
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible metathesis of ammonia in an acyclic germylene–Ni0 complex†
Philip M.
Keil
a,
Tibor
Szilvási
b and
Terrance J.
Hadlington
 *a
*a
aDepartment of Chemistry, Technical University Munich, Lichtenbergstraße 4, 85747 Garching, Germany. E-mail: terrance.hadlington@tum.de
bDepartment of Chemical and Biological Engineering, University of Alabama, Tuscaloosa, AL 35487, USA
First published on 5th March 2021
Abstract
Carbenes, a class of low-valent group 14 ligand, have shifted the paradigm in our understanding of the effects of supporting ligands in transition-metal reactivity and catalysis. We now seek to move towards utilizing the heavier group 14 elements in effective ligand systems, which can potentially surpass carbon in their ability to operate via ‘non-innocent’ bond activation processes. Herein we describe our initial results towards the development of scalable acyclic chelating germylene ligands (viz.1a/b), and their utilization in the stabilization of Ni0 complexes (viz.4a/b), which can readily and reversibly undergo metathesis with ammonia with no net change of oxidation state at the GeII and Ni0 centres, through ammonia bonding at the germylene ligand as opposed to the Ni0 centre. The DFT-derived metathesis mechanism, which surprisingly demonstrates the need for three molecules of ammonia to achieve N–H bond activation, supports reversible ammonia binding at GeII, as well as the observed reversibility in the overall reaction.
Introduction
Beginning with the inception of transition metal (TM) carbene complexes by E. O. Fischer,1 research involving the synthesis and utility of metal carbene complexes has long stood as a pillar of modern organometallic chemistry.2 This has borne numerous classes of carbene complex, a vast number of which have catalytic implications, from highly reactive alkylidene complexes capable of double-bond metathesis,3 to those involving N-heterocyclic carbene (NHC) spectator ligands.4 Whilst considerable efforts have also been given to the study of the heavier tetrylenes in TM complexes, this field has largely focused on synthetic access, and less so on reactivity and utilisation in broader synthetic protocols.5 The heavier tetrylenes have the capacity to act as ‘single centre ambiphiles’, this effect amplified on descending group 14 due to reduced sp-mixing,6 with a σ-donating lone electron pair and a Lewis acidic vacant p-orbital. Although heavier tetrylenes have been employed as ligands in catalysis,5c,7 and notably so amidinato silylenes,7 these, like NHCs, are typically spectator ligands due to common examples being N-heterocyclic in nature. Chelating ligands employing an acyclic, low-coordinate germylene have seen some attention in the literature, and are typically doubly substituted by a ligand bearing a phosphine arm; a number of examples of such systems have been reported by Cabeza and co-workers,8,9 which have been combined with first-row TM halides so as to access TMII complexes, often with tetrylene insertion into TM–X bonds (X = halide).10 Related (carbonyl-free) first-row TM0 complexes bearing acyclic, two-coordinate germylene ligands are rare,11 and should allow for metal–ligand cooperativity (MLC) due to their amphiphilic nature imparting them the potential to remain Lewis acidic when bound to a metal centre. MLC, whereby a metal-bound ligand plays an active role in bond activation,12 is a powerful concept in catalysis,13,14 and has allowed for the facile activation of bonds which are otherwise challenging to cleave, a prime example being the N–H bonds in ammonia.15 In a key demonstration of this process, the phosphino-carbene Ni0 complex A can readily bind ammonia at Ni in the displacement of its Ph3P ligand, with the nucleophilic carbene ligand accepting a proton in N–H bond cleavage (Fig. 1).15c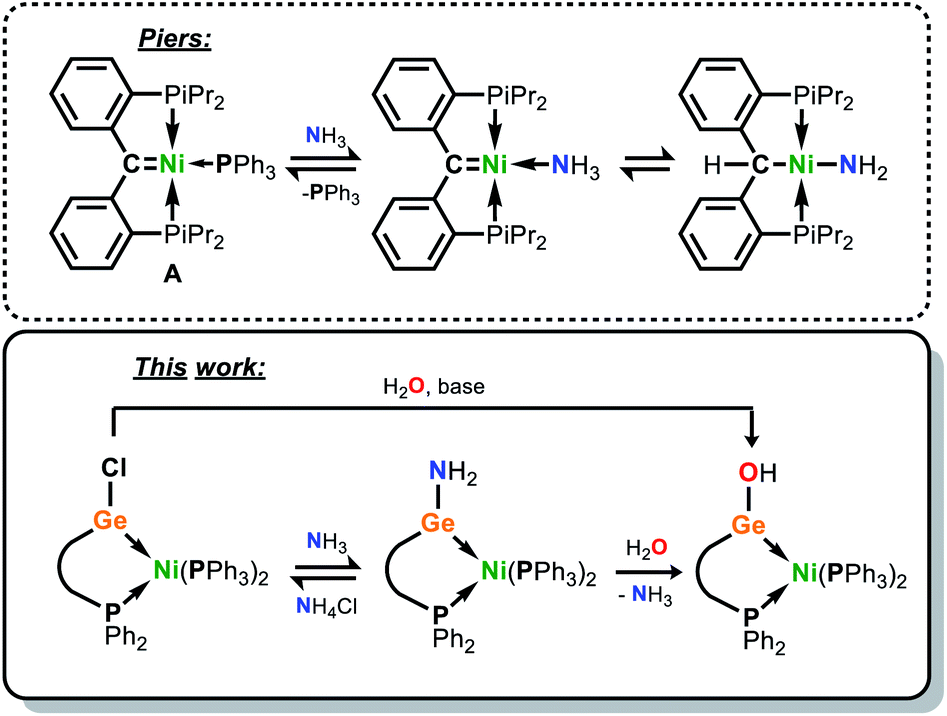 | ||
| Fig. 1 The nucleophilic role of a carbene ligand in reversible ammonia activation (Piers), and the electrophilic role of a germylene ligand in reversible ammonia activation (this work). | ||
Given the potentially Lewis acidic nature of heavier tetrylenes, we envisage that such ligands can reverse the polarity of this MLC process, and act as electrophilic binding sites in TM complexes. This would open a new avenue in the dual-centre activation of small molecules, and indeed in catalysis. In order to investigate the capacity of a tetrylene to remain electrophilic in the coordination sphere of a TM, and perhaps more importantly to be more electrophilic than the TM, we sought to develop a chelating ligand incorporating an acyclic, low-coordinate heavier tetrylene. To this end, we have developed novel phosphine-functionalised amine pro-ligands, PhPhDippNH and PhiPDippNH (PhPhDippNH = Ph2PCH2Si(Ph)2N(H)Dipp; PhiPDippNH = Ph2PCH2Si(iPr)2N(H)Dipp; Dipp = 2,6-iPr2-C6H3), which can be used to generate phosphine-functionalised (amido)(chloro)germylenes which satisfy the targeted acylic, low-coordinate ligand characteristics when combined with a low-valent TM centre (Fig. 1). Herein we report the synthesis of these compounds, specifically as their Ni0 complexes, in which the single centre ambiphile ligand centre (i.e. GeII) maintains its Lewis acidity, and is capable of binding and activating NH3 and H2O, in the former case reversibly.
Results and discussion
The phosphine-functionalised germylene ligands feature a novel ancillary ligand scaffold, namely a phosphine-functionalised amide. The amine pro-ligands are readily accessed through an initial in situ synthesis of phosphine-functionalised chlorosilanes, (Ph2PCH2)(R)2SiCl (R = Ph, iPr),16 which can then be reacted with the lithium anilide, DippN(H)Li, with loss of LiCl. Although the formed pro-ligands, PhiPDippNH and PhPhDippNH, were not isolated in their pure forms, 1H and 31P NMR analyses of crude reaction mixtures suggest they are formed near quantitatively.17 These crude products are readily deprotonated with a suspension of KH in THF, yielding the potassium amides PhiPDippNK and PhPhDippNK in good isolated yields of ∼70 to 75% after work up, based on the Ph2PCH2Li·TMEDA starting material. GeII chloride complexes PhPhDippNGeCl and PhiPDippNGeCl (1a and 1b, respectively; (Scheme 1)) are generated through combination of these potassium amides with GeCl2·dioxane in either THF or toluene, yielding the desired germylene ligands in high yields. Both 1a and 1b show somewhat complex 1H NMR spectra, when compared with potassium amides PhiPDippNK and PhPhDippNK, due to the asymmetric coordination at their GeII centres. Nevertheless, the presence of a single peak in the respective 31P{1H} NMR spectra confirms the presence of a single ligand environment in these compounds. A single crystal X-ray diffraction analysis of the two germylenes confirms their monomeric nature (viz.Fig. 2), and shows that the Ph2P moiety of the amide ligands binds the GeII centre in both species (1a: dPGe = 2.4547(9) Å; 1b: dPGe = 2.458(1) Å). Complexes 1a/b bear some similarity to complexes reported by Wesemann, incorporating bulky aryl ligands at GeII,18 and Baceiredo, who also developed GeII chloride species bearing phosphine-functionalised amide ligands.19 In these cases, Ge–P distances are similar to those in 1a/b.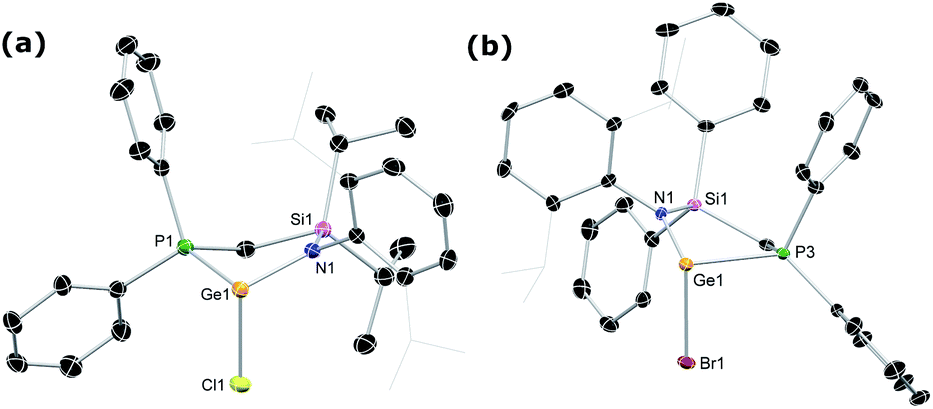 | ||
| Fig. 2 The molecular structures of (a) 1b and (b) 3a, with thermal ellipsoids at 40% probability. Hydrogen atoms omitted for clarity. Selected bond distances (Å) and angles (°) for 1b: Ge1–Cl1 2.355(1); Ge1–P1 2.472(1); Ge1–N1 1.925(3); N1–Ge1–Cl1 100.69(1); Cl1–Ge1–P1 85.98(4); N1–Ge1–P1 85.85(1). For 3a: Ge1–Br1 2.512(1); Ge1–P3 2.455(2); Ge1–N1 1.913(4); N1–Ge1–Br1 103.00(1); Br1–Ge1–P3 84.74(4); N1–Ge1–P3 84.98(1). | ||
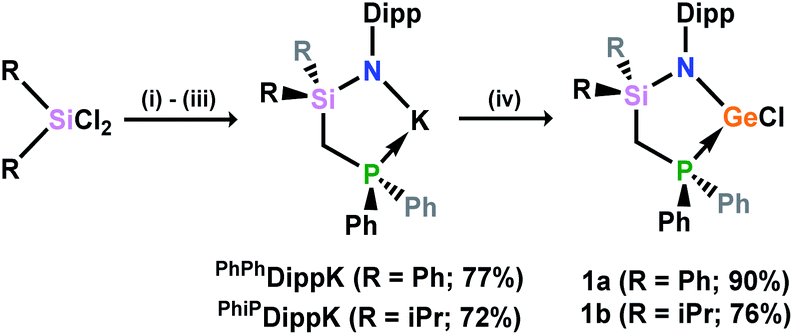 | ||
| Scheme 1 The synthesis of phosphine-functionalised amine pro-ligands, their deprotonation, and subsequent synthesis of chloro-germylene complexes. (i) Ph2PCH2Li·TMEDA, hexane; (ii) DippN(H)Li, THF; (iii) KH, THF; (iv) GeCl2·dioxane, THF (yields in parentheses). Dipp = 2,6-iPr2-C6H3. | ||
With (amido)(chloro)germylene ligands 1a/b in hand, we began our investigations into the complexation chemistry of these pro-chelating ligands towards nickel dihalides. We found that both complexes do not readily react with NiCl2, even after heating, despite a dark red colouration of reaction solutions. However, reactions with an excess of NiBr2·DME in toluene with a small amount of THF led to dark red-brown reaction mixtures, from which dimeric complex 2 could be isolated as large red-brown crystals after filtration (Scheme 2). X-ray structural analysis of these crystals (Scheme 2, inset) revealed that Ni inserts into the Ge–P bond of 1a, forming a chelating ligand motif incorporating P and Ge as we had hoped. Formal oxidative addition of one Ni–Br bond at GeII is also observed, akin to previous examples reported by Cabeza and co-workers.10 Each NiII centre sits in a square planar geometry, bound by two bridging Br ligands, Ge, and P. Compound 2 represents a surprisingly uncommon example of a germyl-nickel complex, and is unstable in solution in the absence of an excess of NiBr2. This was clear in an attempt to obtain NMR spectroscopic data for 2, where the only observable species is the bromo germylene PhPhDippGeBr 3a, presumably through elimination of the nickel dihalide.20 Reassessing the reaction of chloro germylene 1a with NiBr2·DME, we found that addition of a single equivalent leads to a ∼50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture of 1 and 3, i.e. partial Cl/Br exchange (Fig. S24 and S28†). From such a reaction mixture the formation of one or two single crystals of the germyl-nickel mixed halide complex 2′, the acetonitrile-coordinated monomeric form of 2, allowed for structural analysis of this ‘intermediate’ (Fig. S80 in ESI†). Addition of 6 equiv. of NiBr2·DME allows for full conversion to bromo germylene 3,21 whilst a vast excess (∼10 equiv.) of NiBr2·DME affects the crystallisation of small amounts of germyl-nickel bromide complex 2. We presume that the presence of an excess of NiBr2·DME is required to maintain the stability of 2, as in all cases NMR analyses showed only the free chloro/bromo-germylene ligands. Taken as a whole, this demonstrates that NiBr2 can act as a facile halide exchange reagent for the synthesis of bromo germylenes, which are challenging to access via conventional routes due to the lack of readily available GeBr2 reagents.22
:50 mixture of 1 and 3, i.e. partial Cl/Br exchange (Fig. S24 and S28†). From such a reaction mixture the formation of one or two single crystals of the germyl-nickel mixed halide complex 2′, the acetonitrile-coordinated monomeric form of 2, allowed for structural analysis of this ‘intermediate’ (Fig. S80 in ESI†). Addition of 6 equiv. of NiBr2·DME allows for full conversion to bromo germylene 3,21 whilst a vast excess (∼10 equiv.) of NiBr2·DME affects the crystallisation of small amounts of germyl-nickel bromide complex 2. We presume that the presence of an excess of NiBr2·DME is required to maintain the stability of 2, as in all cases NMR analyses showed only the free chloro/bromo-germylene ligands. Taken as a whole, this demonstrates that NiBr2 can act as a facile halide exchange reagent for the synthesis of bromo germylenes, which are challenging to access via conventional routes due to the lack of readily available GeBr2 reagents.22
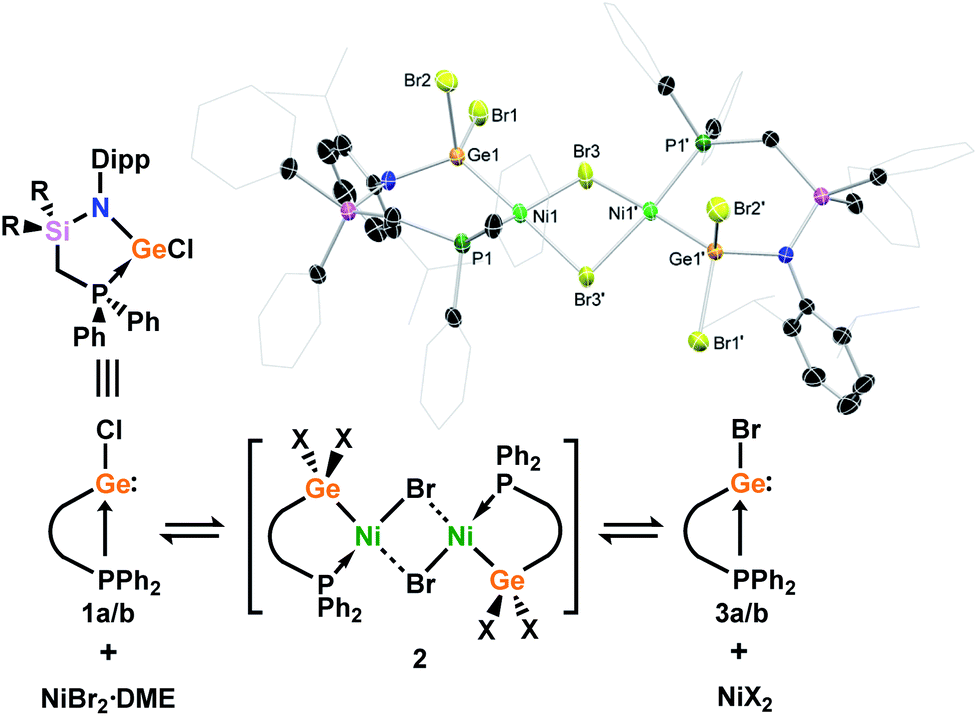 | ||
| Scheme 2 The reaction of chloro-germylenes 1a/b with NiBr2·DME, leading to reversible complexation and Cl/Br exchange at GeII. Inset: the molecular structure of dimeric germyl-nickel complex 2. | ||
Although the isolation of useful quantities of nickel complex 2 was not possible, the observation that it exists within the reaction mixture seemed a promising start. We found that in situ reduction of this reaction mixture, in the presence of Ph3P, gave facile access to chelating phosphino-germylene complexes of Ni0. Room temperature addition of THF to a mixture of 1a/b, NiBr2·DME, Ph3P, and an excess of Zn powder allowed for the clean formation of Ni0 complexes 4a/b after 2 h, albeit as a mixture of the chloro- and bromo-germylene ligated species. Employing NiCl2·DME in place of NiBr2·DME led to considerably extended reaction times, but gave clean access to the chloro-germylene species (Scheme 3). The pure chloro-germylene analogue can also be accessed through combination of 1, Ni(COD)2, and PPh3 in the stoichiometric ratio 1:1:2 (Fig. S31†). Satisfyingly, 31P NMR analysis of crude reaction mixtures in all cases suggest the formation of these novel Ni0 complexes as the sole products, with the crystalline compounds isolated in good yields from diethyl ether extracts of the crude reaction mixtures. A single crystal X-ray structural analysis of dark red-brown crystals of 4b reveal a central Ni0, bound by two Ph3P ligands, and one chelating phosphino-germylene ligand (Fig. 4(a)). Notably, the chloride moiety in the germylene ligand remains intact. The three-coordinate GeII centre holds a trigonal planar geometry, and has a relatively short Ge–Ni contact when compared with previously reported examples, indicative of some back-bonding from Ni to Ge (vide supra). The Ni0 centre holds a tetrahedral geometry, with the three Ni–P distances being as expected when compared with reported phosphine-coordinated Ni0 complexes. Complex 4a is essentially isostructural to 4b.23 The UV/vis spectra of these species show two clear absorption bands (4a: λmax = 484 nm (ε = 1370 L cm−1 mol−1) and 362 nm (6735 L cm−1 mol−1); 4b: 473 nm (1685 L cm−1 mol−1) and 362 nm (9160 L cm−1 mol−1)), in-keeping with related absorptions for Ni-based d–d transitions.24 Likely due to hindered rotation brought about by the sterics of the chelating ligand system, resonances relating the ligand are broadened in the 1H NMR spectrum of both Ni0 species. The 31P NMR spectra show one broadened peak for the Ph3P ligands, centred at δ = 39.8 (4a and 4b) ppm, whilst the flanking phosphine arm of the chelating ligand presents as a well resolved triplet centred at δ = 7.1 (4a) and 9.6 (4b) ppm (Fig. 3(a)). For the mixed bromide/chloride samples, a second set of peaks relating to the bromide species is observed in the 31P NMR spectrum, overlapping with the first (Fig. S32 and S40†).25 Separation of the two compounds proved impossible in our hands, although using the mixture in subsequent chemistry discussed herein did not pose any issues.
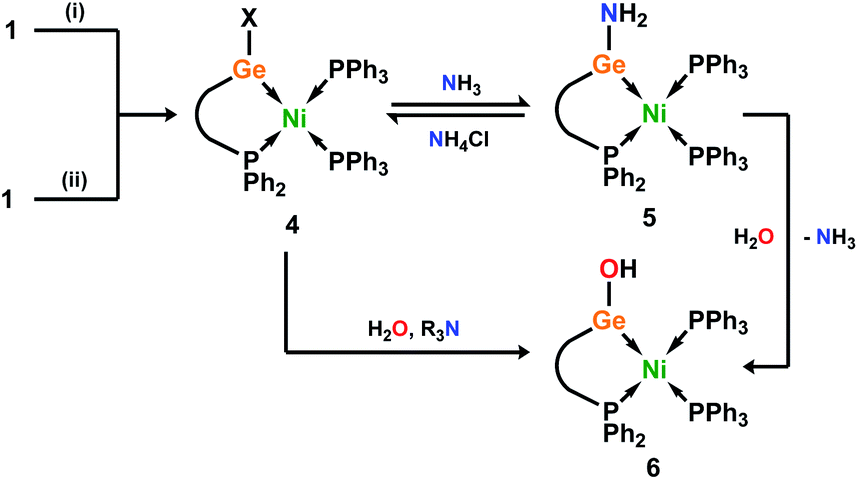 | ||
| Scheme 3 Synthesis of halo-germylene Ni0 complexes 4 and subsequent reactions with NH3 and H2O. (i) 1.1NiX2·DME, 2Ph3P, 6Zn, THF, 2 h (X = Br); ∼24 h (X = Cl); X = Cl and/or Br; (ii) 1Ni(cod)2, 2Ph3P, toluene, 1 h; X = Cl. | ||
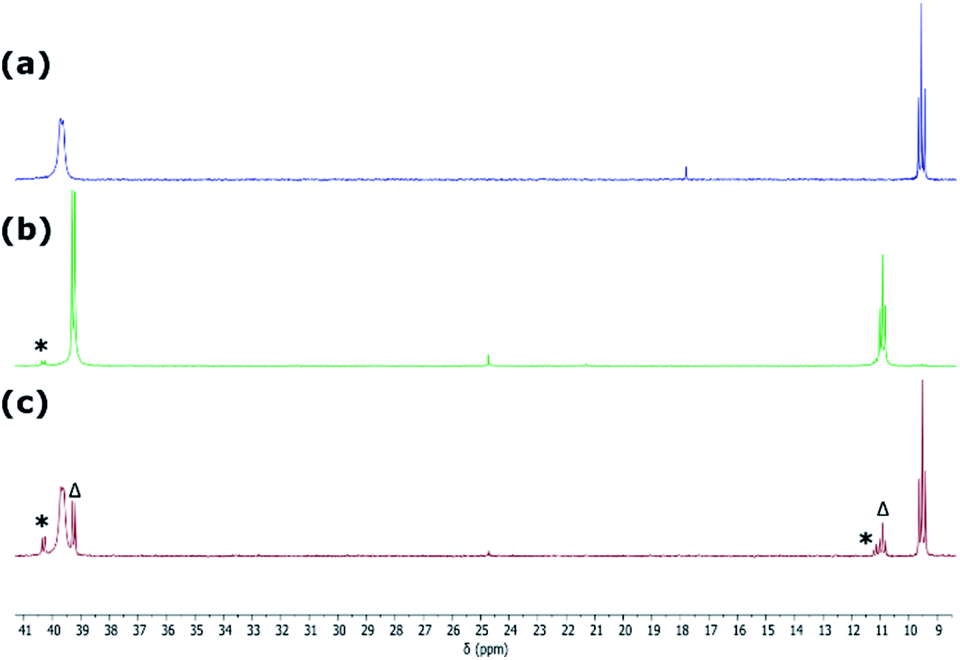 | ||
| Fig. 3 The 31P{1H} NMR spectrum of (a) 4b, (b) in situ addition of NH3 to 5b, and (c) 4b regenerated after sonication and degassing the sample used for spectrum (b). * = small amounts of 6b; Δ = small amounts of residual 5b. | ||
Given the ambiphilic nature of tetrylenes, particularly in acyclic derivatives, the design of complexes 4a/b aims to retain a degree of Lewis acidity at GeII. This forms the central idea of non-innocent single centre ambiphile ligands. This characteristic was probed by Density Functional Theoretical (DFT) analysis of the frontier orbitals in model complex 4′, employing PhMeXylN in place of PhiPDippN/PhPhDipp (PhMeXylN = (Ph2PCH2SiMe2)(Xyl)N). We found that the LUMO of 4′ is located on the Ge centre (Fig. 4(a)), and mainly constitutes a vacant p-orbital which is expected to be Lewis acidic, particularly given the overall NPA charge at Ge of +1.13. Still, Natural Bond Orbital (NBO) analysis of the Ge–Ni bond indicates strong polarisation toward the Ge centre (Table S4†), whilst the HOMO in 4′ (Fig. S85 in ESI†) shows some degree of π-bonding, pointing towards a donor–acceptor description of the Ge–Ni bond. In line with the Lewis acidity of the GeII centre in 4a/b, deep red-brown solutions of these compounds readily react with ammonia at 1 atm pressure to form slightly turbid bright orange reaction mixtures containing ammonia activation products 5a/b. In situ31P{1H} NMR spectroscopic analysis indicated the formation of a single new product for both systems (Fig. 3(b)), whilst complimentary 1H NMR spectra indicated new singlet 2H resonances at δ = 3.13 (5a) and 2.96 (5b) ppm, which were tentatively assigned to an NH2 fragment. The IR spectrum of reaction products is in keeping with this, with two weak N–H stretching bands observed at ν = 3354 and 3461 (5a and 5b) cm−1. Structural analysis of large orange crystals of the product of the reaction of 4a with ammonia reveal that the Ge–Cl moiety in this species has in fact undergone a σ-metathesis reaction with one N–H bond of ammonia, yielding the bis(amido)germylene Ni0 complex 5a (Scheme 3 and Fig. 4(b)).26 Presumably, the colourless solid in reaction mixtures is ammonium chloride, through the net loss of HCl in this reaction (vide supra). As such, removal of the ammonia atmosphere from reaction mixtures, and sonication of amides 5a/b over the precipitated NH4Cl with intermittent degassing leads to regeneration of starting materials 4a/b (Fig. 3), with the loss of ammonia (Scheme 3).27 Such a reversible activation of ammonia is, to the best of our knowledge extremely rare, as is the more general redox-innocent metathesis reaction at a tetrylene centre. Beyond the reaction of 4a/b with ammonia, these species also readily undergo a similar metathesis reaction with water in the presence of nitrogen bases to facilitate HCl abstraction (i.e. CyNH2), to yield (amido)(hydroxyl)germylene Ni0 complexes 6a/b (Scheme 3 and Fig. 4(c)). These species can also be accessed by the reaction of amide complexes 5a/b with water, with concomitant loss of ammonia. The formation of 6a/b can be clearly observed by 1H NMR and IR spectroscopy, the former containing new 1H resonances pertaining to OH residues (δ = 3.13 (6a) and 2.96 (6b) ppm), and the latter clear OH stretching bands (ν = 3547 (6a) and 3541 (6b) cm−1). In recent years a flurry of examples of ammonia activation at low-valent group 14 centres have been forthcoming, but lead almost exclusively to oxidative addition reactions, thus yielding EIV compounds (E = C–Sn).28 That oxidative addition is circumvented in reactions of 4a/b with both NH3 and H2O likely stems from the binding of the Ge lone electron pair to Ni0. Further, it is surprising that the Ni0 centre in these complexes remains unchanged, given previous examples of MLC in ammonia activation involving Ph3P-ligated Ni0 complexes.15c,d Overall, the metathesis reactions of ammonia demonstrated here highlight the targeted high Lewis acidity of the developed acyclic germylene ligands, and are an exciting step towards employing these ligands in complexes which can operate via MLC bond activation processes, where a lower coordinate TM centre is utilised.
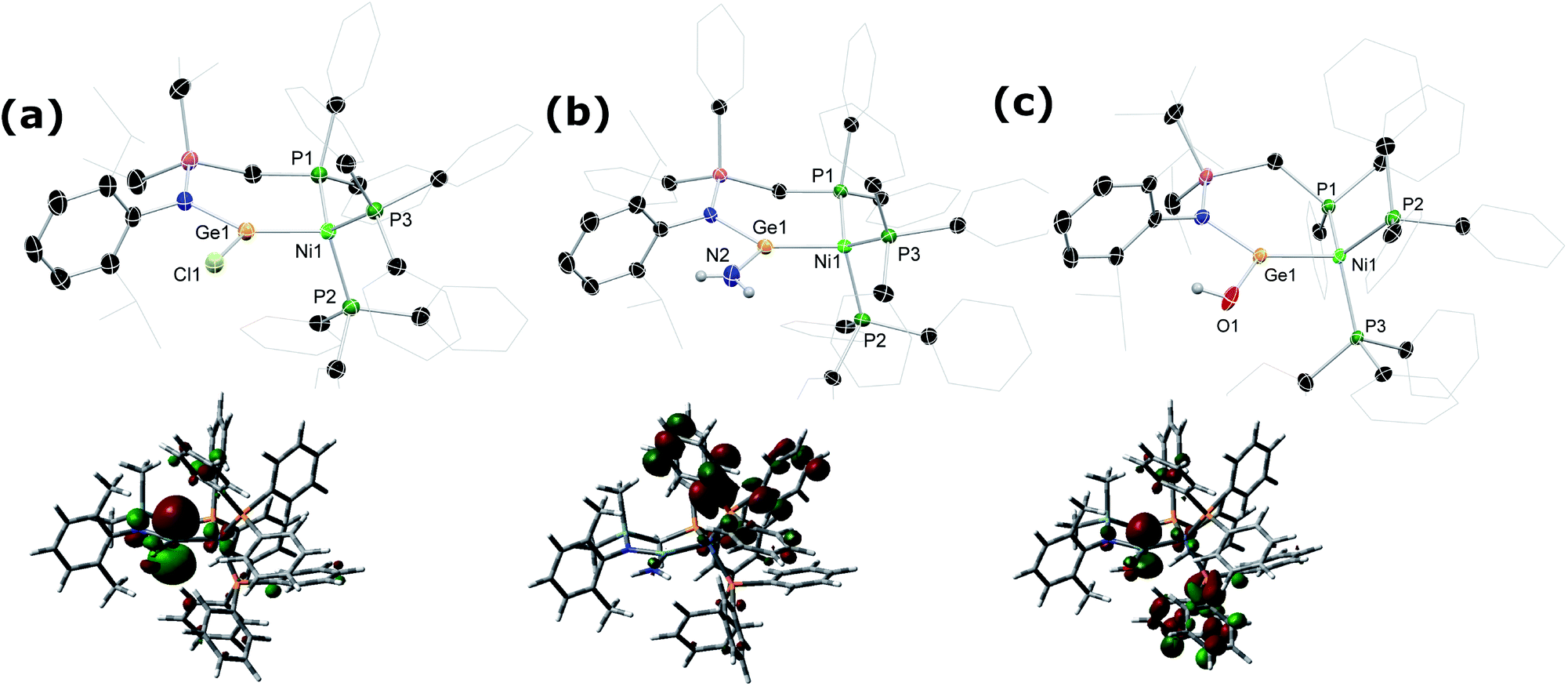 | ||
| Fig. 4 Molecular structures of compounds (a) 4b, (b) 5a, and (c) 6b, with thermal ellipsoids at 40% probability. Hydrogen atoms omitted for clarity, aside from those at N2 and O1 in 5a and 6b, respectively. The LUMO of each compound is inset below the respective structure. Selected bond distances (Å) and angles (°) for 4b: Ge1–Ni1 2.1877(7); N1–Ge1 1.869(2); P1–Ni1 2.201(1); P2–Ni1 2.2079(8); P3–Ni1 2.2055(8); N1–Ge1–Cl1 99.57(7); Ni1–Ge1–N1 133.09(7); Ni1–Ge1–Cl1 126.89(3). For 5a: Ge1–Ni1 2.217(1); N1–Ge1 1.890(2); N2–Ge1 1.819(2); P1–Ni1 2.210(1); P2–Ni1 2.201(1); P3–Ni1 2.1892(9); N1–Ge1–N2 99.07(9); Ni1–Ge1–N1 128.63(6); Ni1–Ge1–N2 132.29(7). For 6b: Ge1–Ni1 2.2077(7); N1–Ge1 1.885(3); O1–Ge1 1.874(2); P1–Ni1 2.210(1); P2–Ni1 2.202(1); P3–Ni1 2.104(1); N1–Ge1–O1 100.98(1); Ni1–Ge1–O1 128.47(7); Ni1–Ge1–N1 130.49(8). | ||
So as to compare the effects of the differing fragments at GeII in the described Ni0 complexes, we performed frontier orbital, NBO, Wiberg Bond Index (WBI), Mayer Bond Order (MBO), and Natural Population analyses on 4′, 5′, and 6′ (Tables S4–S7†), as model complexes of the real systems, again employing PhMeXylN in place of PhiPDippN/PhPhDipp. We found that 4′, 5′, and 6′ all show very similar characteristics in general; the main difference is the interaction of the vacant p-orbital on the GeII centre and the lone pair of the Cl/NH2/OH moiety. Bond analysis showed the highest bond order for the Ge–NH2 in 5′ (MBO: 1.10) which is due to the efficient hybridization of the N lone pair and the vacant p-orbital of Ge that are well-described for low-valent germanium compounds.29 The LUMO of 5′ is the only example in this series which is not represented by a vacant p-orbital at Ge (Fig. 4), most likely due to the described N → Ge donation. Consequently, the Ge–Ni bond order decreases and the Ge–Ni bond length increases (vide infra) due to the smaller back donation from the Ni d-orbital to the Ge vacant orbital. All systems show some degree of multiple bond character between the GeII and Ni0 centres (WBI/MBO for 4′ 1.20/1.13; for 5′: 1.11/0.98; for 6′: 1.17/1.07), with predicted Ge–Ni distances (4′: 2.161 Å; 5′: 2.203 Å; 6′: 2.177 Å) only slightly contracted relative to those observed experimentally (e.g. dGeNi for 4b: 2.1877(7) Å; 5a: 2.217(1) Å; 6a: 2.1956(7) Å), possibly due to the increased steric encumbrance in the real complexes. An NBO analysis indicates a large degree of s-character in these bonds, indicative of the greatest contributing factor being Ge → Ni lone-pair donation, but, as already described for 4′, a high degree of polarisation towards Ge is also apparent, making a donor–acceptor interaction the best description for the Ge–Ni interaction in these complexes.
Further, the mechanism for the amination of model complex 4′ was investigated by means of DFT (Fig. 5), given the reversible nature of this interesting process. The most favourable mechanism begins with binding of NH3 at GeII, highlighting the Lewis acidity of this centre. This process is near thermoneutral (IM1 in Fig. 5, +1.7 kcal mol−1), which corroborates reversibility in this key step. Surprisingly, it was found that two further NH3 molecules are subsequently required to drive the metathesis, which form a H-bond network between the Ge-bound NH3 and Cl ligands. The most challenging step in the overall process is the ion pair formation (TS4 in Fig. 5, +14.5 kcal mol−1), which is only possible on the potential energy surface (PES) if the H-bond network is present, which stabilises the formation of the NH4+ ion. Then, the NH4+ and the additional NH3 molecule facilitate the removal of Cl−. The overall reaction is very close to thermoneutral (5′ in Fig. 5, −0.9 kcal mol−1), making this process reversible simply by changing the reaction conditions, again corroborating the observed experimental results. Finally, we also considered (i) the potential involvement of the Ni centre in NH3 activation, and (ii) oxidative addition of an N–H bond at the Ge centre. Both of these mechanisms can be excluded from the active PES; the Ni–H complex, formed in (i), is relatively unstable (TS2′ in Fig. 5, 19.5 kcal mol−1), whilst the Ge oxidative addition product is not a minimum on the PES (route (ii)).
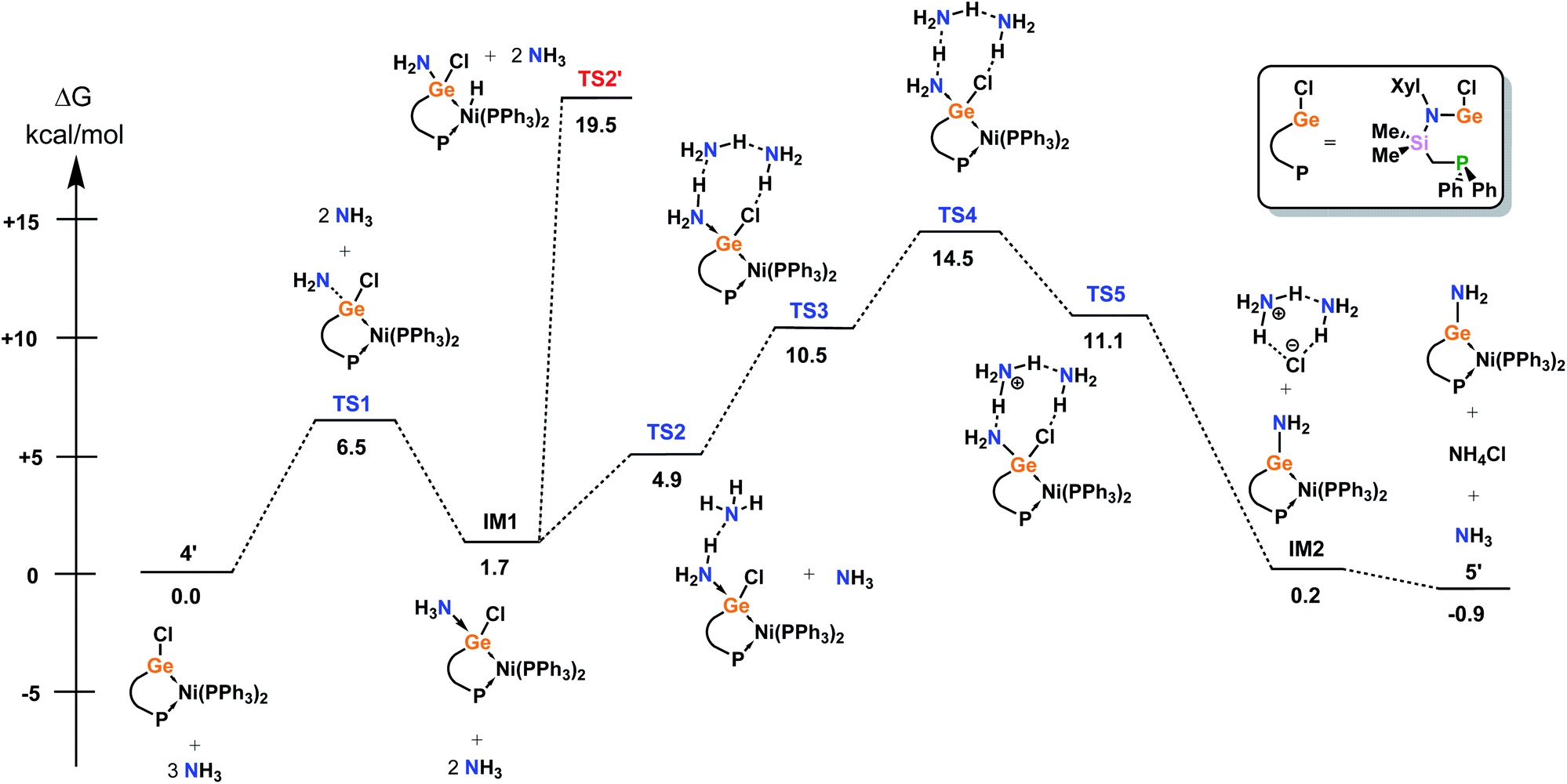 | ||
| Fig. 5 DFT-derived mechanism for the σ-metathesis of ammonia in 4′ leading to 5′. | ||
Conclusions
In conclusion, we have developed a chelating phosphino-germylene ligand scaffold, and a facile route to Ni0 complexes bearing these ligands in a low-cost, one pot synthetic preparation. These ligands, which are centred around an acyclic (amido)(chloro)germylene, remain Lewis acidic when bound to Ni0. This has been demonstrated by the facile and reversible activation of ammonia, as well as the complimentary irreversible reaction with water, both of which lead to GeII products through σ-metathesis of the Ge–Cl bond, thus circumventing oxidation at both germanium and nickel. These results form an initial basis for the single centre ambiphile ligand concept, which we are currently expanding to further low-valent main group species, and further first row transition metals, moving towards cooperative bond activation involving both the ligand and the metal centre.Experimental
General considerations
All experiments and manipulations were carried out under a dry oxygen free argon atmosphere using standard Schlenk techniques, or in a MBraun inert atmosphere glovebox containing an atmosphere of high purity argon. THF and diethyl ether were dried by distillation over a sodium/benzophenone mixture and stored over activated 4 Å mol sieves. C6D6 was dried and stored over a potassium mirror. All other solvents were dried over activated 4 Å mol sieves and degassed prior to use. NiBr2·DME,30 NiCl2·DME,31 DippN(H)Li,32 and PPh2CH2Li·TMEDA33 were synthesized according to known literature procedures. All other reagents were used as received. Commercial CyNH2, when used as received, contained enough residual moisture to allow for the synthesis of 6a. NMR spectra were recorded on a Bruker AV 400 or 500 Spectrometer. The 1H and 13C{1H} NMR spectra were referenced to the residual solvent signals as internal standards. 29Si NMR spectra were externally calibrated with SiMe4. 31P NMR spectra were externally calibrated with H3PO4. LIFDI MS spectra were measured at a Waters Micromass LCT TOF mass spectrometer equipped with an LIFDI ion source (LIFDI 700) from Linden CMS GmbH. The samples were dissolved in dry toluene and filtered using a syringe filter under an inert atmosphere. The TOF setup was externally calibrated using polystyrene. ESI-MS was performed on an exactive plus orbitrap spectrometer from Thermo Fischer Scientific. Infrared spectra were measured with the Alpha FT IR from Bruker containing a platinum diamond ATR device. The compounds were measured as solids under inert conditions in a glovebox. For the ammonia activation experiments water free ammonia 5.0 was used.Representative procedures for the preparation of compounds 1, 3, and 4–6 are given below. Further details for the synthesis and characterisation of all novel compounds are given in the ESI.†
PhPhDippNK
A yellow suspension of PPh2CH2Li·TMEDA (6.0 g, 18.6 mmol) in 100 mL hexane was cooled to −78 °C. The mixture was stirred vigorously and Ph2SiCl2 (3.9 mL, 18.6 mmol) was added. The mixture was allowed to warm to RT overnight. All volatiles were subsequently removed in vacuo, leaving a yellow oil. DippN(H)Li (3.4 g, 18.6 mmol) was added to the residue, and the flask cooled to −78 °C, followed by the addition of 50 mL THF. The mixture was stirred until dissolution of all solids was observed. The cold bath was then removed and the reaction allowed to warm to RT, leading to an orange solution. All volatiles were removed in vacuo and the oily residue extracted with 50 mL hexane, and filtered. The solvent was removed in vacuo and KH (0.9 g, 28.3 mmol) was added. After addition of 50 mL THF, gas started to evolve, and the mixture was vigorously stirred for a further 16 h. The dark brown suspension was filtered, and all volatiles were removed in vacuo. To the resulting oil 50 mL hexane was added, and the mixture treated in an ultrasonic bath causing the precipitation of copious pale brown powder, which was filtered and washed multiple times with hexane, and subsequently dried in vacuo to yield PhPhDippNK as an off-white powder (8.5 g, 14.3 mmol, 77%). Colourless crystals suitable for X-ray diffraction analysis were obtained after two days from a concentrated THF/TMEDA solution layered with hexane stored at −32 °C. 1H NMR (THF-d8, 400 MHz, 298 K): δ = 0.91 (d, 12H, 3JHH = 6.9 Hz, Dipp-Pri-CH3), 1.97 (d, 2H, 2JHP = 5.4 Hz, Ph2P–CH2), 3.94 (hept, 2H, 3JHH = 6.9 Hz, Dipp-Pri-CH), 6.24 (t, 1H, 3JHH = 7.4 Hz, Ar–CH), 6.74 (d, 2H, 3JHH = 7.4 Hz, Ar–CH), 7.00 (m, 6H, Ar–CH), 7.10 (m, 6H, Ar–CH), 7.27 (m, 4H, Ar–CH), 7.48 (m, 4H, Ar–CH). 13C{1H} NMR (THF-d8, 101 MHz, 298 K): δ = 19.7 (d, 1JCP = 29.2 Hz, Ph2P–CH2), 24.9 (Dipp-Pri-CH3), 28.1 (Dipp-Pri-CH), 112.6, 123.00, 127.3, 127.5, 128.3, 128.7, 133.4, 133.6, 135.8, 141.4, 143.9, 144.0, 146.7, 146.7 and 156.2 (Ar–C). 31P{1H} NMR (THF-d8, 162 MHz, 298 K): δ = −19.1 (s, CH2–PPh2). 29Si{1H} NMR (THF-d8, 99 MHz, 298 K): δ = −47.4 (d, 2JSiP = 14.7 Hz, SiPh2).PhPhDippGeCl, 1a
A pale brown solution of PhPhDippNK (7.0 g, 11.7 mmol) in 20 mL THF was added dropwise to a stirring solution of GeCl2·dioxane (2.7 g, 11.7 mmol) in 10 mL THF at −78 °C, and subsequently allowed to warm to RT, resulting in the formation of an orange solution. All volatiles were removed in vacuo, the residue extracted in 20 mL DCM, and filtered. The solvent was removed in vacuo and the residue washed with hexane to yield 1a as an off-white powder (7.0 g, 10.5 mmol, 90%). Colourless crystals suitable for X-ray diffraction analysis were obtained from a concentrated diethyl ether at RT. 1H NMR (C6D6, 400 MHz, 298 K): δ = 0.09 (d, 3H, 3JHH = 6.4 Hz, Dipp-Pri-CH3), 0.68 (d, 3H, 3JHH = 6.6 Hz, Dipp-Pri-CH3), 0.86 (d, 3H, 3JHH = 6.5 Hz, Dipp-Pri-CH3), 1.43 (d, 3H, 3JHH = 6.5 Hz, Dipp-Pri-CH3), 2.54 (m, 3H, Ph2P–CH2/Dipp-Pri-CH), 4.28 (m, 1H, Dipp-Pri-CH), 7.02 (m, 14H, Ar–CH), 7.21 (m, 3H, Ar–CH), 7.46 (m, 2H, Ar–CH), 7.65 (m, 2H, Ar–CH), 8.26 (d, 2H, 3JHH = 7.1 Hz, Ar–CH). 13C{1H} NMR (C6D6, 101 MHz, 298 K): δ = 8.1 (Ph2P–CH2), 21.4, 22.6 and 28.0 (Dipp-Pri-CH3), 28.2 and 28.6 (Dipp-Pri-CH), 28.8 (Dipp-Pri-CH3), 123.9, 124.6, 125.6, 129.0, 129.1, 129.6, 129.7, 131.1, 131.5, 132.8, 132.9, 133.3, 133.4, 134.8, 135.4, 137.3, 137.8, 140.8, 140.9, 147.8 and 149.7 (Ar–C). 31P{1H} NMR (C6D6, 162 MHz, 298 K): δ = 4.0 (s, CH2–PPh2). 29Si{1H} NMR (C6D6, 79 MHz, 298 K): δ = −4.0 (d, 2JSiP = 13.4 Hz, SiPh2). MS/LIFDI-HRMS found (calcd) m/z: 665.1466 (665.1490) for [M]+.PhPhDippGeBr, 3a
To a mixture of 1a (200 mg, 0.30 mmol) and NiBr2·DME (540 mg, 1.8 mmol) was added 5 mL toluene and 1 mL THF, and the resulting mixture stirred for 1 h. All volatiles were removed in vacuo and the residue extracted with 5 mL toluene. The solution was concentrated and layered with hexane yielding colourless crystals of 3a suitable for X-ray diffraction analysis (127 mg, 0.18 mmol, 54%). 1H NMR (C6D6, 400 MHz, 298 K): δ = 0.06 (d, 3H, 3JHH = 6.6 Hz, Dipp-Pri-CH3), 0.67 (d, 3H, 3JHH = 6.7 Hz, Dipp-Pri-CH3), 0.93 (d, 3H, 3JHH = 6.6 Hz, Dipp-Pri-CH3), 1.46 (d, 3H, 3JHH = 6.6 Hz, Dipp-Pri-CH3), 2.55 (m, 3H, Ph2P–CH2/Dipp-Pri-CH), 4.26 (hept, 1H, 3JHH = 6.9 Hz, Dipp-Pri-CH), 6.99 (m, 14H, Ar–CH), 7.21 (m, 3H, Ar–CH), 7.46 (m, 2H, Ar–CH), 7.60 (m, 2H, Ar–CH), 8.30 (d, 2H, 3JHH = 7.3 Hz, Ar–CH). 13C{1H} NMR (C6D6, 101 MHz, 298 K): δ = 8.4 (Ph2P–CH2), 21.4 and 22.6 (Dipp-Pri-CH3), 28.1 and 28.2 (Dipp-Pri-CH), 28.5 and 29.0 (Dipp-Pri-CH3), 123.1, 124.0, 124.8, 125.7, 128.9, 129.0, 129.1, 129.6, 129.8, 131.0, 131.6, 132.7, 132.8, 133.3, 133.4, 134.5, 135.4, 137.6, 140.5, 140.6, 147.8 and 149.9 (Ar–C). 31P{1H} NMR (C6D6, 162 MHz, 298 K): δ = 1.9 (s, CH2–PPh2). 29Si{1H} NMR (C6D6, 99 MHz, 298 K): δ = −3.9 (d, 2JSiP = 13.9 Hz, SiPh2); MS/LIFDI-HRMS found (calcd) m/z: 709.1012 (709.0984) for [M]+.[PhPhDippGe(Cl)]Ni(PPh3)2, 4a
To a mixture of 1a (1.60 g, 2.4 mmol), NiCl2·DME (0.53 g, 2.4 mmol), PPh3 (1.26 g, 4.8 mmol), and Zn (0.94 g, 14.4 mmol) was added 10 mL THF, and the resulting mixture stirred for 24 h at RT resulting in a deep red reaction mixture. All volatiles were removed in vacuo and the residue extracted with 20 mL diethyl ether. Dark red crystals of 4a, which were suitable for X-ray diffraction analysis, were obtained after storing the solution at RT overnight (1.55 g, 1.8 mmol, 52%). 1H NMR (C6D6, 400 MHz, 298 K): δ = 0.49 (d, 6H, 3JHH = 6.6 Hz, Dipp-Pri-CH3), 1.28 (bs, 6H, Dipp-Pri-CH3), 3.03 (s, 2H, Ph2P–CH2), 3.72 (hept, 2H, 3JHH = 6.6 Hz, Dipp-Pri-CH), 6.63 (m, 3H, Ar–CH), 6.94 (m, 36H, Ar–CH), 7.32 (d, 5H, 3JHH = 6.2 Hz, Ar–CH), 7.52 (s, 9H, Ar–CH). 13C{1H} NMR (C6D6, 101 MHz, 298 K): δ = 20.0 (Ph2P–CH2), 23.8 and 26.1 (Dipp-Pri-CH3), 29.2 (Dipp-Pri-CH), 124.0, 125.6, 127.2, 128.8, 128.8, 129.1, 132.3, 134.5, 134.6, 135.3, 136.0, 138.6, 138.9, 142.3 and 145.5 (Ar–C). 31P{1H} NMR (C6D6, 81 MHz, 298 K): δ = 7.1 (t, 2JPP = 18.2 Hz, Ph2P–Ni–(PPh3)2), 39.8 (bs, Ph2P–Ni–(PPh3)2). 29Si{1H} NMR (C6D6, 99 MHz, 298 K): δ = −13.5 (d, 2JSiP = 5.5 Hz, SiPh2). MS/LIFDI-HRMS found (calcd) m/z: 985.1705 (985.1755) for [M − PPh3]+.[PhPhDippGe(NH2)]Ni(PPh3)2, 5a
Compound 4a (20 mg, 0.016 mmol) was dissolved in 0.4 mL C6D6 in an NMR tube. An excess of ammonia was added to the NMR tube, which was then closed and shaken leading to an immediate colour change from deep red to bright orange, with concomitant formation of a colourless solid (NH4Cl). The solution was filtered, and volatiles removed in vacuo to yield 5a as an orange powder (15 mg, 0.013 mmol, 83%). Dark orange crystals suitable for X-ray diffraction analysis were obtained by storage of a concentrated toluene solution layered with hexane at −32 °C. 1H NMR (C6D6, 400 MHz, 298 K): δ = 0.50 (d, 6H, 3JHH = 6.6 Hz, Dipp-Pri-CH3), 1.14 (m, 6H, Dipp-Pri-CH3), 2.97 (s, 2H, Ph2P–CH2), 3.14 (s, 2H, Ge–NH2), 3.79 (hept, 2H, 3JHH = 7.3 Hz, Dipp-Pri-CH), 6.67 (m, 3H, Ar–CH), 6.81 (m, 6H, Ar–CH), 7.00 (m, 31H, Ar–CH), 7.32 (d, 4H, 3JHH = 6.6 Hz, Ar–CH), 7.48 (s, 9H, Ar–CH). 13C{1H} NMR (C6D6, 101 MHz, 298 K): δ = 20.1 (Ph2P–CH2), 23.6 and 26.1 (Dipp-Pri-CH3), 28.8 (Dipp-Pri-CH), 124.4, 125.2, 127.1, 131.6, 132.3, 134.3, 134.5, 136.1, 136.3, 140.2, 141.1 and 146.3 (Ar–C). 31P{1H} NMR (C6D6, 81 MHz, 298 K): δ = 9.3 (t, 2JPP = 15.6 Hz, Ph2P–Ni–(PPh3)2), 39.8 (d, 2JPP = 14.2 Hz, Ph2P–Ni–(PPh3)2). 29Si{1H} NMR (C6D6, 99 MHz, 298 K): δ = −14.8 (d, 2JSiP = 3.2 Hz, SiPh2); IR, ν/cm−1 (ATR): 3354 and 3461 (br, w, Ge–NH2); MS/LIFDI-HRMS found (calcd) m/z: 966.2156 (966.2253) for [M − PPh3]+.[PhPhDippGe(OH)]Ni(PPh3)2, 6a
‘Wet’ cyclohexylaniline (100 mg, 1.00 mmol) was added to a stirring solution of 4a (200 mg, 0.16 mmol) in 5 mL toluene leading to an immediate colour change from deep red to light red-orange. The mixture was stirred for 30 min. All volatiles were then removed in vacuo and the residue extracted with diethyl ether, and filtered. Orange-red crystals of 6a suitable for X-ray diffraction analysis formed over the course of two hours at ambient temperature (133 mg, 0.11 mmol, 68%). 1H NMR (C6D6, 400 MHz, 298 K): δ = 0.47 (d, 6H, 3JHH = 6.3 Hz, Dipp-Pri-CH3), 1.10 (m, 6H, Dipp-Pri-CH3), 2.99 (s, 2H, Ph2P–CH2), 3.77 (hept, 2H, 3JHH = 6.4 Hz, Dipp-Pri-CH), 4.15 (s, 1H, Ge–OH), 6.67 (m, 3H, Ar–CH), 6.83 (m, 6H, Ar–CH), 7.01 (m, 30H, Ar–CH), 7.34 (d, 4H, 3JHH = 7.0 Hz, Ar–CH), 7.54 (s, H, Ar–CH). 13C{1H} NMR (C6D6, 101 MHz, 298 K): δ = 19.4 (Ph2P–CH2), 23.3 and 26.2 (Dipp-Pri-CH3), 28.9 (Dipp-Pri-CH), 124.5, 125.7, 127.2, 128.9, 132.3, 134.5, 134.5, 134.6, 135.7, 135.8, 136.0, 138.8, 139.7, 139.9, 140.0 and 146.6 (Ar–C). 31P{1H} NMR (C6D6, 81 MHz, 298 K): δ = 9.0 (t, 2JPP = 15.8 Hz, Ph2P–Ni–(PPh3)2), 40.6 (d, 2JPP = 15.8 Hz, Ph2P–Ni–(PPh3)2). 29Si{1H} NMR (C6D6, 99 MHz, 298 K): δ = −14.4 (d, 2JSiP = 4.6 Hz, SiPh2); IR, ν/cm−1 (ATR): 3547 (m, Ge–OH); MS/LIFDI-HRMS found (calcd) m/z: 967.2057 (967.2093) for [M − PPh3]+.Author contributions
PMK carried out the vast majority of the chemical synthesis and characterisation. TS carried out all computational work. TJH designed and planned the project, carried out preliminary experimental work, and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TJH gratefully thanks the Fonds der Chemischen Industrie (FCI) for generous funding of this research through a Liebig Stipendium, and the Technical University Munich for the generous endowment of TUM Junior Fellow Funds. TS thanks the University of Alabama and the Office of Information Technology for providing high performance computing resources and support. This computational work was made possible in part by a grant of high-performance computing resources and technical support from the Alabama Supercomputer Authority. We also thank M. Muhr, P. Heiß, and A. Heidecker for the measurement of LIFDI-MS and IR spectra.Notes and references
- E. O. Fischer and A. Maasböl, Angew. Chem., Int. Ed., 1964, 8, 580–581 CrossRef.
- For a broad overview, see the following collections and reviews, and references therein: (a) D. J. Cardin, B. Cetinkaya and M. F. Lappert, Chem. Rev., 1972, 72, 545–574 CrossRef CAS; (b) A. J. Arduengo and G. Bertrand, Chem. Rev., 2009, 109, 3209–3210 CrossRef CAS PubMed; (c) H. G. Raubenheimer, Dalton Trans., 2014, 43, 16959–16973 RSC; (d) F. Ekkehardt Hahn, Chem. Rev., 2018, 118, 9455–9456 CrossRef PubMed.
- (a) R. R. Schrock and A. H. Hoveyda, Angew. Chem., Int. Ed., 2003, 42, 4592–4633 CrossRef CAS PubMed; (b) D. Astruc, Metal–Carbene and –Carbyne Complexes and Multiple Bonds with Transition Metals, in Organometallic Chemistry and Catalysis, Springer, Berlin, Heidelberg, 2007 Search PubMed.
- (a) F. Glorius, N-Heterocyclic Carbenes in Transition Metal Catalysis, Springer, Berlin, Heidelberg, 2007 CrossRef; (b) S. Diez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed; (c) E. Peris, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed.
- (a) R. Waterman, P. G. Hayes and T. D. Tilley, Acc. Chem. Res., 2007, 40, 712–719 CrossRef CAS PubMed; (b) J. Baumgartner and C. Marschner, Rev. Inorg. Chem., 2013, 34, 119–152 Search PubMed; (c) L. Álvarez-Rodríguez, J. A. Cabeza, P. García-Álvarez and D. Polo, Coord. Chem. Rev., 2015, 300, 1–28 CrossRef.
- T. J. Hadlington, M. Driess and C. Jones, Chem. Soc. Rev., 2018, 47, 4176–4197 RSC.
- Y.-P. Zhou and M. Driess, Angew. Chem., Int. Ed., 2019, 58, 3715–3728 CrossRef CAS PubMed.
- (a) L. Álvarez-Rodríguez, J. Brugos, J. A. Cabeza, P. García-Álvarez, E. Pérez-Carreño and D. Polo, Chem. Commun., 2017, 53, 893–896 RSC; (b) L. Álvarez-Rodríguez, J. Brugos, J. A. Cabeza, P. García-Álvarez and E. Pérez-Carreño, Chem.–Eur. J., 2017, 23, 15107–15115 CrossRef PubMed; (c) J. Brugos, J. A. Cabeza, P. García-Álvarez and E. Pérez-Carreño, Organometallics, 2018, 37, 1507–1514 CrossRef CAS; (d) J. A. Cabeza, P. García-Álvarez, C. J. Laglera-Gándara and E. Pérez-Carreño, Chem. Commun., 2020, 56, 14095–14097 RSC.
- A related bis(phosphine) functionalised N-heterocyclic germylene ligand has also been reported by Goicoechea et al., alongside its group 11 halide and Pt0 complexes: S. Bestgen, N. H. Rees and J. M. Goicoechea, Organometallics, 2018, 37, 4147–4155 CrossRef CAS.
- (a) J. A. Cabeza, I. Fernández, J. M. Fernández-Colinas, P. García-Álvarez and C. J. Laglera-Gándara, Chem.–Eur. J., 2019, 25, 12423–12430 CrossRef CAS PubMed; (b) J. A. Cabeza, I. Fernández, P. García-Álvarez and C. J. Laglera-Gándara, Dalton Trans., 2019, 48, 13273–13280 RSC; (c) A. Arauzo, J. A. Cabeza, I. Fernández, P. García-Álvarez, I. García-Rubio and C. J. Laglera-Gándara, Chem.–Eur. J., 2021 DOI:10.1002/chem.202005289.
- (a) K. E. Litz, J. E. Bender IV, J. W. Kampf and M. M. Banaszak Holl, Angew. Chem., Int. Ed. Engl., 1997, 36, 496–498 CrossRef CAS; (b) J. E. Bender IV, A. J. Shusterman, M. M. Banaszak Holl and J. W. Kampf, Organometallics, 1999, 18, 1547–1552 CrossRef; (c) C. Gendy, A. Mansikkamäki, J. Valjus, J. Heidebrecht, P. C.-Y. Hui, G. M. Bernard, H. M. Tuononen, R. E. Wasylishen, V. K. Michaelis and R. Roesler, Angew. Chem., Int. Ed., 2019, 58, 15–158 CrossRef PubMed; (d) T. Watanabe, Y. Kasai and H. Tobita, Chem.–Eur. J., 2019, 25, 13491–13495 CrossRef CAS PubMed; (e) Z. Feng, Y. Jiang, H. Ruan, Y. Zhao, G. Tan, L. Zhang and X. Wang, Dalton Trans., 2019, 48, 14975–14978 RSC; (f) M. Zhong, J. Wei, W.-X. Zhang and Z. Xi, Organometallics, 2021, 40, 310–313 CrossRef CAS.
- (a) H. Grützmacher, Angew. Chem., Int. Ed., 2008, 47, 1814 CrossRef PubMed; (b) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236 CrossRef CAS PubMed; (c) M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC.
- (a) V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270 CrossRef CAS; (b) J. van der Vlugt, Eur. J. Inorg. Chem., 2012, 363 CrossRef CAS; (c) S. Schneider, J. Meiners and B. Askevold, Eur. J. Inorg. Chem., 2012, 412 CrossRef CAS.
- It should be noted that the concept of “Synergistic Catalysis” is perhaps also fitting here, given the generally high reactivity of low-coordinate tetrylenes in their own right. For a review on this concept and its implications, see: U. B. Kim, D. J. Jung, H. J. Jeon, K. Rathwell and S. gi Lee, Chem. Rev., 2020, 120, 13382–13433 CrossRef CAS PubMed.
- (a) A. L. Casalnuovo, J. C. Calabrese and D. Milstein, Inorg. Chem., 1987, 26, 971–973 CrossRef CAS; (b) E. Khaskin, M. A. Iron, L. J. W. Shimon, J. Zhang and D. Milstein, J. Am. Chem. Soc., 2010, 132, 8542–8543 CrossRef CAS PubMed; (c) D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11776–11779 CrossRef CAS PubMed; (d) R. M. Brown, J. Borau Garcia, J. Valjus, C. J. Roberts, H. M. Tuononen, M. Parvez and R. Roesler, Angew. Chem., Int. Ed., 2015, 54, 6274–6277 CrossRef CAS PubMed.
- Procedures modified from those previously reported for related compounds were used. See: R. D. Holmes-Smith, R. D. Osei and S. R. Stobart, J. Chem. Soc., Perkin Trans. 1, 1983, 861–866 RSC.
- The free ligands typically form oils at ambient temperature, but a few single crystals sometimes deposit from decomposed reaction mixtures of the metalled ligands. See ESI† for details..
- J. Schneider, K. M. Krebs, S. Freitag, K. Eichele, H. Schubert and L. Wesemann, Chem.–Eur. J., 2016, 22, 9812–9826 CrossRef CAS PubMed.
- J. Berthe, J. Manuel Garcia, E. Ocando, T. Kato, N. Saffon-Merceron, A. De Cózar, F. P. Cossío and A. Baceiredo, J. Am. Chem. Soc., 2011, 133, 15930–15933 CrossRef CAS PubMed.
- The bromo-germylene, PhiPDippGeBr (3b), can be accessed using an identical method (see ESI† for details)..
- Yields of 50–55% are obtained after recrystallisation..
- As an example, GeBr2·dioxane, used as is GeCl2·dioxane, can be synthesised from GeBr4, which is a high-cost starting material: N. Hayakawa, T. Sugahara, Y. Numata, H. Kawaai, K. Yamatani, S. Nishimura, S. Goda, Y. Suzuki, T. Tanikawa, H. Nakai, D. Hashizume, T. Sasamori, N. Tokitoh and T. Matsuo, Dalton Trans., 2018, 47, 814–822 RSC.
- Note that single crystals of pure 4a reproducibly gave poor X-ray diffraction data, not of a publishable standard. However, 4a co-crystallised with ∼19% of 4-Br gave data of a publishable standard. See ESI† for details..
- C. A. Tolman, W. C. Seidel and D. H. Gerlach, J. Am. Chem. Soc., 1972, 94, 2669–2676 CrossRef CAS.
- Attempts to synthesise pure samples of the bromo-germylene Ni0 complex, 4-Br, are outlined in the ESI.†.
- Despite attempts to recrystallise the corresponding amide complex 5b from a range of solvents, only very fine needles could be isolated which diffracted too poorly to attain meaningful X-ray data..
- As ascertained by 1H and 31P NMR spectroscopic analysis. Due to the extremely high sensitivity of 5a/b towards moisture, the degassing cycles led to the formation of small amounts of hydroxide complexes 6a/b..
- For some recent examples, see: (a) T. J. Hadlington, J. A. B. Abdalla, R. Tirfoin, S. Aldridge and C. Jones, Chem. Commun., 2016, 52, 1717–1720 RSC; (b) A. V. Protchenko, J. I. Bates, L. M. A. Saleh, M. P. Blake, A. D. Schwarz, E. L. Kolychev, A. L. Thompson, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 4555–4564 CrossRef CAS PubMed; (c) D. C. H. Do, A. V. Protchenko, M. Á. Fuentes, J. Hicks, P. Vasko and S. Aldridge, Chem. Commun., 2020, 56, 4684–4687 RSC.
- Z. Benedek and T. Szilvási, Organometallics, 2017, 36, 1591–1600 CrossRef CAS.
- H. Bauer, J. Weismann, D. Saurenz, C. Färber, M. Schär, W. Gidt, I. Schädlich, G. Wolmershäuser, Y. Sun, S. Harder and H. Sitzmann, J. Organomet. Chem., 2016, 809, 63–73 CrossRef CAS.
- A. Boudier, P.-A. R. Breuil, L. Magna, H. Olivier-Bourbigou and P. Braunstein, J. Organomet. Chem., 2012, 718, 31–37 CrossRef CAS.
- J. T. Patton, M. M. Bokota and K. A. Abboud, Organometallics, 2002, 21, 2145–2148 CrossRef CAS.
- N. E. Schore, L. S. Benner and B. E. LaBelle, Inorg. Chem., 1981, 20, 3200–3208 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data for all new compounds, full details of computational studies, summarised crystallography data, and complete CIFs. CCDC 2057295–2057309. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00450f |
| This journal is © The Royal Society of Chemistry 2021 |