
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NHC-catalyzed covalent activation of heteroatoms for enantioselective reactions
Runjiang
Song
b,
Zhichao
Jin
a and
Yonggui Robin
Chi
 *ab
*ab
aLaboratory Breeding Base of Green Pesticide and Agricultural Bioengineering, Key Laboratory of Green Pesticide, Agricultural Bioengineering Ministry of Education, Guizhou University, Huaxi District, Guiyang 550025, China
bDivision of Chemistry & Biological Chemistry, School of Physical & Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: robinchi@ntu.edu.sg
First published on 2nd March 2021
Abstract
Covalent activation of heteroatoms enabled by N-heterocyclic carbene (NHC) organic catalysts for enantioselective reactions is evaluated and summarized in this review. To date, sulfur, oxygen, and nitrogen atoms can be activated in this manner to react with another substrate to construct chiral carbon–heteroatom bonds with high optical enantioselectivities. The activation starts with addition of an NHC catalyst to the carbonyl moiety (aldehyde or imine) of substrates that contain heteroatoms. The key in this approach is the formation of intermediates covalently bound to the NHC catalyst, in which the heteroatom of the substrate is activated as a nucleophilic reactive site.
 Runjiang Song | Runjiang Song received his BS degree from the East China University of Science and Technology in 2016, and MSc degree in 2019 from Nanyang Technological University. He is now pursuing his PhD degree under the guidance of Prof. Robin Chi. His research interests include asymmetric catalysis and functional molecule synthesis. |
 Zhichao Jin | Prof. Zhichao Jin received his MSc degree from the East China University of Science and Technology in 2011 under the guidance of Prof. Jinxing Ye, and PhD from Nanyang Technological University in 2015 under the guidance of Prof. Robin Chi. After a two-year postdoctoral stay with Prof. Yixin Lu at the National University of Singapore, he started his independent career at Guizhou University in 2017. His current research interest includes the development of novel catalytic activation modes and bioactive molecules for agricultural applications. |
 Yonggui Robin Chi | Prof. Robin Chi received his undergraduate training from Tsinghua University and Hong Kong Baptist University during 1998–2002. He obtained PhD in Chemistry from the University of Wisconsin-Madison under the guidance of Prof. Sam Gellman in 2007. After a two-year postdoctoral stay at the University of California-Berkeley with Prof. Jean Frechet, he started his independent career at Nanyang Technological University and was quickly promoted to full professor. His current research, supported by students and researchers from Nanyang Technological University and Guizhou University, focuses on the development of new basic activation modes and reactions mediated by organic catalysts, efficient transformations for rapid construction of sophisticated molecules, selective modification of Chinese medicines and biomacromolecules, green reactions and processes for industrial applications, and functional molecules for applications in medicines, agricultural chemicals, and materials. |
1. Introduction
Heteroatoms are essential to life of all kinds and ubiquitous in nearly all functional molecules with medicinal uses. Asymmetric construction of carbon–heteroatom (C–X) bonds has therefore been an active research topic in chemical synthesis. In the last two decades, catalysis enabled by N-heterocyclic carbenes (NHCs) has been developed as a versatile approach to prepare both carbon–carbon and carbon–heteroatom bonds in highly enantioselective manners.1 In the subdomain of NHC-catalyzed asymmetric C–X bond formation, the dominant approach is to activate carbon atoms to react with heteroatoms or substrates containing heteroatoms such as amines, ureas and thioureas (Fig. 1a). The carbon atoms can be activated as electrophilic reaction centres via NHC-derived (unsaturated) acyl azolium intermediates or their analogues to react with nucleophilic heteroatoms.2 Alternatively, the carbon atoms are activated as nucleophilic reaction centers (via acyl anion and enolate intermediate or their analogues). The NHC-derived nucleophilic intermediates then react with heteroatoms (in their oxidized states) or heteroatom-containing substrates (such as imines) as the electrophiles.3Fig. 1b shows representative substrates with carbon atoms activated by NHC catalysts as the reactive centres. The heteroatoms involved in these reactions, at least by designing, are not activated by NHC catalysts. These types of reactions have now been covered in many review articles concerning NHC catalysis from different perspectives.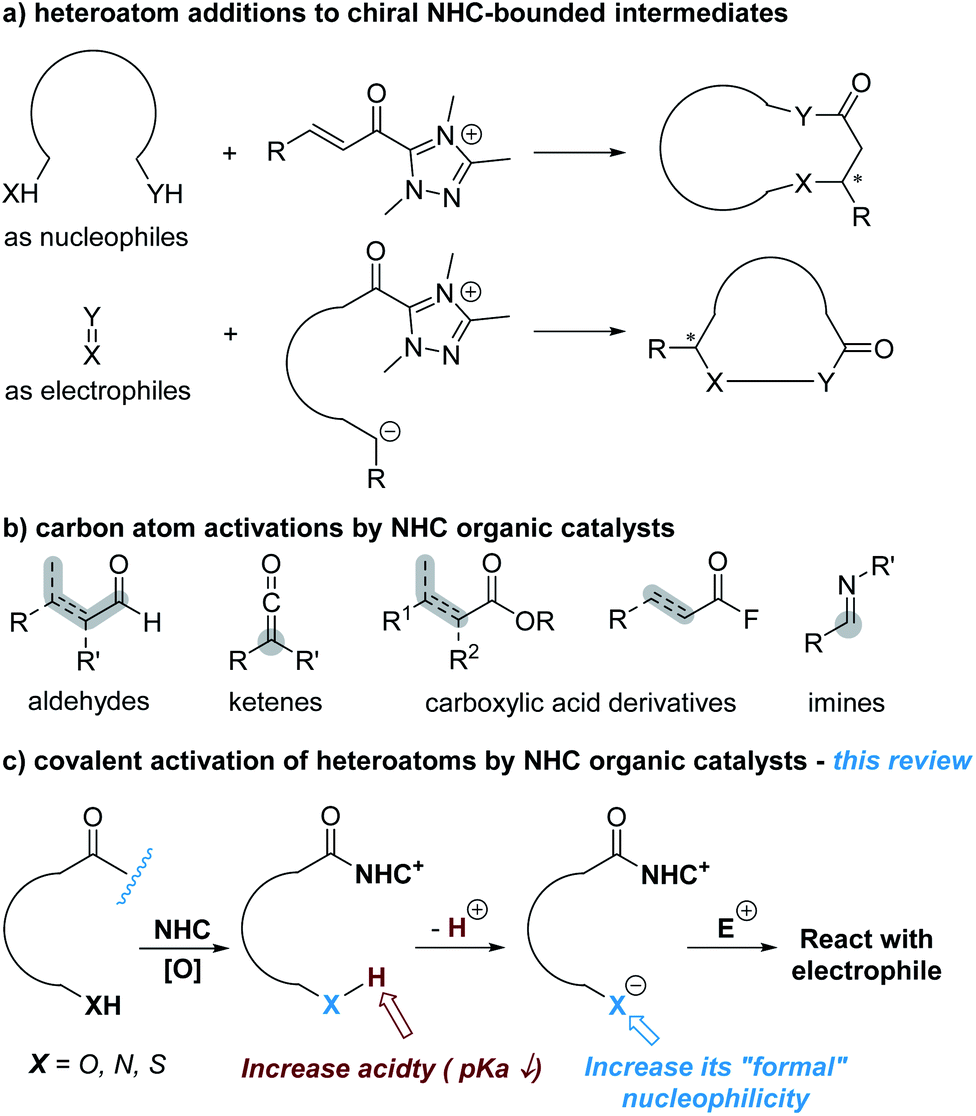 | ||
| Fig. 1 NHC-catalyzed activation of carbon and heteroatoms. | ||
A much less-developed approach is to activate heteroatoms through addition of NHC catalysts to the respective substrates for enantioselective reactions. The basic design, as illustrated in Fig. 1c, is to increase the reactivity of heteroatoms by forming covalent bonds between NHC catalysts and substrates. This review is to summarize the development of this direction that is still at its infant stage.
It is worth noting that activation of heteroatoms for asymmetric reactions via non-covalent interactions using azolium salts (precursor of N-heterocyclic carbene), pioneered by Huang and co-workers, is not covered in this article.4 It is also important to note that in those reactions designed via NHC-catalyzed activation of carbon atoms as the key step, heteroatoms in the substrates or intermediates may also be activated as a result of catalyst–substrate bonding. These studies are also not discussed in this article.
With the scope defined, this review article will discuss NHC-catalyzed activation of oxygen and nitrogen atoms for enantioselective reactions. The use of NHC catalysts to cleave the N–S or C–S bond to release nucleophilic sulfur atoms for asymmetric reactions will also be described.
2. Covalent activation of oxygen atoms of phenols
Aromatic aldehydes are routinely activated by NHC catalysts and participate in Benzoin condensations, Stetter reactions and acylation reactions. In 2013, the Chi lab found that the benzylic C(sp3)–H bond of an indole aldehyde can be activated by NHC catalysts under oxidative conditions through the formation of an o-QDM intermediate.5 This study suggests that the activating power of NHC catalysts can go across the conjugated aromatic ring system.Encouraged by this discovery, the Chi group moved to explore the activation of heteroatoms attached to aromatic scaffolds. In 2017, they disclosed that phenol OH can be enantioselectively functionalized through activation of salicylaldehyde by NHC organic catalysts (Fig. 2).6 In a model reaction using salicylaldehyde (1) and trifluoroacetophenone (2) as substrates, the use of A as the NHC pre-catalyst (5 mol%) in the presence of I as an additive (20 mol%) and an oxidant gave the chiral acetal product (3) in 99% yield, 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 er. The urea additive was critical and behaved as a co-catalyst. The product er dropped to 73:27 in the absence of the urea additive. The reaction appears to be general with good functional group tolerance, as shown in 28 examples. The NHC catalyst loading can be decreased in 1 mol% for gram scale synthesis. The authors also found that several of their enantiomerically enriched products showed encouraging antifungal activities against Eggplant Verticillium, Phytophthora infestans and Fusarium oxysporum.
:6 er. The urea additive was critical and behaved as a co-catalyst. The product er dropped to 73:27 in the absence of the urea additive. The reaction appears to be general with good functional group tolerance, as shown in 28 examples. The NHC catalyst loading can be decreased in 1 mol% for gram scale synthesis. The authors also found that several of their enantiomerically enriched products showed encouraging antifungal activities against Eggplant Verticillium, Phytophthora infestans and Fusarium oxysporum.
 | ||
| Fig. 2 Covalent activation of phenol OH by chiral NHC. | ||
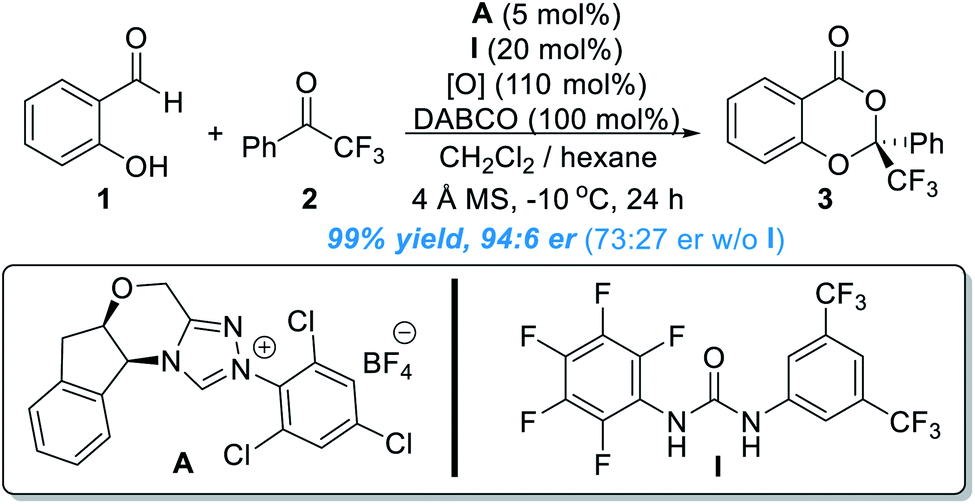
Based on experimental observations and DFT calculations, the reaction was believed to proceed through a pathway as illustrated in Fig. 3. Addition of the NHC catalyst to salicylaldehyde 1 gives the Breslow intermediate 4, which can be oxidized to produce the acylazolium intermediate 5. Deprotonation of 5 under basic conditions leads to the formation of the zwitter ionic phenoxide 6, which is stabilized through resonance structures including the o-QMs 6′. The asymmetric oxa-[4 + 2] reaction between 6 (6′) and 2 gives the chiral acetal product 3 and releases the free NHC catalyst for additional catalytic cycles.
 | ||
| Fig. 3 Proposed mechanism of the NHC-catalyzed phenol OH activation reaction. | ||
3. Covalent activation of nitrogen atoms
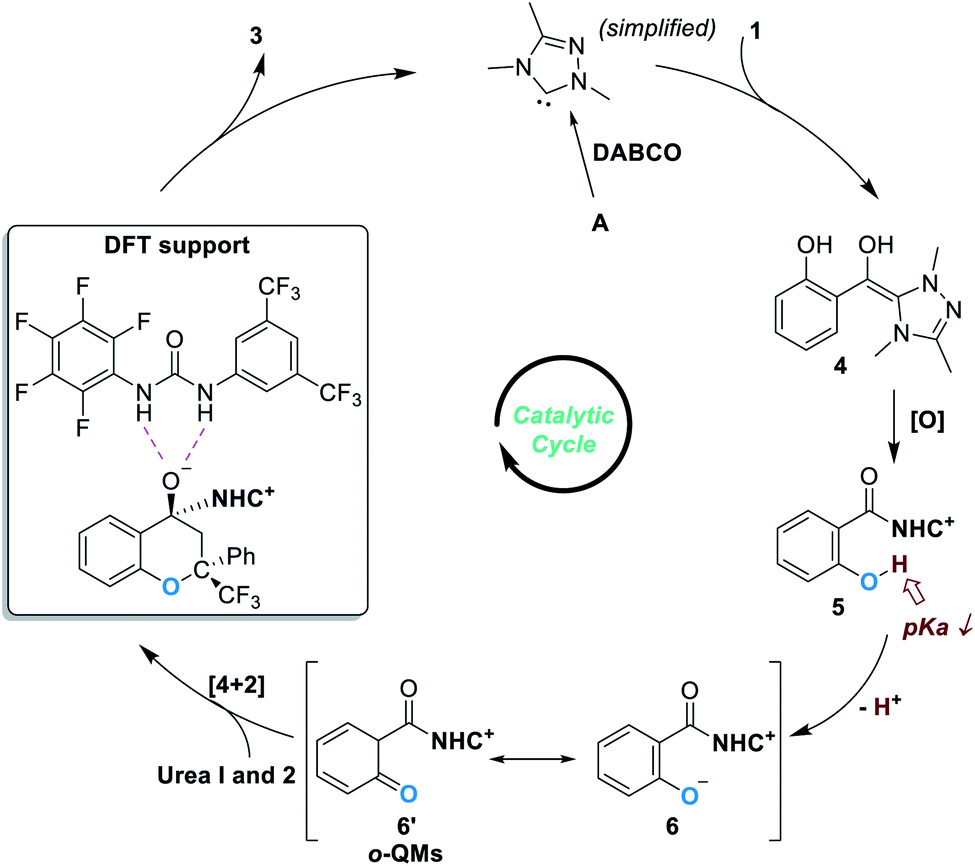
The oxygen atom in phenols above can in principle be replaced by other heteroatoms such as nitrogen and sulphur atoms for NHC-catalyzed enantioselective reactions. An elegant design in this direction was disclosed by Scheidt and co-workers in 2019 (Fig. 4).7 Addition of a carbene catalyst (B) to an N-methylisatoic anhydride (7) gives zwitter ionic acylazolium intermediate 8 with elimination of one equivalent of CO2. Intermediate 8 is a resonance structure of the aza-o-QMs intermediate 8′. DFT studies suggest the intermediate 8/8′ reacts with trifluoroacetophenone 2 through a concerted enantioselective [4 + 2] annulation pathway. The target product (9) is obtained with an excellent enantioselectivity. Common functional groups are well tolerated in this reaction, and the authors demonstrated 19 examples in their substrate scope evaluation.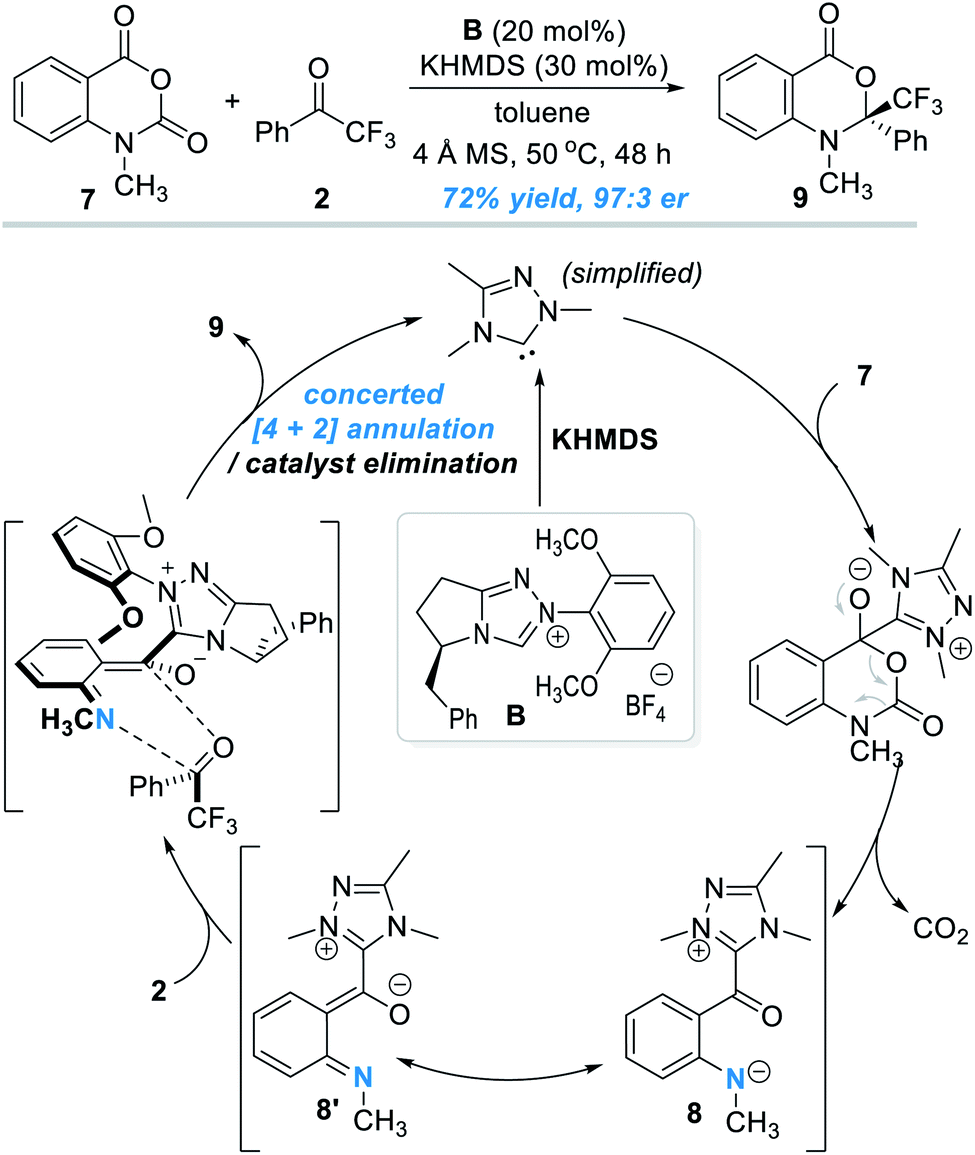 | ||
| Fig. 4 Covalent activation of the nitrogen atom of N-methylisatoic anhydride by the chiral NHC catalyst. | ||
Nitrogen atoms that are part of the aromatic scaffold can also be activated by NHC catalysts for enantioselective reactions. In 2020, Chi and Jin disclosed that the aromatic N atoms of the 2-carbaldehyde derivatives of indole or pyrrole structures can be effectively activated via the formation of an unprecedented NHC-bounded aza-fulvene intermediate 10 (Fig. 5).8 The N atoms of the indole or pyrrole molecules are selectively activated to react with various ketone substrates through nucleophilic addition reactions.
 | ||
| Fig. 5 NHC-catalyzed nucleophilic activation of aromatic N atoms of indole-2-carbaldehydes or pyrrole-2-carbaladehydes. | ||
Up to 75 enantiomerically enriched N,O-acetal compounds have been efficiently prepared through this protocol under the catalysis of several chiral NHC organic catalysts (Fig. 6a). For example, with chiral NHC C used as the reaction catalyst, spiro-cyclic compounds 11 can be obtained in moderate to excellent yields with excellent optical purities. Switching the NHC catalyst to D, α-ketoesters can be applied as suitable electrophiles to react with substituted indole-2-carbaldehydes (12). Pyrrole-2-carbaldehydes bearing different substituents can also work well in this process, although a urea co-catalyst II is needed for better enantioselectivity control over the NHC-catalyzed [3 + 2] cycloaddition reaction (13).
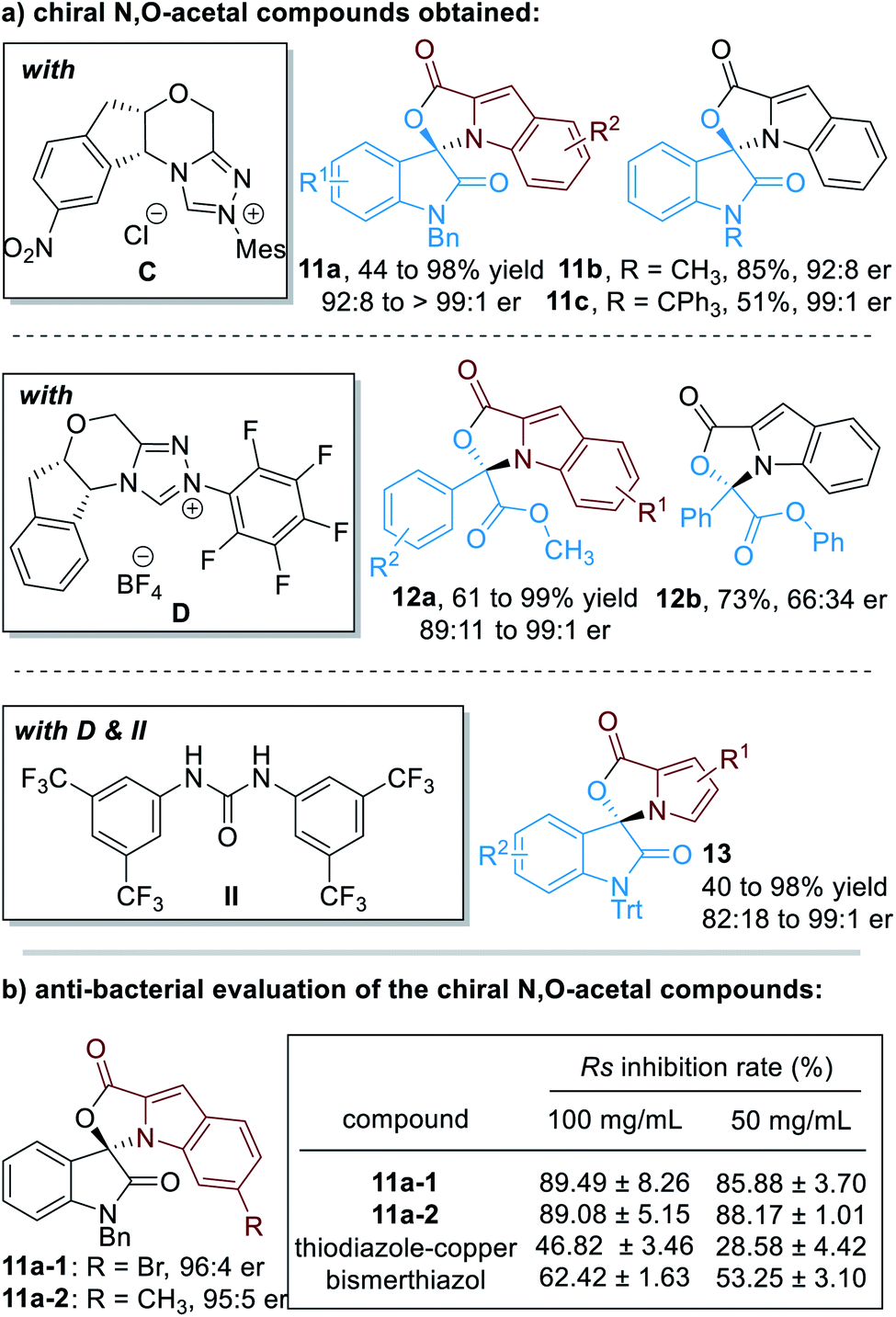 | ||
| Fig. 6 Chiral N,O-acetals prepared through NHC organocatalytic reactions and their bioactive evaluations. | ||
It is worth noting that several of these spiro-cyclic compounds have shown promising anti-microbial activities (Fig. 6b). For instance, compounds 11a-1 and 11a-2 exhibit much better anti-bacterial activities against Ralstonia solanacearum (abbreviated as Rs) than the commercial agrichemicals thiodiazole–copper and bismerthiazol.
A short time later, Biju,9 Hui10 and co-workers independently reported similar transformations to this protocol. DFT studies towards the NHC-catalyzed aromatic N activation mechanism and additional electrophile investigations are provided in their studies.
When the aldehyde group was installed on the 7-position of the indole structure, the aromatic N atom can also be functionalized through covalent activations with NHC organic catalysts (Fig. 7).11 In this case, various electron-deficient ketone substrates can be used as electrophiles. With the catalysis of NHC E or F, a variety of chiral [1,3]oxazino[5,4,3-hi]indole derivatives 14 can be afforded in good to excellent yields and enantioselectivities.
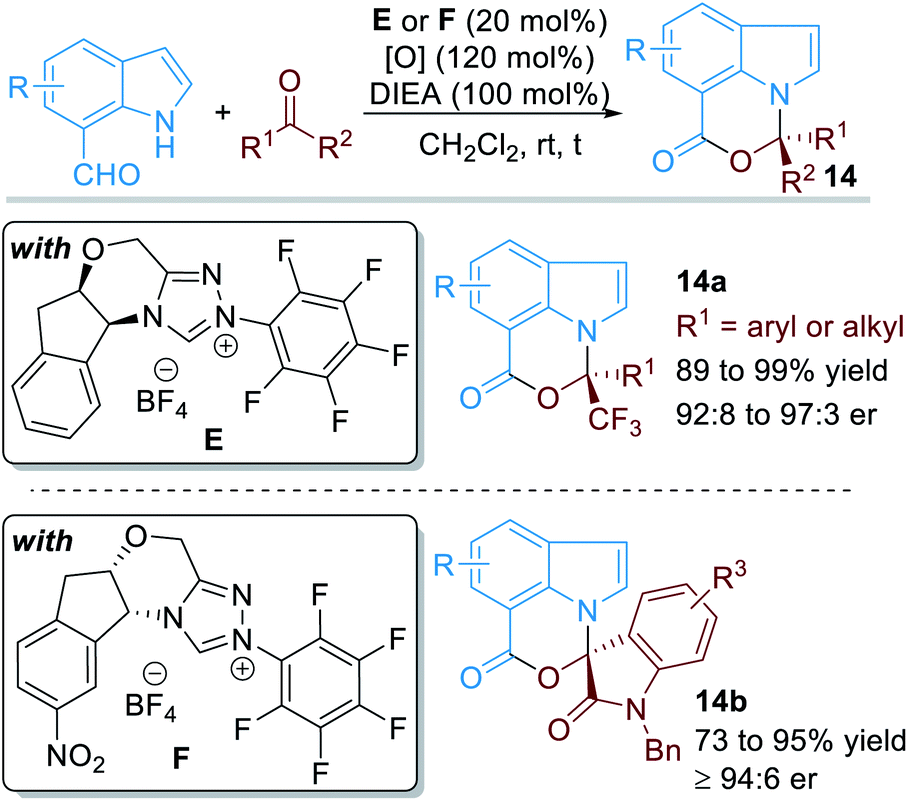 | ||
| Fig. 7 NHC-catalyzed enantioselective activation of indole-7-carbaldehydes. | ||
Very recently, the groups of Chi12 and Fu13 independently found that the remote N atom of the guanidine-derived aldimine 15 can be activated by NHC catalyst G under oxidative conditions to react with isatin 16 and gave spiocyclic product 17 in an excellent yield and enantioselectivity (Fig. 8). Addition of the NHC catalyst G to 15 leads to the formation of the aza-Breslow intermediate 18, which can be transformed to give the azolium intermediate 19 under oxidative conditions. Deprotonation of 19 gives the triaza-diene intermediate 20, which then reacts with isatin 16 through a concerted asynchronous addition process to give the intermediate 21. Finally, the target chiral spirocyclic product 17 can be afforded after elimination of the NHC catalyst.
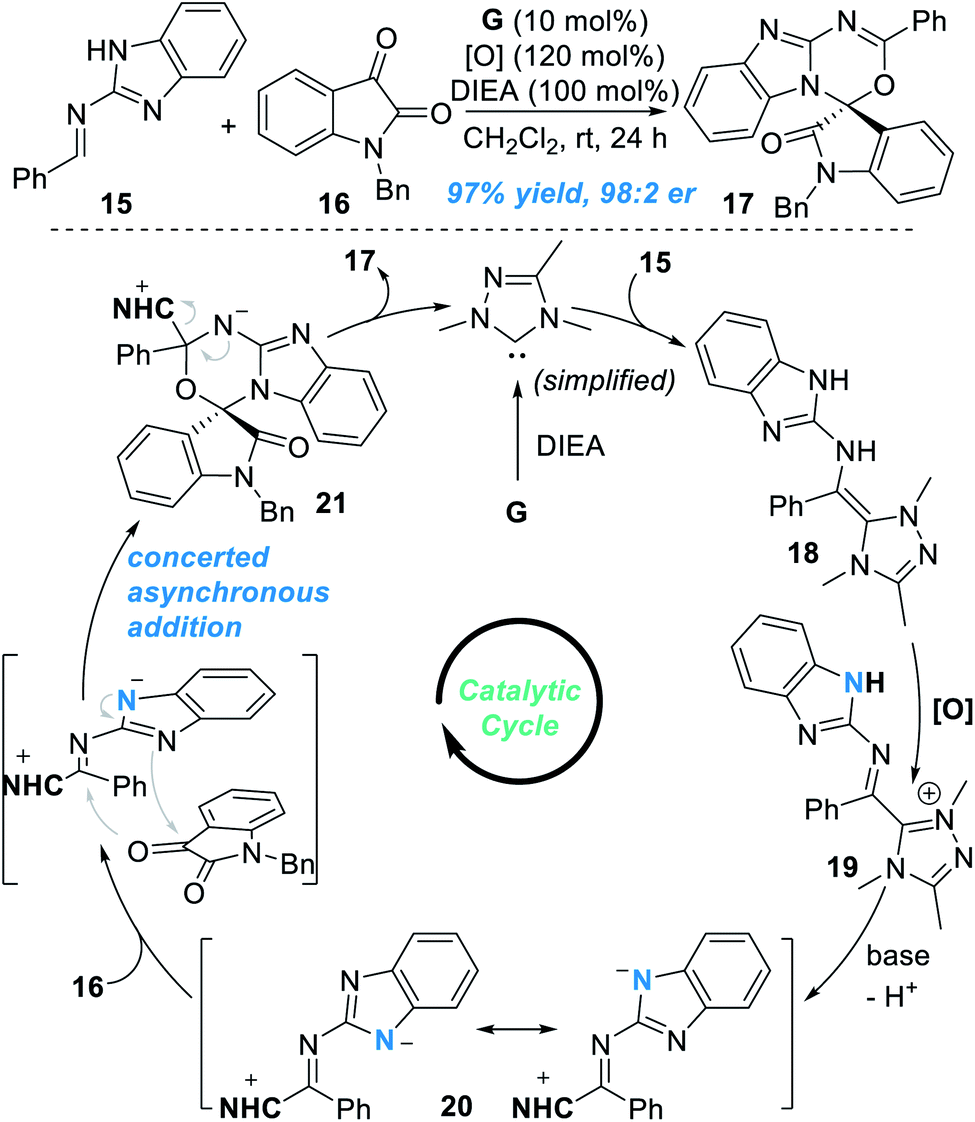 | ||
| Fig. 8 NHC-catalyzed enantioselective activation of the guanidine-derived aldimine. | ||
Another interesting study in this direction is activation of the nitrogen atoms of azirines (Fig. 9).14 In this study, formyl azirine 22 was successfully activated by the NHC catalyst and a novel azolium aza-dienolate intermediate 23 was generated. Aza-[4 + 2] cycloaddition between 23 and the electron-deficient ketone 24 gave the oxazinone product 25 in moderate to excellent yields. Although a wide range of substrates with various substitution patterns worked well in this process, the asymmetric version of this transformation remains undisclosed.
 | ||
| Fig. 9 Activation of azirines by the NHC organic catalyst. | ||
4. NHC-catalyzed C–S and N–S bond cleavage to release sulphur atoms for enantioselective reactions
The N–S bond in aldehyde-derived sulfonimide (26) was cleavable as a result of NHC-catalyzed activation, as disclosed by Hou and co-workers in 2008.15 This N–S bond cleavage releases nucleophilic sulfinic anions that can react with other electrophiles. In these reactions, since the NHC catalyst is not bound to the sulfinic anions, enantioselectivity control cannot be induced by chiral NHC catalysts. In 2013, Chi and co-workers introduced a multi-catalytic approach combining NHC and a thiourea/tertiary amine multifunctional catalyst to achieve enantioselective transformations (Fig. 10a).16Aromatic enone substrates 27 worked well in this co-operative catalytic process, with the β-sulfinic ketone 28 afforded in moderate to good yields and good to excellent enantioselectivities (Fig. 10b, right part). NHC pre-catalyst H and the chiral amino-thiourea II were used as the co-operative catalysts for this asymmetric transformation. β-Alkyl substituted enones 29 were also promising substrates for this reaction, although the yields and er values of the final products 30 were only moderate even after additional condition optimization investigations (Fig. 10b, left part).
 | ||
| Fig. 10 NHC-catalyzed sulfonimide N–S bond cleavage. | ||
The NHC catalyst is believed to first attack the sulfonimide 26 through nucleophilic addition and give the aza-Breslow intermediate 31. The aza-Breslow intermediate 31 readily cracks into the sulfinic anion 32 and the azolium cation 33. Thio-Michael addition reactions of 32 and enones (27 or 29) and intermolecular proton transfer lead to the formation of the β-sulfinic ketone products (28 or 30) together with the side-product benzonitrile 34. The free NHC catalyst is regenerated during this process and can participate in further catalytic cycles (Fig. 10c).
The NHC-catalyzed N–S bond cleavage strategy has recently been used in the late-stage functionalization of bio-active complex sulfonamides.17 The key intermediate of the sulfonimide can be formed in situ from benzaldehyde and the corresponding sulfonamide. Therefore, various sulfonamide substrates can be de-aminated and transformed to other sulfone-containing functional molecules through a one-pot protocol in the presence of benzaldehyde and an NHC organic catalyst.
The C–S bond of thioesters derived from thiophenols can be broken to release thiophenol with the sulphur atom reacting as a nucleophile, as reported by Xu and co-workers (Fig. 11).18 In this reaction, the enantioselectivity is controlled by the other reacting partner (α,β-unsaturated acyl azolium ester intermediate) connected with the NHC catalyst.
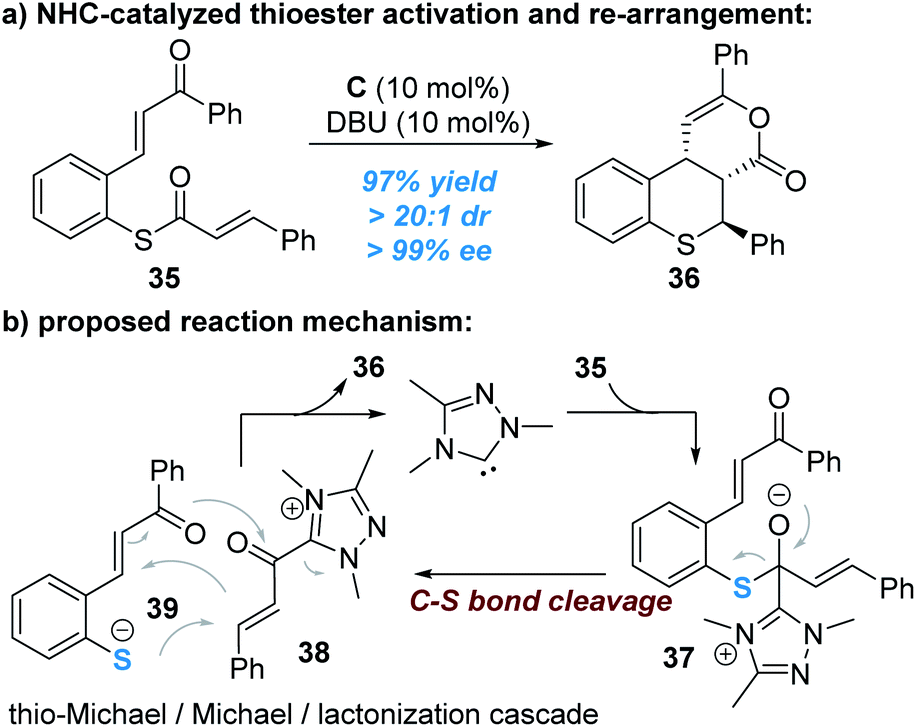 | ||
| Fig. 11 NHC-catalyzed N–S bond cleavage of thioesters. | ||
Nucleophilic attack of the thioester 35 by the NHC catalyst generates the zwitter ion 37, which can be broken up to give the α,β-unsaturated acylazolium intermediate 38 and the thio anion 39. A stereoselective cascade thio-Michael addition/Michael addition/lacton formation process between the ionic intermediates 38 and 39 finally affords the chiral product 36 and liberates the free NHC catalyst for additional catalytic cycles (Fig. 11b).
5. Summary and outlook
NHC organic catalysis has been proven to be a versatile strategy in asymmetric synthesis. The extraordinary development in the past two decades, on the other hand, has mainly focused on activation of carbon atoms for enantioselective reactions. Activation of heteroatoms by NHCs for asymmetric reactions is less explored. This review highlights the impressive yet still very limited studies in this direction. Specifically, oxygen, nitrogen, and sulphur atoms in relevant substrates have been activated by NHCs as nucleophiles for a number of highly enantioselective reactions. These reactions involve previously unexplored NHC-bound intermediates and present interesting reactivities. It is expected that this new approach will allow for quick construction of functional molecules, especially those bearing multiple heteroatoms, in a much simpler manner that is not achievable with the more matured approach of carbon atom activation. At present, the potential in activating heteroatoms by NHCs does not seem to be realized to any considerable level. We hope that this short review will encourage further development in this arena.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (21772029, 21801051, 21961006, 82360589, and 81360589), The 10 Talent Plan (Shicengci) of Guizhou Province ([2016]5649), the Guizhou Province Returned Oversea Student Science and Technology Activity Program [(2014)-2], the Science and Technology Department of Guizhou Province ([2018]2802, [2019]1020), the Program of Introducing Talents of Discipline to Universities of China (111 Program, D20023) at Guizhou University, Frontiers Science Center for Asymmetric Synthesis and Medicinal Molecules, Department of Education, Guizhou Province [Qianjiaohe KY (2020)004], and Guizhou University. We also acknowledge Singapore National Research Foundation under its NRF Investigatorship (NRF-NRFI2016-06) and Competitive Research Program (NRF-CRP22-2019-0002), the Ministry of Education, Singapore, under its MOE AcRF Tier 1 Award (RG108/16, RG5/19, RG1/18), MOE AcRF Tier 2 (MOE2019-T2-2-117), MOE AcRF Tier 3 Award (MOE2018-T3-1-003), the Agency for Science, Technology and Research (A*STAR) under its A*STAR AME IRG Award (A1783c0008, A1783c0010), Nanyang Research Award Grant, and Nanyang Technological University.Notes and references
- For selected recent reviews on NHC catalysis: (a) D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534–541 CrossRef CAS; (b) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS; (c) A. T. Biju, N. Kuhl and F. Glorius, Acc. Chem. Res., 2011, 44, 1182–1195 CrossRef CAS; (d) D. T. Cohen and K. A. Scheidt, Chem. Sci., 2012, 3, 53–57 RSC; (e) A. Grossmann and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 314–325 CrossRef CAS; (f) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC; (g) J. Douglas, G. Churchill and A. D. Smith, Synthesis, 2012, 44, 2295–2309 CrossRef CAS; (h) J. Izquierdo, G. E. Hutson, D. T. Cohen and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 51, 11686–11698 CrossRef CAS; (i) H. U. Vora, P. Wheeler and T. Rovis, Adv. Synth. Catal., 2012, 354, 1617–1639 CrossRef CAS; (j) S. J. Ryan, L. Candish and D. W. Lupton, Chem. Soc. Rev., 2013, 42, 4906–4917 RSC; (k) S. J. Connon, Angew. Chem., Int. Ed., 2014, 53, 1203–1205 CrossRef CAS; (l) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS; (m) J. Mahatthananchai and J. W. Bode, Acc. Chem. Res., 2014, 47, 696–707 CrossRef CAS; (n) R. S. Menon, A. T. Biju and V. Nair, Chem. Soc. Rev., 2015, 44, 5040–5052 RSC; (o) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS; (p) M. H. Wang and K. A. Scheidt, Angew. Chem., Int. Ed., 2016, 55, 14912–14922 CrossRef CAS; (q) X. Y. Chen, Q. Liu, P. Chauhan and D. Enders, Angew. Chem., Int. Ed., 2018, 57, 3862–3873 CrossRef CAS; (r) Z. Wang, D. Pan, T. Li and Z. Jin, Chem.–Asian J., 2018, 13, 2149–2163 CrossRef CAS; (s) R. Song and Y. R. Chi, Angew. Chem., Int. Ed., 2019, 58, 8628–8630 CrossRef CAS; (t) X.-Y. Chen, Z.-H. Gao and S. Ye, Acc. Chem. Res., 2020, 53, 690–702 CrossRef CAS; (u) X. Chen, H. Wang, Z. Jin and Y. R. Chi, Chin. J. Chem., 2020, 38, 1167–1202 CrossRef CAS; (v) H. Ohmiya, ACS Catal., 2020, 10, 6862–6869 CrossRef CAS; for a recent book on NHC organocatalysis, see: (w) A. T. Biju, N-Heterocyclic carbenes in organocatalysis, Wiley-VCH, Weinheim, Germany, 2019 Search PubMed.
- For selected examples, see: (a) H. R. Zhang, Z. W. Dong, Y. J. Yang, P. L. Wang and X. P. Hui, Org. Lett., 2013, 15, 4750–4753 CrossRef CAS; (b) N. A. White and T. Rovis, J. Am. Chem. Soc., 2014, 136, 14674–14677 CrossRef CAS; (c) Y. Zhang, Y. Du, Z. Huang, J. Xu, X. Wu, Y. Wang, M. Wang, S. Yang, R. D. Webster and Y. R. Chi, J. Am. Chem. Soc., 2015, 137, 2416–2419 CrossRef CAS; (d) X. Wu, B. Liu, Y. Zhang, M. Jeret, H. Wang, P. Zheng, S. Yang, B. A. Song and Y. R. Chi, Angew. Chem., Int. Ed., 2016, 55, 12280–12284 CrossRef CAS; (e) X. Wu, L. Hao, Y. Zhang, M. Rakesh, R. N. Reddi, S. Yang, B. A. Song and Y. R. Chi, Angew. Chem., Int. Ed., 2017, 56, 4201–4205 CrossRef CAS; (f) C. Fang, T. Lu, J. Zhu, K. Sun and D. Du, Org. Lett., 2017, 19, 3470–3473 CrossRef CAS; (g) S. Mukherjee, S. Shee, T. Poisson, T. Besset and A. T. Biju, Org. Lett., 2018, 20, 6998–7002 CrossRef CAS; (h) C. Fang, J. Cao, K. Sun, J. Zhu, T. Lu and D. Du, Chem.–Eur. J., 2018, 24, 2103–2108 CrossRef CAS; (i) M. Lang and J. Wang, Eur. J. Org. Chem., 2018, 2958–2962 CrossRef CAS; (j) Y. Liu, Q. Chen, C. Mou, L. Pan, X. Duan, X. Chen, H. Chen, Y. Zhao, Y. Lu, Z. Jin and Y. R. Chi, Nat. Commun., 2019, 10, 1675 CrossRef; (k) H. Wang, Y. Wang, X. Chen, C. Mou, S. Yu, H. Chai, Z. Jin and Y. R. Chi, Org. Lett., 2019, 21, 7440–7444 CrossRef CAS; (l) A. Ghosh, S. Barik and A. T. Biju, Org. Lett., 2019, 21, 8598–8602 CrossRef CAS; (m) C. Liu, S. Wu, J. Xu, L. Chen, P. Zheng and Y. R. Chi, Org. Lett., 2019, 21, 9493–9496 CrossRef CAS.
- (a) A. Chan and K. A. Scheidt, J. Am. Chem. Soc., 2008, 130, 2740–2741 CrossRef CAS; (b) F. T. Wong, P. K. Patra, J. Seayad, Y. Zhang and J. Y. Ying, Org. Lett., 2008, 10, 2333–2336 CrossRef CAS; (c) X. L. Huang, L. He, P. L. Shao and S. Ye, Angew. Chem., Int. Ed., 2009, 48, 192–195 CrossRef CAS; (d) X. L. Huang, X. Y. Chen and S. Ye, J. Org. Chem., 2009, 74, 7585–7587 CrossRef CAS; (e) P. L. Shao, X. Y. Chen and S. Ye, Angew. Chem., Int. Ed., 2010, 49, 8412–8416 CrossRef CAS; (f) P.-L. Shao, X.-Y. Chen, L.-H. Sun and S. Ye, Tetrahedron Lett., 2010, 51, 2316–2318 CrossRef CAS; (g) T. Wang, X. L. Huang and S. Ye, Org. Biomol. Chem., 2010, 8, 5007–5011 RSC; (h) T. Y. Jian, L. He, C. Tang and S. Ye, Angew. Chem., Int. Ed., 2011, 50, 9104–9107 CrossRef CAS; (i) Z. X. Sun and Y. Cheng, Org. Biomol. Chem., 2012, 10, 4088–4094 RSC; (j) X. Y. Chen, F. Xia, J. T. Cheng and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 10644–10647 CrossRef CAS; (k) R. Han, J. Qi, J. Gu, D. Ma, X. Xie and X. She, ACS Catal., 2013, 3, 2705–2709 CrossRef CAS; (l) X. Y. Chen, F. Xia and S. Ye, Org. Biomol. Chem., 2013, 11, 5722–5726 RSC; (m) S. B. Poh, J. Y. Ong, S. Lu and Y. Zhao, Angew. Chem., Int. Ed., 2018, 57, 1645–1649 CrossRef CAS.
- (a) L. Wang, J. Chen and Y. Huang, Angew. Chem., Int. Ed., 2015, 54, 15414–15418 CrossRef CAS; (b) J. Chen and Y. Huang, Sci. China: Chem., 2015, 59, 251–254 CrossRef; (c) S. Santra, U. Maji and J. Guin, Org. Lett., 2020, 22, 468–473 CrossRef CAS.
- X. Chen, S. Yang, B. A. Song and Y. R. Chi, Angew. Chem., Int. Ed., 2013, 52, 11134–11137 CrossRef CAS.
- X. Chen, H. Wang, K. Doitomi, C. Y. Ooi, P. Zheng, W. Liu, H. Guo, S. Yang, B. A. Song, H. Hirao and Y. R. Chi, Nat. Commun., 2017, 8, 15598 CrossRef.
- A. Lee, J. L. Zhu, T. Feoktistova, A. C. Brueckner, P. H. Cheong and K. A. Scheidt, Angew. Chem., Int. Ed., 2019, 58, 5941–5945 CrossRef CAS.
- Y. Liu, G. Luo, X. Yang, S. Jiang, W. Xue, Y. R. Chi and Z. Jin, Angew. Chem., Int. Ed., 2020, 59, 442–448 CrossRef CAS.
- K. Balanna, K. Madica, S. Mukherjee, A. Ghosh, T. Poisson, T. Besset, G. Jindal and A. T. Biju, Chem.–Eur. J., 2020, 26, 818–822 CrossRef CAS.
- C. Wang, Z. Li, J. Zhang and X.-P. Hui, Org. Chem. Front., 2020, 7, 1647–1652 RSC.
- X. Yang, G. Luo, L. Zhou, B. Liu, X. Zhang, H. Gao, Z. Jin and Y. R. Chi, ACS Catal., 2019, 9, 10971–10976 CrossRef CAS.
- X. Yang, Y. Xie, J. Xu, S. Ren, B. Mondal, L. Zhou, W. Tian, X. Zhang, L. Hao, Z. Jin and Y. R. Chi, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202016506.
- G. Wang, Q. Zhang, C. Wei, Y. Zhang, L. Zhang, J. Huang, D. Wei, Z. Fu and W. Huang, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202017017.
- Q. Peng, B. Zhang, Y. Xie and J. Wang, Org. Lett., 2018, 20, 7641–7644 CrossRef CAS.
- D. D. Chen, X. L. Hou and L. X. Dai, J. Org. Chem., 2008, 73, 5578–5581 CrossRef CAS.
- Z. Jin, J. Xu, S. Yang, B. A. Song and Y. R. Chi, Angew. Chem., Int. Ed., 2013, 52, 12354–12358 CrossRef CAS.
- P. S. Fier and K. M. Maloney, J. Am. Chem. Soc., 2019, 141, 1441–1445 CrossRef CAS.
- H. Lu, J.-L. Zhang, J.-Y. Liu, H.-Y. Li and P.-F. Xu, ACS Catal., 2017, 7, 7797–7802 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |