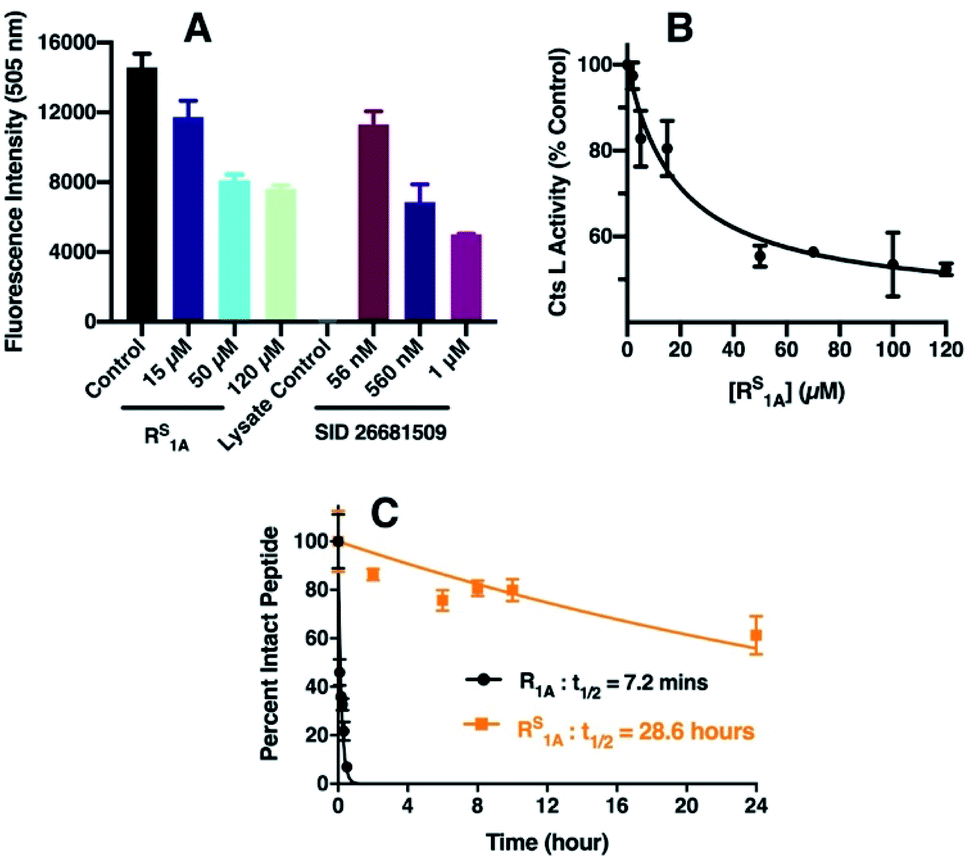 | ||
| Fig. 4 Inhibition of Cts L activity by RS1A peptide in HepG2 whole cell lysate. (A) Cts L activity in HepG2 cell lysate monitored by fluorescence intensity at 505 nm. The control (black bar) was done without the inhibitor. The colored bars show fluorescence signals in the presence of select RS1A peptide concentrations. SID 26681509, a known Cts L inhibitor,59,60 was used as the positive control. The lysate control was a background control for any inherent fluorescence signals from the cell lysate. (B) Cts L activity at different inhibitor concentrations calculated by taking the % of the average fluorescence signal from the control (without inhibitor). Error bars represent the standard deviations from three trials. The IC50 value was obtained from fitting to a sigmoidal dose–response equation in GraphPad Prism 8 (details of fitting are in the ESI†). (C) Stability of the all-amide peptide (R1A) and thioamide-peptide RS1A in HepG2 lysate as monitored by HPLC. | ||
Computational modeling
In order to rationalize the specific inhibitory effects of our peptides, we utilized computational modeling to flexibly dock the longer peptide RS3A and the truncated peptide RS1A with Cts L and the other four cathepsins investigated in this project. Interestingly, exclusively in the Cts L simulations, we observe that the P1 thioamide bond N–H of the RS3A peptide can interact with His163, which is part of the Cts L catalytic triad (Fig. 5A). Hydrogen bonding in this manner would prevent His163 from efficiently deprotonating Cys25, thereby attenuating the proteolytic activity of Cts L and making the RS3A peptide a good inhibitor. Similarly, with the truncated peptide RS1A, only with Cts L, did we observe the interaction between the P1 thioamide N–H group of the peptide and His163 (Fig. 5B). This hydrogen bond would be expected to be stronger for the thioamide than for the amide.5,65 This may explain why both the RS3A and RS1A peptides can effectively inhibit Cts L.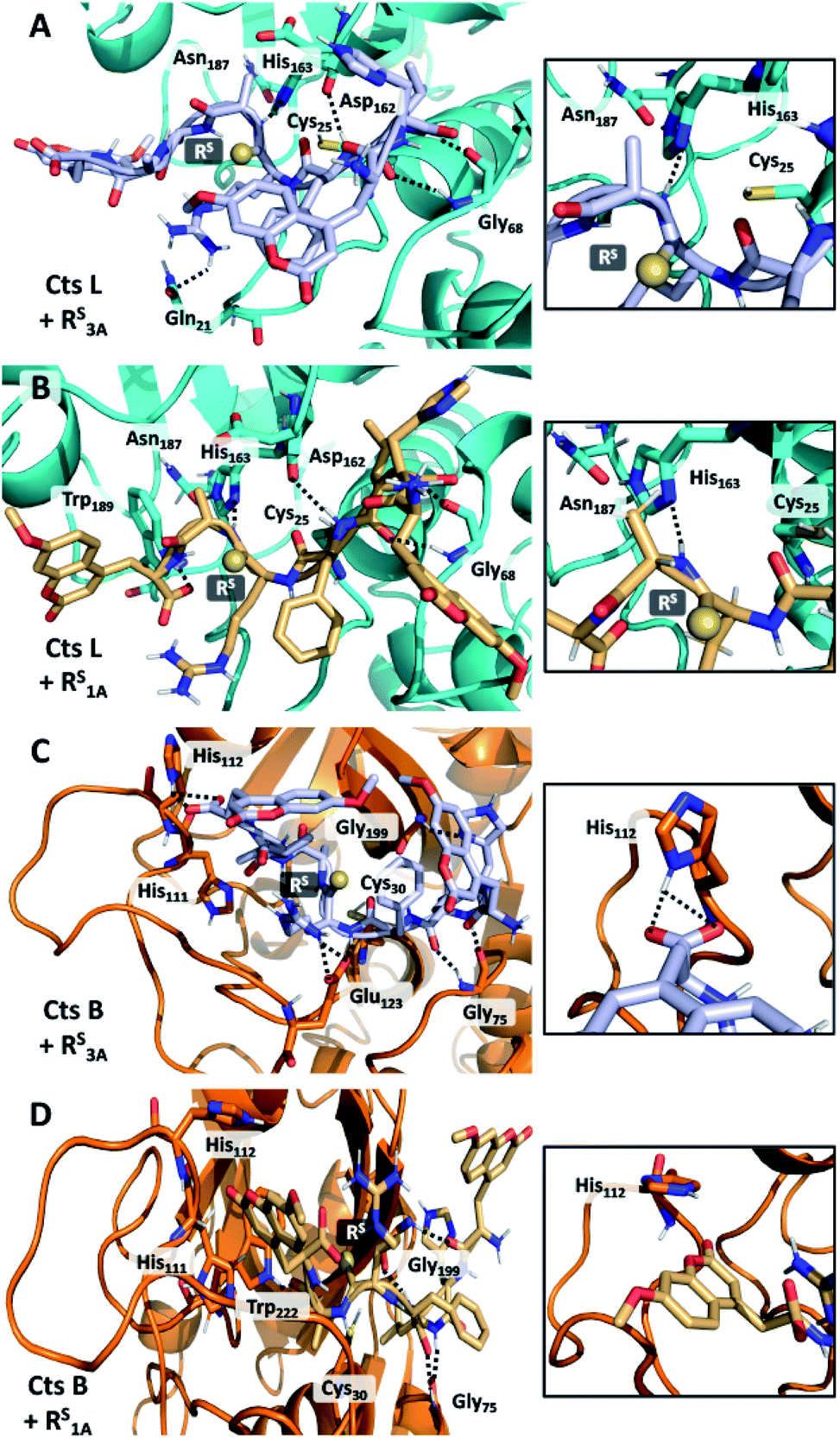 | ||
| Fig. 5 Interactions of RS3A and RS1A peptides in cathepsin L and cathepsin B active sites. (A and B) Docked structures of RS3A (A) and RS1A (B) with Cts L. The N–H group of Ala of either peptide forms hydrogen bond with His163 of the Cts L's catalytic triad. (C) Structure shows interaction of the C-terminal μ of RS3A peptide with His112 on the occluding loop of Cts B, allowing the peptide to be cleaved in a carboxydipeptidase manner and may explain why this peptide is not stabilized against Cts B. (D) The truncated peptide RS1A no longer possess this interaction with His112, which may protect it against proteolysis by Cts B in addition to other proteases. | ||
Our computational modeling can also be used to reasonably explain our other experimental data. From the steady-state protease assays, we found that the only exception to the P1 thioamide stabilization effect was with Cts B, where the RS3A peptide was cleaved at the last two C-terminal alanine residues (μHLFRSAA/Aμ), which is consistent with the fact that Cts B is both an endopeptidase and a carboxydipeptidase.28 Upon examination of the docked structure of the RS3A and Cts B, we found that the carboxylic acid of the C-terminal μ of the peptide interacts with His112 on the occluding loop, which is one of the two histidines (His111/His112 or His110/His111) known in the literature to anchor the C-terminal carboxylate of substrates to give Cts B its carboxydipeptidase properties (Fig. 5C).66,67 The truncated peptide RS1A eliminates this interaction, thus protecting the peptide from proteolysis by Cts B and making it inert to all five cathepsins L, V, K, S, and B while specifically inhibiting Cts L (Fig. 5D).
In an effort to further rationalize why incorporation of a thioamide at the P1 position imbues inhibitory effects for both the longer peptide RS3A and shorter peptide RS1A with Cts L and not the other cathepsins, we performed the following two analyses. The first analysis investigated the change in distances observed between the active site cysteine sulfur and the scissile bond carbonyl carbon upon incorporation of the thioamide. We detected large increases of up to 1.2 Å in this distance (placing the active site residue outside the range for nucleophilic attack) for the two Cts L peptides of interest (Table S25†). Importantly, despite this change in backbone geometry, the key histidine hydrogen bonding interactions were preserved. Our second retrospective analysis utilized unsupervised machine learning (KMeans Clustering) of residue-level energy differences between amide and thioamide peptide complexes from our structural models. Energy feature clustering analysis demonstrated that the Cts L peptides were clustered with each other, but separately from all other clusters (Fig. 6). These data indicate that the changes in energy associated with thioamidation in Cts L complexes are distinct when compared with thioamidation energy changes for the other cathepsins. Taken together, our identification of relevant hydrogen bonding interactions, tolerance of the complexes to the incorporation of a P1 thioamide (change in distance upon removal of constraints), and energy feature clustering identify distinct aspects of the RS3A and RS1A complexes with Cts L that can explain the mechanism of their specific inhibition: thioamidation disrupts binding of the peptides to other proteases while it strengthens a hydrogen bonding interaction with Cts L that keeps RS3A or RS1A tightly bound.
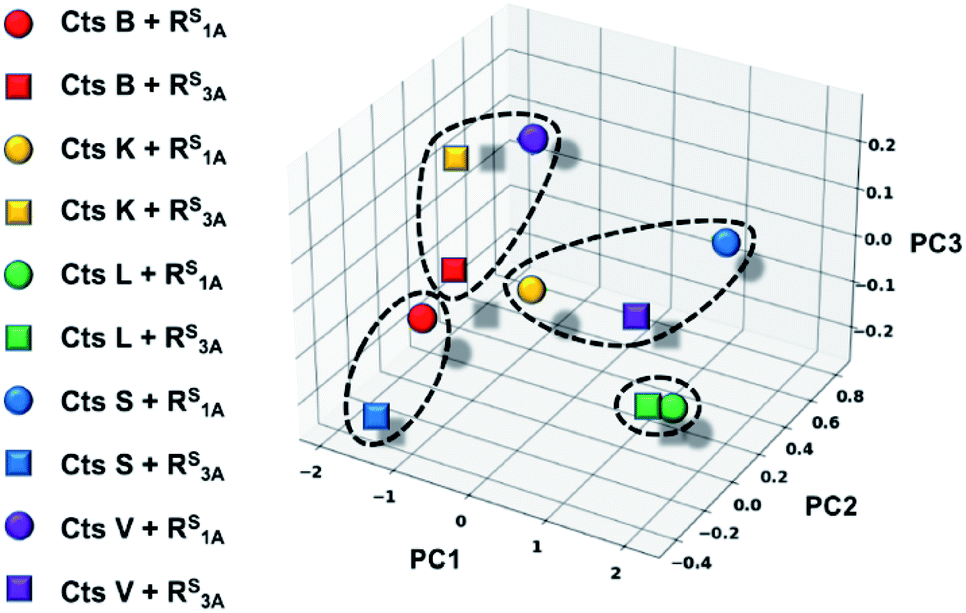 | ||
| Fig. 6 Three-dimensional plot displaying energetic clustering of the cathepsin–peptide complexes. The x, y, and z axes represent the condensed energy vectors of the complexes from principle component analysis (PC1, PC2, PC3). The solid colored shapes correspond to each of the ten protease–peptide complexes simulated in this study, as indicated. The dotted lines surrounding data points indicate the four clusters, including one that comprises the two Cts L complexes. | ||
Conclusions
In summary, we have examined several thioamide peptide scaffolds and identified one peptide, RS1A, that is not only resistant to proteolysis by all five cathepsins (Cts L, Cts V, Cts K, Cts S, and Cts B), but is also a potent, specific inhibitor of Cts L. This peptide can reversibly inhibit Cts L without degradation in HepG2 liver cancer cell lysate and shows promising activity in MDA-MB-231 breast cancer cells. Such a peptide is desirable since peptide-based agents, especially those targeting proteases, are often subject to degradation in vivo. Furthermore, reversible inhibitors like this could potentially address the safety concerns from lack of specificity and potential elicitation of immune responses with irreversible, covalent inhibitors.49,50 While the selectivity against other cathepsins is not as high as some previously reported peptidomimetics (primarily covalent inhibitors),45 this has not been our focus here. Rather, we sought to demonstrate that one can rationally design a potent reversible protease inhibitor by strategic modification of amino acid sidechains and thioamide position based on sensor data from our own work and others. More detailed mechanistic studies, as well as further optimization of this peptide for higher affinity and selectivity will be pursued and reported subsequently. Our studies show the potential of utilizing thioamides as stabilized peptide inhibitors and reaffirm the value of thioamides in the peptide drug design toolbox. In future studies, we will further optimize the thioamide peptide scaffolds by exploring substitutions with unnatural amino acids as well as more carefully examining the role of the N- and C-terminal coumarin groups, removal of which led to a 13-fold decrease in KI. More rigorous biological studies, including assessment of cell permeability, are also warranted to more fully assess the utility of these compounds for in vivo studies of Cts L and possible therapeutic advancement. Given our previous success in machine learning approach to predict thioamide effects and the existing database of sequence effects on cathepsin activity, we may be able to computationally design peptide-based inhibitors for cathepsins as well as for other targets.23 Taken together these approaches can form a paradigm for developing thioamide-stabilized peptides as enzyme inhibitors.Experimental
Protease assays with sensor peptides
For a typical trial, a 7.5 μM peptide solution was incubated in the absence or presence of the appropriate concentration of Cts L (30.3 nM), Cts V (20.5 nM), Cts K (42.6 nM), Cts S (21.6 nM), or Cts B (37.6 nM) in 100 mM sodium acetate, 100 mM NaCl, 1 mM EDTA, 5 mM DTT, and pH 5.5 buffer at 27 °C. The fluorescence was monitored as a function of time at 390 nm with an excitation wavelength of 325 nm using a Tecan M1000 plate reader. Three independent trials were conducted for each assay to ensure reproducibility. More details of the assays, along with the raw data and analysis, are included in the ESI.†Inhibition assays with cathepsins L, V, K, S, and B
For Cts L, various concentrations of its substrate Z-Phe-Arg-AMC (Z-FR-AMC; 1 μM, 2 μM, 3 μM, 5 μM, 7 μM, 10 μM, 15 μM, and 20 μM) were reacted with 37.93 nM Cts L. For Cts V, different concentrations of its substrate Z-Leu-Arg-AMC (Z-LR-AMC; 1 μM, 3 μM, 5 μM, 7 μM, 10 μM, 15 μM, 20 μM, and 25 μM) were reacted with 19.3 nM Cts V. For Cts K, different concentrations of the substrate Z-LR-AMC (10 μM, 15 μM, 20 μM, 30 μM, 40 μM, 60 μM, 80 μM, and 100 μM) were reacted with 53.2 nM Cts K. For Cts S, various concentrations of the substrate Z-LR-AMC (15 μM, 20 μM, 25 μM, 30 μM, 40 μM, 60 μM, 80 μM, and 100 μM) were reacted with 33.8 nM Cts S. Lastly, for Cts B, various concentrations of the substrate Z-Arg-Arg-AMC (Z-RR-AMC; 40 μM, 100 μM, 200 μM, 400 μM, 600 μM, 800 μM, 1000 μM, and 1200 μM) were reacted with 40.9 nM Cts B. All assays were performed in an assay buffer consisted of 100 mM sodium acetate, 100 mM NaCl, 1 mM EDTA, 5 mM DTT, and pH 5.5 in a 96-well plate at 27 °C. The peptide inhibitors were pre-incubated with the appropriate proteases in the assay buffer for 10 min to ensure full interactions prior being added to the fluorogenic substrates. The fluorescence of the reaction was monitored as a function of time at 460 nm with an excitation wavelength of 380 nm by a Tecan M1000 plate reader. Each assay was done in triplicates to ensure reproducibility. Details of the analysis for these assays are outlined in the ESI.†Cathepsin L activity assay with HepG2 whole cell lysate
Cts L activity in human hepatocellular carcinoma HepG2 whole cell lysate (200 μg at 2.5 mg mL−1; ab166833; Abcam, Cambridge, MA) was evaluated using a fluorometric Cathepsin L Activity Assay Kit (ab65306; Abcam, Cambridge, MA, USA) following the manufacturer's protocols. Briefly, in each well of a 96-well plate, 50 μL of the HepG2 cell lysate diluted in CL buffer (to a final concentration of 0.05 mg mL−1 HepG2) was incubated with 50 μL of CL buffer without (Control) or with the presence of different peptide inhibitor RS1A concentrations (15 μM, 20 μM, 30 μM, 50 μM, 70 μM, 80 μM, 100 μM, and 120 μM). The cell lysate and the peptide inhibitor were incubated at room temperature for 10 min. A total of 2 μL of 10 mM CL substrate Ac-FR-AFC substrate (to a final concentration of 200 μM) was then added to each well, except the Lysate Background Control wells. Different concentrations (56 nM, 560 nM, and 1 μM) of SID 26681509, a known Cts L inhibitor, were used as positive controls. The samples were mixed; the plate was sealed to avoid evaporation and incubated at 37 °C for 1 h. The fluorescence of each sample was measured at 505 nm with an excitation wavelength of 400 nm on the Tecan plate reader. More details of the assay and data fitting are included in the ESI.† Stability assays of the peptides in HepG2 whole cell lysate are also detailed in the ESI.†Computational modeling
In order to simulate the protease/peptide complexes from this study, the structure of the papain protease (PDB ID: 1BP4) which contains a peptide-like covalent inhibitor68 was used as a template in order to provide a reasonable starting structure for docking. Manual docking was performed by replacing the native covalent inhibitor with the WHLFRAAAW peptide which was prepared using PyRosetta.69 The cathepsin proteases of interest, Cts B (PDB ID 1GMY),70 Cts K (PDB ID 1BGO),71 Cts L (PDB ID 3HHA),72 Cts S (PDB ID 1MS6),73 and Cts V (PDB ID 1FH0),74 were aligned to the manually docked papain complex using PyMOL. The cathepsin protease WHLFRAAAW starting complexes were formally docked by performing the FlexPepDock protocol in Rosetta in order to optimize the binding interaction between the proteases and peptides of interest.75 The tryptophan residues in WHLFRAAAW were mutated to 7-methoxycoumarinyl alanine (μ) residues using the MutateResidue tool in PyRosetta toolbox with a params and rotamer library generated previously.23 Next, a constrained FastRelax was performed in PyRosetta in order to accommodate the newly mutated 7-methoxycoumarinyl alanine residues. A flat harmonic constraint was used to maintain proximity of the scissile bond to the active site cysteine residue. Thioamides were introduced into the relaxed complexes through patches written previously.23,76 The thioamide containing peptides were then simulated with five independent local relax trajectories without any constraints.Machine learning
Unsupervised machine learning was performed by clustering energy features from PyRosetta modeling with scikit-learn.77 Specifically, score differences (termed deltas) between the residue total energies (energy in thioamide peptide complex minus energy in all-amide peptide complex) of the three residues of the protease catalytic triad as well as the P1 and P1′ residues of the peptide were computed from all of our FlexPepDock models. These five energy score deltas were reduced into three dimensions with Principal Component Analysis.77 The three principal component axes were then clustered with the KMeans algorithm utilizing four clusters which was derived by maximizing the Silhouette heuristic.78 The three-dimensional data were plotted and visualized with matplotlib.79Data availability
Kinetic and inhibition data with associated fitting as well as structural models of the peptides in complex with proteases are available as ESI. A key file is provided with descriptions of each data file.Author contributions
H. A. T. P. and E. J. P. designed the experiments. H. A. T. P. performed all of the experiments with some guidance and preliminary inhibition data from T. M. B. and C. L. S. G. G. performed computational modeling and machine learning analysis. H. A. T. P. and E. J. P. performed the experimental data analysis and structural model analysis. H. A. T. P. and E. J. P. wrote the manuscript with input from all other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Pennsylvania and the National Science Foundation (NSF CHE-1708759 to E. J. P.). T. M. B. thanks the NIH for funding through the Chemistry Biology Interface Training Program (T32 GM071339). S. G. G. thanks the NSF for funding through the Graduate Research Fellowship Program (DGE-1845298). We are grateful to Youngchool Choe and Charles Craik for generously sharing their positional data on cysteine protease activity.Notes and references
- K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122 CrossRef CAS PubMed.
- A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382 CrossRef CAS PubMed.
- A. C.-L. Lee, J. L. Harris, K. K. Khanna and J.-H. Hong, Int. J. Mol. Sci., 2019, 20, 2383 CrossRef PubMed.
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136 CrossRef CAS.
- T. M. Barrett, K. E. Fiore, C. Liu and E. James Petersson, in Chemistry of Thioamides, ed. T. Murai, Springer, Singapore, 2019, p. 193 Search PubMed.
- V. Frey, J. Viaud, G. Subra, N. Cauquil, J.-F. Guichou, P. Casara, G. Grassy and A. Chavanieu, Eur. J. Med. Chem., 2008, 43, 966 CrossRef CAS PubMed.
- M. T. Weinstock, J. N. Francis, J. S. Redman and M. S. Kay, Pept. Sci., 2012, 98, 431 CrossRef CAS PubMed.
- K. Wiśniewski, R. Galyean, H. Tariga, S. Alagarsamy, G. Croston, J. Heitzmann, A. Kohan, H. Wiśniewska, R. Laporte, P. J. M. Rivière and C. D. Schteingart, J. Med. Chem., 2011, 54, 4388 CrossRef PubMed.
- J. Mcelroy, D. J. S. Guthrie, N. M. Hooper and C. H. Williams, Biochem. Soc. Trans., 1998, 26, S31 CrossRef CAS PubMed.
- M. Schutkowski, M. Jakob, G. Landgraf, I. Born, K. Neubert and G. Fischer, Eur. J. Biochem., 1997, 245, 381 CrossRef CAS.
- H. Verma, B. Khatri, S. Chakraborti and J. Chatterjee, Chem. Sci., 2018, 9, 2443 RSC.
- S. Yao, R. Zutshi and J. Chmielewski, Bioorg. Med. Chem. Lett., 1998, 8, 699 CrossRef CAS.
- B. Asboth and L. Polgar, Biochemistry, 1983, 22, 117 CrossRef CAS.
- P. A. Bartlett, K. L. Spear and N. E. Jacobsen, Biochemistry, 1982, 21, 1608 CrossRef CAS PubMed.
- R. E. Beattie, D. T. Elmore, C. H. Williams and D. J. S. Guthrie, Biochem. J., 1987, 245, 285 CrossRef CAS PubMed.
- M. D. Bond, B. Holmquist and B. L. Vallee, J. Inorg. Biochem., 1986, 28, 97 CrossRef CAS PubMed.
- P. Campbell and N. T. Nashed, J. Am. Chem. Soc., 1982, 104, 5221 CrossRef CAS.
- K. Cho, Anal. Biochem., 1987, 164, 248 CrossRef CAS.
- K. L. Foje and R. P. Hanzlik, Biochim. Biophys. Acta, Gen. Subj., 1994, 1201, 447 CrossRef.
- L. Maziak, G. Lajoie and B. Belleau, J. Am. Chem. Soc., 1986, 108, 182 CrossRef CAS.
- X. Chen, E. G. Mietlicki-Baase, T. M. Barrett, L. E. Mcgrath, K. Koch-Laskowski, J. J. Ferrie, M. R. Hayes and E. J. Petersson, J. Am. Chem. Soc., 2017, 139, 16688 CrossRef CAS.
- T. M. Barrett, X. S. Chen, C. Liu, S. Giannakoulias, H. a. T. Phan, J. Wang, E. K. Keenan, R. J. Karpowicz and E. J. Petersson, ACS Chem. Biol., 2020, 15, 774 CrossRef CAS.
- S. Giannakoulias, S. R. Shringari, C. Liu, H. a. T. Phan, T. M. Barrett, J. J. Ferrie and E. J. Petersson, J. Phys. Chem. B, 2020, 124, 8032 CrossRef CAS PubMed.
- C. Liu, T. M. Barrett, X. Chen, J. J. Ferrie and E. J. Petersson, ChemBioChem, 2019, 20, 2059 CrossRef CAS.
- X. S. Puente, L. M. Sánchez, C. M. Overall and C. López-Otín, Nat. Rev. Genet., 2003, 4, 544 CrossRef CAS.
- B. Turk, Nat. Rev. Drug Discovery, 2006, 5, 785 CrossRef CAS.
- O. C. Olson and J. A. Joyce, Nat. Rev. Cancer, 2015, 15, 712 CrossRef CAS.
- V. Turk, V. Stoka, O. Vasiljeva, M. Renko, T. Sun, B. Turk and D. Turk, Biochim. Biophys. Acta, Proteins Proteomics, 2012, 1824, 68 CrossRef CAS.
- T. Nakagawa, W. Roth, P. Wong, A. Nelson, A. Farr, J. Deussing, J. A. Villadangos, H. Ploegh, C. Peters and A. Y. Rudensky, Science, 1998, 280, 450 CrossRef CAS PubMed.
- B. Friedrichs, C. Tepel, T. Reinheckel, J. Deussing, K. Von Figura, V. Herzog, C. Peters, P. Saftig and K. Brix, J. Clin. Invest., 2003, 111, 1733 CrossRef CAS PubMed.
- L. Funkelstein, M. Beinfeld, A. Minokadeh, J. Zadina and V. Hook, Neuropeptides, 2010, 44, 457 CrossRef CAS PubMed.
- S. Yasothornsrikul, D. Greenbaum, K. F. Medzihradszky, T. Toneff, R. Bundey, R. Miller, B. Schilling, I. Petermann, J. Dehnert, A. Logvinova, P. Goldsmith, J. M. Neveu, W. S. Lane, B. Gibson, T. Reinheckel, C. Peters, M. Bogyo and V. Hook, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9590 CrossRef CAS PubMed.
- J. Dennemärker, T. Lohmüller, S. Müller, S. V. Aguilar, D. J. Tobin, C. Peters and T. Reinheckel, Biol. Chem., 2010, 391, 913 Search PubMed.
- J. Reiser, B. Adair and T. Reinheckel, J. Clin. Invest., 2010, 120, 3421 CrossRef CAS PubMed.
- D. Spira, J. Stypmann, D. J. Tobin, I. Petermann, C. Mayer, S. Hagemann, O. Vasiljeva, T. Günther, R. Schüle, C. Peters and T. Reinheckel, J. Biol. Chem., 2007, 282, 37045 CrossRef CAS PubMed.
- J. Stypmann, K. Gläser, W. Roth, D. J. Tobin, I. Petermann, R. Matthias, G. Mönnig, W. Haverkamp, G. Breithardt, W. Schmahl, C. Peters and T. Reinheckel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6234 CrossRef CAS PubMed.
- Q. Tang, J. Cai, D. Shen, Z. Bian, L. Yan, Y.-X. Wang, J. Lan, G.-Q. Zhuang, W.-Z. Ma and W. Wang, J. Mol. Med., 2009, 87, 249 CrossRef CAS PubMed.
- C. Jedeszko and B. F. Sloane, Biol. Chem., 2004, 385, 1017 CAS.
- B. Sobotič, M. Vizovišek, R. Vidmar, P. V. Damme, V. Gocheva, J. A. Joyce, K. Gevaert, V. Turk, B. Turk and M. Fonović, Mol. Cell. Proteomics, 2015, 14, 2213 CrossRef PubMed.
- X. Zheng, F. Chu, P. M. Chou, C. Gallati, U. Dier, B. L. Mirkin, S. A. Mousa and A. Rebbaa, Am. J. Physiol.: Cell Physiol., 2009, 296, C65 CrossRef CAS PubMed.
- X. Ou, Y. Liu, X. Lei, P. Li, D. Mi, L. Ren, L. Guo, R. Guo, T. Chen, J. Hu, Z. Xiang, Z. Mu, X. Chen, J. Chen, K. Hu, Q. Jin, J. Wang and Z. Qian, Nat. Commun., 2020, 11, 1620 CrossRef CAS PubMed.
- M.-M. Zhao, W.-L. Yang, F.-Y. Yang, L. Zhang, W.-J. Huang, W. Hou, C.-F. Fan, R.-H. Jin, Y.-M. Feng, Y.-C. Wang and J.-K. Yang, Signal Transduction Targeted Ther., 2021, 6, 134 CrossRef CAS PubMed.
- G. Das, S. Ghosh, S. Garg, S. Ghosh, A. Jana, R. Samat, N. Mukherjee, R. Roy and S. Ghosh, RSC Adv., 2020, 10, 28243 RSC.
- C. Gil, T. Ginex, I. Maestro, V. Nozal, L. Barrado-Gil, M. Á. Cuesta-Geijo, J. Urquiza, D. Ramírez, C. Alonso, N. E. Campillo and A. Martinez, J. Med. Chem., 2020, 63, 12359 CrossRef CAS PubMed.
- T. Liu, S. Luo, P. Libby and G.-P. Shi, Pharmacol. Ther., 2020, 213, 107587 CrossRef CAS PubMed.
- D. Dana and S. K. Pathak, Molecules, 2020, 25, 698 CrossRef CAS PubMed.
- M. Drag and G. S. Salvesen, Nat. Rev. Drug Discovery, 2010, 9, 690 CrossRef CAS PubMed.
- B. Walker and J. F. Lynas, CMLS, Cell. Mol. Life Sci., 2001, 58, 596 CrossRef CAS PubMed.
- D. S. Johnson, E. Weerapana and B. F. Cravatt, Future Med. Chem., 2010, 2, 949 CrossRef CAS PubMed.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307 CrossRef CAS PubMed.
- J. M. Goldberg, S. Batjargal, B. S. Chen and E. J. Petersson, J. Am. Chem. Soc., 2013, 135, 18651 CrossRef CAS PubMed.
- J. M. Goldberg, X. Chen, N. Meinhardt, D. C. Greenbaum and E. J. Petersson, J. Am. Chem. Soc., 2014, 136, 2086 CrossRef CAS PubMed.
- Y. Choe, F. Leonetti, D. C. Greenbaum, F. Lecaille, M. Bogyo, D. Brömme, J. A. Ellman and C. S. Craik, J. Biol. Chem., 2006, 281, 12824 CrossRef CAS PubMed.
- M. L. Biniossek, D. K. Nägler, C. Becker-Pauly and O. Schilling, J. Proteome Res., 2011, 10, 5363 CrossRef CAS PubMed.
- M. Schutkowski, K. Neubert and G. Fischer, Eur. J. Biochem., 1994, 221, 455 CrossRef CAS.
- GraphPad Prism 8 Curve Fitting Guide, https://www.graphpad.com/guides/prism/latest/curve-fitting/index.htm.
- R. A. Copeland, Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, John Wiley & Sons, Incorporated, Somerset, United States, 2013 Search PubMed.
- H. A. Elshabrawy, J. Fan, C. S. Haddad, K. Ratia, C. C. Broder, M. Caffrey and B. S. Prabhakar, J. Virol., 2014, 88, 4353 CrossRef PubMed.
- M. C. Myers, P. P. Shah, S. L. Diamond, D. M. Huryn and A. B. Smith, Bioorg. Med. Chem. Lett., 2008, 18, 210 CrossRef CAS.
- P. P. Shah, M. C. Myers, M. P. Beavers, J. E. Purvis, H. Jing, H. J. Grieser, E. R. Sharlow, A. D. Napper, D. M. Huryn, B. S. Cooperman, A. B. Smith and S. L. Diamond, Mol. Pharmacol., 2008, 74, 34 CrossRef CAS PubMed.
- J. A. Joyce, A. Baruch, K. Chehade, N. Meyer-Morse, E. Giraudo, F. Y. Tsai, D. C. Greenbaum, J. H. Hager, M. Bogyo and D. Hanahan, Cancer Cell, 2004, 5, 443 CrossRef CAS.
- J. Ruan, H. Zheng, W. Fu, P. Zhao, N. Su and R. Luo, PLoS One, 2014, 9, e112136 CrossRef PubMed.
- N. Ueki, S. Lee, N. S. Sampson and M. J. Hayman, Nat. Commun., 2013, 4, 2735 CrossRef.
- D. Dana, J. Garcia, A. I. Bhuiyan, P. Rathod, L. Joo, D. A. Novoa, S. Paroly, K. R. Fath, E. J. Chang and S. K. Pathak, Bioorg. Chem., 2019, 85, 505 CrossRef CAS PubMed.
- N. Mahanta, D. M. Szantai-Kis, E. J. Petersson and D. A. Mitchell, ACS Chem. Biol., 2019, 14, 142 CrossRef CAS PubMed.
- D. Musil, D. Zucic, D. Turk, R. A. Engh, I. Mayr, R. Huber, T. Popovic, V. Turk, T. Towatari and N. Katunuma, EMBO J., 1991, 10, 2321 CrossRef CAS PubMed.
- D. Turk, M. Podobnik, T. Popovic, N. Katunuma, W. Bode, R. Huber and V. Turk, Biochemistry, 1995, 34, 4791 CrossRef CAS PubMed.
- J. M. Lalonde, B. Zhao, W. W. Smith, C. A. Janson, R. L. Desjarlais, T. A. Tomaszek, T. J. Carr, S. K. Thompson, H.-J. Oh, D. S. Yamashita, D. F. Veber and S. S. Abdel-Meguid, J. Med. Chem., 1998, 41, 4567 CrossRef CAS PubMed.
- S. Chaudhury, S. Lyskov and J. J. Gray, Bioinformatics, 2010, 26, 689 CrossRef CAS PubMed.
- P. D. Greenspan, K. L. Clark, R. A. Tommasi, S. D. Cowen, L. W. Mcquire, D. L. Farley, J. H. Van Duzer, R. L. Goldberg, H. Zhou, Z. Du, J. J. Fitt, D. E. Coppa, Z. Fang, W. Macchia, L. Zhu, M. P. Capparelli, R. Goldstein, A. M. Wigg, J. R. Doughty, R. S. Bohacek and A. K. Knap, J. Med. Chem., 2001, 44, 4524 CrossRef CAS PubMed.
- R. L. Desjarlais, D. S. Yamashita, H.-J. Oh, I. N. Uzinskas, K. F. Erhard, A. C. Allen, R. C. Haltiwanger, B. Zhao, W. W. Smith, S. S. Abdel-Meguid, K. D'alessio, C. A. Janson, M. S. Mcqueney, T. A. Tomaszek, M. A. Levy and D. F. Veber, J. Am. Chem. Soc., 1998, 120, 9114 CrossRef CAS.
- N. Asaad, P. A. Bethel, M. D. Coulson, J. E. Dawson, S. J. Ford, S. Gerhardt, M. Grist, G. A. Hamlin, M. J. James, E. V. Jones, G. I. Karoutchi, P. W. Kenny, A. D. Morley, K. Oldham, N. Rankine, D. Ryan, S. L. Wells, L. Wood, M. Augustin, S. Krapp, H. Simader and S. Steinbacher, Bioorg. Med. Chem. Lett., 2009, 19, 4280 CrossRef CAS PubMed.
- Y. D. Ward, D. S. Thomson, L. L. Frye, C. L. Cywin, T. Morwick, M. J. Emmanuel, R. Zindell, D. Mcneil, Y. Bekkali, M. Hrapchak, M. Deturi, K. Crane, D. White, S. Pav, Y. Wang, M.-H. Hao, C. A. Grygon, M. E. Labadia, D. M. Freeman, W. Davidson, J. L. Hopkins, M. L. Brown and D. M. Spero, J. Med. Chem., 2002, 45, 5471 CrossRef CAS PubMed.
- J. R. Somoza, H. Zhan, K. K. Bowman, L. Yu, K. D. Mortara, J. T. Palmer, J. M. Clark and M. E. Mcgrath, Biochemistry, 2000, 39, 12543 CrossRef CAS PubMed.
- B. Raveh, N. London and O. Schueler-Furman, Proteins, 2010, 78, 2029 CrossRef CAS PubMed.
- J. Yoon, J. J. Ferrie and E. J. Petersson, J. Phys. Chem. B, 2020, 124(47), 10653 CrossRef CAS PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and É. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825 Search PubMed.
- P. J. Rousseeuw, J. Comput. Appl. Math., 1987, 20, 53 CrossRef.
- J. D. Hunter, Comput. Sci. Eng., 2007, 9, 90 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Descriptions of materials and methods, including peptide synthesis, characterization, and purification, protease assays, validation of protease assays with HPLC and MS, details on cell lysate assay and inhibition assays. See DOI: 10.1039/d1sc00785h |
| This journal is © The Royal Society of Chemistry 2021 |