
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiocompatible metal–organic frameworks for the storage and therapeutic delivery of hydrogen sulfide†
Faith E.
Chen‡
 a,
Ruth M.
Mandel‡
a,
Joshua J.
Woods
ab,
Jung-Hoon
Lee
c,
Jaehwan
Kim
a,
Jesse H.
Hsu
a,
José J.
Fuentes-Rivera
a,
Justin J.
Wilson
a and
Phillip J.
Milner
*a
a,
Ruth M.
Mandel‡
a,
Joshua J.
Woods
ab,
Jung-Hoon
Lee
c,
Jaehwan
Kim
a,
Jesse H.
Hsu
a,
José J.
Fuentes-Rivera
a,
Justin J.
Wilson
a and
Phillip J.
Milner
*a
aDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14850, USA. E-mail: pjm347@cornell.edu
bRobert F. Smith School for Chemical and Biomolecular Engineering, Cornell University, Ithaca, NY 14850, USA
cComputational Science Research Center, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea
First published on 30th April 2021
Abstract
Hydrogen sulfide (H2S) is an endogenous gasotransmitter with potential therapeutic value for treating a range of disorders, such as ischemia-reperfusion injury resulting from a myocardial infarction or stroke. However, the medicinal delivery of H2S is hindered by its corrosive and toxic nature. In addition, small molecule H2S donors often generate other reactive and sulfur-containing species upon H2S release, leading to unwanted side effects. Here, we demonstrate that H2S release from biocompatible porous solids, namely metal–organic frameworks (MOFs), is a promising alternative strategy for H2S delivery under physiologically relevant conditions. In particular, through gas adsorption measurements and density functional theory calculations we establish that H2S binds strongly and reversibly within the tetrahedral pockets of the fumaric acid-derived framework MOF-801 and the mesaconic acid-derived framework Zr-mes, as well as the new itaconic acid-derived framework CORN-MOF-2. These features make all three frameworks among the best materials identified to date for the capture, storage, and delivery of H2S. In addition, these frameworks are non-toxic to HeLa cells and capable of releasing H2S under aqueous conditions, as confirmed by fluorescence assays. Last, a cellular ischemia-reperfusion injury model using H9c2 rat cardiomyoblast cells corroborates that H2S-loaded MOF-801 is capable of mitigating hypoxia-reoxygenation injury, likely due to the release of H2S. Overall, our findings suggest that H2S-loaded MOFs represent a new family of easily-handled solid sources of H2S that merit further investigation as therapeutic agents. In addition, our findings add Zr-mes and CORN-MOF-2 to the growing lexicon of biocompatible MOFs suitable for drug delivery.
Introduction
Hydrogen sulfide (H2S) is a colorless, flammable gas with a distinctive “rotten-egg” odor that is pervasive in industrial processes such as petroleum refining, natural gas processing, and wastewater treatment.1,2 Because H2S is an irritant to respiratory linings and a chemical asphyxiant, it has a recommended Immediately Dangerous to Life and Health (IDLH) value of 100 ppm, above which prolonged exposure can be fatal. In addition to being toxic to humans at high concentrations, H2S impurities in gas streams are highly corrosive to piping and other equipment.3,4 In spite of its toxic and corrosive nature, H2S is also an important gasotransmitter in mammals with proposed roles in calcium homeostasis, long-term potentiation, and mediation of oxidative stress.5–7 As such, H2S has been invoked as a potential therapeutic for numerous disorders, including bone disease, brain injury, inflammation, Parkinson's disease, high blood pressure, cancer, and ischemia-reperfusion injury.8–15The toxicity and flammability of H2S limit the viability of inhalation as a therapeutic delivery pathway.9 Indeed, studies regarding the potential biological role and therapeutic value of H2S rarely employ this gas directly. Instead, researchers have designed small molecule donors that decompose to generate H2S or related species under physiological conditions.9,10,12,13,16,17 However, the application of these donors is complicated by the concomitant production of reactive byproducts and other sulfur-based species upon H2S release.9 A promising alternative strategy is to design biocompatible porous solids capable of storing and controllably releasing gaseous H2S, avoiding the formation of biologically-active byproducts. In particular, metal–organic frameworks (MOFs), which are porous materials constructed from organic “linkers” and inorganic “nodes,” are promising materials for the controlled delivery of bioactive molecules,18–20 such as the gasotransmitter nitric oxide (NO).21 The chemical tunability of MOFs achievable through linker modification and surface functionalization is a promising feature which could potentially enable control over the rate and location of H2S delivery.22 Nonetheless, the potential application of MOFs as H2S donors in medicine remains almost completely unexplored.22,23
To be useful for H2S delivery in vivo, MOF-based platforms must meet several criteria, including: (1) construction from non-toxic metals and linkers; (2) stability towards H2S; (3) high H2S capacities with controlled release under physiological conditions; and (4) stability under physiological conditions (e.g., serum at 37 °C). However, none of the MOFs that have been studied for H2S adsorption to date fulfill all of these criteria, as many are limited by framework destruction upon irreversible H2S binding,22–30 low capacities,26,31 poor biocompatibility, or harsh H2S desorption conditions.2,23,24,30,32 In addition, many previously studied MOFs are constructed from toxic metals (e.g., Ni, Cr)22,30 or complex organic linkers33 that could lead to unwanted side effects in vivo. For example, the only MOF that has been explored for therapeutic H2S delivery to date, namely, Zn-MOF-74, fails to meet several of these criteria.23 We have found that H2S adsorption in Zn-MOF-74 is completely irreversible, which precludes its application for H2S delivery. This framework was also found to be highly toxic to HeLa cells (see ESI or ESI Section 15† for details). Therefore, there remains a critical need to identify biocompatible MOFs that display strong yet reversible H2S adsorption for application as next-generation H2S donors.
A promising strategy to identify new MOFs capable of reversibly adsorbing H2S is to leverage its properties as a hydrogen-bond donor and weak hydrogen-bond acceptor, similar to water.34 As such, we were inspired by the recent deluge of MOFs designed for reversible water capture35–38 to identify potential frameworks for H2S storage. In particular, we hypothesized that the surprisingly strong binding of water (−ΔHads ≈ 60 kJ mol−1) in Zr6O4(OH)4(fumarate)6 or MOF-801 (Fig. 1, left), which is due to ordered hydrogen-bonding interactions within the tetrahedral cavities,39 may translate to strong binding of H2S within this MOF as well. In addition, Zr is a non-toxic metal40 and fumaric acid occurs naturally as part of the Krebs cycle,41,42 suggesting that MOF-801 and closely related frameworks should also be biocompatible.43 Here, through combined experimental and computational analyses we demonstrate that MOF-801, as well as the related biocompatible mesaconate-based framework Zr-mes44,45 and itaconate-based framework CORN-MOF-2 (CORN = Cornell University)46,47 undergo strong but reversible adsorption of H2S with minimal framework degradation (Fig. 1). In addition, these frameworks are capable of delivering H2S under physiologically relevant conditions, allowing for the therapeutic benefits of H2S to be accessed using easily-handled solids. Taken together, these features make the frameworks described here promising H2S donors for biomedical applications.
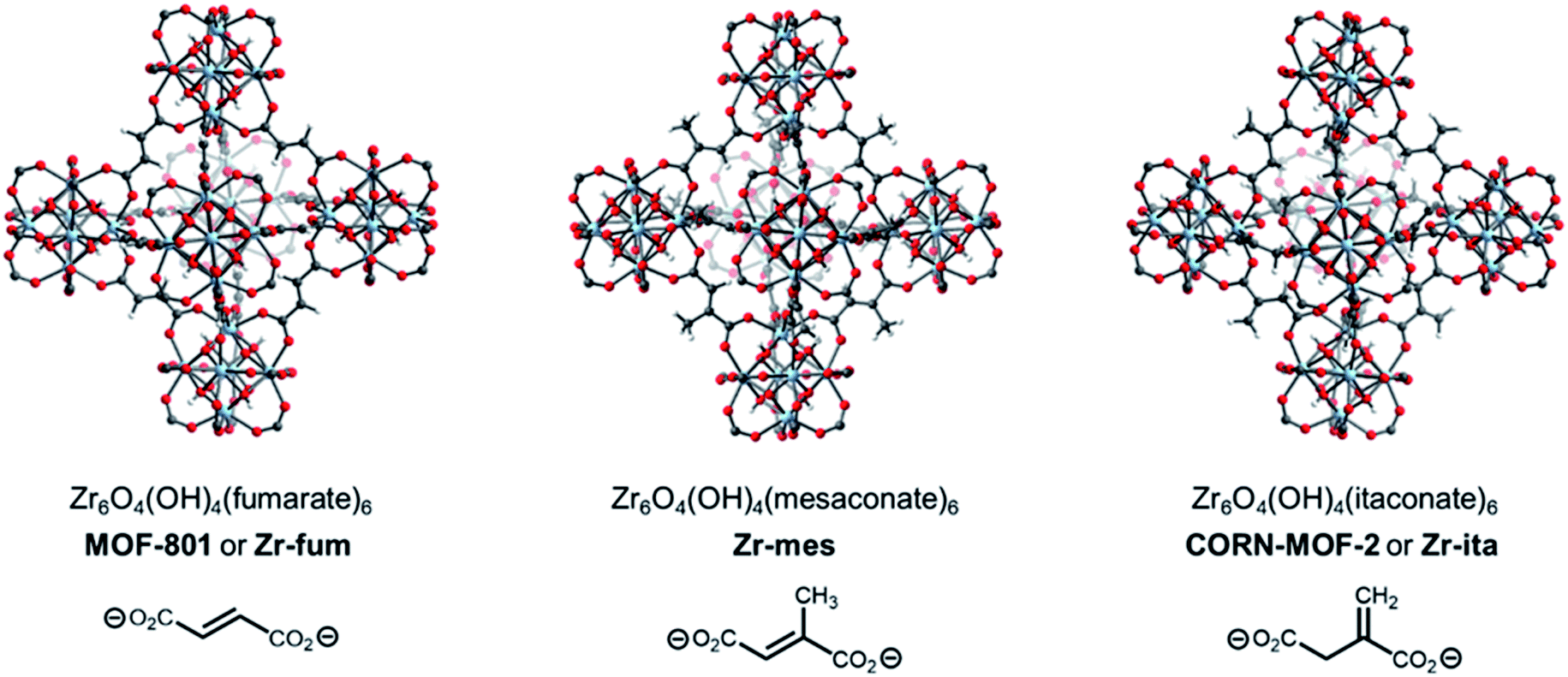 | ||
| Fig. 1 Density functional theory-calculated ideal structures of known and new sustainably-derived metal–organic frameworks prepared from fumaric, mesaconic, and itaconic acid as part of this work. Gray, white, red, and pale blue spheres correspond to carbon, hydrogen, oxygen, and zirconium, respectively. | ||
Results and discussion
Preparation of MOFs
The MOFs Zr6O4(OH)4(fumarate)6 and Zr6O4(OH)4(mesaconate)6 are also known as MOF-801 (ref. 39) and Zr-mes,44 respectively. Herein, we refer to these frameworks as Zr-fum and Zr-mes, respectively, for clarity. Following procedures adapted from the literature, we prepared Zr-fum and Zr-mes under solvothermal conditions on 10 mmol scale (see ESI Sections 3–6† for details). Specifically, these frameworks were synthesized from ZrOCl2·8H2O and the corresponding dicarboxylic acid in either N,N-dimethylformamide (Zr-fum-DMF,39 Zr-mes-DMF44) or water (Zr-fum-H2O,48 Zr-mes-H2O48) using formic acid as a modulator. After optimization, 30 equivalents of formic acid were found to produce Zr-fum and Zr-mes with high crystallinity, as determined by powder X-ray diffraction (PXRD; ESI Fig. S1 and S2†). Notably, higher concentrations of formic acid led to the formation of significant impurities.The PXRD patterns of solvated Zr-fum and Zr-mes prepared in DMF and in H2O are shown in Fig. 2. The PXRD patterns for Zr-fum-DMF and Zr-fum-H2O match the predicted pattern corresponding to the single-crystal X-ray diffraction (SCXRD) structure of MOF-801.39 Likewise, the patterns for Zr-mes match the predicted pattern based on the density functional theory (DFT)-calculated ideal structure of this framework (see ESI Section 14† for details). The Langmuir surface areas determined from 77 K N2 adsorption isotherms of all four frameworks are in good agreement with previously reported values (ESI Fig. S7, S13, S19, and S25†). Unlike Zr-fum-H2O, Zr-mes-H2O was found to undergo partial amorphization upon complete desolvation under high vacuum (<10 μbar) at 100 °C, although this did not impact its porosity. Notably, synthesis under aqueous conditions produced nanocrystals of Zr-fum-H2O less than 100 nm in size (ESI Fig. S15†), suitable for applications in medicine,49 whereas synthesis in DMF produced larger crystallites approximately 500 nm in size (ESI Fig. S9†). The framework Zr-mes was obtained as crystallites of intermediate size (100–200 nm) under both sets of conditions (ESI Fig. S21 and S27†). Owing to the similar properties of frameworks prepared in water and in DMF, we employed Zr-fum-H2O and Zr-mes-H2O for subsequent studies to bypass the use of toxic DMF.
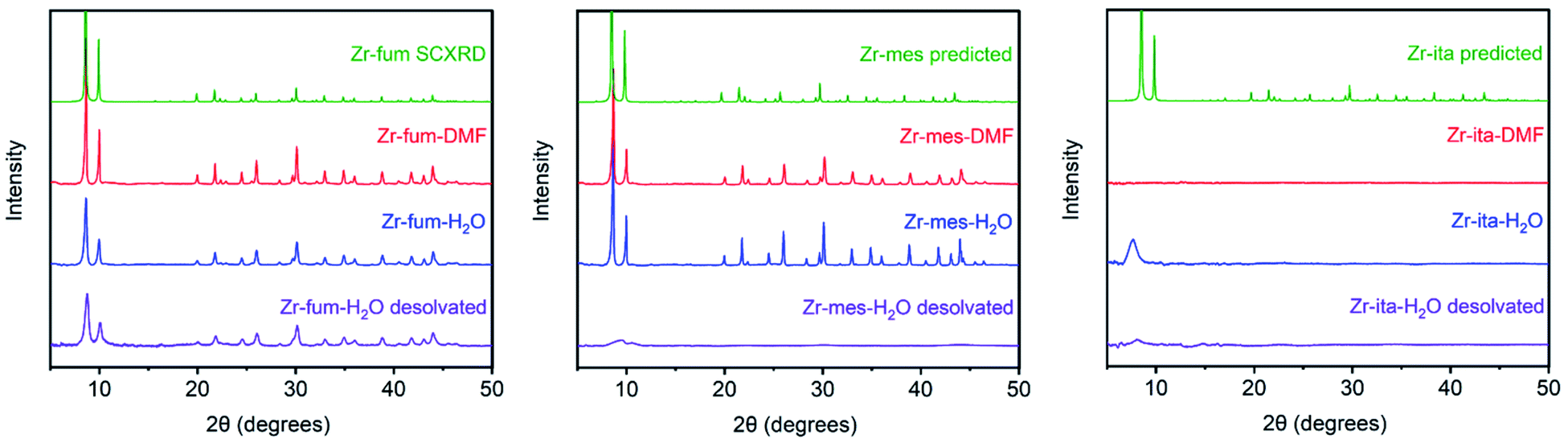 | ||
| Fig. 2 Comparison of PXRD patterns of Zr-fum, Zr-mes, and Zr-ita prepared in DMF and in water. The patterns for MOFs prepared in DMF (red) and H2O (blue) were obtained after activation at 100 °C under vacuum (<100 mbar) using a Schlenk line and were found to still contain ethanol by 1H-NMR digestion. Further desolvation at 100 °C under stronger vacuum (<10 μbar) led to partial amorphization of Zr-mes and Zr-ita, but not Zr-fum (purple). The predicted patterns based on the previously reported single crystal X-ray diffraction pattern of Zr-fum and from the DFT-calculated ideal structures of Zr-mes and Zr-ita are included for reference (green). | ||
After preparing Zr-fum-H2O and Zr-mes-H2O, we next investigated whether the naturally-derived and inexpensive dicarboxylic acid itaconic acid46,47 can be used to synthesize an isostructural MOF, which we term CORN-MOF-2 and refer to as Zr-ita herein for consistency (Fig. 1; see ESI Sections 7 and 8† for details). Produced via the fermentation of carbohydrates by fungi from the genus Aspergillus, itaconic acid is a desirable linker because of its very low cost and lack of toxicity.46 Unfortunately, solvothermal synthesis using ZrCl4 and itaconic acid in DMF produced amorphous material, as confirmed by PXRD (Fig. 2). Several acid modulators were evaluated to enhance the crystallinity of Zr-ita-DMF, including acetic acid, L-proline, concentrated HCl, pivalic acid, and formic acid, but none led to crystalline MOF. In addition, N2 sorption isotherms of Zr-ita-DMF prepared without an acid modulator revealed minimal porosity and a Brunauer–Emmett–Teller (BET) surface area of only 33 m2 g−1 (ESI Fig. S30†). Insight into the poor quality of amorphous Zr-ita-DMF was obtained from 1H-NMR spectra of samples digested using a saturated K3PO4/D2O solution in DMSO-d6 (ESI Fig. S31†). Peaks due to itaconate and residual solvent were observed as expected. However, additional peaks ascribed to the isomeric mesaconate were also observed. We hypothesize that basic N,N-dimethylamine generated by the hydrolysis of DMF mediates the isomerization of itaconate to mesaconate during framework self-assembly, leading to an amorphous structure. In addition, residual DMF was observed by 1H-NMR even after extensive soaking in tetrahydrofuran. Therefore, the significant linker isomerization and difficulty in removing DMF from the pores together account for the low surface area of Zr-ita-DMF.
We hypothesized that the key to preparing high-quality Zr-ita was to avoid the use of base during MOF synthesis. Consistently, employing water as the solvent instead of DMF produced semi-crystalline Zr-ita-H2O for the first time (Fig. 2). The modest crystallinity of Zr-ita-H2O compared to Zr-fum-H2O and Zr-mes-H2O is likely due to the sp3-hybridized carbon in the backbone of this flexible linker. Because of these characteristics, Zr-ita-H2O is likely an amorphous MOF (aMOF), which have found application for toxic gas capture and drug delivery.50 Because the DFT-calculated structure of Zr-ita-H2O is idealized and does not account for linker flexibility, the PXRD pattern simulated from this structure in Fig. 1 does not reflect the amorphous nature of Zr-ita-H2O. When prepared on 10 mmol scale, Zr-ita-H2O exhibited improved porosity compared to Zr-ita-DMF, with a 77 K N2 BET surface area of 235 ± 2 m2 g−1 and a Langmuir surface area of 487 ± 22 m2 g−1 (ESI Fig. S35†). Marked hysteresis was also observed upon N2 desorption in Zr-ita-H2O. Analysis of the pore size distribution from the N2 sorption isotherm revealed micropores approximately 6.6 Å in diameter (ESI Fig. S39†), as expected based on the idealized structure, along with mesopores 20–100 Å in size. Thus, we attribute the observed hysteresis to mesoporous defects or cavities between particles. Consistent with the hypothesis outlined above, the 1H-NMR spectra of Zr-ita-H2O digested using a saturated K3PO4/D2O solution in DMSO-d6 revealed only resonances attributed to the itaconate linker, residual formate from defect sites, and solvent (ESI Fig. S36†). To further illustrate the scalability of Zr-ita-H2O, we prepared this new framework on 0.5 mol scale, resulting in a total yield of approximately 87 g (73% yield) of activated framework (see ESI Section 8† for details). Thus, Zr-ita-H2O was employed for subsequent H2S adsorption studies to compare with Zr-fum-H2O and Zr-mes-H2O.
H2S adsorption in MOFs
After preparing Zr-fum-H2O, Zr-mes-H2O, and Zr-ita-H2O, we next evaluated the reversible H2S capture performance of all three frameworks by carrying out isothermal adsorption measurements at 25 °C, 40 °C, and 55 °C (Fig. 3; see ESI Section 10† for details). At 25 °C and 1 bar of H2S, Zr-fum-H2O displayed the highest H2S uptake (4.0 mmol g−1, 11 wt%) followed by Zr-mes-H2O (3.3 mmol g−1, 10 wt%) and Zr-ita-H2O (1.3 mmol g−1, 4.0 wt%), which trends with the frameworks' respective surface areas. These measurements indicate that Zr-fum-H2O and Zr-mes-H2O possess capacities comparable to those in MOFs that undergo (partially) reversible H2S adsorption under similar conditions, such as UiO-66 (∼3.0 mmol g−1; UiO = Universitetet i Oslo),21 MIL-53 (Al) (∼3.5 mmol g−1),13 MIL-53 (Cr) (∼3.5 mmol g−1),13 and Ga-soc-MOF-1a (∼4.5 mmol g−1; soc = square octahedral).22 Moreover, the relatively smooth overlay of the adsorption and desorption data suggests that H2S binds reversibly at every temperature (see below for further discussion). Although H2S adsorption is readily reversible at 25 °C in all three frameworks, they were regenerated at 100 °C under high vacuum (<10 μbar) between experiments for consistency.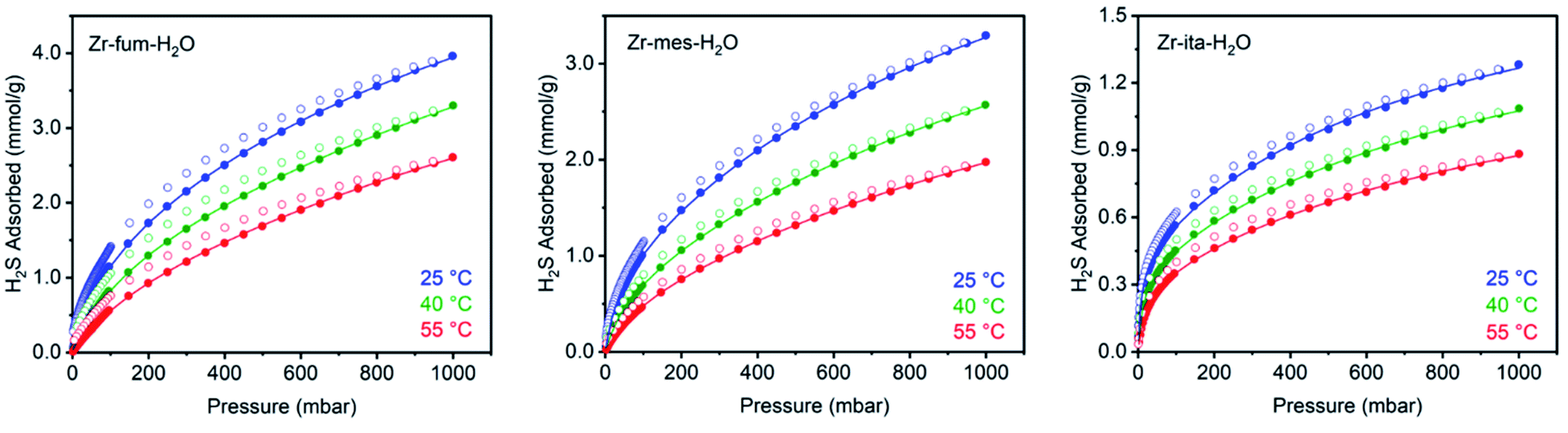 | ||
| Fig. 3 H2S adsorption (closed circles) and desorption (open circles) isotherms at 25 °C (blue), 40 °C (green), and 55 °C (red) of activated Zr-fum-H2O, Zr-mes-H2O, and Zr-ita-H2O. Solid lines represent fits of the adsorption data to a Langmuir–Freundlich model. A data point was considered equilibrated after <0.01% pressure change occurred over a 45 s interval. | ||
To investigate the thermodynamics of H2S binding in Zr-fum-H2O, Zr-mes-H2O, and Zr-ita-H2O, we fit the adsorption data in Fig. 3 using both dual-site Langmuir and Langmuir–Freundlich models (ESI Fig. S46, S49, S52 and Tables S2–S4†). Although both models provided adequate fits to the data, the empirical Langmuir–Freundlich fits (Fig. 3) were found to be superior and thus were employed for subsequent analysis. The differential enthalpies of adsorption were calculated as a function of isosteric H2S loading using the Clausius–Clapeyron equation (eqn (1)), in which PQ is the pressure at a constant uptake Q, ΔHads is the differential enthalpy of adsorption, R is the ideal gas constant, T is the temperature, and c is a constant.
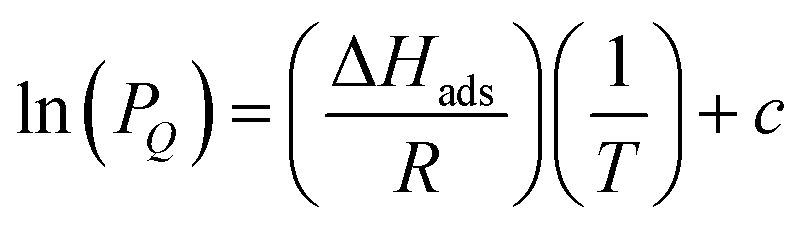 | (1) |
The differential enthalpies of adsorption (−ΔHads) for all three frameworks are included in Fig. 4. The low-coverage adsorption enthalpies follow the trend Zr-ita-H2O (53.2 ± 0.8 kJ mol−1) > Zr-mes-H2O (44.7 ± 0.6 kJ mol−1) > Zr-fum-H2O (32.2 ± 5.6 kJ mol−1). The same trend was observed when the data were fit using dual-site Langmuir models as well (ESI Fig. S46, S49, and S52†). This difference is likely due to the smaller pores of Zr-ita-H2O and Zr-mes-H2O, which enable enhanced hydrogen-bonding interactions between H2S and the framework.31,33,51–53 Consistently, preliminary in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements of H2S-dosed Zr-fum-H2O are indicative of hydrogen-bonding interactions between H2S and the framework upon adsorption (ESI Fig. S106; see ESI Section 16† for details).52 The data in Fig. 4 are among the first experimentally-derived differential enthalpies of H2S adsorption in MOFs, and they are comparable to or higher than those predicted computationally for previously-reported frameworks.24,31,33 The comparatively strong binding of H2S in Zr-fum-H2O, Zr-mes-H2O, and Zr-ita-H2O bodes well for their ability to store H2S prior to release upon exchange with water under physiological conditions.
 | ||
| Fig. 4 Differential enthalpies of adsorption (−ΔHads) for H2S as a function of uptake for activated Zr-fum-H2O (blue), Zr-mes-H2O (red), and Zr-ita-H2O (green), as determined using the Clausius–Clapeyron equation (eqn (1)) and Langmuir–Freundlich fits in Fig. 3. A data point was considered equilibrated after <0.01% pressure change occurred over a 45 s interval. | ||
The reversibility of H2S adsorption and long-term stability towards H2S are also critical criteria for prospective H2S-storage materials. As such, we carefully evaluated the stability of Zr-fum-H2O, Zr-mes-H2O, and Zr-ita-H2O towards H2S in both the gas and solution phases. To characterize the stability of Zr-fum-H2O, Zr-mes-H2O, and Zr-ita-H2O to H2S in solution, we soaked each MOF in a 0.8 M solution of H2S in THF at 50 °C for 48 h. Negligible changes in the PXRD patterns, infrared spectra, and 1H-NMR spectra after digestion with a saturated K3PO4/D2O solution were observed for the MOFs after exposure to H2S in solution, suggesting that, in contrast to Zn-MOF-74, little-to-no framework degradation occurred under these conditions (see ESI Section 11† for details).
Consistent with the desorption data in Fig. 3, the 77 K N2 Langmuir surface areas of all three MOFs also remained essentially unchanged after exposure to gaseous H2S at 25 °C (Fig. 5). In contrast, water vapor binds strongly and only semi-reversibly to all three frameworks, leading to a partial reduction in their Langmuir surface areas after exposure to H2O vapor at 35 °C (see ESI Section 9† for details). To corroborate the reversibility of H2S adsorption in Zr-fum-H2O, Zr-mes-H2O, and Zr-ita-H2O, we also carried out cycling experiments in which the MOFs were repeatedly exposed to approximately 1 bar of H2S at 30 °C followed by desorption at 100 °C under vacuum (Fig. 6). The H2S cycling capacities of all three frameworks remained relatively constant over ten cycles, decreasing by only 3% for Zr-fum-H2O, 8% for Zr-mes-H2O, and 6% for Zr-ita-H2O, respectively. In addition, 77 K N2 adsorption measurements confirmed that the Langmuir surface areas of all three frameworks remained high after repeated cycling of H2S, and characterization by PXRD and 1H-NMR after digestion with a saturated K3PO4/D2O solution confirmed that minimal degradation occurred during H2S cycling (see ESI Section 12† for details). Last, the lack of residual sulfur species in Zr-fum-H2O that had been exposed to H2S and subsequently activated under vacuum was confirmed by X-ray photoelectron spectroscopy (ESI Fig. S102 and S103†). Together, these experimental measurements confirm that H2S adsorbs reversibly and non-destructively within Zr-fum-H2O, Zr-mes-H2O, and Zr-ita-H2O, in contrast to Zn-MOF-74.22 Indeed, the excellent performance of these materials upon H2S cycling places them among the upper echelon of frameworks that have been studied for H2S capture and/or delivery to date. Importantly, the lack of degradation upon H2S adsorption in these materials indicates that they should be capable of cleanly delivering pure H2S that is uncontaminated by any other sulfur-containing species.9
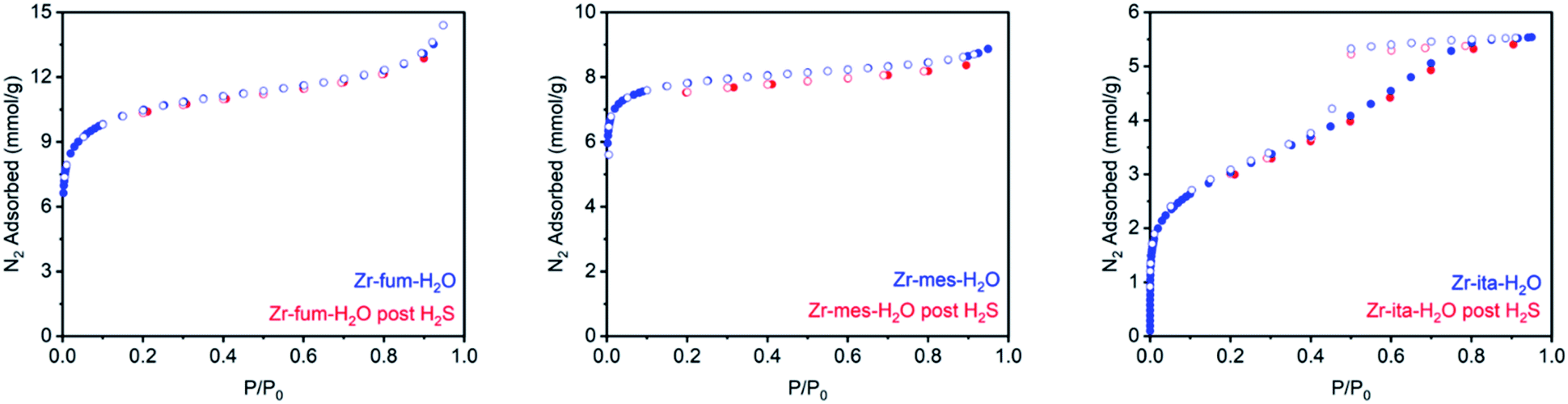 | ||
| Fig. 5 77 K N2 adsorption isotherms of activated Zr-fum-H2O, Zr-mes-H2O, and Zr-ita-H2O before (blue) and after treatment with H2S at 25 °C (red). After H2S adsorption, the MOFs were regenerated at 100 °C under vacuum (<10 μbar) for 48 h. | ||
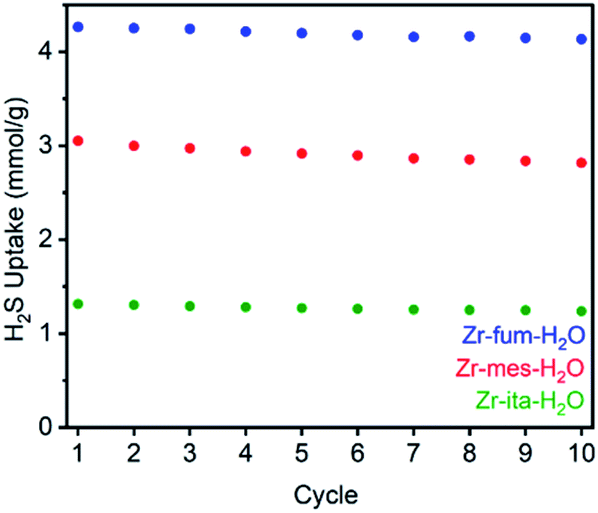 | ||
| Fig. 6 Cycling capacities for H2S adsorption (1 bar, 30 °C) in Zr-fum-H2O, Zr-mes-H2O, and Zr-ita-H2O. The samples were regenerated at 100 °C under high vacuum (<10 μbar) between cycles. | ||
Due to the microcrystalline nature of the MOFs studied herein, we were unable to study the binding modes of H2S within each MOF by single-crystal X-ray diffraction. Thus, we turned to density functional theory (DFT) calculations to probe the structure of H2S bound within Zr-fum, Zr-mes, and Zr-ita (Fig. 7; see ESI Section 14† for details). Calculations were carried out using a plane-wave basis and projector augmented-wave (PAW) pseudopotentials and corrected for dispersive interactions.54–56 The fidelity of our calculations was confirmed by comparing the calculated structure for Zr-fum with the previously reported single-crystal X-ray diffraction structure (ESI Fig. S86†).39 As expected, H2S was found to preferentially bind within the tetrahedral cavities of all three frameworks (Fig. 7, top; see ESI Fig. S89† for space-filling model). Comparing the activated and H2S-bound structures yielded 0 K adsorption energies (−ΔEads) of 41, 44, and 43 kJ mol−1 for Zr-fum, Zr-mes, and Zr-ita, respectively, which are comparable to the experimental enthalpies of adsorption (Fig. 4). The H2S molecules are predicted to be anchored within the tetrahedral cavities by O–H⋯S interactions with the OH− groups on the Zr6 clusters (2.16–2.43 Å in length). Additional hydrogen-bonding interactions between adjacent H2S molecules are predicted, with S–H⋯S distances ranging from 2.38 Å to 2.54 Å. Notably, these distances are slightly shorter than the S–H⋯S distance in (H2S)2 (2.78 Å),34 likely due to the increased polarization of H2S molecules interacting with the framework pores. The favorable packing of H2S within the tetrahedral cavities accounts for its strong yet reversible adsorption in all three frameworks.
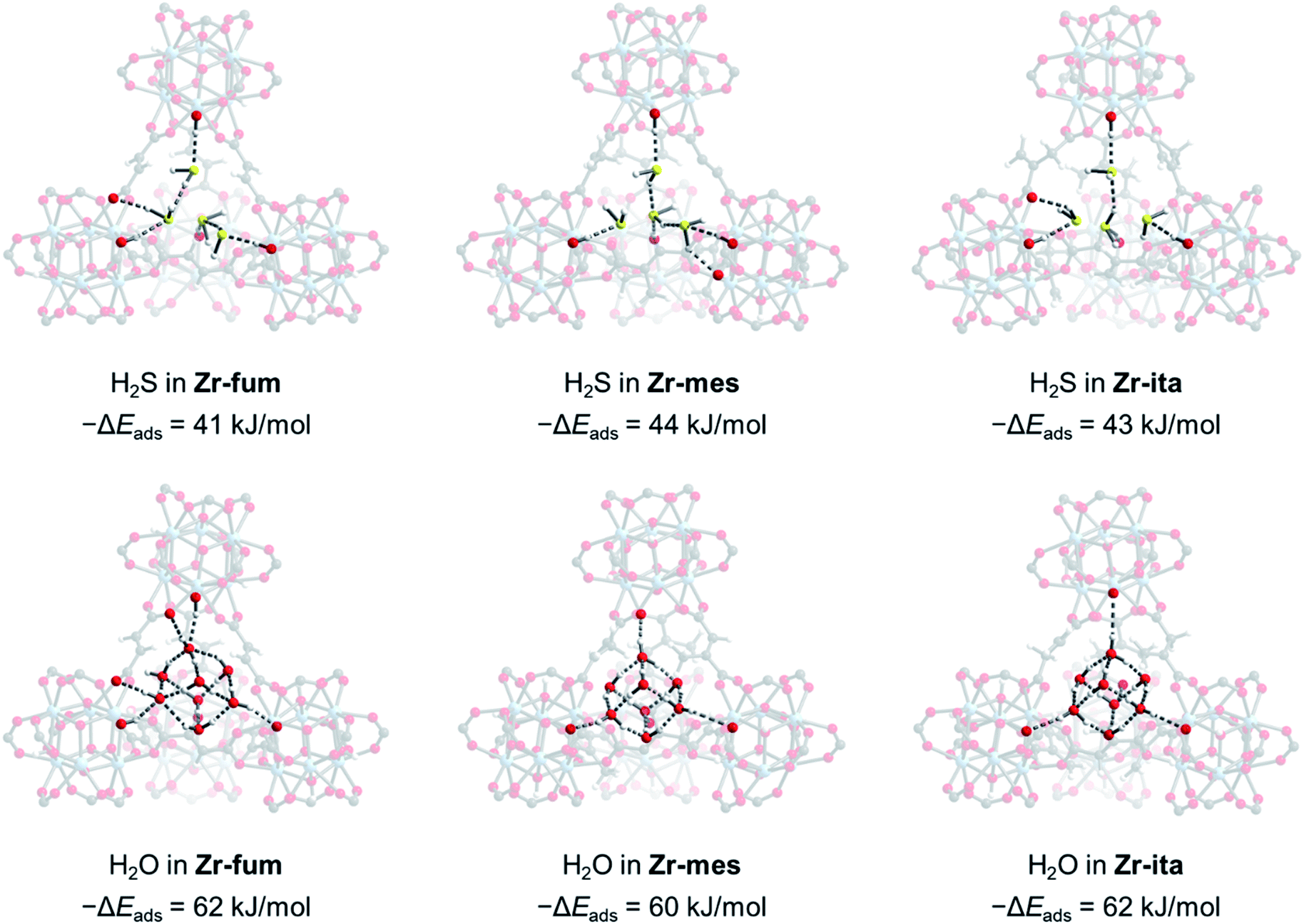 | ||
| Fig. 7 DFT-calculated structures of H2S binding in Zr-fum, Zr-mes, and Zr-ita with calculated energies of adsorption (−ΔEads) indicated. The DFT-calculated structures and energies of H2O adsorption in all three MOFs are included for comparison. Gray, white, red, yellow, and pale blue spheres correspond to carbon, hydrogen, oxygen, sulfur, and zirconium, respectively. | ||
For comparison, we also predicted the preferred binding modes for H2O in Zr-fum, Zr-mes, and Zr-ita (Fig. 7, bottom). Similar to H2S, H2O was found to preferentially bind within the tetrahedral cavities of all three frameworks, albeit with more favorable binding energies in every case. The calculated binding energy for water in Zr-fum (−ΔEads = 62 kJ mol−1) is similar to the previously reported experimental enthalpy of adsorption (−ΔHads = 60 kJ mol−1),39 validating our predictions. Notably, the stronger nature of O–H⋯O interactions compared to S–H⋯S interactions favors binding additional water molecules within the tetrahedral cavities, leading to a higher predicted uptake in addition to more thermodynamically favorable binding for H2O over H2S. The stronger overall binding of H2O suggests that it should be able to displace H2S from the tetrahedral cavities of MOFs, providing an avenue to trigger H2S release under physiological conditions.
H2S delivery from MOFs
Prior to evaluating the ability of H2S-loaded Zr-fum, Zr-mes, and Zr-ita to release H2S under physiologically relevant conditions, we evaluated the stability of these frameworks under aqueous conditions as well as their biocompatibility. Similar to other Zr-MOFs,57,58 all three frameworks were found to be stable in neutral deionized water (pH 7.0) and cell culture medium (pH 7.4) prepared from Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) over extended periods (3–10 days) (ESI Fig. S73–S75; see ESI Section 13† for details). Based on its high H2S capacity and stability under physiological conditions, we selected Zr-fum-H2O as the most promising framework to study further for H2S delivery. The biocompatibility of Zr-fum-H2O was assessed by exposing HeLa cells to varying amounts of activated framework suspended in DMEM supplemented with 10% FBS for 72 h at 37 °C (Fig. 8; see ESI Section 13† for details). Viabilities were determined by incubating the exposed cells with (4,5-dimethylthiazol-2-yl)-2,5-diephenyltetrazolium bromide (MTT) followed by colorimetric quantification in comparison with cells that were not treated with MOF (100% viability).59 Near 100% viabilities were observed up to concentrations of 0.2 mg mL−1, and >90% viability was observed for Zr-fum-H2O at concentrations as high as 0.5 mg mL−1. This is in contrast to other MOFs such as Zn-MOF-74, which was found to be highly toxic to HeLa cells at concentrations greater than 0.1 mg mL−1 (ESI Fig. S101†).60 Assuming Zr-fum-H2O is capable of delivering 4 μmol of H2S per mg of MOF (Fig. 3), a concentration of 0.1 mg mL−1 corresponds to a deliverable concentration of H2S of up to 400 μM, which is comparable to or above endogenous concentrations (e.g., 50–160 μM in the brain).7 This highlights the potential benefit in using a porous solid to deliver H2S: low concentrations (<0.1 mg mL−1) of framework can be employed to release therapeutic levels of H2S, minimizing any potential side effects from the framework itself. Promisingly, our results suggest that Zr-mes and Zr-ita are also relatively non-toxic to HeLa cells (ESI Fig. S80†).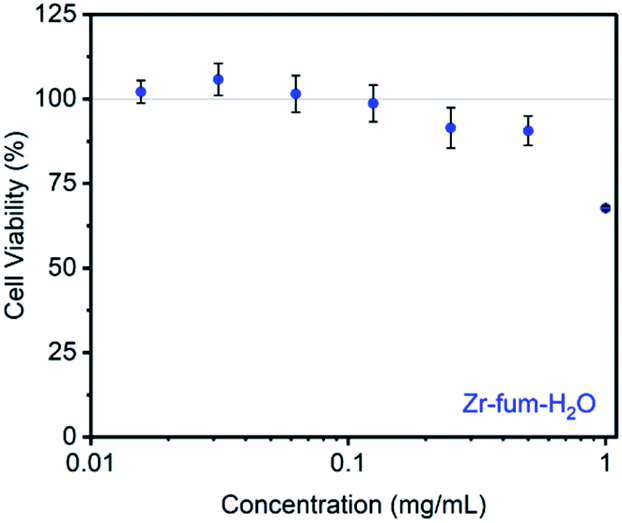 | ||
| Fig. 8 Viability of HeLa cells upon exposure to suspensions of activated Zr-fum-H2O at a range of concentrations in DMEM supplemented with 10% FBS for 72 h at 37 °C. Viabilities were determined by incubating the cells with MTT followed by colorimetric analysis using a microplate reader. Results are reported as the average cell viability of 6 wells/concentration compared to untreated cells from three independent trials, with the standard deviation (SD) reported as the error (±SD). | ||
Having established the biocompatibility of Zr-fum-H2O, we next evaluated its ability to release H2S upon subjection to aqueous solution (Fig. 9 and 10). First, a known amount of H2S-dosed MOF was submerged into a large volume of phosphate buffered saline (PBS) with stirring (∼1 mg MOF per mL PBS). Aliquots from the suspension were quenched with a large excess (>20 equiv.) of the fluorescent probe 1,5-dansyl azide (DNS-az), which exhibits rapid turn-on fluorescence (λmax = 520 nm) upon reaction with H2S.61 The release of H2S from H2S–Zr-fum-H2O into PBS was reproducibly confirmed by fluorescence spectroscopy (Fig. 9, upper; ESI Fig. S78†). The concentration of H2S in solution reached saturation in approximately 30 seconds; given the large excess of free DNS-az present in solution, this suggests that the release of H2S from Zr-fum-H2O is complete in less than 30 s. Importantly, a control experiment in which H2S was dosed into a tube lacking MOF and then “poured” into PBS following the same procedure produced no detectable amounts of H2S, confirming that all of the H2S was released from Zr-fum-H2O (black line, Fig. 9). We also reproducibly confirmed the release of H2S from H2S–Zr-ita-H2O using the same assay (Fig. 9, lower; Fig. S79†). In contrast to Zr-fum-H2O, H2S release from Zr-ita-H2O took 120 s to reach saturation, indicating that H2S release from Zr-ita-H2O may be slower than from Zr-fum-H2O. Unfortunately, due to unavoidable loss of gaseous H2S to air this assay cannot be used to reliably quantify the concentration of H2S released into solution.
 | ||
| Fig. 9 Fluorescence assay (excitation at 340 nm) using the turn-on fluorescence probe dansyl azide (DNS-az) to confirm the release of H2S from Zr-fum-H2O (upper) and Zr-ita-H2O (lower) upon submersion in phosphate buffered saline (PBS). The black line corresponds to a control experiment lacking MOF. The data were smoothed by adjacent-averaging (15 points). | ||
Notably, while the release of H2S from Zr-fum-H2O and Zr-ita-H2O into PBS is relatively rapid, H2S release under ambient conditions from all three MOFs is quite slow. As assessed by exposure to strips of filter paper soaked in lead chloride solution, all three frameworks were found to still contain H2S after standing for at least four days in humid air, with Zr-fum-H2O still containing H2S after six days in humid air (ESI Fig. S81–S83†). In contrast, both Na2S·9H2O and NaSH were found to completely hydrolyze and degrade in less than 5 h under ambient conditions (Fig. S84†). The clean release of H2S into water and the long-term stability of these MOFs in humid air makes them ideal solid sources of H2S that can be easily handled in the laboratory.9
The ability of H2S–Zr-fum-H2O to deliver H2S to cells under physiologically-relevant conditions was further probed using confocal fluorescence microscopy (Fig. 10). Specifically, HeLa cells were incubated first with DMEM containing WSP5 (WSP = Washington State Probe), a selective fluorescence probe for intracellular H2S,62 followed by rinsing and exposure to water (Fig. 10a), H2S–Zr-fum-H2O (Fig. 10b), or Na2S·9H2O (Fig. 10c). For both H2S–Zr-fum-H2O and Na2S·9H2O, a statistically significant enhancement in corrected total cellular fluorescence (CTCF) was observed compared to the control experiment, validating the release and cellular uptake of H2S in both cases. Together, these fluorescence experiments (Fig. 9 and 10) confirm the ability of H2S–Zr-fum-H2O to release H2S under physiologically relevant conditions for subsequent uptake by cells.
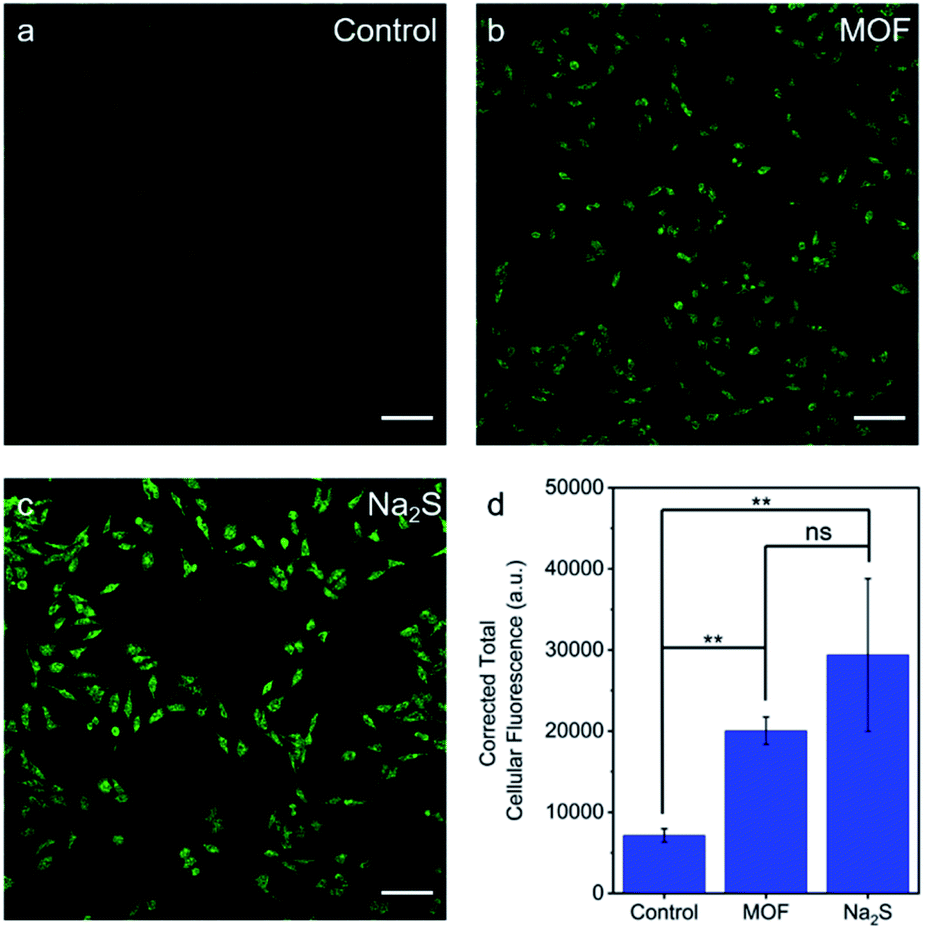 | ||
| Fig. 10 Representative fluorescence microscopy images of H2S release in WSP5-loaded HeLa cells treated with (a) water, (b) 0.05 mg mL−1 H2S–Zr-fum-H2O, or (c) 200 μM Na2S for 1 h at 37 °C. (d) Quantification of total cellular fluorescence of HeLa cells incubated under the conditions described. The scale bar indicates 100 μm. ns indicates not significant, ** indicates p < 0.01. | ||
One of the primary proposed therapeutic modes of H2S is its ability to mediate oxidative stress within cells.13,63 For example, H2S has been shown to limit the oxidative damage to tissue that occurs following the temporary blockage of blood flow during a stroke or myocardial infarction when a period of ischemia or hypoxia is followed by a return of normoxic blood flow, known as ischemia-reperfusion injury (R).63,64 We evaluated the ability of Zr-fum-H2O to deliver H2S and mitigate reperfusion injury using an in vitro model of this condition, hypoxia-reoxygenation, in H9c2 rat cardiomyoblast cells (Fig. 11). As expected, cardiomyoblast cells incubated under normoxic conditions in the absence (Norm) or presence of desolvated Zr-fum-H2O (Norm + M) demonstrated similar viabilities (96 ± 6% and 94 ± 1%, respectively). The latter result is consistent with the lack of toxicity observed for this material with HeLa cells (Fig. 9). In contrast, cells incubated under hypoxic conditions (5% CO2 in N2 for 10 minutes) followed by a return to normoxic conditions, simulating a reperfusion injury (R), demonstrated significantly reduced viabilities (44 ± 1%). However, subjecting cells to this reperfusion injury model in the presence of H2S–Zr-fum-H2O (R + S–M) led to a significant improvement in their viabilities (67 ± 1%), confirming the potential of Zr-fum-H2O as a delivery vessel for therapeutic H2S. Importantly, the presence of activated MOF (R + M) did not lead to an increase in viability compared to cells in the absence of MOF (41 ± 1%), corroborating that the therapeutic value of H2S–Zr-fum-H2O is likely due to H2S release. Likewise, a statistically identical improvement in cell viabilities (73 ± 7%) was observed using Na2S·9H2O in place of H2S–Zr-fum-H2O (R + Na2S), confirming that H2S-loaded MOFs provide similar therapeutic value as traditional sources of H2S but with enhanced stability in the solid state. Overall, our results suggest that Zr-fum-H2O, as well as Zr-mes-H2O and Zr-ita-H2O, represent promising vehicles for the delivery of biologically active H2S under physiological conditions.
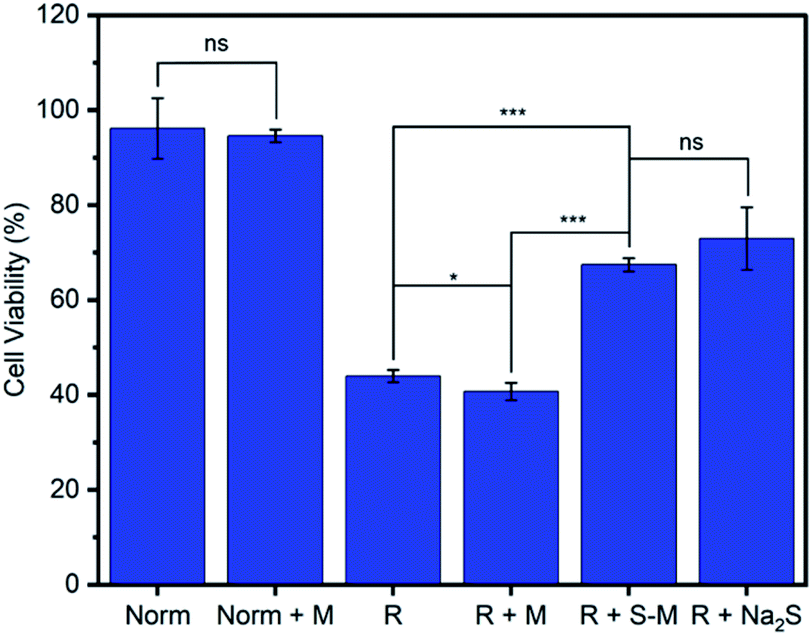 | ||
| Fig. 11 Hypoxia-reoxygenation model of ischemia-reperfusion injury using H9c2 rat cardiomyoblast cells to confirm the ability of Zr-fum-H2O to delivery H2S under physiological conditions. Norm = normoxic conditions; Norm + M = normoxic conditions with activated MOF; R = hypoxia followed by normoxic conditions, simulating reperfusion injury; R + MOF = hypoxia followed by normoxic conditions with activated MOF; R + S–M = hypoxia followed by normoxic conditions with H2S-loaded Zr-fum-H2O; R + Na2S = hypoxia followed by normoxic conditions with Na2S·9H2O. * indicates p < 0.05, *** indicates p < 0.001, and ns indicates not significant. | ||
Conclusions
The controlled delivery of H2S is an unsolved challenge in medicine and biology.9 Here, we demonstrate that biocompatible MOFs that are completely stable to H2S, including Zr-fum, Zr-mes, and Zr-ita, present a promising new direction for the design of vehicles for H2S release under physiological conditions. In particular, given its excellent stability, high H2S capacity, lack of toxicity, ambient stability, and performance in a cellular hypoxia-reoxygenation injury assay, Zr-fum-H2O represents a next-generation platform for the therapeutic delivery of H2S. In addition to Zr-fum-H2O, this work also adds Zr-mes-H2O and Zr-ita-H2O to the growing lexicon of biocompatible MOFs for drug delivery applications. Future work will focus on functionalizing the surface of these frameworks to further control the rate and location of H2S delivery under physiological conditions.Author contributions
P. J. M., J. J. Wilson, R. M. M., F. E. C., and J. J. Woods conceived the project. R. M. M. and F. E. C. synthesized and characterized MOFs and carried out all gas adsorption measurements. J. J. Woods carried out all cellular experiments with assistance from R. M. M. J.-H. L. carried out DFT calculations. J. K. designed the DRIFTS cell and carried out experiments with assistance from R. M. M. J. H. H. preliminarily optimized the synthesis of MOFs. J. J. F.-R. carried out SEM measurements. The final version of the manuscript was written with contributions from all co-authors.Conflicts of interest
P. J. M. is listed as a co-inventor on several patents that include functionalized metal–organic frameworks.Acknowledgements
We thank Cornell University for initial financial support of this work. Further support for this work was provided by the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM138165 (F. E. C., R. M. M., J. K., J. J. F. R., P. J. M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We are grateful to Cornell University for providing a Summer Experience Grant to F. E. C. We thank the American Heart Association for providing a predoctoral fellowship to J. J. Woods (20PRE35120390). Computational work and resources were supported by the KIST Institutional Program (Project No. 2E30460) and KISTI Supercomputing Centre (Project No. KSC-2020-CRE-0189) (J.-H. L). This work made use of the Cornell Center for Materials Research Shared Facilities, which are supported through the NSF MRSEC program (DMR-1719875). 1H NMR data were collected on a Bruker INOVA 500 MHz spectrometer that was purchased with support from the NSF (CHE-1531632). Imaging data was acquired through the Cornell University Biotechnology Resource Center, which is supported in part by the NIH (S10RR025502). The purchase of the diffuse reflectance IR cell used was supported by the Kavli Institute at Cornell for Nanoscale Science.Notes and references
- M. Khabazipour and M. Anbia, Ind. Eng. Chem. Res., 2019, 58, 22133–22164 CrossRef CAS.
- M. S. Shah, M. Tsapatsis and J. I. Siepmann, Chem. Rev., 2017, 117, 9755–9803 CrossRef CAS PubMed.
- X. Zhang, Y. Tang, S. Qu, J. Da and Z. Hao, ACS Catal., 2015, 5, 1053–1067 CrossRef CAS.
- V. Garcia-Arriaga, J. Alvarez-Ramirez, M. Amaya and E. Sosa, Corros. Sci., 2010, 52, 2268–2279 CrossRef CAS.
- K. Qu, S. W. Lee, J. S. Bian, C.-M. Low and P. T.-H. Wong, Neurochem. Int., 2008, 52, 155–165 CrossRef CAS PubMed.
- G. Yang, L. Wu, B. Jiang, W. Yang, J. Qi, K. Cao, Q. Meng, A. K. Mustafa, W. Mu, S. Zhang, S. H. Snyder and R. Wang, Science, 2008, 322, 587–590 CrossRef CAS PubMed.
- K. Abe and H. Kimura, J. Neurosci., 1996, 16, 1066–1071 CrossRef CAS PubMed.
- X. Cao, L. Ding, Z. Xie, Y. Yang, M. Whiteman, P. K. Moore and J.-S. Bian, Antioxid. Redox Signaling, 2019, 31, 1–38 CrossRef CAS PubMed.
- C. R. Powell, K. M. Dillon and J. B. Matson, Biochem. Pharmacol., 2018, 149, 110–123 CrossRef CAS PubMed.
- S. Sestito, G. Nesi, R. Pi, M. Macchia and S. Rapposelli, Front. Chem., 2017, 5, 72 CrossRef PubMed.
- X. Cao, L. Cao, L. Ding and J. Bian, Mol. Neurobiol., 2017, 55, 3789–3799 Search PubMed.
- J. L. Wallace and R. Wang, Nat. Rev. Drug Discovery, 2015, 14, 329–345 CrossRef CAS PubMed.
- Z. J. Song, M. Y. Ng, Z.-W. Lee, W. Dai, T. Hagen, P. K. Moore, D. Huang, L.-W. Deng and C.-H. Tan, MedChemComm, 2014, 5, 557–570 RSC.
- D. J. Lefer, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17907–17908 CrossRef CAS PubMed.
- C. Szabó, Nat. Rev. Drug Discovery, 2007, 6, 917–935 CrossRef PubMed.
- J. J. Woods and J. J. Wilson, Angew. Chem., Int. Ed., 2021, 60, 1588–1592 CrossRef CAS PubMed.
- J. J. Woods, J. Cao, A. R. Lippert and J. J. Wilson, J. Am. Chem. Soc., 2018, 140, 12383–12387 CrossRef CAS PubMed.
- Y. Sun, L. Zheng, Y. Yang, X. Qian, T. Fu, X. Li, Z. Yang, H. Yan, C. Cui and W. Tan, Nano-Micro Lett., 2020, 12, 103 CrossRef CAS PubMed.
- L. Wang, M. Zheng and Z. Xie, J. Mater. Chem. B, 2018, 6, 707–717 RSC.
- R. C. Huxford, J. Della Rocca and W. Lin, Curr. Opin. Chem. Biol., 2010, 14, 262–268 CrossRef CAS PubMed.
- N. J. Hinks, A. C. McKinlay, B. Xiao, P. S. Wheatley and R. E. Morris, Microporous Mesoporous Mater., 2010, 129, 330–334 CrossRef CAS.
- P. K. Allan, P. S. Wheatley, D. Aldous, M. I. Mohideen, C. Tang, J. A. Hriljac, I. L. Megson, K. W. Chapman, G. De Weireld, S. Vaesen and R. E. Morris, Dalton Trans., 2012, 41, 4060 RSC.
- K. Vikrant, V. Kumar, Y. S. Ok, K.-H. Kim and A. Deep, TrAC, Trends Anal. Chem., 2018, 105, 263–281 CrossRef CAS.
- J. Liu, Y. Wei, P. Li, Y. Zhao and R. Zou, J. Phys. Chem. C, 2017, 121, 13249–13255 CrossRef CAS.
- H. Jo, W. R. Lee, N. W. Kim, H. Jung, K. S. Lim, J. E. Kim, D. W. Kang, H. Lee, V. Hiremath, J. G. Seo, H. Jin, D. Moon, S. S. Han and C. S. Hong, ChemSusChem, 2017, 10, 541–550 CrossRef CAS PubMed.
- O. Shekhah, Y. Belmabkhout, K. Adil, P. M. Bhatt, A. J. Cairns and M. Eddaoudi, Chem. Commun., 2015, 51, 13595–13598 RSC.
- B. Supronowicz, A. Mavrandonakis and T. Heine, J. Phys. Chem. C, 2013, 117, 14570–14578 CrossRef CAS.
- C. Petit and T. J. Bandosz, Dalton Trans., 2012, 41, 4027 RSC.
- C. Petit, B. Mendoza and T. J. Bandosz, ChemPhysChem, 2010, 11, 3678–3684 CrossRef CAS PubMed.
- L. Hamon, C. Serre, T. Devic, T. Loiseau, F. Millange, G. Férey and G. D. Weireld, J. Am. Chem. Soc., 2009, 131, 8775–8777 CrossRef CAS PubMed.
- S. Vaesen, V. Guillerm, Q. Yang, A. D. Wiersum, B. Marszalek, B. Gil, A. Vimont, M. Daturi, T. Devic, P. L. Llewellyn, C. Serre, G. Maurin and G. De Weireld, Chem. Commun., 2013, 49, 10082 RSC.
- A. J. Rieth, A. M. Wright and M. Dincă, Nat. Rev. Mater., 2019, 4, 708–725 CrossRef CAS.
- E. Sánchez-González, P. G. M. Mileo, M. Sagastuy-Breña, J. R. Álvarez, J. E. Reynolds, A. Villarreal, A. Gutiérrez-Alejandre, J. Ramírez, J. Balmaseda, E. González-Zamora, G. Maurin, S. M. Humphrey and I. A. Ibarra, J. Mater. Chem. A, 2018, 6, 16900–16909 RSC.
- A. Das, P. K. Mandal, F. J. Lovas, C. Medcraft, N. R. Walker and E. Arunan, Angew. Chem., Int. Ed., 2018, 57, 15199–15203 CrossRef CAS PubMed.
- A. J. Rieth, A. M. Wright, G. Skorupskii, J. L. Mancuso, C. H. Hendon and M. Dincă, J. Am. Chem. Soc., 2019, 141, 13858–13866 CrossRef CAS PubMed.
- H. Kim, S. Yang, S. R. Rao, S. Narayanan, E. A. Kapustin, H. Furukawa, A. S. Umans, O. M. Yaghi and E. N. Wang, Science, 2017, 356, 430–434 CrossRef CAS PubMed.
- J. Canivet, A. Fateeva, Y. Guo, B. Coasne and D. Farrusseng, Chem. Soc. Rev., 2014, 43, 5594–5617 RSC.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- I. C. Smith and B. L. Carson, Trace metals in the environment, Ann Arbor Science Publishers, Ann Arbor, MI, 1978, vol. 3 – Zirconium Search PubMed.
- G. Aquilina, A. Bach, V. Bampidis, M. D. Lourdes Bastos, G. Flachowsky, J. Gasa-Gasó, M. Gralak, C. Hogstrand, L. Leng, S. López-Puente, G. Martelli, B. Mayo, D. Renshaw, G. Rychen, M. Saarela, K. Sejrsen, P. V. Beelen, R. J. Wallace and J. Westendorf, EFSA J., 2013, 11, 14 Search PubMed.
- C. W. Song, D. I. Kim, S. Choi, J. W. Jang and S. Y. Lee, Biotechnol. Bioeng., 2013, 110, 2025–2034 CrossRef CAS.
- I. Abánades Lázaro, S. Haddad, J. M. Rodrigo-Muñoz, R. J. Marshall, B. Sastre, V. del Pozo, D. Fairen-Jimenez and R. S. Forgan, ACS Appl. Mater. Interfaces, 2018, 10, 31146–31157 CrossRef PubMed.
- M. Sk, S. Bhowal and S. Biswas, Eur. J. Inorg. Chem., 2015, 2015, 3317–3322 CrossRef CAS.
- J. Wang and K. Zhang, Metab. Eng., 2015, 30, 190–196 CrossRef CAS.
- T. Cordes, A. Michelucci and K. Hiller, Annu. Rev. Nutr., 2015, 35, 451–473 CrossRef CAS PubMed.
- T. Klement and J. Büchs, Bioresour. Technol., 2013, 135, 422–431 CrossRef CAS.
- H. Reinsch, S. Waitschat, S. M. Chavan, K. P. Lillerud and N. Stock, Eur. J. Inorg. Chem., 2016, 2016, 4490–4498 CrossRef CAS.
- S. K. Murthy, Int. J. Nanomed., 2007, 2, 129–141 CAS.
- T. D. Bennett and A. K. Cheetham, Acc. Chem. Res., 2014, 47, 1555–1562 CrossRef CAS PubMed.
- C. E. Bien, K. K. Chen, S.-C. Chien, B. R. Reiner, L.-C. Lin, C. R. Wade and W. S. W. Ho, J. Am. Chem. Soc., 2018, 140, 12662–12666 CrossRef CAS PubMed.
- J. A. Zárate, E. Sánchez-González, T. Jurado-Vázquez, A. Gutiérrez-Alejandre, E. González-Zamora, I. Castillo, G. Maurin and I. A. Ibarra, Chem. Commun., 2019, 55, 3049–3052 RSC.
- L. Hamon, H. Leclerc, A. Ghoufi, L. Oliviero, A. Travert, J.-C. Lavalley, T. Devic, C. Serre, G. Férey, G. De Weireld, A. Vimont and G. Maurin, J. Phys. Chem. C, 2011, 115, 2047–2056 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- D. Bůžek, J. Demel and K. Lang, Inorg. Chem., 2018, 57, 14290–14297 CrossRef PubMed.
- C. G. Piscopo, A. Polyzoidis, M. Schwarzer and S. Loebbecke, Microporous Mesoporous Mater., 2015, 208, 30–35 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- À. Ruyra, A. Yazdi, J. Espín, A. Carné-Sánchez, N. Roher, J. Lorenzo, I. Imaz and D. Maspoch, Chem.–Eur. J., 2015, 21, 2508–2518 CrossRef PubMed.
- H. Peng, Y. Cheng, C. Dai, A. L. King, B. L. Predmore, D. J. Lefer and B. Wang, Angew. Chem., Int. Ed., 2011, 50, 9672–9675 CrossRef CAS PubMed.
- B. Peng, W. Chen, C. Liu, E. W. Rosser, A. Pacheco, Y. Zhao, H. C. Aguilar and M. Xian, Chem.–Eur. J., 2014, 20, 1010–1016 CrossRef CAS PubMed.
- J. W. Calvert, W. A. Coetzee and D. J. Lefer, Antioxid. Redox Signaling, 2010, 12, 1203–1217 CrossRef CAS PubMed.
- D. Wu, J. Wang, H. Li, M. Xue, A. Ji and Y. Li, Oxid. Med. Cell. Longevity, 2015, 2015, 1–16 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic, computational, and biological assay details. See DOI: 10.1039/d1sc00691f |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |