
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCHIP-mediated hyperubiquitylation of tau promotes its self-assembly into the insoluble tau filaments†
Ji Hyeon
Kim‡
 a,
Jeeyoung
Lee‡
a,
Won Hoon
Choi‡
a,
Seoyoung
Park‡
b,
Seo Hyeong
Park
a,
Jung Hoon
Lee
b,
Sang Min
Lim
c,
Ji Young
Mun
d,
Hyun-Soo
Cho
e,
Dohyun
Han
f,
Young Ho
Suh
ab and
Min Jae
Lee
*ab
a,
Jeeyoung
Lee‡
a,
Won Hoon
Choi‡
a,
Seoyoung
Park‡
b,
Seo Hyeong
Park
a,
Jung Hoon
Lee
b,
Sang Min
Lim
c,
Ji Young
Mun
d,
Hyun-Soo
Cho
e,
Dohyun
Han
f,
Young Ho
Suh
ab and
Min Jae
Lee
*ab
aDepartment of Biomedical Sciences, Seoul National University Graduate School, Seoul 03080, Korea. E-mail: minjlee@snu.ac.k; Fax: +82 2-744-4534; Tel: +82 2-740-8254
bDepartment of Biochemistry & Molecular Biology, Neuroscience Research Institute, Seoul National University College of Medicine, Seoul 03080, Korea
cConvergence Research Center for Diagnosis, Treatment and Care System of Dementia, Korea Institute of Science and Technology, Seoul 02792, Korea
dNeural Circuit Research Group, Korea Brain Research Institute, Daegu 41062, Korea
eDepartment of Systems Biology, College of Life Science and Biotechnology, Yonsei University, Seoul 03722, Korea
fProteomics Core Facility, Biomedical Research Institute, Seoul National University Hospital, Seoul 03080, Korea
First published on 17th March 2021
Abstract
The tau protein is a highly soluble and natively unfolded protein. Under pathological conditions, tau undergoes multiple post-translational modifications (PTMs) and conformational changes to form insoluble filaments, which are the proteinaceous signatures of tauopathies. To dissect the crosstalk among tau PTMs during the aggregation process, we phosphorylated and ubiquitylated recombinant tau in vitro using GSK3β and CHIP, respectively. The resulting phospho–ub-tau contained conventional polyubiquitin chains with lysine 48 linkages, sufficient for proteasomal degradation, whereas unphosphorylated ub-tau species retained only one–three ubiquitin moieties. Mass-spectrometric analysis of in vitro reconstituted phospho–ub-tau revealed seven additional ubiquitylation sites, some of which are known to stabilize tau protofilament stacking in the human brain with tauopathy. When the ubiquitylation reaction was prolonged, phospho–ub-tau transformed into insoluble hyperubiquitylated tau species featuring fibrillar morphology and in vitro seeding activity. We developed a small-molecule inhibitor of CHIP through biophysical screening; this effectively suppressed tau ubiquitylation in vitro and delayed its aggregation in cultured cells including primary cultured neurons. Our biochemical findings point to a “multiple-hit model,” where sequential events of tau phosphorylation and hyperubiquitylation function as a key driver of the fibrillization process, thus indicating that targeting tau ubiquitylation may be an effective strategy to alleviate the course of tauopathies.
Introduction
Intraneuronal cytosolic inclusions of tau proteins are a pathological hallmark of corticobasal neurodegeneration (CBD), Alzheimer's disease (AD), and other neurodegenerative diseases that are collectively termed as tauopathies as a nosological entity. Initially, tau accumulation was thought to be a consequence of tauopathies, instead of a causal process. Nevertheless, many studies now clearly indicate that tau has a direct and pivotal role in the onset of tauopathies.1,2 Tau is encoded by a single gene, microtubule-associated protein tau (MAPT), which contains 16 exons, among which exons 2, 3, and 10 are alternatively spliced thus yielding six isoforms.3 The longest tau isoform (tau40) consists of the following four functional domains: an N-domain, proline-rich domain (PRD), microtubule-binding domain (MBD), and C-terminal region. Among these, the MBD repeats comprise 18 amino acids with a Lys–Ile/Cys–Gly–Ser motif, through which tau electrostatically interacts with negatively charged axonal microtubules, stabilizing microtubule networks in neurons.4 Tau phosphorylation may weaken this interaction5 although most of the phosphorylation sites of tau are located in the isotropically flexible “fuzzy coat”.6,7Tau is an extraordinarily hydrophilic (average hydropathicity of −0.87) and basic polypeptide (net pI = 8.2).8 Despite these biophysical features, in pathological states, otherwise soluble tau monomers self-assemble into β-sheet-enriched straight or paired helical filaments.9 Genetic mutations found in autosomal dominant tauopathies, e.g., G272V, P301L, V337M, and E342V, may reduce tau affinity toward microtubules, thereby contributing its discharge, self-assembly and fibrillar aggregation.10 On the other hand, evidence indicates that these mutations alone might not be sufficient to induce substantial conformational changes and that a wide range of post-translational modifications (PTMs) of tau plays a prerequisite part in the autonomous fibrillization propensity.11 Nonetheless, biochemical crosstalk among the PTMs has not yet been clearly characterized. Regarding proteostatic regulation, the primary effect of tau ubiquitylation on proteasomal degradation is a matter of controversy, partly because the disordered structure of tau enables this protein to be recognized and degraded by proteasomes even in the absence of ubiquitin.12,13 Moreover, a recent study by Arakhamia et al. on human CBD and AD brains reveals that polyubiquitin (polyUb) chains on tau are also involved in inter-protofilament packing of tau.14
Phosphorylation has been considered the rate-limiting step of tau fibrillization since hyperphosphorylated tau was identified in neurofibrillary tangles and filamentous inclusions in postmortem brain tissues from patients with AD.15 Despite this notion, pharmacological inhibition of this PTM event has resulted in only a limited benefit in various clinical trials.16 Although these negative outcomes do not necessarily invalidate the strategy of targeting tau kinases, we presumed that the results instead reflect complex consequences of tau phosphorylation coordinated with other PTMs. Here, we report an unforeseen function of tau phosphorylation: promotion of tau ubiquitylation. This sequential enzymatic process generated more readily degradable phospho–ub-tau species. By contrast, when ubiquitylation was prolonged, phospho–ub-tau became hyperubiquitylated and transformed into insoluble filament cores. We also developed a small-molecule inhibitor of tau ubiquitylation, which effectively blocked tau aggregate formation in rat primary cortical and hippocampal neurons. These data for the first time uncover the biochemical crosstalk between tau phosphorylation and ubiquitylation that determines its fates. This study also suggests a more efficient and possibly mechanism-modifying strategy for delaying the clinical onset of tauopathies by targeting tau ubiquitylation instead of phosphorylation.
Results
In vitro reconstitution of tau PTMs and their combinations
To reconstitute tau PTM reactions in vitro, human tau40 was purified and incubated with GSK3β and ATP (for phosphorylation), p300 and acetyl-coenzyme A (for acetylation), or UBA1, UbcH5b, CHIP, and ubiquitin (for ubiquitylation)17–19 (Fig. S1†). Recombinant tau40 has a calculated molecular weight of ∼46 kDa but showed a ∼65 kDa size with multiple smaller-size bands due to its net positive charge and unstructured conformations as previously reported.20 An immunoblotting (IB) analysis revealed that the tau proteins were effectively chemically modified in the presence of cognate substrates and enzymes. Notably, we found that CHIP-mediated ubiquitylation, regardless of chaperon HSC70, failed to conjugate more than four ubiquitin moieties with tau (thus forming ub-tau; Fig. 1A and S1C†), which are generally required for strong interactions with proteasomes. In sharp contrast, when tau was modified by phosphorylation first (phospho-tau), subsequent in vitro ubiquitylation of tau generated smeared bands of higher-molecular-weight tau species (phospho–ub-tau) during sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) under reducing conditions (Fig. 1A and B); this pattern is a feature of conventional polyubiquitylated proteins. Tau challenged with heat-inactivated GSK3β (Fig. 1A) or CHIP (Fig. S1D†) yielded virtually no adequately ubiquitylated tau species, indicating that the catalytic activities of these enzymes, not unintended cross-reactivities, are directly involved in the outcomes of these tandem reactions. Unlike phosphorylation, precedent acetylation did not affect the immunoblotting patterns of tau ubiquitylation (Fig. 1B).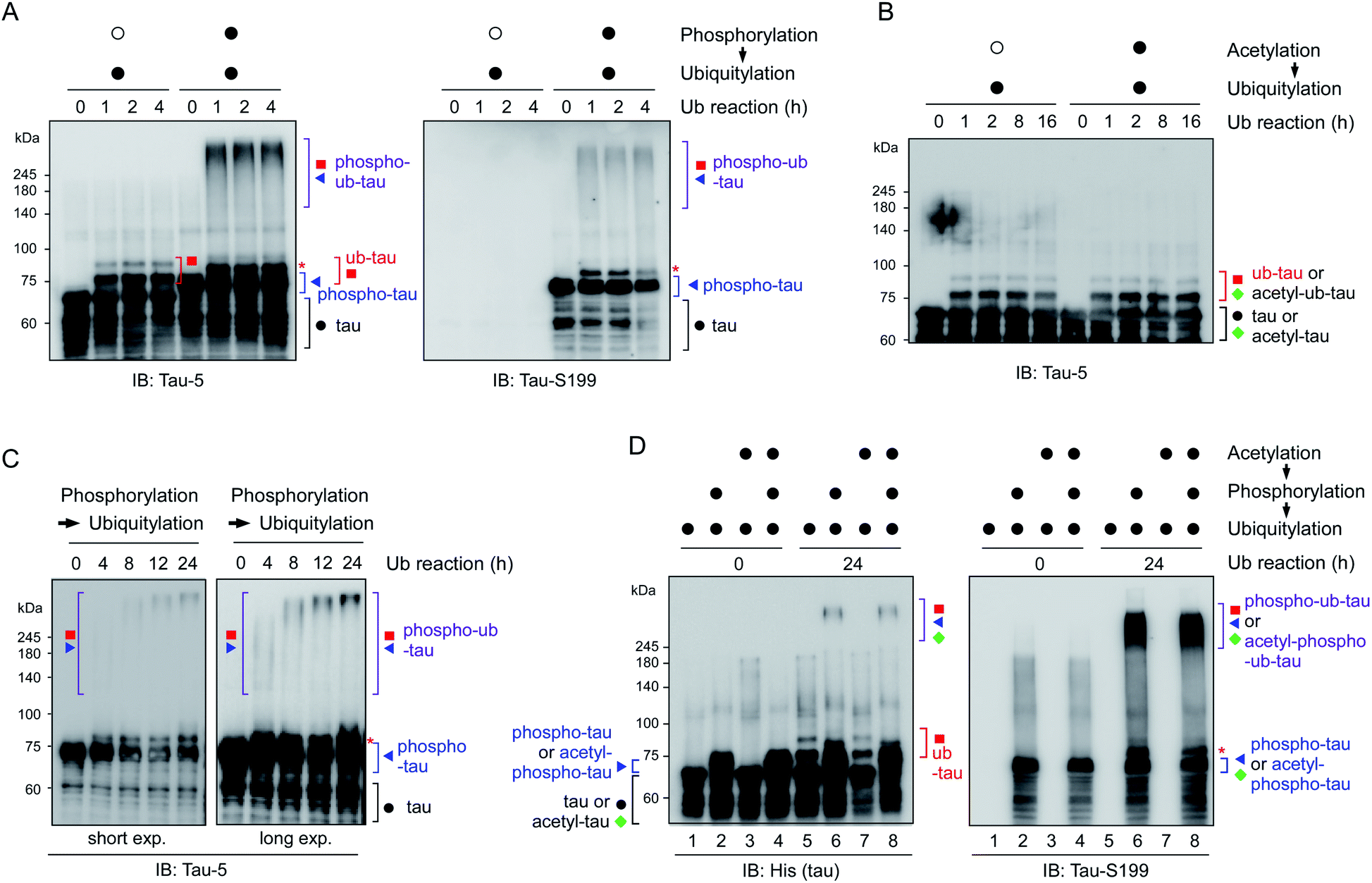 | ||
| Fig. 1 Tau phosphorylation is required for adequate tau ubiquitylation in vitro. (A) Recombinant full-length tau protein (tau40; 0.5 μg) was ubiquitylated in vitro by means of ubiquitin (1 μg) and UBA1 (200 ng), UbcH5b (400 ng), and CHIP (4 μg) as enzymes E1, E2, and E3, respectively, for the indicated time periods. Prior to ubiquitylation, the phosphorylation reaction was carried out using heat-inactivated (open circle) or active (filled circle) GSK3β (1 μg) for 4 h. Reaction products were assayed by SDS-PAGE followed by immunoblotting (IB) with anti-tau-5 and anti-tau-S199 antibodies. Although unmodified tau was decorated with only 1–3 ubiquitin moieties (ub-tau), phosphorylated tau was conjugated with a much larger number of ubiquitin units (phospho–ub-tau) by CHIP. (B) The similar in vitro ubiquitylation reactions were carried out following acetylation using either heat-inactivated (open circle) or active p300 (filled circle) for 4 h. (C) As in (A), except that phosphorylated tau (phospho-tau) was ubiquitylated in vitro for longer periods (up to 24 h), which yielded higher-molecular-weight tau species in a time-dependent manner. (D) Acetylation had only a limited effect on the course of tau phosphorylation and ubiquitylation in vitro. Tau acetylation was performed using p300 (1 μg) for 4 h before other post-translational modification (PTM) reactions were initiated. Total tau and phosphorylated tau were monitored by SDS-PAGE/IB under reducing conditions. *, mono-ubiquitylated tau. | ||
As the ubiquitylation reaction time following phosphorylation was extended, the resultant tau products possessed significantly longer polyUb chains in a time-dependent manner, thereby generating tau species not entering the resolving gel (hyper-ub-tau) after a 24 h reaction (Fig. 1C). In complementary experiments, we additionally modified tau via acetylation in several combinations with phosphorylation and demonstrated that acetylation by p300 essentially did not affect tau phosphorylation or vice versa (Fig. S2A and B†). Regardless of preceding acetylation, tau phosphorylation/ubiquitylation robustly generated slowly migrating phospho–ub-tau species (Fig. 1D) in vitro. Our results clearly indicated a hitherto unidentified role of tau phosphorylation: conversion of ubiquitylation modes of tau (and its potential degradation and aggregation tendency as described below). We anticipate that tau phosphorylation occurring in the flanking regions of MBDs may lead to its conformational change, which gives CHIP greater access to Lys residues in the MBDs and promotes the covalent conjugation of extra ubiquitin moieties.
Polyubiquitylated tau species degraded by 26S proteasomes
To further characterize the phospho–ub-tau products, we first employed methylated ubiquitin that lacks oxidizable NH3+ groups and thus is incapable of forming the ubiquitin–ubiquitin conjugation. We found that the addition of methylated ubiquitin abrogated the formation of polyubiquitylated tau (Fig. 2A), indicating that the observed high-molecular-weight tau species were not oligomeric forms with intermolecular disulfide bonds or tightly stacked β-sheets but were rather covalently linked with multiple polyUb moieties. When challenged with affinity-purified human proteasomes, phospho–ub-tau was completely degraded by 26S proteasomes in less than 2 h but was virtually unaffected by 20S proteasomes (Fig. 2B). Only limited degradation was observed in non-ubiquitylated tau or mono/di-ubiquitylated tau (Fig. S3†). Therefore, cellular degradation of adequately ubiquitylated tau species (phospho–ub-tau) by 26S proteasomes is expected to be more efficient than that of mono- or di-ubiquitylated tau species.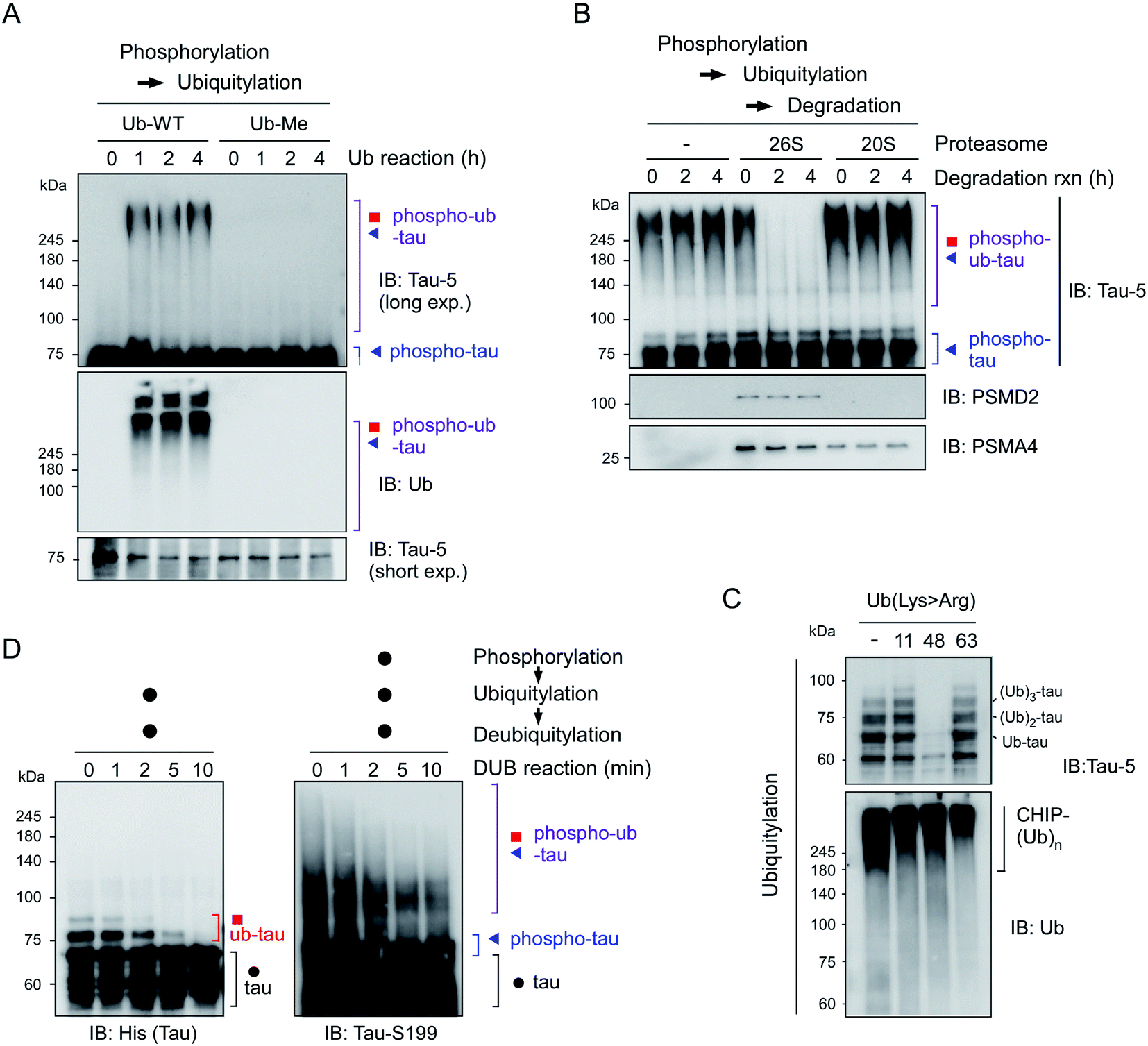 | ||
| Fig. 2 Adequately ubiquitylated phospho–ub-tau species are degraded by 26S proteasomes in vitro. (A) Wild-type ubiquitin (Ub-WT) and methylated ubiquitin (Ub-Me), whose seven internal Lys residues and α-amino group were all methylated, were used for the in vitro ubiquitylation of phospho-tau. (B) Purified human proteasomes (5 nM), either 26S or 20S, were incubated for the indicated time periods with phospho–ub-tau species (100 nM) generated by in vitro phosphorylation/ubiquitylation reactions. (C) The topology of polyubiquitin chain assembly on tau by UBA1, UbcH5b, and CHIP from WT or mutant ubiquitin (Lys > Arg). Ub-Lys48Arg yielded markedly less ubiquitylated tau after the 24 h in vitro reconstitution reaction. The IB analysis against Ub identified autoubiquitylated CHIP species that had higher molecular weights than that of ub-tau. (D) In vitro deubiquitylation of polyUb moieties conjugated with tau or phospho-tau. Recombinant USP2 (50 nM) was reacted with the tau species (100 nM) after PTM reactions for the indicated periods. Samples were analyzed by reducing SDS-PAGE/IB. | ||
To assess the topology of polyUb chains on phospho–ub-tau, we carried out in vitro ubiquitylation reactions with either wild-type (WT) or mutant ubiquitin with a Lys to Arg (K#R) substitution. In the reactions with ubiquitin-K11R and -K63R, similar polyUb chains were generated relative to those of WT, whereas the formation of polyUb chains failed when ubiquitin-K48R was used (Fig. 2C). According to these results, the polyUb chains on phospho–ub-tau seemed to be mainly linked through the canonical degradation signal (Lys48-linked polyUb chain). This notion is consistent with other studies, showing impaired tau degradation in the presence of various proteasome inhibitors.21,22 In addition, the polyUb chains of ub-tau and phospho–ub-tau were both efficiently deubiquitylated by the catalytic activity of ubiquitin-specific protease 2 (USP2; Fig. 2D). Therefore, tau ubiquitylation is a reversible process (counterbalanced by deubiquitylation) before which the ultimate proteolysis by the proteasome is commenced.
Our data collectively indicated that phosphorylation events might be as critical as ubiquitylation for catabolic regulation of tau proteins. Several reports have shown that intrinsically disordered tau could be degraded by both ubiquitin-independent (by 20S proteasomes) and -dependent manners (by 26S).12,13 Considering that the half-life of unmodified tau is longer than that of most of the other unstructured proteins, tau may require a multistep process (phosphorylation-induced ubiquitylation) under stress conditions for more efficient 26S proteasome-mediated proteolysis. The preceding phosphorylation may mask excess positive charges of tau or alleviate structural and physical constraints for the interaction with E3 ubiquitin ligases, such as CHIP.
Identification of ubiquitylated Lys residues in phospho–ub-tau
To assess the changes of PTMs in tau, mass-spectrometric (MS) analysis was performed using recombinant tau40 after in vitro ubiquitylation (ub-tau) and sequential phosphorylation/ubiquitylation (phospho–ub-tau). Mainly four internal Lys residues (K267, K290, K343, and K353; orange text and circles in Fig. 3) within the MBDs were found to be ubiquitylated in ub-tau. Among these, K267, K343, and K353 were recently reported to be polyubiquitylated in the brain tissues of patients with both CBD and AD.14 When phospho-tau was ubiquitylated, markedly increased MS signals were detected not only at the original four ubiquitylation residues but also on seven de novo Lys sites (K257, K259, K274, K281, K321, K375, and K385; red in Fig. 3) within the MBDs and nearby C-terminal tail. These data substantiate the in vitro reconstitution results (Fig. 1), which revealed upregulated tau ubiquitylation after phosphorylation. Lys residues in previously suggested chaperone-mediated autophagy-targeting motifs (336QVEVK340 and 347KDRVQ351) or dimerizing hexapeptide motifs (275VQIINK280 and 306VQIVYK311) turned out to be unmodified. Two tau phosphorylation sites (Ser262 and Ser356) were located in the MBDs, and both were found to be a part of the microtubule-interacting Lys–Ile–Gly–Ser motif, where the Lys residues in the motifs (K259 and K353) were ubiquitylated. A similar MS analysis of in vitro tau acetylation indicated that most of the tau acetylation sites did not overlap with the ubiquitylation sites except for K259 and K274 (Fig. 3A and B). Overall, these results point to the novel function of tau phosphorylation and ubiquitylation which cooperatively modulates its biochemical and biophysical characteristics. Whether and how individual phosphorylation sites in MBDs contribute to subsequent ubiquitylation or aggregation events remain to be investigated.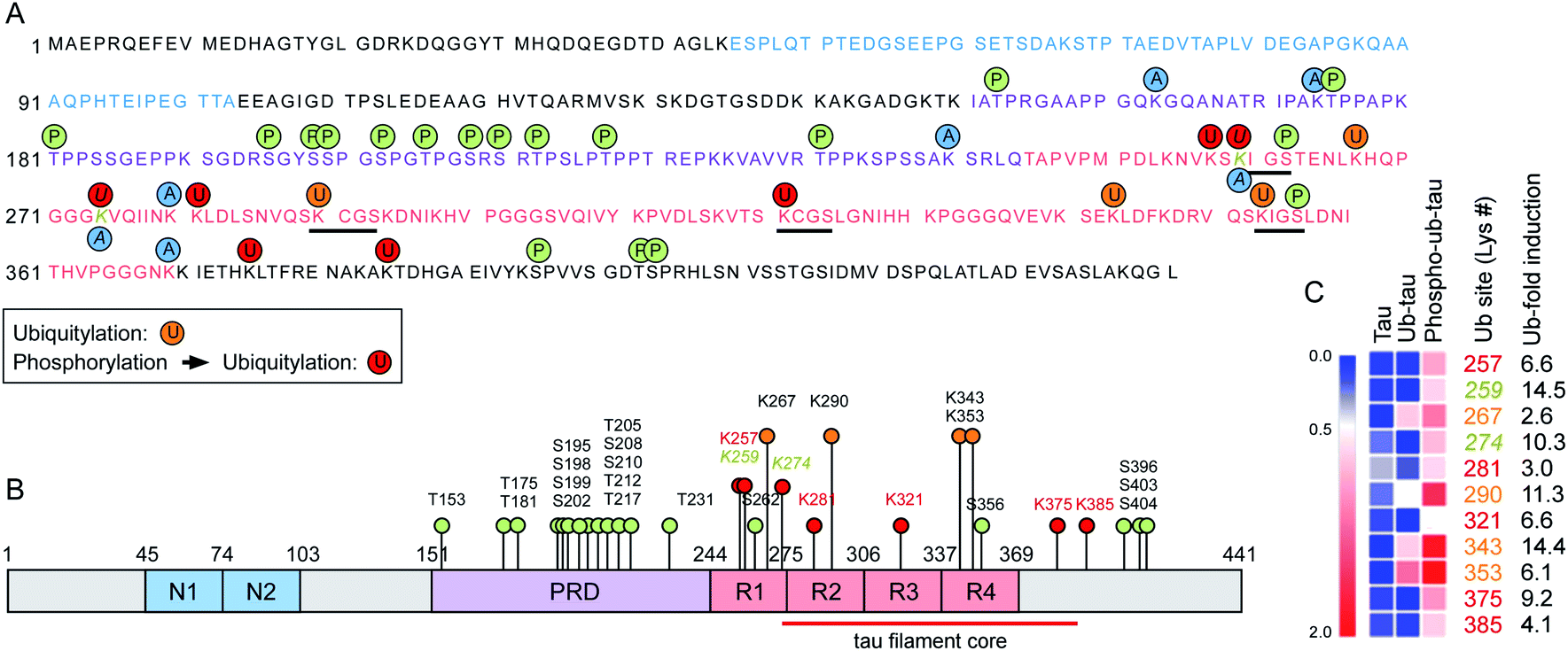 | ||
| Fig. 3 The expanded set of ubiquitylation sites in tau after phosphorylation in vitro were identified with mass spectrometry. (A) Phosphorylation and ubiquitylation sites of recombinant tau after in vitro PTM reactions were identified by mass spectrometry (MS). Orange and red circles depict ubiquitylation sites of ub-tau (ubiquitylation only) and phospho–ub-tau (phosphorylation and subsequent ubiquitylation), respectively. Amino acid residues with single letter codes in N-domains (N1 and N2) are depicted in blue, the proline-rich domain (PRD) in purple, and microtubule-binding domain (MBD) repeats 1 to 4 (R1–R4) in red. Phosphorylation and acetylation sites are indicated with green and blue circles, respectively. Residues modified by both acetylation and ubiquitylation are Lys259 and Lys274 (gold and italic). Black underlining highlights four Lys–Ile/Cys–Gly–Ser motifs. (B) PTMs are pointed out on a schematic diagram of tau40. Color schemes are depicted as in (A). Although phosphorylation was abundant on the PRD, ubiquitylation occurred mostly on the MBD. (C) A distinct change in relative abundance and modification sites of tau ubiquitylation following phosphorylation was revealed by quantitative MS. Degrees of upregulation (red) are explained by the color key of the heat map. Pre-existing ubiquitylation sites in ub-tau and de novo sites in phospho–ub-tau are denoted by the orange and red text, respectively. Fold-inductions (from ub-tau to phospho–ub-tau) of ubiquitin signal intensities at the indicated Lys residues are shown. The red lines indicate the core structure comprising the tau fibrils in corticobasal neurodegeneration (CBD; ref. 14). | ||
Transition of phospho–ub-tau to insoluble fibrils
To further examine the consequences of tau hyperubiquitylation, we incubated tau with different PTMs in the ubiquitylation condition over prolonged periods (>2 d). When the tau species were analyzed by SDS-PAGE followed by IB with phospho-tau antibodies, both phospho-tau and phospho–ub-tau turned out to be transformed into oligomeric species. However, the kinetics were markedly more rapid for hyper-ub-tau (Fig. 4A), showing significant amounts stuck in the stacking gel after 2 d of in vitro ubiquitylation reactions. Once again, acetylation did not affect the formation of less soluble hyper-ub-tau species (Fig. S4†).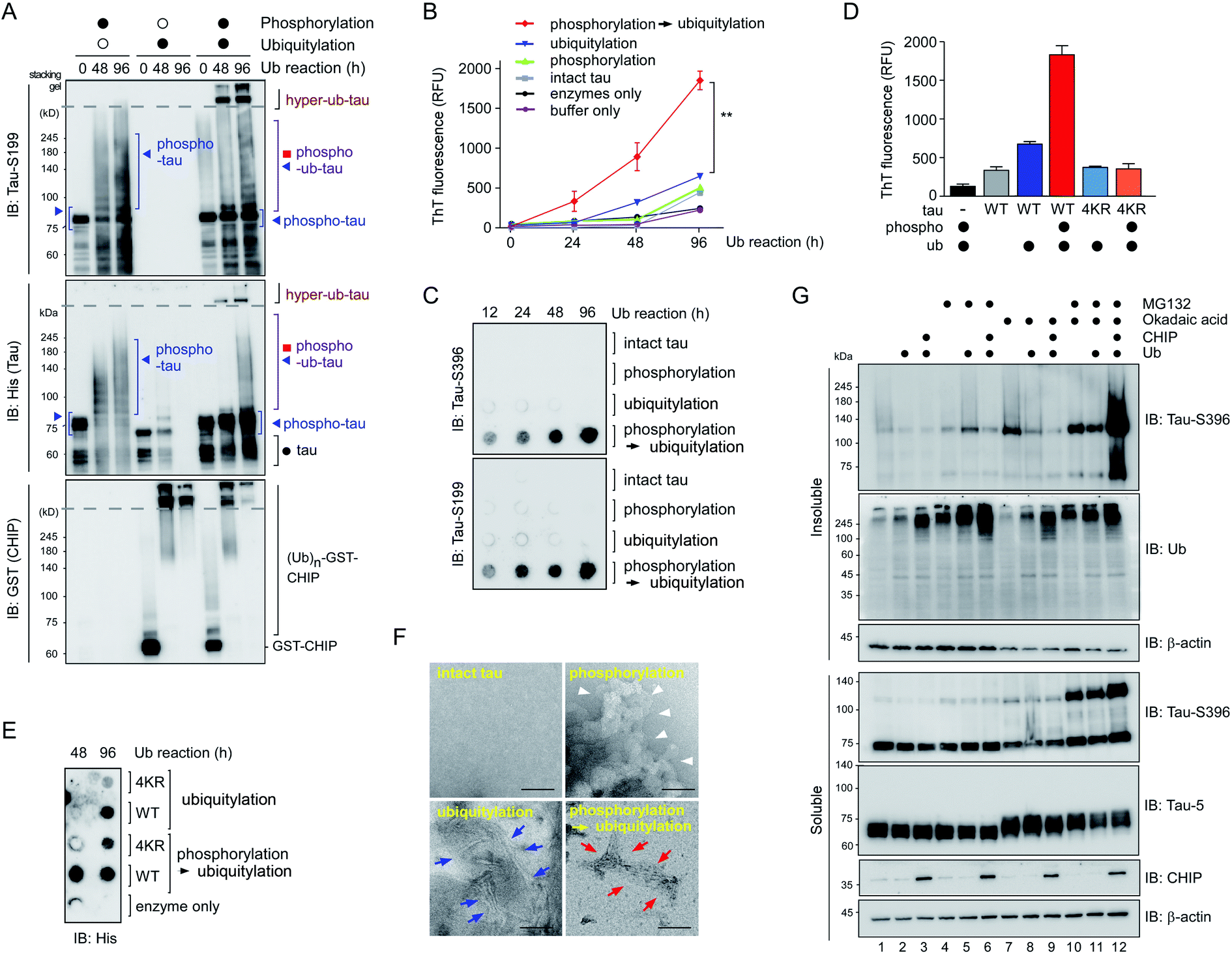 | ||
| Fig. 4 Phospho–ub-tau more readily transforms to fibrillary proteins after prolonged incubation. (A) In vitro phosphorylated tau was ubiquitylated for prolonged incubation periods (up to 4 d), which gave rise to hyperubiquitylated tau species (hyper-ub-tau) observed on top of the separating gel (dotted line). PTM reactions involving inactive GSK3β or CHIP are depicted by open circles, while filled circles indicate reactions with active enzymes. Tau species were detected by SDS-PAGE/IB with anti-His and anti-tau-S199 antibodies. (B) In fibrillization reactions involving intact or PTM-modified tau (3.33 μM), thioflavin T (50 μM) fluorescence intensity at 480 nm increased significantly more with phospho–ub-tau than with either phospho- or ub-tau. The phosphorylation reaction was carried out for 4 h, and ubiquitylation was performed for the indicated periods. **, p < 0.001 (n = 3, two-way analysis of variance). Error bars denote standard error of the mean. (C) Samples were prepared as described in (B) and were subjected to filter trap assays. Intact tau or tau with PTMs (4 μg) were loaded and filtered into each well of a 96-well plate, and the trapped tau oligomers were detected by IB with the indicated anti-tau antibodies. (D) Thioflavin T assays were performed as in (B) using tau-WT and mutant tau (tau-4KR) after 96 h ubiquitylation reactions. In tau-4KR variants, the four Lys sites (K321, K343, K353, and K375), in which ubiquitin modifications were enhanced by preceding phosphorylation, were substituted to Arg. (E) Dot blotting analysis for tau revealed significantly reduced amounts of tau-4KR, compared to tau-WT, trapped in the cellulose acetate membrane after the PTM reactions. (F) Representative images from negative-staining electron micrographs of intact tau and tau with various PTMs. The phosphorylation and ubiquitylation reactions were conducted for 4 h and 2 d, respectively. While phospho-tau formed amorphous aggregates (white arrowheads), hyper-ub-tau was relatively more uniformly fibrillized with shorter filaments and bundled structures (red arrows), as compared with those of ub-tau (blue arrows). Scale bar, 100 nm. Of note, no heparin or aggregation inducers were included in all in vitro assays. (G) The accumulation of phosphorylated tau in the insoluble fraction of HEK293-derived cells stably overexpressing tau when they were cotreated with MG132 (5 μM) and okadaic acid (OA, 30 nM) for 6 h along with overexpression of CHIP and ubiquitin. RIPA-soluble and insoluble (pellet) fractions from whole cell lysates were isolated and subjected to SDS-PAGE/IB analysis. | ||
We hypothesized that these migration-resistant tau species might represent less soluble tau filaments. To directly assess the effect of tau hyperubiquitylation on tau fibrillization, we first used thioflavin T (ThT), which intercalates into β-sheet structures of self-assembled multimeric tau23 without aggregation inducers such as anionic heparin. Intact tau alone did not generate any ThT-positive signals even after 4 d, but, in contrast, hyper-ub-tau proteins emitted significantly stronger fluorescence signals than those of phospho-tau or ub-tau (Fig. 4B). ThT signals of hyper-ub-tau began to be prominent after ∼24 h ubiquitylation reactions and gradually increased until time point 96 h in the reactions. A possible explanation for this observation is the conversion of hyper-ub-tau to the nucleate for tau self-assembly. When we fractionated tau species depending on molecular weights with size-exclusion chromatography and examined their transmission electron microscopy (TEM) images and in vitro seeding activity, we found that fractionated tau species with >250 kDa-size, but not the fractions containing monomeric tau, have clear fibrillar morphologies and potent transformation ability of the monomeric tau into aggregates (Fig. S5A–C†). These data strongly support the notion that hyper-ub-tau not only acts as a nucleate for pathologic tau aggregates but also undergoes facilitated self-assembly into early intermediates of insoluble aggregates. Consistently, a filter trap assay of diverse tau species revealed that hyper-ub-tau proteins were distinctively less membrane-penetrating, compared with other tau species, even in the absence of aggregation inducers (Fig. 4C). Other components of the PTM reactions, such as UBA1 (E1) and CHIP (E3) enzymes, were not trapped in the cellulose acetate membrane (Fig. S5B†).
We have further explored the role of phosphorylation-induced tau ubiquitylation by modifying the four key Lys resides (K321, K343, K353, and K375; Fig. 3), which were identified from the MS analysis. We found that single, double, or triple substitution of these sites to Arg generated little changes in the biochemical behaviors of tau proteins upon sequential phosphorylation and ubiquitylation (data not shown). However, purified tau variants with quadruple Lys mutations (tau-4KR) showed significant reduced in vitro aggregation propensities, observed by ThT analysis or filter trap assays (Fig. 4D and E). These data strongly suggest that the hyperubiquitylation of tau, rather than phosphorylation, may be critical for its fibrillization.
Complementing the biochemical experiments, we examined intact and modified tau proteins by negative-staining TEM and found that hyperubiquitylated tau formed narrower and shorter filament bundles than those of phospho-tau or ub-tau (Fig. 4F). Intact tau did not fibrillize without aggregation inducers, and phospho-tau formed mainly amorphous aggregates. This observation is similar to the results of coarse-grained modeling, where tau phosphorylation facilitates the formation of amorphous aggregates instead of amyloid fibrils.24 The short fibrils might function as a protofilamental seed for further tau aggregation. Taken together, our in vitro data strongly indicate the mediatory function of tau hyperubiquitylation in the conformational transition of tau from a soluble monomer to oligomers and eventually molecular seeds for the pathological self-assembly. Via crosstalk with other PTMs, tau may be differentially ubiquitylated and aggregated into distinct conformations of a tau filament (tau strains), thereby potentially determining the biochemical or symptomatic phenotype of diverse tauopathies.
The crucial function of CHIP in insoluble-tau formation in test tubes and cultured cells
To verify the consequence of cooperative tau PTMs in live cells, we first tested HEK293-derived cells that stably expressed tau, which did not produce substantial amounts of insoluble tau oligomers under normal conditions.25 When okadaic acid (OA), which efficiently blocks tau dephosphorylation by inhibiting protein phosphatase 2A,26 was incubated with the cells, mildly elevated amounts of phosphorylated tau oligomers (with a molecular weight of ∼110 kDa) were detected in a RIPA-insoluble fraction of whole-cell lysates (WCLs; Fig. 4G, lanes 7–9). The high-molecular-weight phospho–ub-tau seen in the in vitro assays was hardly detectable in the WCLs possibly due to the antigen-masking effects.27,28 When the proteasome inhibitor MG132 was combined with OA, significantly elevated levels of tau oligomers were noted both in the soluble and insoluble fractions (Fig. 4G, lanes 10 and 11). Moreover, facilitating tau ubiquitylation using transient overexpression of E3 ubiquitin ligase CHIP led to marked increases in the levels of insoluble tau oligomers (Fig. 4G, lane 12). These results, consistent with our in vitro analysis data, indicate that tau phosphorylation or ubiquitylation alone may not be sufficient to mediate tau aggregation in the cell. Rather, multiple PTM reactions, along with relevant cellular conditions incapable of clearing adequately ubiquitylated tau species, may cause accumulation of pathological hyper-ub-tau species and can facilitate their self-assembly into the insoluble fibrils.CHIP functions as a critical quality controller of cellular proteome implicated in diverse human diseases,29 but, to dates, small-molecule inhibitors suppressing the catalytic activity of CHIP remain essentially unexplored. To validate the role of CHIP in tau hyperubiquitylation and aggregation both in vitro and in vivo, we conducted a biophysical screening of more than 500 curated small-molecule fragments, which was based on the NMR analysis implemented with a saturation transfer difference (STD) protocol (Fig. 5A; see Methods). The fragments that directly bind to purified CHIP were chemically combined and further modified to strengthen the binding affinities. Among them, we have confirmed that compound #153 effectively and specifically blocked the CHIP-mediated ubiquitylation of inhibited proteasomes (IC50 = ∼85 μM; Fig. S6†).30 When their activity was assessed in our standard in vitro tau ubiquitylation reactions, we found that compound #153, but not its structural control #154, had a strong inhibitory effect toward CHIP-mediated tau ubiquitylation (Fig. 5B and C). This CHIP inhibitor also drastically reduced the levels of hyper-ub-tau species that were generated by sequential in vitro phosphorylation/ubiquitylation and were non-migratable to the resolving gel (24 and 48 h; Fig. 5D, lanes 7 and 8). Compound #153 (50 μM) had little effect on tau phosphorylation and manifested no noticeable cytotoxicity even when combined with MG132 (10 μM) for 6 h (Fig. 5E, F, and S7A†).
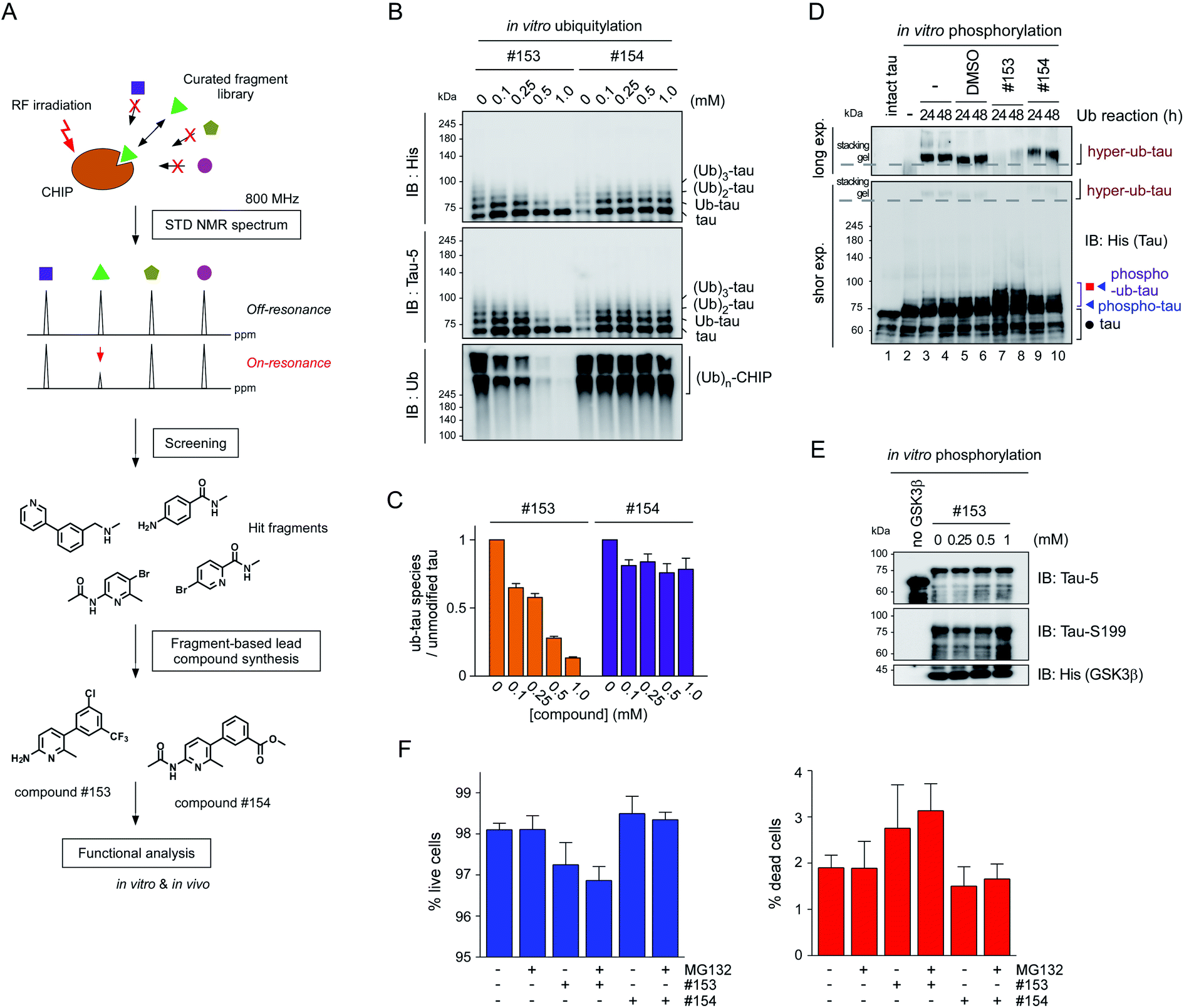 | ||
Fig. 5 The small-molecule inhibitor of CHIP, identified via biophysical screening, potently delays the insoluble tau formation. (A) The screening and synthesis processes of the CHIP inhibitors. (B) Compound #153 and its structural control (compound #154) at 0, 0.1, 0.25, 0.5, or 1 mM were added to the in vitro tau ubiquitylation reaction (without phosphorylation) for 24 h. Ubiquitylated tau proteins were detected by SDS-PAGE/IB with the anti-His or tau-5 antibody. Autoubiquitylated CHIP enzymes were detected higher than 200 kDa size. (C) Quantification of ub-tau species with normalization to the signals of unmodified tau. (D) In vitro phosphorylated tau was ubiquitylated for 24 or 48 h in the presence of DMSO, compounds #153, or #154. Tau species were detected by SDS-PAGE/IB with the anti-His antibody. Long and short exp., long and short exposures, respectively. The dotted lines indicate the border between the stacking and separating gels. (E) Tau was phosphorylated in vitro for 4 h at 37 °C in the presence of the CHIP inhibitor (compound #153) at the indicated concentrations. (F) Cytotoxicity of compounds #153 and #154 (50 μM) in the presence and absence of MG132 (10 μM) for 6 h, determined by high-content cell counting. ∼40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well were imaged by the ImageXpress Micro system after stained with Hoechst 33342 (for total cells) and propidium iodide (for dead cells), and were counted with the automatic analysis module within. No significant difference was found among the live cell (left) and dead cell (right) groups (one-way ANOVA followed by the Bonferroni post hoc test). 000 cells per well were imaged by the ImageXpress Micro system after stained with Hoechst 33342 (for total cells) and propidium iodide (for dead cells), and were counted with the automatic analysis module within. No significant difference was found among the live cell (left) and dead cell (right) groups (one-way ANOVA followed by the Bonferroni post hoc test). | ||
We then proceeded to apply the small molecule to live cells to determine whether the suppression of tau ubiquitylation by CHIP might delay the process of tau aggregation into the insoluble fractions. After transient overexpression of tau in immortalized mouse hippocampal HT22 cells, they were treated with MG132 and OA in the presence or absence of compound #153, for 6 h. A gel-based analysis and subsequent quantification showed markedly reduced levels of monomeric and oligomeric tau from the insoluble fraction of WCLs with compound #153 (Fig. 6A and B). Similar effects of the CHIP inhibitor were observed in nonneuronal HEK293 cells (Fig. S7B†). Next, we isolated rat primary cortical cells and treated them with compound #153 along with OA and/or MG132. We found that the levels of endogenous tau, both phosphorylated and non-phosphorylated forms, in the insoluble fraction were significantly reduced in the presence of the CHIP inhibitor but not by its structural control #154 (Fig. 6C, S7C and D†). When we enriched overexpressed tau from HT22 WCLs using immunoprecipitation, we observed that compound #153 significantly reduced the extent of polyubiquitination of tau (Fig. 6D and E). The correlation between the aggregation propensity of tau in the cultured cells and its degree of polyubiquitylation may implicate the possible role of tau ubiquitylation in the fibrillization process upon the phosphorylation queue.
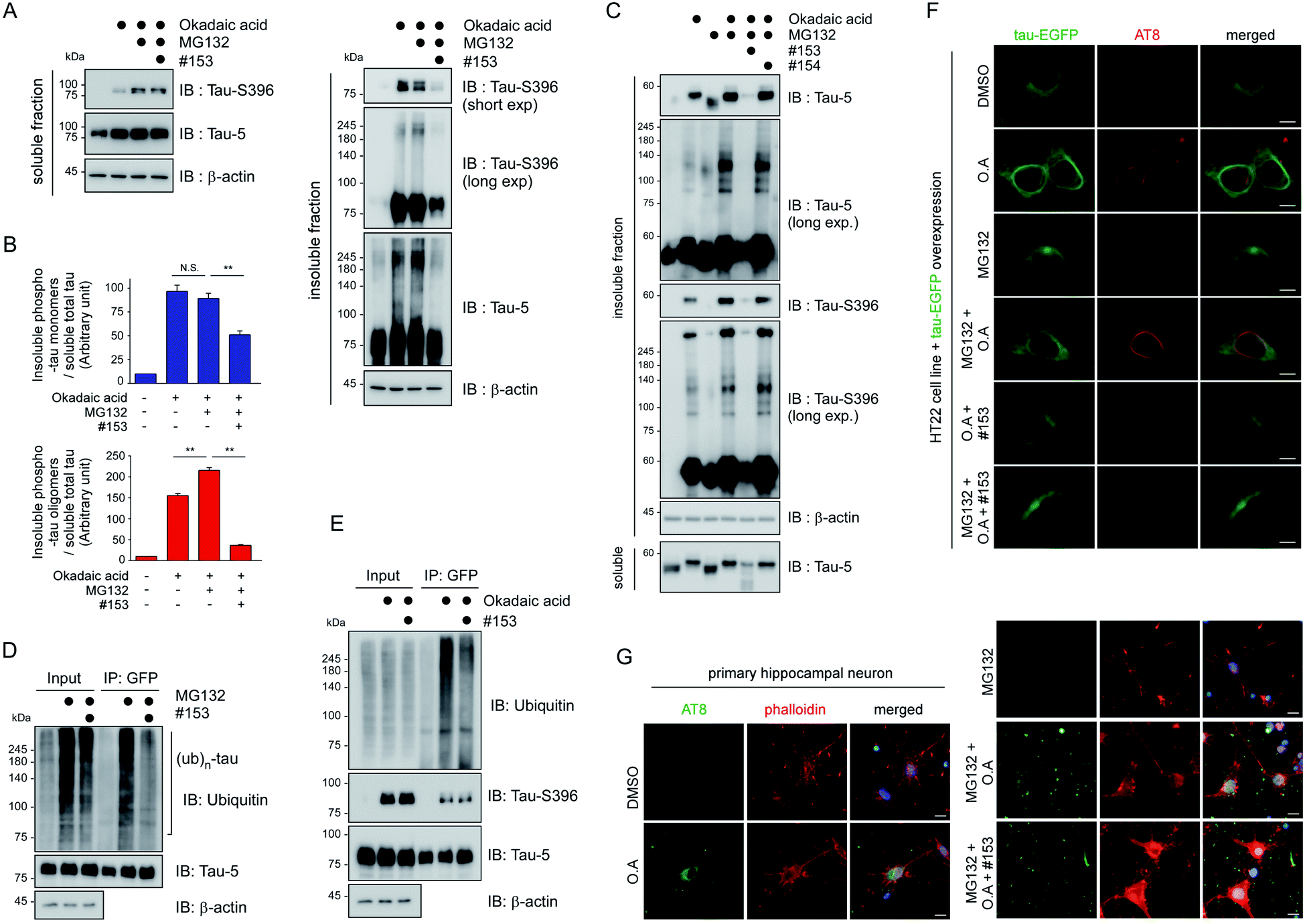 | ||
| Fig. 6 The CHIP inhibitor delays the aggregation propensity of tau in cultured cells. (A) Reduced levels of tau, both monomeric and oligomeric forms, in the detergent-insoluble fraction of HT22 mouse hippocampal cells. Tau40 was transiently overexpressed in HT22 cells in the presence or absence of 30 nM OA, 10 μM MG132, and/or 100 μM compound #153 for 6 h. RIPA-soluble and -insoluble fractions of whole cell lysates (WCLs) were isolated and subjected to SDS-PAGE/IB with indicated antibodies. (B) Relative levels of insoluble monomeric tau (upper) and oligomeric tau (lower) were quantified with normalization to those of soluble total tau and plotted as mean ± SD from three independent experiments as performed in A. N.S, not significant. **, p < 0.01 (one-way ANOVA followed by the Bonferroni post hoc test). (C) Similar to the experiments as in A, except that the assay was performed in rat primary cortical cells with either compound #153 or #154 control (50 μM) to monitor the changes of endogenous tau. Shown is one of the sets of triplicate experiments (Fig. S7C†). (D) Reduced polyubiquitylation of tau in HT22 cells where EGFP-tagged tau was transiently overexpressed and treated with MG132 (10 μM) and/or compound #153 (50 μM) for 6 h. WCLs were subjected to immunoprecipitation with anti-GFP antibodies, and subsequent SDS-PAGE/IB. (E) Co-immunoprecipitation analysis as in D, except that OA (30 nM) was treated instead of MG132. (F) Representative fluorescence images of HT22 cells with AT8 immunostaining, which transiently overexpress tau-EGFP after treated with DMSO, MG132 (10 μM), OA (30 nM), and/or compound #153 (50 μM) for 6 h. Tau-positive signals formed in OA and MG132-treated cells exhibit dense and curvilinear morphologies but the CHIP inhibitor effectively reduced the signal. (G) Reduced tau aggregation tendency in primary rat hippocampal neurons in the presence of 50 μM compound #153. Various combinations of 30 nM OA and 10 μM MG132 were co-treated to the primary neurons in 12-well plates at day 7 in vitro. Immunostaining with AT8 antibodies with DAPI and phalloidin co-counterstaining. Scale bar, 10 μm. | ||
HT22 cells exhibited only basal fluorescent signals when tau-EGFP was transfected under normal conditions (Fig. 6F). In the presence of OA and MG132, however, the thin and hair-like structures of tau appeared in the cytoplasm with one or two weak EGFP-positive inclusions near the microtubule-organization center. In sharp contrast, we observed that the curvilinear phospho-tau signals detected with AT8 immunostaining were effectively depleted when the cells were treated with compound #153 (Fig. 6F). Consistently, primary hippocampal neurons treated with compound #153 appeared to have reduced AT8-positive punctate signals, which were mainly located in the dendritic branches and neurites (Fig. 6G and S8†). With phalloidin staining of assembled F-actin,31 we also observed visibly restored synaptic networks after treatment with the CHIP inhibitors. Therefore, the small-molecule inhibitor of CHIP not only inhibits tau ubiquitylation in vitro but also effectively delays the tau aggregation process in cultured neurons under stress conditions (for example, in the presence of both MG132 and OA). These data collectively indicate that CHIP may play a critical part in tau aggregation in vivo, which was more eminent when tau dephosphorylation and 26S proteasome-mediated clearance were impaired.
Discussion
Accumulating evidence suggests that the fibrillization of oligomeric tau is a critical step in the onset and progression of tauopathies. During the aggregation process, numerous PTMs on tau appear to be complicatedly involved,8,14 but little is known about the biochemical crosstalk among the PTMs. Based on the results of our systematic in vitro reconstitution reactions, we propose a “multiple-hit model” where phosphorylation and ubiquitylation, as the first and second hit, respectively, play a crucial role in the conjugation of adequate numbers of polyUb moieties to tau and facilitate its proteasomal degradation under nonpathological circumstances (Fig. 7). This phenomenon helps the cell to maintain intracellular tau concentration below the critical point. However, when the threshold of the proteasome's capacity to degrade tau is exceeded, excess phospho–ub-tau transforms into hyperubiquitylated tau species and is readily self-assembled into insoluble pathogenic filaments. Thus, our study uncovered diverse consequences of tau ubiquitylation: (1) ubiquitylation alone was not effective enough to form polyubiquitylated tau species for efficient proteasomal clearance; (2) ubiquitylation with preceding phosphorylation generated a more readily processible tau species enabling cells to effectively cope with diverse stressful conditions; and (3) accumulated hyperubiquitylation of tau, owing to impaired proteasomal activity or excessive amounts of phospho–ub-tau in the cell, might lead to the formation of an insoluble filament core, which is implicated in the manifestation of tauopathies (Fig. 7).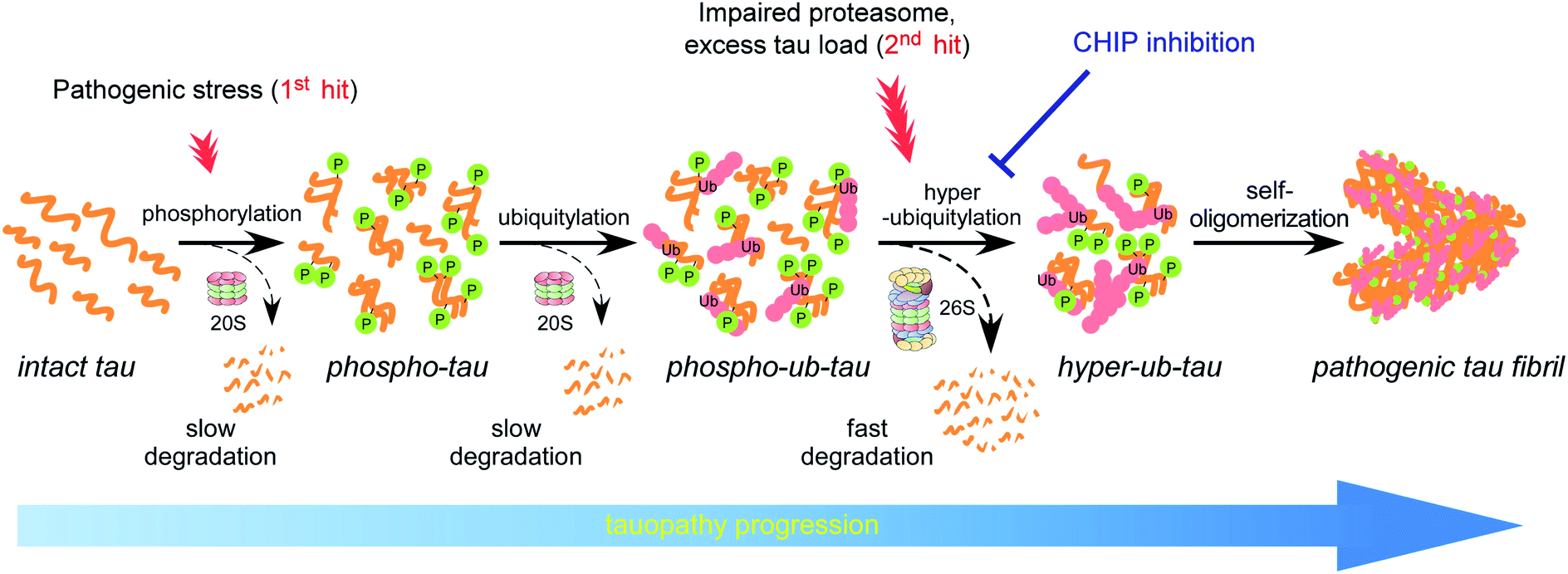 | ||
| Fig. 7 The proposed model illustrating the effect of PTMs on tau fibrillization and tauopathy progression. Under physiological conditions, the tau protein is decorated with a relatively small number of ubiquitin moieties, which are incapable of directly interacting with proteasomes. At an early stage of tauopathies, tau proteins are modified by phosphorylation, and the resulting structural changes may allow tau to be conjugated with adequate numbers of polyUb for proteasomal degradation (dashed arrows). When the clearance of phospho–ub-tau is inefficient—possibly due to a combination of proteasome inactivation and excessive amounts of loads on the proteasome (as observed in late-stage tauopathies)—tau progressively becomes hyperubiquitylated and self-polymerizes into insoluble fibrils (solid arrows). Clinical significance of this in vivo PTM-mediated tau proteostatic pathway remains to be determined. | ||
Among the novel ubiquitin-conjugated Lys residues after phosphorylation (Fig. 3), four residues (from K274 to K375) were found in recently identified tau protofilament repeats in human CBD brains.32 The ubiquitin moieties may provide additional hydrogen bonds between the tau backbone and side chains along the axis of the filaments. Therefore, the ubiquitin chains on tau potentially stabilize parallel β-strand stacking, leading to ordered tau fibrils rather than the formation of amorphous aggregates via random stacking. We also found that most phosphorylation sites (>20) had no significant changes after tau ubiquitylation and were located outside the MBDs in our MS analysis (Fig. 3B), consistent with a previous study reporting little changes in the phosphorylation sites during AD progression.33 The competition between acetylation and ubiquitylation on the same Lys residue seems to need further investigation as monitoring the global dynamics of diverse PTMs will provide an insight into tauopathy pathogenesis.
Our findings may clarify many previously incomprehensible results regarding tau phosphorylation. For example, overexpression of constitutively active GSK3β in transgenic mice does not aggravate tau fibrillization-related pathology but rather alleviates many neuropathological symptoms when these mice are crossed with tau transgenic mice.34,35 Moreover, tau phosphorylation inhibitors showed only minimal therapeutic effectiveness in various clinical trials.36–38 Accumulating evidence suggests that a soluble pool of phospho-tau may not necessarily be the direct etiology of tauopathy. Our study revealed that the phosphorylation of tau is a prerequisite signal for adequate tau ubiquitylation, which leads to tau degradation in physiological states but to tau aggregation in pathological states. Therefore, as is the case for cyclins, cyclin-dependent kinases, and SMAD1, the phosphate groups on tau may be a novel class of “phospho-degrons” that are specifically recognized by CHIP.39–41 This accords with the earlier observations showing preferential binding of CHIP to phosphorylated forms of tau,18,42 whilst expanding the role of CHIP-mediated tau ubiquitylation from proteolysis to fibrillization. It is worth noting that increasing evidence suggests that truncated tau (tau-Asp421) generated by caspase-3 may possess elevated fibrillogenic propensities and stronger binding affinities toward CHIP than the full-length tau protein.43–49 Therefore, altered caspase activity during tauopathy progression could be another functional contributor to complex tau homeostasis. Acetylation, ubiquitylation, and methylation are expected to crosstalk with one another for lysine residues of tau and are subjected to highly complex functional regulation. How tau phosphorylation and ubiquitylation affect its truncation (and vice versa) remains to be elucidated.
In addition, heparin-fibrillized tau exhibits polymorphic structures and none of them are consistent with from monomorphic fibrils derived from patients with AD that have larger cores with different repeat compositions.50 Although heparin may participate in neutralizing positive charges of the filament core, its polar and nonpolar interactions to the crosslinked tau molecules are expected to be global and intrinsically nonspecific. Conversely, our data indicate that the influence of polyUb chains are limited to certain Lys residues, especially in the fourth MBD, which is the integral part of tau filament core in AD or CBD brain but, in heparin-fibrillized tau, shows a disordered structure.8 This also raises questions about the relevance of using the four MBD-containing K18 fragments, which lack the key residues to adopt the same tau structures identified from AD brain although they more readily form heparin-induced filaments than full-length tau.
We observed that tau aggregation into the insoluble fraction of live cells was induced synergistically when both phosphorylation and ubiquitylation of tau were promoted but its proteasomal clearance was inhibited. By developing the potent CHIP inhibitor that effectively antagonizes tau ubiquitylation and fibrillization in test tubes and cultured primary neurons, we demonstrated that the CHIP-mediated ubiquitylation of phospho-tau may serve as a critical rate-limiting step of pathological tau transformation in vivo. As tau proteins can slowly be degraded in a ubiquitin-, ATP-, and 26S proteasome-independent manner,12 it is conceivable that the contribution of CHIP to tau ubiquitylation is more critical after tau phosphorylation (i.e., the first pathogenic hit; Fig. 7). Together, these data suggest that the beneficial effects of inhibiting CHIP enzymes are better observed when proteasomes fail to adequately degrade tau proteins (as seen under proteostatic and oxidative stress conditions). Therefore, reduced proteasome activity or elevated polyUb conjugate levels may serve as a good indication for a CHIP-targeting therapeutic strategy. Small nucleates of hyperubiquitylated tau, which is a non-processable substrate for the proteasome and potentially inhibits its activity via the “clogging” effect,51,52 may initiate a vicious cycle producing excessive amounts of tau aggregates. This may relate tauopathy etiology to the decreased proteasomal activity reported in postmortem human AD brains.53,54 In this regard, the ubiquitin molecules abundantly present in insoluble paired helical filaments are not mere undigested remnants but rather reflect the extensive and autonomous modification of tau during the aggregation process.
To summarize, our biochemical experiments uncovered a novel cooperative, multistep regulation of tau, which is based on PTMs and ultimately has either beneficial (through tau proteostasis) or detrimental (tau fibrillization) consequences, depending on the cellular environment (e.g., cellular proteasomal activity). Only with the preceding phosphorylation, tau proteins are adequately ubiquitylated and eliminated by the 26S proteasome. When the tau-proteolytic system is impaired or when excess phospho–ub-tau species clog proteasomes, tau proteins undergo biochemical transformation into insoluble, ThT-positive short fibrils. We still have a limited understanding of the signature PTMs on tau involved in pathological self-assembly and cellular regulation towards the specificity and activity of the enzymes involved. Nonetheless, our findings provide important insights into molecular pathophysiology of diverse tauopathies and will likely point to novel therapeutic approaches targeting tau ubiquitylation. The application of CHIP inhibitors to animal models of tauopathy, along with longitudinal biochemical and neuropathological analyses, will validate this strategy.
Author contributions
J. H. K., W. H. C., J. L., and S. P. carried out the in vitro experiments. J. H. K., W. H. C., and J. H. L. performed the cell-based assays. S. H. P. measured the cytotoxicity of the compounds and H.-S. C. purified recombinant proteins including GSK3β. S. M. L. and J. Y. M. carried out the STD-based fragment screening for CHIP inhibitors and the electron microscopic works, respectively. D. H. conducted the mass spectrometric analysis. Y. H. S. isolated rat primary neurons and performed the related experiments with them. M. J. L. prepared the manuscript and are responsible for the overall design of the study.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by grants from the National Research Foundation (2021R1A2C2008023 to M. J. L., 2019R1A2C1005987 to J. H. L., 2020R1I1A1A01066946 to S. P., 2019R1A6A3A01094785 to W. H. C, and 2020R1A5A1019023 to D. H., Y. H. S., and M. J. L.), KBRI basic research program (21-BR-01-11 to J. Y. M.), Korea Toray Science Foundation, and the Creative-Pioneering Researchers Program through Seoul National University.References
- D. W. Ethell, Neuroscientist, 2010, 16, 614–617 CrossRef CAS PubMed.
- C. R. Jack Jr, D. S. Knopman, W. J. Jagust, L. M. Shaw, P. S. Aisen, M. W. Weiner, R. C. Petersen and J. Q. Trojanowski, Lancet Neurol., 2010, 9, 119–128 CrossRef.
- A. Andreadis, Prog. Mol. Subcell. Biol., 2006, 44, 89–107 CrossRef CAS PubMed.
- E. M. Mandelkow and E. Mandelkow, Trends Cell Biol., 1998, 8, 425–427 CrossRef CAS.
- A. D. Alonso, T. Zaidi, M. Novak, H. S. Barra, I. Grundke-Iqbal and K. Iqbal, J. Biol. Chem., 2001, 276, 37967–37973 CrossRef CAS PubMed.
- S. Wegmann, I. D. Medalsy, E. Mandelkow and D. J. Muller, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E313–E321 CrossRef CAS PubMed.
- A. J. Dregni, V. S. Mandala, H. Wu, M. R. Elkins, H. K. Wang, I. Hung, W. F. DeGrado and M. Hong, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16357–16366 CrossRef CAS PubMed.
- S. Park, J. H. Lee, J. H. Jeon and M. J. Lee, BMB Rep., 2018, 51, 265–273 CrossRef CAS PubMed.
- M. Goedert, E. S. Cohen, R. Jakes and P. Cohen, FEBS Lett., 1992, 312, 95–99 CrossRef CAS.
- M. S. Wolfe, J. Biol. Chem., 2009, 284, 6021–6025 CrossRef CAS PubMed.
- C. Kontaxi, P. Piccardo and A. C. Gill, Front. Mol. Biosci., 2017, 4, 56 CrossRef PubMed.
- T. Grune, D. Botzen, M. Engels, P. Voss, B. Kaiser, T. Jung, S. Grimm, G. Ermak and K. J. Davies, Arch. Biochem. Biophys., 2010, 500, 181–188 CrossRef CAS PubMed.
- R. Cliffe, J. C. Sang, F. Kundel, D. Finley, D. Klenerman and Y. Ye, Cell Rep., 2019, 26, 2140–2149 CrossRef CAS PubMed e2143.
- T. Arakhamia, C. E. Lee, Y. Carlomagno, D. M. Duong, S. R. Kundinger, K. Wang, D. Williams, M. DeTure, D. W. Dickson, C. N. Cook, N. T. Seyfried, L. Petrucelli and A. W. P. Fitzpatrick, Cell, 2020, 180, 633–644 CrossRef CAS PubMed e612.
- I. Grundke-Iqbal, K. Iqbal, Y. C. Tung, M. Quinlan, H. M. Wisniewski and L. I. Binder, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 4913–4917 CrossRef CAS PubMed.
- A. Schneider, J. Biernat, M. von Bergen, E. Mandelkow and E. M. Mandelkow, Biochemistry, 1999, 38, 3549–3558 CrossRef CAS PubMed.
- K. A. Jellinger, J. Neural Transm., Suppl., 2003, 101–144, DOI:10.1007/978-3-7091-0643-3_7.
- H. Shimura, D. Schwartz, S. P. Gygi and K. S. Kosik, J. Biol. Chem., 2004, 279, 4869–4876 CrossRef CAS PubMed.
- S. W. Min, X. Chen, T. E. Tracy, Y. Li, Y. Zhou, C. Wang, K. Shirakawa, S. S. Minami, E. Defensor, S. A. Mok, P. D. Sohn, B. Schilling, X. Cong, L. Ellerby, B. W. Gibson, J. Johnson, N. Krogan, M. Shamloo, J. Gestwicki, E. Masliah, E. Verdin and L. Gan, Nat. Med., 2015, 21, 1154–1162 CrossRef CAS PubMed.
- R. L. Best, P. J. Chung, S. J. Benbow, A. Savage, N. E. LaPointe, C. R. Safinya and S. C. Feinstein, in Methods in Tau Cell Biology, ed. S. C. Feinstein and N. E. LaPointe, Academic Press, New York, 2017, ch. 1, pp. 3–26 Search PubMed.
- D. C. David, R. Layfield, L. Serpell, Y. Narain, M. Goedert and M. G. Spillantini, J. Neurochem., 2002, 83, 176–185 CrossRef CAS PubMed.
- M. J. Lee, J. H. Lee and D. C. Rubinsztein, Prog. Neurobiol., 2013, 105, 49–59 CrossRef CAS PubMed.
- I. Santa-Maria, M. Perez, F. Hernandez, J. Avila and F. J. Moreno, J. Alzheimer's Dis., 2006, 9, 279–285 CAS.
- X. Chen, M. Chen, N. P. Schafer and P. G. Wolynes, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4125–4130 CrossRef CAS PubMed.
- Y. Jiang, J. Lee, J. H. Lee, J. W. Lee, J. H. Kim, W. H. Choi, Y. D. Yoo, H. Cha-Molstad, B. Y. Kim, Y. T. Kwon, S. A. Noh, K. P. Kim and M. J. Lee, Autophagy, 2016, 12, 2197–2212 CrossRef CAS PubMed.
- F. Liu, I. Grundke-Iqbal, K. Iqbal and C. X. Gong, Eur. J. Neurosci., 2005, 22, 1942–1950 CrossRef PubMed.
- M. Boban, M. Babic Leko, T. Miskic, P. R. Hof and G. Simic, J. Neurosci. Methods, 2019, 319, 60–68 CrossRef CAS PubMed.
- S. S. Kwak, K. J. Washicosky, E. Brand, D. von Maydell, J. Aronson, S. Kim, D. E. Capen, M. Cetinbas, R. Sadreyev, S. Ning, E. Bylykbashi, W. Xia, S. L. Wagner, S. H. Choi, R. E. Tanzi and D. Y. Kim, Nat. Commun., 2020, 11, 1377 CrossRef CAS PubMed.
- V. Joshi, A. Amanullah, A. Upadhyay, R. Mishra, A. Kumar and A. Mishra, Front. Mol. Neurosci., 2016, 9, 93 Search PubMed.
- W. H. Choi, Y. Yun, S. Park, J. H. Jeon, J. Lee, J. H. Lee, S. A. Yang, N. K. Kim, C. H. Jung, Y. T. Kwon, D. Han, S. M. Lim and M. J. Lee, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 19190–19200 CrossRef CAS PubMed.
- J. H. Kim, E. Kim, W. H. Choi, J. Lee, J. H. Lee, H. Lee, D. E. Kim, Y. H. Suh and M. J. Lee, Mol. Pharm., 2016, 13, 2039–2048 CrossRef CAS PubMed.
- A. W. P. Fitzpatrick, B. Falcon, S. He, A. G. Murzin, G. Murshudov, H. J. Garringer, R. A. Crowther, B. Ghetti, M. Goedert and S. H. W. Scheres, Nature, 2017, 547, 185–190 CrossRef CAS PubMed.
- E. Ercan-Herbst, J. Ehrig, D. C. Schondorf, A. Behrendt, B. Klaus, B. Gomez Ramos, N. Prat Oriol, C. Weber and D. E. Ehrnhoefer, Acta Neuropathol. Commun., 2019, 7, 192 CrossRef CAS PubMed.
- K. Spittaels, C. Van den Haute, J. Van Dorpe, K. Bruynseels, K. Vandezande, I. Laenen, H. Geerts, M. Mercken, R. Sciot, A. Van Lommel, R. Loos and F. Van Leuven, Am. J. Pathol., 1999, 155, 2153–2165 CrossRef CAS.
- K. Spittaels, C. Van den Haute, J. Van Dorpe, H. Geerts, M. Mercken, K. Bruynseels, R. Lasrado, K. Vandezande, I. Laenen, T. Boon, J. Van Lint, J. Vandenheede, D. Moechars, R. Loos and F. Van Leuven, J. Biol. Chem., 2000, 275, 41340–41349 CrossRef CAS PubMed.
- O. V. Forlenza, I. Aprahamian, V. J. de Paula and T. Hajek, Curr. Alzheimer Res., 2016, 13, 879–886 CrossRef CAS PubMed.
- M. K. King, M. Pardo, Y. Cheng, K. Downey, R. S. Jope and E. Beurel, Pharmacol. Ther., 2014, 141, 1–12 CrossRef CAS PubMed.
- S. Matsunaga, T. Kishi, P. Annas, H. Basun, H. Hampel and N. Iwata, J. Alzheimer's Dis., 2015, 48, 403–410 CAS.
- X. L. Ang and J. W. Harper, Sci. STKE, 2004, 242, pe31 Search PubMed.
- M. Tyers and P. Jorgensen, Curr. Opin. Genet. Dev., 2000, 10, 54–64 CrossRef CAS PubMed.
- R. F. Li, Y. Shang, D. Liu, Z. S. Ren, Z. Chang and S. F. Sui, J. Mol. Biol., 2007, 374, 777–790 CrossRef CAS PubMed.
- C. A. Dickey, A. Kamal, K. Lundgren, N. Klosak, R. M. Bailey, J. Dunmore, P. Ash, S. Shoraka, J. Zlatkovic, C. B. Eckman, C. Patterson, D. W. Dickson, N. S. Nahman Jr, M. Hutton, F. Burrows and L. Petrucelli, J. Clin. Invest., 2007, 117, 648–658 CrossRef CAS PubMed.
- M. Ravalin, P. Theofilas, K. Basu, K. A. Opoku-Nsiah, V. A. Assimon, D. Medina-Cleghorn, Y. F. Chen, M. F. Bohn, M. Arkin, L. T. Grinberg, C. S. Craik and J. E. Gestwicki, Nat. Chem. Biol., 2019, 15, 786–794 CrossRef CAS PubMed.
- J. J. Jarero-Basulto, J. Luna-Munoz, R. Mena, Z. Kristofikova, D. Ripova, G. Perry, L. I. Binder and F. Garcia-Sierra, J. Neuropathol. Exp. Neurol., 2013, 72, 1145–1161 CrossRef CAS PubMed.
- P. J. Dolan and G. V. Johnson, J. Biol. Chem., 2010, 285, 21978–21987 CrossRef CAS PubMed.
- C. A. Dickey, M. Yue, W. L. Lin, D. W. Dickson, J. H. Dunmore, W. C. Lee, C. Zehr, G. West, S. Cao, A. M. Clark, G. A. Caldwell, K. A. Caldwell, C. Eckman, C. Patterson, M. Hutton and L. Petrucelli, J. Neurosci., 2006, 26, 6985–6996 CrossRef CAS PubMed.
- R. A. Rissman, W. W. Poon, M. Blurton-Jones, S. Oddo, R. Torp, M. P. Vitek, F. M. LaFerla, T. T. Rohn and C. W. Cotman, J. Clin. Invest., 2004, 114, 121–130 CrossRef CAS.
- L. Petrucelli, D. Dickson, K. Kehoe, J. Taylor, H. Snyder, A. Grover, M. De Lucia, E. McGowan, J. Lewis, G. Prihar, J. Kim, W. H. Dillmann, S. E. Browne, A. Hall, R. Voellmy, Y. Tsuboi, T. M. Dawson, B. Wolozin, J. Hardy and M. Hutton, Hum. Mol. Genet., 2004, 13, 703–714 CrossRef CAS PubMed.
- E. Nachman, A. S. Wentink, K. Madiona, L. Bousset, T. Katsinelos, K. Allinson, H. Kampinga, W. A. McEwan, T. R. Jahn, R. Melki, A. Mogk, B. Bukau and C. Nussbaum-Krammer, J. Biol. Chem., 2020, 295, 9676–9690 CrossRef CAS PubMed.
- W. Zhang, B. Falcon, A. G. Murzin, J. Fan, R. A. Crowther, M. Goedert and S. H. Scheres, Elife, 2019, 8, e43584 CrossRef PubMed.
- R. Gupta, M. Lan, J. Mojsilovic-Petrovic, W. H. Choi, N. Safren, S. Barmada, M. J. Lee and R. Kalb, eNeuro, 2017, 4, e0249-16 CrossRef PubMed.
- S. K. Shin, J. H. Kim, J. H. Lee, Y. H. Son, M. W. Lee, H. J. Kim, S. A. Noh, K. P. Kim, I. G. Kim and M. J. Lee, Exp. Mol. Med., 2017, 49, e287 CrossRef CAS PubMed.
- J. N. Keller, K. B. Hanni and W. R. Markesbery, J. Neurochem., 2000, 75, 436–439 CrossRef CAS PubMed.
- J. H. Lee, S. K. Shin, Y. Jiang, W. H. Choi, C. Hong, D. E. Kim and M. J. Lee, Sci. Rep., 2015, 5, 10757 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00586c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |