
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient metal-free organic room temperature phosphors
Aakash D.
Nidhankar†
ab,
Goudappagouda†
ab,
Vivek C.
Wakchaure
 ab and
Sukumaran Santhosh
Babu
*ab
ab and
Sukumaran Santhosh
Babu
*ab
aOrganic Chemistry Division, National Chemical Laboratory (CSIR-NCL), Dr Homi Bhabha Road, Pune-411008, India. E-mail: sb.sukumaran@ncl.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
First published on 4th March 2021
Abstract
An innovative transformation of organic luminescent materials in recent years has realised the exciting research area of ultralong room-temperature phosphorescence. Here the credit for the advancements goes to the rational design of new organic phosphors. The continuous effort in the area has yielded wide varieties of metal-free organic systems capable of extending the lifetime to several seconds under ambient conditions with high quantum yield and attractive afterglow properties. The various strategies adopted in the past decade to manipulate the fate of triplet excitons suggest a bright future for this class of materials. To analyze the underlying processes in detail, we have chosen high performing organic triplet emitters that utilized the best possible ways to achieve a lifetime above one second along with impressive quantum yield and afterglow properties. Such a case study describing different classes of metal-free organic phosphors and strategies adopted for the efficient management of triplet excitons will stimulate the development of better candidates for futuristic applications. This Perspective discusses the phosphorescence features of single- and multi-component crystalline assemblies, host–guest assemblies, polymers, and polymer-based systems under various classes of molecules. The various applications of the organic phosphors, along with future perspectives, are also highlighted.
 Aakash D. Nidhankar | Aakash D. Nidhankar obtained his M.Sc. (Chemistry) from Yeshwant Mahavidyalaya, SRTM University, Nanded, India in 2015. He is pursuing his Ph.D. in Chemical Science at the National Chemical Laboratory (CSIR-NCL), Pune, India, under the supervision of Dr Santhosh B. Sukumaran. His research interests are mainly focused on functional organic materials with ultralong phosphorescence. |
 Goudappagouda | Dr Goudappagouda obtained his M.Sc. (Chemistry) from Sahyadri Science College, Kuvempu University, Karnataka, India in 2012. Later, he received his Ph.D. in Chemical Science under the guidance of Dr Santhosh Babu Sukumaran from CSIR-National Chemical Laboratory (CSIR-NCL), Pune, India, in 2020. His research work mainly focused on the design and synthesis of donor–acceptor small organic molecules for lighting and light-harvesting applications. Besides, he worked on charge carrier mobility and conductivity of donor–acceptor co-assemblies for electronic applications. |
 Vivek C. Wakchaure | Vivek C. Wakchaure completed his M.Sc. (Organic Chemistry) in 2013 from Savitribai Phule Pune University, Pune, India. In 2015, he started his doctoral study under the supervision of Dr Santhosh Babu Sukumaran at the National Chemical Laboratory (CSIR-NCL) Pune, India. His thesis mainly focuses on strategies to develop processable 2D-polymers using organic solvent-free functional liquids. The thesis describes the synthesis of small organic molecules and two-dimensional polymers for various applications such as sensing, tunable emission, and electrochemical charge storage. |
 Sukumaran Santhosh Babu | Dr Sukumaran Santhosh Babu obtained his Ph.D. on oligo(p-phenylenevinylene) self-assembly from the National Institute for Interdisciplinary Science and Technology (CSIR-NIIST), Trivandrum, India. After several years of postdoctoral studies at the Max Planck Institute of Colloids and Interfaces, Germany, National Institute for Materials Science (NIMS), Japan, and the University of Namur, Belgium, he joined the Organic Chemistry Division of the National Chemical Laboratory (CSIR-NCL) in 2014 as a Senior Scientist. His research interests include self-assembly of luminescent organic materials, phosphorescence, and 2D-polymers for catalytic and energy applications. |
1. Introduction
One of the areas where organic molecules excelled in the recent past is room temperature phosphorescence (RTP).1–30 RTP organic molecules have attracted scientific interest due to the large Stokes shift, long lifetime, and strong afterglow that enable applications in bio-imaging, organic optoelectronics, anti-counterfeiting, sensing, etc.24,25,33,34 For the past many decades, heavy metal complexes were explored as phosphors because of the presence of ligand–metal and metal–ligand charge transfer (CT) and strong spin–orbit coupling, which improves the intersystem crossing (ISC) and controls the triplet decay rate.27 Despite many advantages, organometallic complexes have the drawbacks of high toxicity, limited resources, and high cost. Hence this situation promoted the search for metal-free organic molecules as alternative phosphors. However, for organic materials, the nonradiative (knr) and quenching (kq) rates of the triplet states are much larger than the radiative decay rate (kp) under ambient conditions. Moreover, the triplet states are vulnerable to quenching by molecular oxygen, and hence RTP from metal-free organic molecules under ambient conditions continues to be challenging.1 Nonetheless, the recent past has witnessed a massive jump in the exploration of metal-free organic phosphors through innovative molecular designs and control over the radiative and nonradiative decay processes associated with triplet excitons. The critical parameters that are being considered while designing organic RTP (ORTP) molecules include (1) populating the triplet state by efficient singlet-to-triplet ISC, (2) minimizing the nonradiative relaxation pathways, and (3) delaying the radiative decay. In accordance with that, heteroatoms such as N, O, S, Te, and halogens are incorporated as an integral part of the molecular design of organic phosphors.4–27 Besides, nonradiative relaxation pathways are controlled by molecular packing in the crystalline state and with the help of supportive media such as polymers and cavitands. The compact molecular packing with the assistance of various intermolecular interactions provided extra stabilization of the triplet excitons to extend the lifetimes beyond seconds. In this way, a synergistic effect of all the supportive features resulted in enhanced RTP of metal-free organic phosphors.5–9 Besides, stabilization of the triplet excitons is found to be a conceptually new and exciting strategy to achieve newer heights for RTPs (Fig. 1).20 Many new concepts such as suppression of the nonradiative deactivation pathways of the triplet state through hydrogen bonding, H-aggregation, helical arrays, and excited-state manipulations such as CT, energy transfer (ET), radical ion pair formation, energy migration, etc. have been introduced to stabilize the triplet state.20,21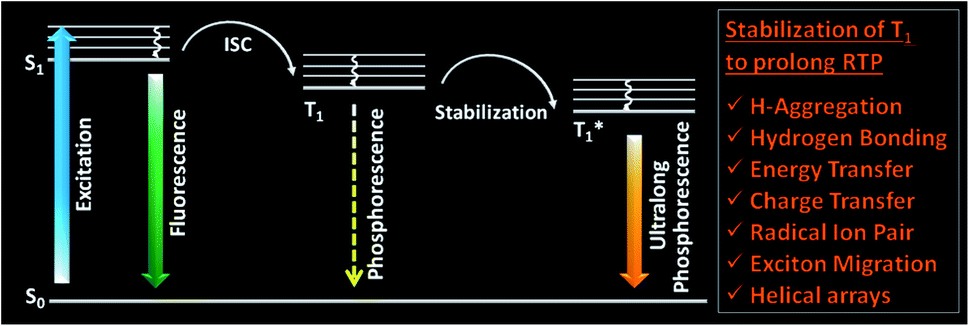 | ||
| Fig. 1 Schematic of stabilization of the triplet state, leading to ultralong phosphorescence in organic molecules. | ||
Until now, many attempts to understand the relationship between ultralong RTP (URTP) and molecular packing have been reported.8–10 Those studies uncovered the vital role of ordered molecular arrays in tailoring lifetime,14–16 emission efficiency,18 luminescent colour,19 and even realizing the unique dynamic URTP features. Even though the structure–property correlation of organic phosphors has been achieved to a certain extent, the involvement of complex factors limits a complete understanding of the underlying mechanism of URTP. Hence a deeper understanding of the supportive role of adequate molecular packing, rigidification by polymers and hosts, and related controlling factors is of great significance. Even though URTP is a fascinating concept, a slow radiative decay leading to a long-lived lifetime is not supportive for any device applications, especially in light-emitting diodes (LEDs).31,32 However, compared to short-lived fluorescent materials, the advantages of an ultralong lifetime and large Stokes shifts make URTP materials promising candidates for applications such as bioimaging, information storage, data encryption, anti-counterfeiting, sensing, and photodynamic therapy.24,25,33,34 Besides, the most critical aspect of URTP is that the triplet state excitons are susceptible to deactivation by molecular motions, oxygen, and humidity.1,3,35 Hence it has been extremely challenging to prolong the phosphorescent emission of organic materials at room temperature by overcoming all these hurdles. In this context, URTP of small molecule-based metal-free ORTPs has prime importance as a fundamental challenge. The numerous design strategies and self-assembly methods have been found successful in achieving an ultralong lifetime. Here, we firmly believe that a case study describing the different classes of organic phosphors and how to manage the triplet excitons to prolong the lifetime will inspire us to design better candidates for futuristic applications.
Recently, many reviews have appeared on RTP as an update of the field.20–25,29,30 However, the present study will mainly focus on the strategies adopted to achieve high performing metal-free organic triplet emitters, including phosphors having lifetimes of more than one second, high quantum yields, and attractive afterglow properties. More importantly, we have provided a comparison of the influence of radiative and nonradiative decay rates on the lifetime and quantum yield of each category of phosphors. Besides, the applications of ORTPs, along with future perspectives, are highlighted at the end.
2. Ways to improve RTP lifetime and quantum yield
The main challenges in metal-free ORTPs include weak spin–orbit coupling (SOC) (<0.1 cm−1) resulting in inefficient ISC, enhanced knr due to many deactivation pathways, and the susceptibility of the T1 state to oxygen and temperature.1,3,35 Hence, to design excellent ORTPs and estimate the performance, the following discussion can be helpful. The important parameters, i.e. quantum efficiency of ISC (ϕisc) (from S1 to T1), quantum yield (ϕp), lifetime (τp), and radiative decay (kp), of phosphorescence can be defined as| ϕisc = kisc/(kf + kic + kisc) | (1) |
| ϕp = ϕisckpτp | (2) |
| τp = 1/kp + knr | (3) |
| kp = (64π4/3h4c2)ΔE3T1→S0∣μT1→S0∣2 | (4) |
2.1. Rate of intersystem crossing kisc
There have been several attempts to obtain an enhanced kisc necessary for efficient RTP, such as effective SOC and small ΔEST.36–38 In this direction, the major directive for a high kisc through effective SOC has come from El-Sayed's rule, which states that effective orbital overlapping is possible in a singlet to triplet transition with different molecular–orbital configurations.39 In other words, compared to the transition from 1(n,π*) to 3(n,π*) or from 1(π,π*) to 3(π,π*), effective ISC can be observed in transitions from 1(n,π*) to 3(π,π*) or from 1(π,π*) to 3(n,π*) states. Many recent successful designs showed that the presence of n orbitals perpendicular to π orbitals is beneficial to facilitate a strong SOC and thereby promote the ISC from singlet to triplet excited states.12,14 Hence the presence of hybrid (n,π*) and (π,π*) configurations is supportive for URTP. An enhanced kisc through SOC significantly contributed to achieve high ϕp. However, a long τp heavily depends on the stabilization of triplet excitons through multiple pathways.2.2. Energy gap ΔEST
One of the reasons for the inefficient RTP in organic molecules is the large ΔEST due to inappropriate molecular designs. It has been noticed that kisc can be significantly promoted by small ΔEST, which depends on the spatial overlap of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) wavefunctions. A strong CT interaction between the donor and acceptor building units of a phosphor can be influential because CT interaction can control the spatial overlap. A CT state can be introduced either by bridging donor–acceptor (D–A) groups or through the assistance of intermolecular interactions between donor and acceptor molecules. Such examples for enhanced RTP through small ΔEST driven by the CT state38 and through aggregation controlled management of ΔEST (ref. 40) are available in the literature. The formation of various aggregates can dictate ΔEST values and thereby influence ISC.2.3. Rate of nonradiative decay knr
The current era of organic phosphors and the sudden developments are attributed to crystallization to a greater extent. The crystallization of phosphors using intermolecular interactions such as π–π stacking, van der Waals, halogen, and hydrogen bonding suppresses the nonradiative decay pathways to facilitate RTP.41 Besides, host–guest assemblies, immobilization of phosphors in frameworks, and polymers also protected the phosphors from nonradiative decay. In this case, the most celebrated “H-aggregation” was found to be effective in stabilizing the triplet excitons to achieve long-lived RTP.42 As per eqn (2) and (3), a variation in knr also significantly alters the values of both ϕp and τp. Hence the design strategies should consider the incorporation of corresponding functional moieties to impart various noncovalent interactions to regulate knr. Another vital strategy adopted to reduce knr by decreasing reorganization energy is deuteration, which blocks the molecular vibrations.43 The high ΔET1→S0 value was also found to be effective in suppressing the nonradiative decay.2,3,392.4. Rate of radiative decay kp
Eqn (4) shows that kp is complicated and mainly determined by many factors involving both the singlet and triplet states, such as SOC, transition dipole moment μT1→S0, and energy gap ΔET1→S0. A high ΔET1→S0 accelerates kp to facilitate RTP with a reduced lifetime and it is the same with the transition dipole moment as well.2,3,39 It has been found that in ORTPs showing excellent features, an exceptional stabilization of the triplet excitons through various supports delays the radiative decay. In such systems, kp is suppressed to achieve a long lifetime over seconds. A slow kp, in turn, enhances τp to realize URTP, while it inversely affects ϕp.2.5. Rate of quenching kq
Along with other factors controlling the efficiency of RTP, the suppression of kq is also equally important. Even though recent studies have employed crystalline assemblies and the support of polymers, hosts, and porous structures to avoid the effect of moisture and oxygen, the quenching rate of the triplet excitons is found to be crucial to deliver excellent RTP features under ambient conditions. Hence the deactivation patterns of the triplet excitons require more attention. The current studies on ORTPs lack a deeper understanding of the effect of kq, assuming that stabilization through self-assembly or other supports will control kq.202.6. Triplet exciton diffusion
Recent studies have shown the importance of triplet exciton diffusion length on persistent RTP.44 It has been suggested that short-range triplet exciton diffusion can suppress kq by stopping the excitons from reaching the trap sites. The importance of exciton diffusion length is more dominant in the case of crystalline arrays with more traps. However, a very recent demonstration indicated that the triplet exciton diffusion with the assistance of helical arrays of phosphor played an essential role in delaying the triplet radiative decay to extend lifetime beyond 4 s.45 Hence the quality of the generated crystals or thin films and management of triplet excitons can impart excellent RTP features. More studies in this direction are required for detailed understanding and further development. In addition, hyperfine-coupling (HFC) driven intersystem crossing in CT complexes and radical ion pairs (RIPs),46 singlet fission,47etc. were also found to be useful in controlling the RTP features.The above sections describe the possible ways for organic phosphors to achieve enhanced RTP features. The implemented molecular designs have been found to be successful to a certain extent and thus delivered some exceptional RTP candidates exhibiting a lifetime longer than one second. In this Perspective, we attempt to reveal the special effects in molecular design and ground state arrangements of metal-free organic phosphors to exhibit prodigious RTP qualities. The recent progress in the lifetime, quantum yield, and afterglow properties of these unusual candidates promoted metal-free ORTPs as a capable material in many functional applications. We want to mention that the dedicated efforts of various research groups worldwide made significant contributions in this area to gain an admirable position for ORTPs in current research.1–30
3. Efficient organic phosphors
RTP of metal-free organic molecules fascinated the scientific community as early as the 1940s, and the exciting demonstrations in the early stages have been summarised in many reviews.39,48–50 The initial experiments were limited in solutions, that too under cryogenic and oxygen-free conditions.51–54 Later, the developments were focused on RTP of salts of aromatic carboxylic acids, phenols, amines, and sulfonic acids in rigid media including boric acid glass,52–54 and filter paper, silica or alumina,55–59 micelles,60 and hosts such as cyclodextrins,61 zeolites,62 hemicarcerand,63etc. The rigid matrices prepared using glucose, sucrose, citric acid, and tartaric acid have supported the RTP of organic molecules.51,64 Similarly, plastics such as poly(methylmethacrylate) and poly(vinyl alcohol) also provided a rigid medium for organic phosphors to excel.65–67 The subsequent investigations on ORTP over a few decades emerged the area and found it useful for many potential applications.68,69 In between, RTP of crystals was reported in 1939 by Clapp and coworkers.70 Crystals of tetraphenylsilane and tetraphenylmethane (Scheme 1) showed RTP afterglow emission up to 23 s visible to the bare eye. Later, in 1978, Bilen and coworkers studied the afterglow of carbazole, dibenzothiophene, dibenzofuran, triphenylene (Scheme 1), etc., having a lifetime up to 4.85 s in the crystal state.71 After a long gap, the technological improvement in materials chemistry during the last decade resulted in a drastic development in RTP of metal-free organic molecules.20–25,29,30 Many new molecular designs and self-assembly models have been experimented with and eventually URTP was realized. The following sections detail the recent advancements of ORTPs.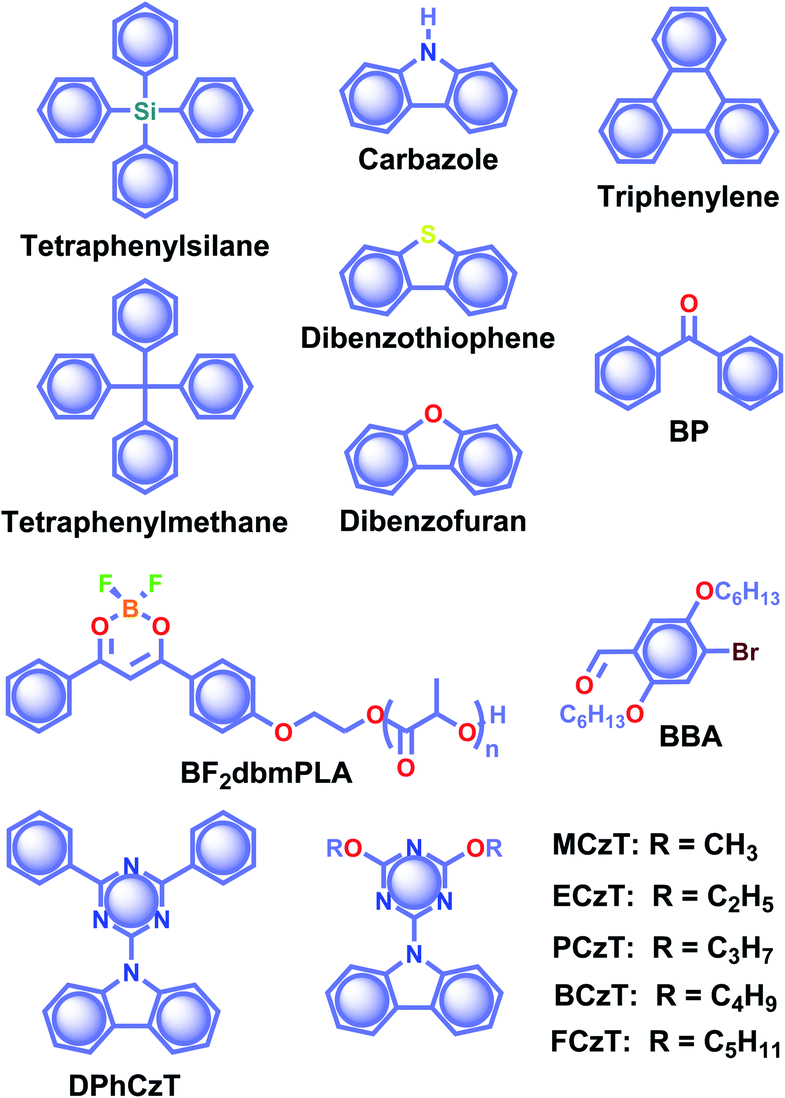 | ||
| Scheme 1 Chemical structure of phosphors tetraphenylsilane, tetraphenylmethane, carbazole, dibenzothiophene, dibenzofuran, triphenylene, BP, BF2dbmPLA, BBA, DPhCzT, MCzT, ECzTPCzT, BCzT, and FCzT. | ||
In 2007, the research group of Fraser came up with boron difluoride dibenzoylmethane (BF2dbm) conjugated poly(lactic acid) BF2dbmPLA (Scheme 1) having oxygen-sensitive RTP.72 Later in 2010, Tang and coworkers reported RTP of a series of benzophenone BP derivatives (Scheme 1) and found that the presence of a carbonyl group or halogen atom, and nonplanar conformation are supportive factors in RTP.73 RTP of 2,5-dihexyloxy-4-bromobenzaldehyde BBA (Scheme 1) was reported by Kim and coworkers in 2011.74 This molecule shows weak fluorescence in solution; however, it exhibits a green phosphorescence emission with a lifetime of 5.4 ms in the crystal state. Single-crystal XRD analysis revealed that RTP is due to the presence of close contact between the bromine and the carbonyl oxygen of the neighbouring molecule (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯Br–C, 2.86 Å). RTP retains a lifetime longer than 100 ms with an afterglow approaching seconds after cessation of excitation. The above three studies greatly attracted the attention towards organic phosphors and hence resulted in a visible change in the area.
O⋯Br–C, 2.86 Å). RTP retains a lifetime longer than 100 ms with an afterglow approaching seconds after cessation of excitation. The above three studies greatly attracted the attention towards organic phosphors and hence resulted in a visible change in the area.
3.1 Single and two-component crystalline organic phosphors
Recently, significant developments have occurred in single-component crystalline assemblies that exhibit long τp along with high ϕp.75 In general, RTP of such single-component assemblies is enhanced by crystallization mainly because of the following reasons: (1) availability of specific intermolecular interaction in the crystal state to improve ISC through SOC; (2) intact molecular packing suppresses the molecular motions and eventually helps to minimize knr of triplet excitons, (3) crystalline assemblies will provide protection from triplet quenching by oxygen. The recent exciting development of URTPs is strongly supported by molecular organization in the crystal state, which stabilizes the excited triplet state by trapping triplet excitons and controls both radiative and nonradiative decays.In 2015, Chen, Liu, Huang, and coworkers synthesized a series of pure organic molecules of carbazole and triazine containing O, N, and P atoms (Scheme 1).42 Notably, the presence of heteroatoms facilitates the spin-forbidden transfer of singlet-to-triplet excited states through n–π* transitions to populate triplet excitons. In this series, 9-(4,6-diphenyl-1,3,5-triazin-2-yl)-9H-carbazole DPhCzT and 9-(4,6-diethoxy-1,3,5-triazin-2-yl)-9H-carbazole ECzT (Scheme 1) exhibited τp up to 1.35 s and 1.05 s, respectively. The enhanced ISC in these molecules is due to the increased number of energy transition channels supported by the H-aggregated dimers of DPhCzT and ECzT in the crystals. The strong coupling via π–π stacking in the H-aggregate dimers with a large transition dipole moment provides stabilization and thus protects the triplet excitons. The stabilized triplet excited state 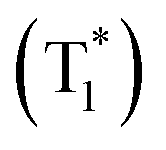 functions as an energy trap at a lower energy level, offering suppressed radiative and nonradiative deactivation decay rates in favour of long-lived phosphorescence. As a continuation, by varying the alkyl chain length on the triazine unit, the same group achieved photoactivation assisted smart URTP materials.76 Among the molecules, the lifetime of BCzT (Scheme 1) drastically increased from 1.8 ms to 1.33 s upon photoirradiation for 10 min. The prolonged irradiation of molecules with UV light suppresses knr by controlling the molecular motion to enhance both RTP emission intensity and lifetime. A significant difference in the distances of intermolecular interactions between adjacent molecules is observed after photoactivation. As the length of the alkoxy chain increased, both the lifetime of photoactivation and deactivation for URTP decreased drastically from MCzT to FCzT. This study points to the importance of photo-irradiation assisted control of nonradiative transition in ORTPs.
functions as an energy trap at a lower energy level, offering suppressed radiative and nonradiative deactivation decay rates in favour of long-lived phosphorescence. As a continuation, by varying the alkyl chain length on the triazine unit, the same group achieved photoactivation assisted smart URTP materials.76 Among the molecules, the lifetime of BCzT (Scheme 1) drastically increased from 1.8 ms to 1.33 s upon photoirradiation for 10 min. The prolonged irradiation of molecules with UV light suppresses knr by controlling the molecular motion to enhance both RTP emission intensity and lifetime. A significant difference in the distances of intermolecular interactions between adjacent molecules is observed after photoactivation. As the length of the alkoxy chain increased, both the lifetime of photoactivation and deactivation for URTP decreased drastically from MCzT to FCzT. This study points to the importance of photo-irradiation assisted control of nonradiative transition in ORTPs.
In 2015, Yuasa and his team explained the nuclear spin magnetism-assisted spin-exchange of a radical ion pair (RIP).46 In this study, the RTP afterglow of benzoic acid derivatives such as isophthalic acid IPA and pyromellitic acid PMA (Scheme 2) was observed for several seconds. Phosphorescence measurements of IPA and PMA revealed the presence of bands at 532 and 533 nm with 0.970 and 1.1 s lifetime, respectively. Photoexcitation of these carboxylic acid derivatives leads to the generation of singlet and further triplet RIPs through hyperfine-coupling (HFC) induced spin conversion, mediated through the magnetic field of neighbouring 1H nuclear spins. Here, deuterium labeling of carboxylic acids suppressed the phosphorescence intensity, confirming the HFC mechanism and a nuclear spin magnetism-assisted spin conversion (1RIP–3RIP) responsible for URTP. In another report, Yan and coworkers reported the phosphorescence lifetime of IPA in the crystal state as 1.11 s and explained that the presence of only one type of hydrogen bonding interaction in IPA stabilizes the carboxylic acid dimers.77
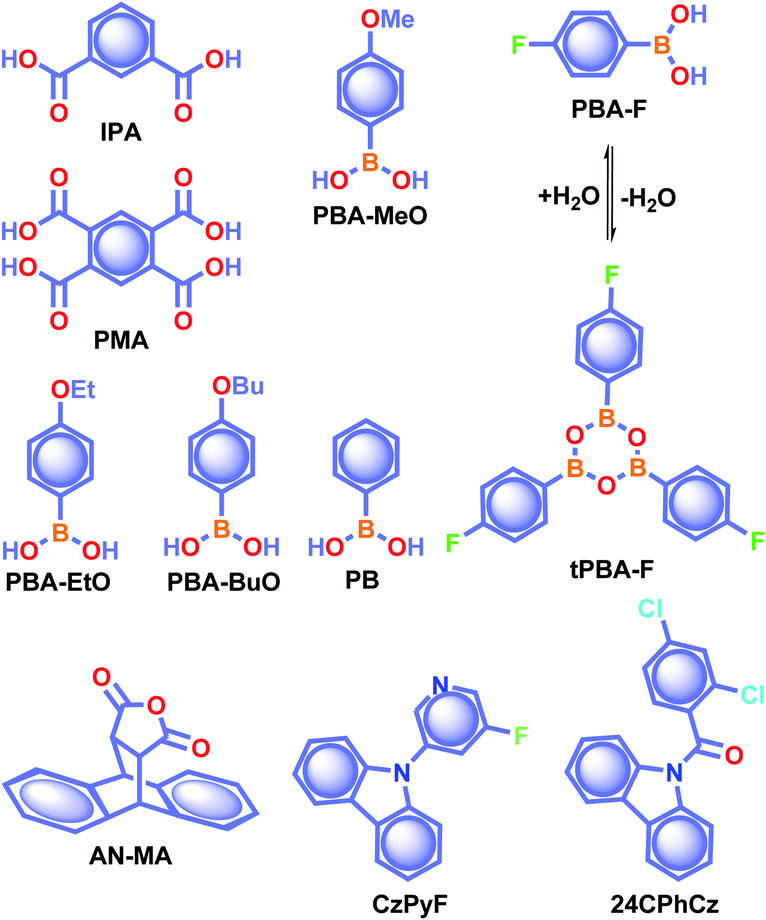 | ||
| Scheme 2 Chemical structure of benzoic acids IPA and PMA, boron-containing phosphors PBA-MeO, PBA-EtO, PBA-BuO, PBA-F, tPBA-F, and PB, and AN-MA, CzPy, and 24CPhCz. | ||
Recently, many aryl boronic acids and esters with stable and extended RTP have been reported. In one of the first reports, Nakai, Fukushima, and coworkers reported long-lived RTP of a series of aryl boronic esters BE-1–5 (Fig. 2a).78BE-1 displayed RTP in the solid-state with a green afterglow that lasted for several seconds (Fig. 2b and c). In this series BE-1–6, the value of τp varied as 1.85, 1.79, 1.65, 1.73, 1.57, and 1.39 s, respectively (Fig. 2c). A combined experimental and theoretical study revealed that an out-of-plane distortion is introduced at the (pinacol) B–Cipso moiety in the 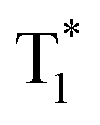 state, and it enables the mixing of π and σ orbitals to enhance SOC and thereby lead to URTP. Later, Li and coworkers studied the RTP of many commercially available phenylboronic acids and their thermally prepared triphenylborazine derivatives.41 The phosphorescence spectrum of 4-methoxyphenyl boronic acid PBA-MeO (Scheme 2) revealed two resolved emission peaks at 457 and 488 nm with τp of 2.24 and 2.19 s, respectively. The long lifetime is due to rigid conformation and strong intermolecular interactions via hydrogen bonds, which decrease knr. An effective π–π stacking stabilizing the triplet excitons also contributes to bright and prolonged RTP. The importance of π–π stacking interactions was further analyzed by theoretical calculations of dimers, indicating that strong π–π stacking decreases ΔEST to favour ISC. The authors studied RTP by varying the length of the alkyl chain and found that PBA-EtO and PBA-BuO also showed RTP with τp of 1.11 s and 1.28 s, respectively. In this series of boronic acids, the support of H-bonds through C–H⋯F interactions enabled PBA-F and the thermally prepared triphenylborazine tPBA-F derivative (Scheme 2) to show a long lifetime of 1.34 and 1.96 s, respectively.
state, and it enables the mixing of π and σ orbitals to enhance SOC and thereby lead to URTP. Later, Li and coworkers studied the RTP of many commercially available phenylboronic acids and their thermally prepared triphenylborazine derivatives.41 The phosphorescence spectrum of 4-methoxyphenyl boronic acid PBA-MeO (Scheme 2) revealed two resolved emission peaks at 457 and 488 nm with τp of 2.24 and 2.19 s, respectively. The long lifetime is due to rigid conformation and strong intermolecular interactions via hydrogen bonds, which decrease knr. An effective π–π stacking stabilizing the triplet excitons also contributes to bright and prolonged RTP. The importance of π–π stacking interactions was further analyzed by theoretical calculations of dimers, indicating that strong π–π stacking decreases ΔEST to favour ISC. The authors studied RTP by varying the length of the alkyl chain and found that PBA-EtO and PBA-BuO also showed RTP with τp of 1.11 s and 1.28 s, respectively. In this series of boronic acids, the support of H-bonds through C–H⋯F interactions enabled PBA-F and the thermally prepared triphenylborazine tPBA-F derivative (Scheme 2) to show a long lifetime of 1.34 and 1.96 s, respectively.
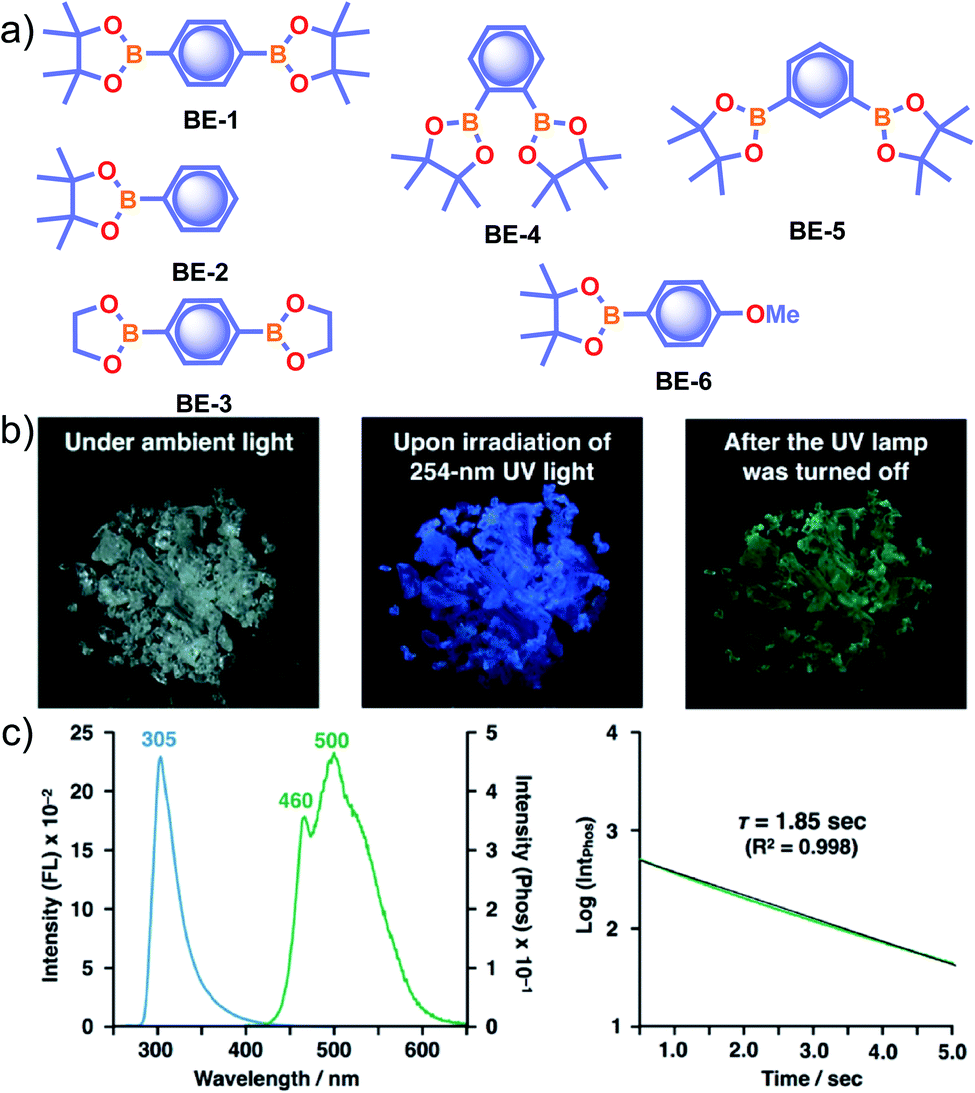 | ||
| Fig. 2 (a) Chemical structure of aryl boronic esters BE-1–6. (b) Photographs of BE-1 under ambient light (left) and irradiation with 254 nm UV light in the dark (middle), and after the UV light was turned off in the dark (right). (c) Fluorescence and phosphorescence spectra (left) and the corresponding phosphorescence decay profile of BE-1 (right). Reproduced with permission from ref. 78. Copyright 2017, American Chemical Society. | ||
In the same year, HFC driven ISC in CT complexes was reported by Yuasa and coworkers using phenylboronic acid derivatives (PBs) such as phenyl-mono-boronic acid PB (Scheme 2) and p-phenylenediboronic acid ethyleneglycol ester BE-3 (Fig. 2a) as examples.79 The phosphorescence intensities of PBs mainly depend on the magnetic-field and spin-isotope effects controlled by HFC. Interestingly, PB showed a RT afterglow for several seconds with τp of 1.2 s. Furthermore, the authors investigated the effect of steric bulkiness on phosphorescence by taking BE-3 as an example, which showed a pale blue afterglow for about 12 s with τp of 1.6 s. The luminescence quantum yield of PB and BE-1 was found to be 18 and 77%, respectively. The phosphorescence mechanism was summarized to follow the transitions S0 → 1CT → 3CT → T1 → S0, indicating the direct involvement of both singlet and triplet CT states. Furthermore, studies on the effect of halogen on RTP of PBs showed that among the series, only 2-(4-fluorophenyl)-1,3,2-dioxaborolane PBA-F exhibited long τp of 1.7 s. In this line, Huang and his group enhanced τp by introducing multiple fluorine atoms on the phenylboronic acid 24FPB (Fig. 3a and b).80 The maximum lifetime of 2.50 s was exhibited by 2,4-difluorophenylboronic acid 24FPB crystals (Fig. 3c). The prolonged lifetime is because of the stabilization of the triplet excited state by H-aggregation and intramolecular O–H⋯F hydrogen bonding (2.22 Å) in the crystal. The hydrogen bonding in crystals fixed the dihedral angle (Θ) between the benzene ring and the boronic acid group, resulting in rotation confinement leading to a longer lifetime. Interestingly, a persistent green luminescence for 24FPB was observed for more than 20 s after the UV light was turned off (Fig. 3d).
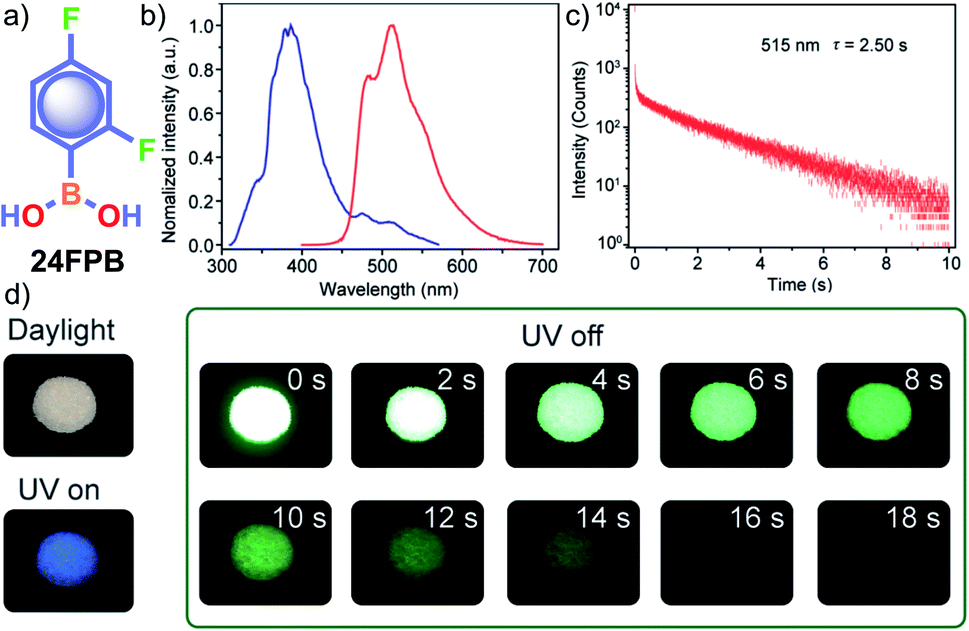 | ||
| Fig. 3 (a) Chemical structure of 24FPB and (b) steady-state photoluminescence (blue line) and phosphorescence (red line) spectra of 24FPB. (c) Phosphorescence decay profile of the emission band at 515 nm. (d) Photographs of the 24FPB crystal taken at different time intervals before and after removing the excitation source (a 365 nm UV lamp). Reproduced with permission from ref. 80. Copyright 2019, WILEY-VCH. | ||
Since intermolecular interaction assisted packing in the crystalline assembly plays a critical role in ORTP, the examples of polymorphs need a special mention.81 Polymorphs offer the opportunity to develop crystals with varying sizes, shapes, and optical properties through tuning the cultivation conditions. It has been noticed that even slight variations in the molecular packing impart significant changes in the excited state properties. In this context, the benefit of polymorphism is the informative correlation between molecular packing and the resulting functional properties. In another way, it encourages rational molecular design to improve the optical properties and hence RTP polymorph is an interesting topic of research.
Yuan and coworkers reported a long lifetime for the cycloaddition product AN-MA of anthracene and maleic anhydride (Scheme 2).82 Out of the two polymorphs form A and form B, afterglow emission of the latter one lasted for several seconds with τp of 1.6 s and ϕp of 8%, because of its much stronger intramolecular π–π interactions. The presence of the carbonyl group and oxygen atoms with lone pairs, together with the effective intra- and intermolecular interactions, helps AN-MA to achieve bright green RTP emission. The relatively low knr value resulting from the more rigid conformations significantly contributed towards the long lifetime. Cai et al. reported that out of the three polymorphs formed by hydrogen-bonded frameworks of benzene-1,3,5-triyltris((9H-carbazol-9-yl)methanone), only one with two different types of tetragonal pore in the crystal packing exhibited URTP of 198 ms. The presence of strong intralayer π–π interactions between carbazole units in the framework stabilized the triplet excitons to achieve URTP.83 Similarly, Yang et al. demonstrated how the different molecular conformations in the polymorphs of 4-(4a,10a-dihydro-10H-phenothiazin-10-yl)benzonitrile control RTP.84 In the series of three polymorphs generated, the one with the more intermolecular interactions has the more extended RTP lifetime of 266 ms. These studies showed that the various modes of packing arising from the different molecular configurations greatly contribute to the incomparable RTP features of the polymorphs. Due to the lower number of reports and nominal performance, RTP polymorphs and the related discussion is restricted in this Perspective.
To understand the effect of a heavy atom to obtain longer RTP, Sasabe, Kido and coworkers studied the RTP features of a series of 3-pyridylcarbazole derivatives with H, F, Cl, Br, and I as substituents on the pyridine ring.85 The crystals of the fluorine-substituted derivative CzPyF (Scheme 2) showed an ultralong τp of 1.1 s and ϕp of 1.2%. Theoretical and experimental data revealed the crucial role of n orbital on the central pyridine ring in enhancing the intersystem crossing between 1CT* and the locally excited triplet (3LE*) states. X-ray crystallographic studies indicated that both the pyridine ring and fluorine atom contributed to the enhancement of RTP through restricted motion owing to weak C–H⋯N and H⋯F hydrogen-bonding interactions. The presence of a halogen atom with larger electronegativity enabled a longer RTP lifetime in this series. Similarly, in 2019, Shi, An, Huang, and coworkers provided a detailed study related to the critical role of molecular stacking in generating triplet excitons by using a series of carbazole derivatives having chlorine substitution at different positions.86 The combined experimental and calculated results revealed that 24CPhCz (Scheme 2) with robust intermolecular coupling between carbazoles exhibited long τp of 1.06 s and ϕp of 2.5%. A detailed crystal structure analysis of 24CPhCz showed that the existence of intermolecular interactions played an essential role in enhancing the lifetime. The molecules were restricted by abundant CCl⋯π, CO⋯Cl, CCl⋯H–C, Cl⋯Cl, and C–H⋯π interactions. The restricted nonradiative transitions through molecular packing in the crystal state prolong the lifetime. However, a weak interchromophoric coupling between carbazoles resulted in weak phosphorescence for the other derivatives in the series.
Tunable emission organic phosphors are rare and are difficult to achieve in a single-component phosphor. A tunable phosphorescence under different excitation wavelengths was reported by Gu et al. utilizing the available multiple emitting centers in a phosphor (Fig. 4a).87 A triazine derivative, 2-chloro-4,6-dimethoxy-1,3,5-triazine DMOT (Fig. 4a and b), contains various heteroatoms that improve the kisc to boost the population of triplet excitons. The planar structure of the molecule strongly supports H-aggregation through multiple intermolecular interactions such as N⋯C, CH⋯C, and π–π interactions with the surrounding six molecules (Fig. 4a). Besides, H-aggregation assisted restricted molecular motion in the crystal ensures excellent phosphorescence features with τp of 2.45 s and ϕp of 31.2% (Fig. 4b–d). Interestingly, upon changing the excitation wavelength from 250 to 400 nm, the emission colour of DMOT was tuned from violet to sky blue, owing to single-molecule and H-aggregate phosphorescence, respectively (Fig. 4c). Such tunable emission smart RTP material will be useful for displays, sensors, and imaging applications.
 | ||
| Fig. 4 (a) Top-view crystal structure of DMOT showing detailed information on the intermolecular interactions. (b) Proposed mechanism and molecular design for excitation-dependent colour-tunable URTP. (c) Excitation-phosphorescence mapping of DMOT. (d) Phosphorescence decay profiles of emission bands at 430 and 470 nm for DMOT. Reproduced with permission from ref. 87. Copyright 2019, Springer Nature Limited. | ||
Similarly, Yuan and coworkers worked on an unexplored pyrimidine molecule, hydantoin (HA) (Scheme 3), with tunable emission colour in response to the excitation wavelength.88 The synergistic effect of through-space conjugation between carbonyls (CO) and nitrogen (N) heteroatoms, and intermolecular interactions through multiple hydrogen-bonds enabled HA to be an excellent RTP emitter with τp of 1.74 s and ϕp up to 21.8%. Crystals of HA displayed sky-blue and yellowish-green afterglows lasting for over 10 s upon excitation with 312 and 365 nm UV lights, respectively. A stable self-assembled network with the adjacent molecules using multiple H-bonds, CO⋯H and OC⋯CO (π–π) interactions at a relatively shorter distance strengthens the assembly and implies extended through-space delocalization. A dimer of HA, 1,1′-methylenedihydantoin MDHA (Scheme 3), also exhibited tunable URTP, but with comparatively lower efficiency having τp of 1.27 and ϕp of 3.6%. The tunable RTP feature is supported by a clustering-triggered emission mechanism, where the presence of different clusters with through-space conjugation and conformation rigidification resulted in tunable RTP.
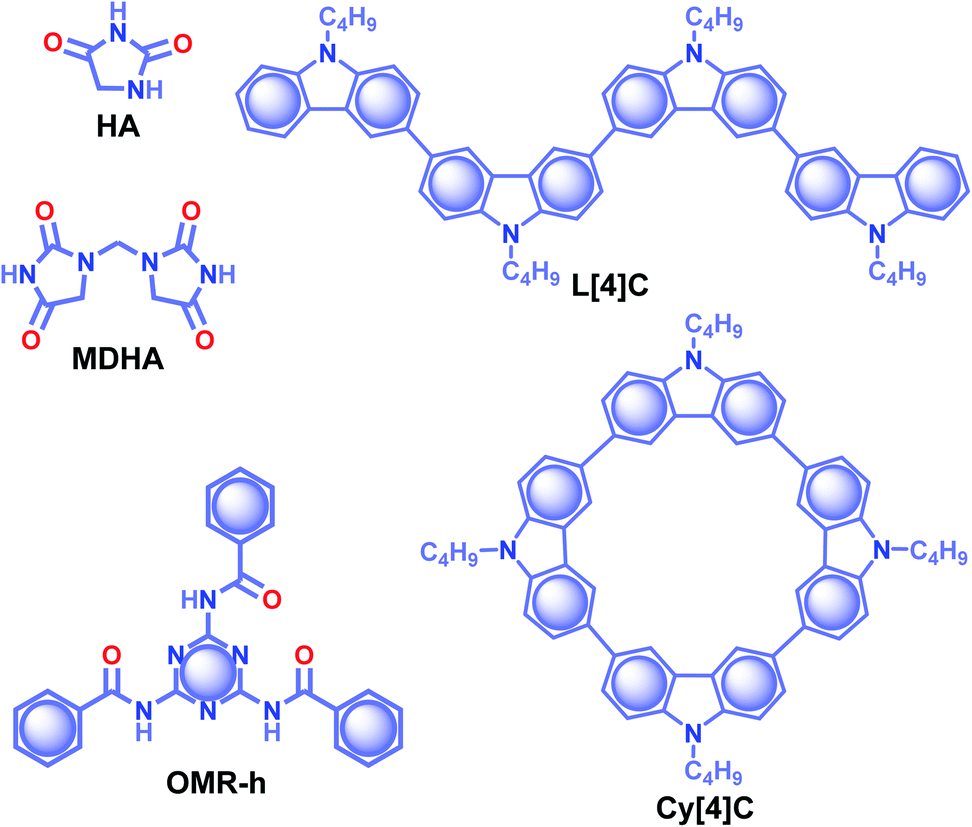 | ||
| Scheme 3 Chemical structure of HA, MDHA, OMR-h, L[4]C and Cy[4]C. | ||
Very recently, Babu and coworkers came up with a new strategy of stabilizing triplet excitons by helical molecular packing (Fig. 5).45 An N-alkylated carbazole decorated with phenylmethanone units PCz (Fig. 5a) exhibited URTP with high efficiency (τp > 4.1 s and ϕp of 11%) (Fig. 5b and c). A helical molecular array of PCz in the crystal state enabled the singlet–triplet states to be mixed up to enhance ISC. A right-handed helical molecular array of PCz acts as a trap and exhibits triplet exciton migration to deliver the exceptionally long lifetime (Fig. 5d and e). An extended molecular array was formed by the arrangement of molecules mainly through π–π interaction (3.34 Å) between carbazole and phenylmethanone units of adjacent molecules. The thus-formed 1D-helical array is stabilized by CH⋯π interaction between the alkyl chain on carbazole and phenylmethanone unit in the adjacent helical columns. Space filled packing rigidifies the molecular conformations and remarkably blocks the nonradiative decay pathways. A combined experimental and theoretical study sheds light on the stabilization of the triplet state by the helical arrays. The micro rods of PCz exhibit triplet exciton migration that prolongs RTP lifetime (Fig. 5f). In contrast to other carbazole based small molecule phosphors, PCz failed to show afterglow emission under ambient conditions.
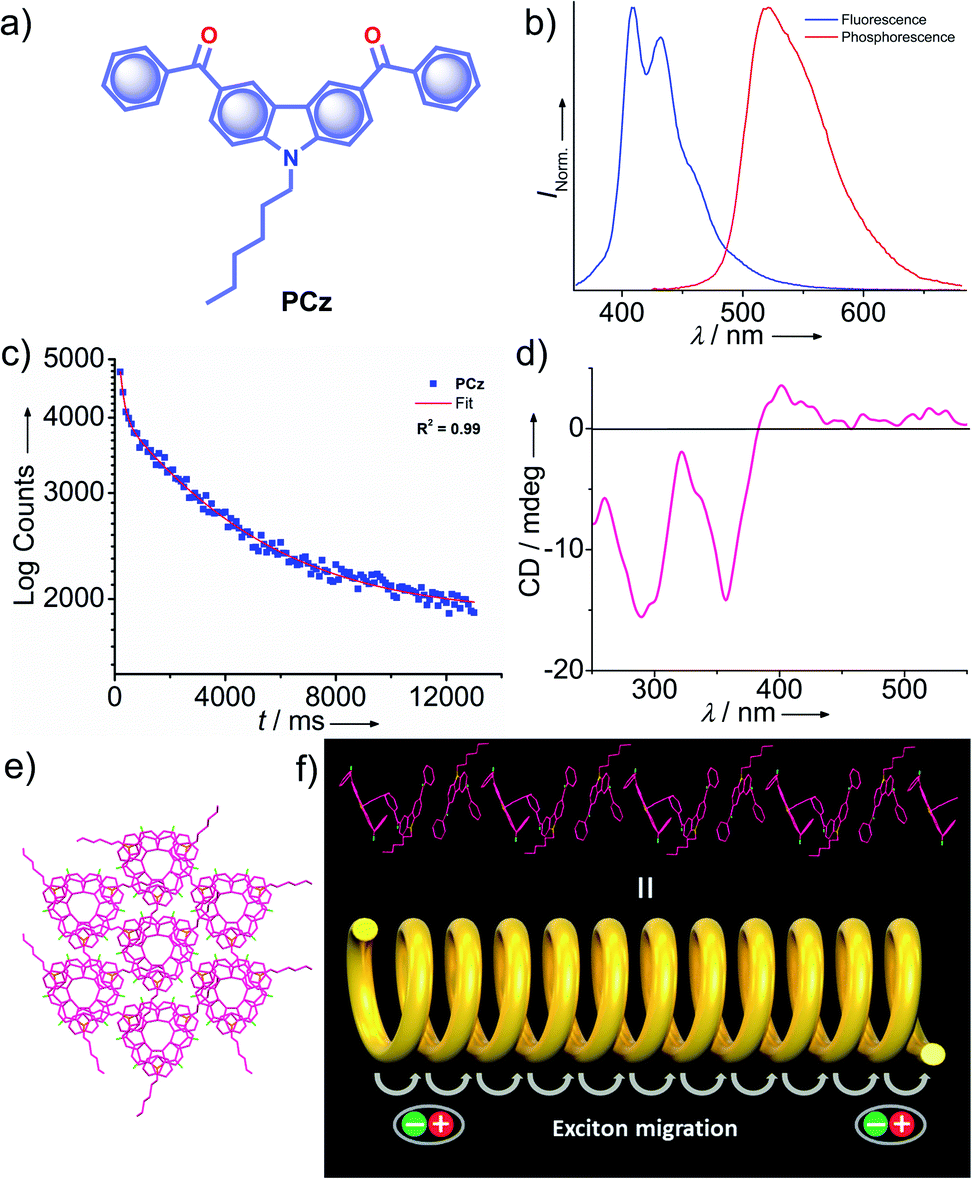 | ||
| Fig. 5 (a) Chemical structure of PCz. (b) Steady-state photoluminescence (blue line) in solution and phosphorescence (red line) spectra in the crystal state of PCz. (c) Phosphorescence decay profile of PCz crystals. (d) The solid-state CD spectrum of PCz crystals. (e) Six adjacent helical arrays of PCz leading to extended columnar packing in the c-axis. (f) Schematic of the helical array of PCz leading to triplet exciton migration. Reproduced with permission from ref. 45. Copyright 2020, WILEY-VCH. | ||
In 2020, Shan and coworkers reported URTP of organic micro-rods of OMR-h, which is synthesized by heating a mixture of melamine and benzoic acid in an aqueous solution (Scheme 3).89 In the presence of water, the micro-rods form a hydrogen-bonded network that rigidifies the molecular motion. A significant enhancement in RTP features with τp of 1.64 s and ϕp of 11.4% was observed under the wet conditions. The observed lifetime of the hydrogen-bonded structure of OMR-h is one of the extended lifetimes of ORTP materials in water. A cyclization-driven enhancement of a less RTP active candidate, N-butyl carbazole (τp of 1.45 ms), is reported by Zhu et al.90 An increase in the conjugation resulted in an efficient ISC for both linear L[4]C and cyclic Cy[4]C derivatives of N-butyl carbazole (Scheme 3) and helped to achieve longer lifetimes of 2.24 and 3.41 s, respectively. The prolonged lifetime is correlated with the significantly lower ΔEST for Cy[4]C with a near-planar structure. Moreover, the synergistic effect of rigidification also contributed to suppress the nonradiative decay. The obtained value is highest among the lifetimes for an organic phosphor without any heavy atom or a carbonyl group.
Similar to single component RTPs, many attempts have been reported to stabilize the triplet state through multiple interactions between two different structural units with complementary recognition parts. This section primarily summarizes the developments in the area of two-component RTP systems. An, Huang and coworkers reported that cocrystals formed by the assembly of melamine ME and IPA resulted in a stable framework via multiple interactions (Fig. 6).91 The two-component assembly ME–IPA exhibited URTP with τp of 1.91 s and ϕp of 24.3% under ambient conditions (Fig. 6c and d). The rigid framework confined the molecules in a three-dimensional network (Fig. 6e) and thus helped to limit knr of the triplet excitons and improved kisc. Similarly, the cocrystals of ME–TPA also presented excellent RTP features with τp of 1.09 s and ϕp of 19.4%. The RTP of the cocrystal is confirmed by the faster kisc (9.3 × 107 s−1) of the ME–IPA framework than that of the individual components ME (1.7 × 106 s−1) and IPA (8.7 × 106 s−1). It has to be noted that the SOC ξ(S1,Tn) of ME–TPA and ME–IPA increased to 16.1 cm−1 and 33.9 cm−1, respectively, compared to the relatively lower values of monomers. If we compare ME–IPA and ME–TPA, the knr and kp varied as 0.13 and 0.18 s−1, and 0.4 and 0.74 s−1, respectively. However, the co-assembly of ME–PA exhibited comparatively less RTP efficiency with τp of 0.68 s and ϕp of 0.82%. The advantages of the two-component phosphor enabled a simultaneous enhancement of τp and ϕp.
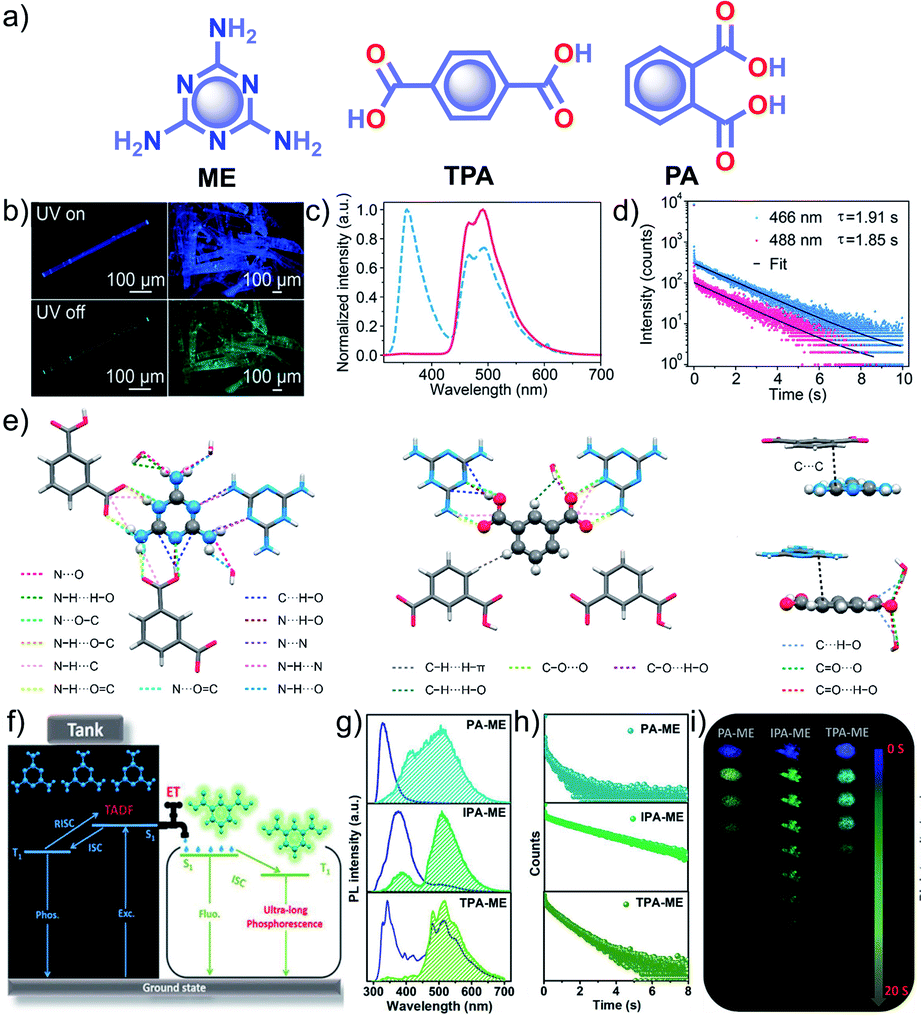 | ||
| Fig. 6 (a) Chemical structure of ME, TPA and PA. (b) Photographs of emission (top) and phosphorescence (bottom) of the ME–IPA cocrystal. (c) Steady-state photoluminescence (blue dotted line) and phosphorescence (red line) spectra along with (d) phosphorescence decay of the emission bands at 466 and 488 nm, respectively, of ME–IPA. (e) Molecular packing of ME–IPA showing intermolecular interactions from the same plane with ME as the center and IPA as the center along with ME and IPA with adjacent planes. (f) Schematic of TADF-assisted transfer from ME to IPA enhancing the RTP lifetime. (g) Prompt and delayed fluorescence spectra and corresponding (h) time-resolved fluorescence decay profiles, and (i) photographs showing the long-afterglow of ME–PA, ME–IPA, and ME–TPA. Reproduced with permission from (a–e) ref. 91 (copyright 2018, American Chemical Society) and (f–h) ref. 77 (copyright 2019, WILEY-VCH). | ||
A detailed mechanistic aspect of this significantly high RTP performance of the acid–amine cocrystals was explained by Yan and coworkers through TADF-assisted Förster resonance ET from the energy donor ME to the phosphor acceptor acids leading to a longer lifetime (Fig. 6f).77 Density functional theory calculations of the D–A assembly showed that the HOMO is located on ME, while the LUMO is on PA/IPA/TPA for ME–PA, ME–IPA, and ME–TPA, respectively. Furthermore, the possibility of ET was supported by the spectral overlap between the absorption of the acceptors (PA, IPA, and TPA) and the emission of ME. The emission peaks at 318, 373, and 342 nm have been shifted to 540, 524, and 554 nm in the delayed spectra with a lifetime of 0.43, 2.00, and 0.77 s, respectively, for ME–PA, ME–IPA, and ME–TPA (Fig. 6g and h). The enhanced RTP lifetime is strictly due to the TADF assisted efficient ET (76%) in the cocrystals. Interestingly, the cocrystals exhibited a strong afterglow lasting for many seconds (Fig. 6i). Two-component assembly promotes triplet state stabilization by additional charge-mediated hydrogen bonding and π–π stacking, resulting in enhanced RTP. A new strategy of multi-component URTP material utilizing both TADF and ET raises hope for further improvement.
The above section pointed out the recent developments in the area of organic crystalline phosphorescent materials. Many single- and multi-component assemblies have been examined to understand the underlying design principles for achieving high quantum yield and extended lifetime up to seconds. However, crystalline RTP materials lack processability and require tedious optimizations for practical applications. This situation demands alternative candidates to troubleshoot the existing barriers in such materials. Hence the organic materials chemists took up this challenge and introduced various new methods to improve RTP features, and the recent developments will be discussed in the following sections.92
3.2 Host–guest based organic phosphors
The effective control of the host and guest units to create a wide variety of soft materials drew much attention to supramolecular chemistry. Recently, researchers have successfully achieved RTP of pure organic host–guest systems by managing various intermolecular interactions.93 The host–guest interactions are highly selective because multiple factors such as size, shape, charge, polarity, etc. limit the host's inclusion. Therefore the selection of appropriate host and guest combinations is very critical. In general, the cavity of the host molecule specifically recognizes the guest molecule and provides a rigid environment to confine the guest molecules. The support is obtained not only from cavitands but other hosts such as small molecules and frameworks, also strongly supported phosphors. Organic host–guest based persistent RTP materials are mainly developed by minimizing knr of triplet excitons and keeping it smaller than the small kp.93 Since the fate of the triplet state is heavily dependent on nonradiative deactivation pathways of the guest and quenching by the diffusional motion of the host as well as molecular oxygen, the selection of a suitable host–guest combination remains challenging. Herein, the research progress of ORTP systems based on host–guest interactions is reviewed.Alfimov and coworkers achieved long-lived RTP from arene-β-cyclodextrin (β-CD) cage–hydrocarbon complexes in the presence of oxygen.94 Among the different combinations, the complex naphthalene-d8-β-CD cage (Scheme 4) with various hydrocarbons showed variable RTP lifetime of 11.9 s for diadamantyl, 9.4 s for diamantine, and 10.3 s for adamantine in the presence of oxygen. However, the RTP lifetime of these complexes further increased in the absence of oxygen. The ternary complexes aggregate in water to form micro-particles, which prevent molecular motions and reduce the quenching effect from oxygen to achieve a longer lifetime. In another attempt, the same group demonstrated URTP from the supramolecular complex of naphthalene-d8-β-CD-cyclohexane (I) with a lifetime of around 16 s while the naphthalene-d8-β-CD-cyclohexane-benzophenone (II) complex showed 14.7 s.95 More interestingly, the long lifetime of complex II is attributed to the triplet–triplet (T–T) ET from benzophenone (donor) to naphthalene-d8 (acceptor).
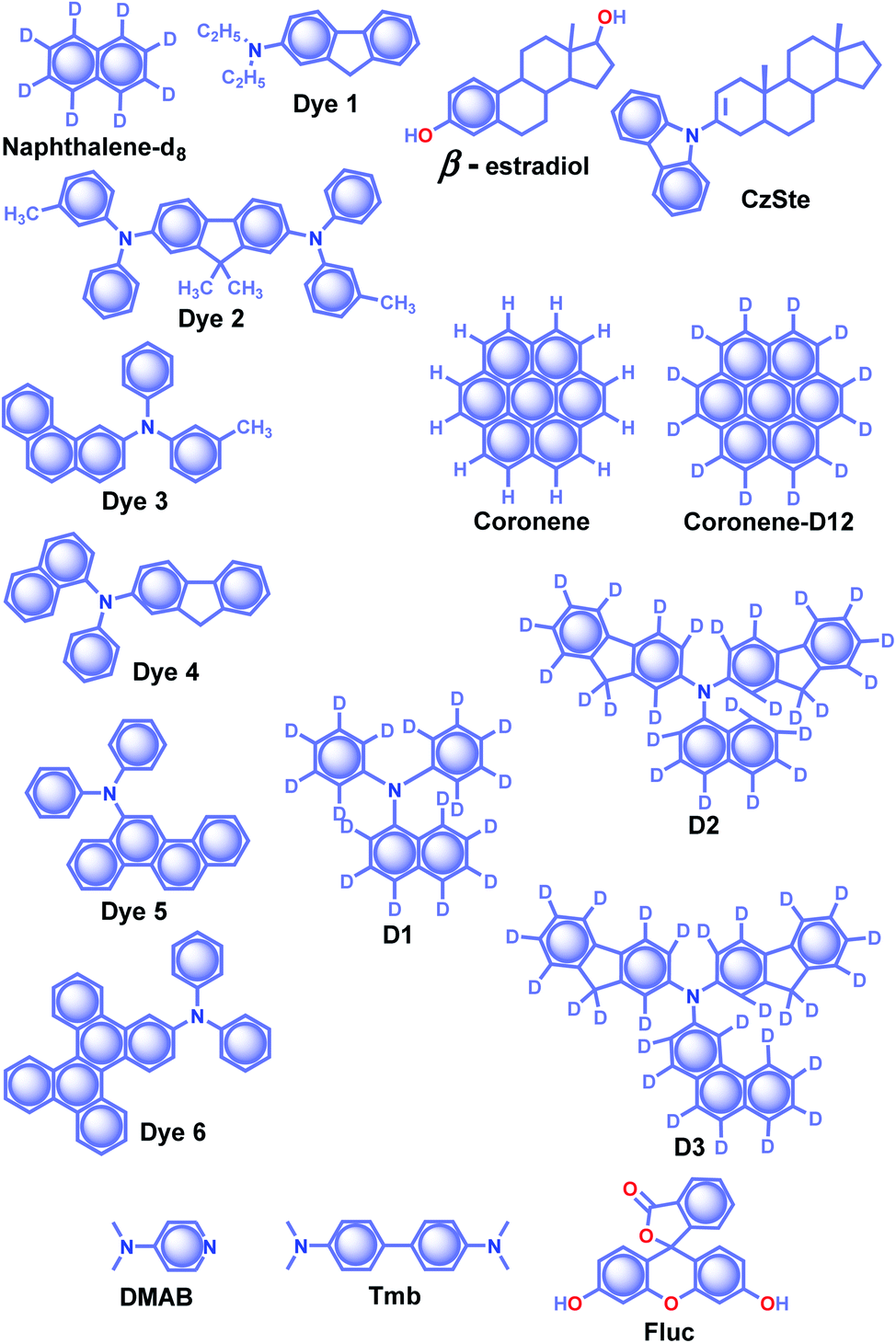 | ||
| Scheme 4 Chemical structure of β-estradiol, CzSte and DMAB (host) and naphthalene-d8, Dyes1–6, coronene, coronene-d12, D1–3, and Fluc (acceptor) molecules. | ||
Another work in the field of host–guest supramolecular systems was reported by Liu and coworkers using cucurbit[6]uril (CB[6]) as the host and a heavy-atom-free phenylmethylpyridinium as the guest (Fig. 7a).96 The PBC/CB[6] complex formed by grinding showed a phosphorescence peak at 510 nm with an ultralong τp of 2.62 s and ϕp of 9.7% (Fig. 7b–d). The encapsulation in CB[6] promotes ISC in phenylmethylpyridinium, which in turn boosts the population of triplet excitons. Moreover, CB[6] provides a rigid matrix for the guest molecule to suppress molecular motions such as vibrations, rotations, and inter-collisions as well as to provide protection from oxygen. Eventually, the successful PBC/CB[6] complex prolonged the phosphorescence lifetime. Notably, the distinct lifetime and robust phosphorescence properties of PBC/CB[6] enabled the triple lifetime-encoding for information encryption and anti-counterfeiting applications.
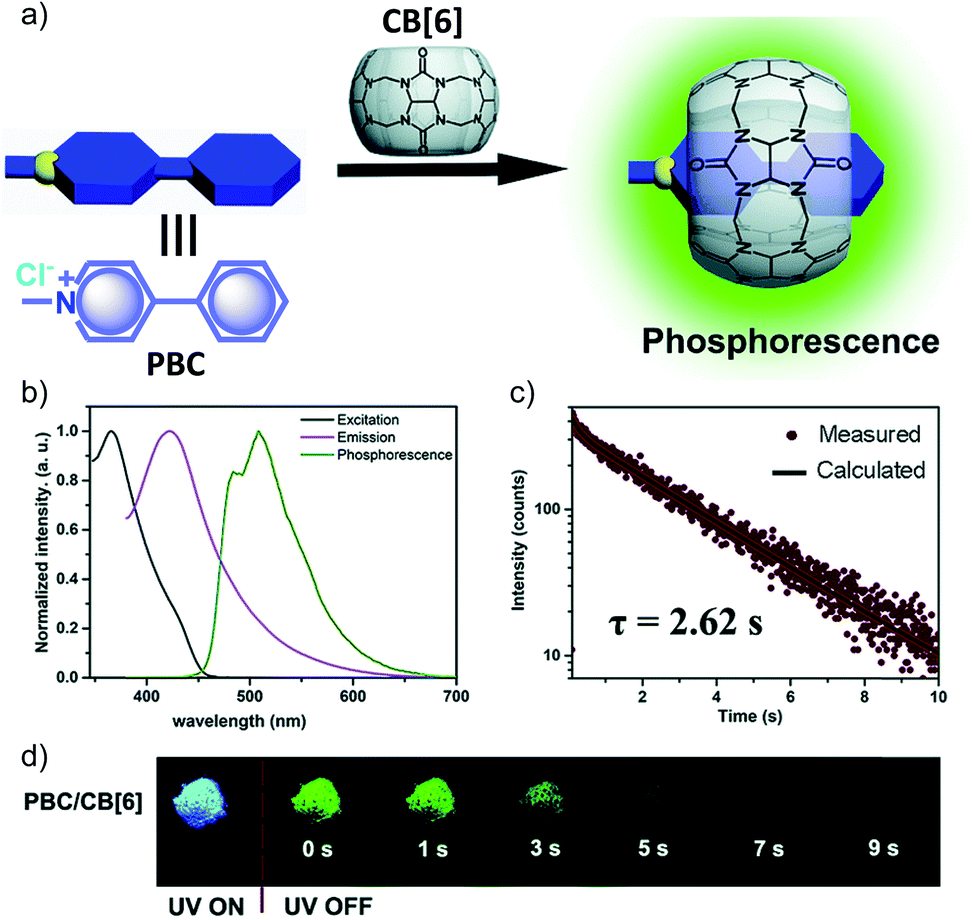 | ||
| Fig. 7 (a) Schematic illustration of the solid-state supramolecular strategy of PBC and CB[6] for URTP. (b) Excitation, emission, and phosphorescence spectra of PBC/CB[6] in the solid state. (c) Phosphorescence decay of PBC/CB[6] at 510 nm. (d) Photographs of PBC/CB[6] powder under 365 nm UV irradiation and at different time intervals after removal of the ultraviolet lamp. Reproduced with permission from ref. 96. Copyright 2019, Royal Society of Chemistry. | ||
The salient features of a steroidal compound, β-estradiol (Scheme 3), such as rigidity, oxygen barrier, high T1 energy, etc. motivated the research group of Adachi to use it as a host material to suppress the triplet quenching of phosphors.97,98 Besides, the use of a deuterated aromatic hydrocarbon as the guest minimized nonradiative deactivation. Red-green-blue persistent RTP with τp > 1 s, ϕp > 10% and a persistent RTP afterglow for several seconds was realized for dyes 1–6 (Scheme 4).97 In 2016, the same group introduced a new host molecule, 3-(N-carbazolyl)-androst-2-ene (CzSte) (Scheme 4), to enhance the performance of afterglow LEDs.98 To maximize the lifetime, the authors used coronene-d12 as the emitter and found a significant improvement in RTP features. The planar structure of coronene-d12 was found to be suitable for forming a rigid host matrix through intermolecular CH–π interactions with the steroid moiety of CzSte. The suppression of molecular vibration and nonradiative decay of the guest emitter resulted in an extended τp of 4.7 s and ϕp of 5.3%. Furthermore, the prepared host–guest system was found to be useful in LEDs yielding higher external quantum efficiency and longer afterglow.
Hirata and coworkers reported a new heavy atom-free organic molecular design consisting of a secondary amine as an RTP antenna substituted with different RTP centers D1–3 (Scheme 4) having smaller T1 energy, exhibiting persistent RTP (Scheme 4).99 The notable feature of the molecular design is the steric hindrance introduced between the RTP antenna and the RTP center that decreases kf and enables efficient ϕisc. The authors cleverly extended the conjugation of the RTP antenna in anticipation of obtaining kp > knr. To validate the design strategy, RTP candidates (0.3 wt%) were dispersed in a β-estradiol host and a persistent emission was observed from 470 to 800 nm. The ϕp and τp of the host–guest systems varied as 11, 50, and 46%, and 1.60, 1.00, and 1.40 s for D1, D2, and D3, respectively.
Recently, a thermoresponsive RTP has been reported by taking advantage of ET and intermolecular CT between N,N-dimethylpyridin-4-amine DMAP (host) and N,N,N′,N′-tetramethylbenzidine Tmb (guest) (Scheme 4).100 The cocrystals of DMAP and Tmb with a mass ratio of 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 displayed blue RTP emission with τp and ϕp up to 2.1 s and 13.4%, respectively. In addition, the authors studied the concentration-dependent emission changes by incorporating an additional energy acceptor, fluorescein Fluc (Scheme 4), to form a ternary blend. Upon changing the concentration of Fluc, a colour-tunable afterglow from blue to yellow was realized. More interestingly, on heating, both DMAP–Tmb and DMAP–Tmb–Fluc exhibited turn-on RTP with increasing RTP lifetime from 1.4 s to 1.97 s. Here the enhanced intermolecular interactions in DMAP–Tmb and DMAP–Tmb–Fluc played a significant role in enhancing the phosphorescence lifetime. Furthermore, the thermoresponsive nature of the host–guest RTP materials has been used for multi-colour thermal printing.
:1 displayed blue RTP emission with τp and ϕp up to 2.1 s and 13.4%, respectively. In addition, the authors studied the concentration-dependent emission changes by incorporating an additional energy acceptor, fluorescein Fluc (Scheme 4), to form a ternary blend. Upon changing the concentration of Fluc, a colour-tunable afterglow from blue to yellow was realized. More interestingly, on heating, both DMAP–Tmb and DMAP–Tmb–Fluc exhibited turn-on RTP with increasing RTP lifetime from 1.4 s to 1.97 s. Here the enhanced intermolecular interactions in DMAP–Tmb and DMAP–Tmb–Fluc played a significant role in enhancing the phosphorescence lifetime. Furthermore, the thermoresponsive nature of the host–guest RTP materials has been used for multi-colour thermal printing.
Metal–organic frameworks (MOFs) are capable of encapsulating guest species in their cavities, and the guest confinement can deliver a significant improvement in RTP. Along these lines, Kabe, Adachi, and coworkers demonstrated a long-lived emission from triplet excitons achieved by encapsulating coronene in a zeolitic imidazolate framework (ZIF-8) host.101 It is confirmed that the coronene wholly isolated within the pores of the MOF suppresses the nonradiative decay and molecular vibrations, enabling long-lived RTP. Coronene@ZIF-8 exhibited τp of 7.4 s, while an extended lifetime of 22.4 s was achieved for coronene-d12@ZIF-8 under the same experimental conditions. The vibrational energy of the C–D stretching mode is lower than that of the C–H stretching mode, and hence it helps coronene-d12@ZIF-8 to show enhanced lifetime. Moreover, the temperature-dependent lifetime measurement confirmed the suppression of nonradiative deactivation of coronene.
In 2017, Yan and coworkers explored a phosphorescence ET by incorporating donor and acceptor guest molecules in the interlayer nanogallery of an inorganic graphene-like layered double hydroxide (LDH) host material (Fig. 8).102 The authors used different benzene dicarboxylic acid isomers, namely IPA (Scheme 2), TPA, and PA (Fig. 6a), as potential donors assembled into the interlayer of the Zn–Al-LDH host by the co-precipitation method. Interestingly, among the nanohybrids, the IPA/LDH showed a green phosphorescence emission with the longest τp up to 1.2 s and ϕp of 3.02% (Fig. 8b–d). An H-type aggregation between IPA dimers and LDH nanosheets stabilized the lowest triplet excited state and minimized the nonradiative decay to prolong the RTP lifetime. Besides, IPA/LDH showed thermoresponsive RTP upon varying the temperature from 295 to 335 K. Subsequently, the co-intercalation of eosin Y as an energy acceptor with the IPA energy donor into the nanogalleries of LDH nanosheets imparted excellent triplet–triplet ET (EP = 99.7%) (Fig. 8e).
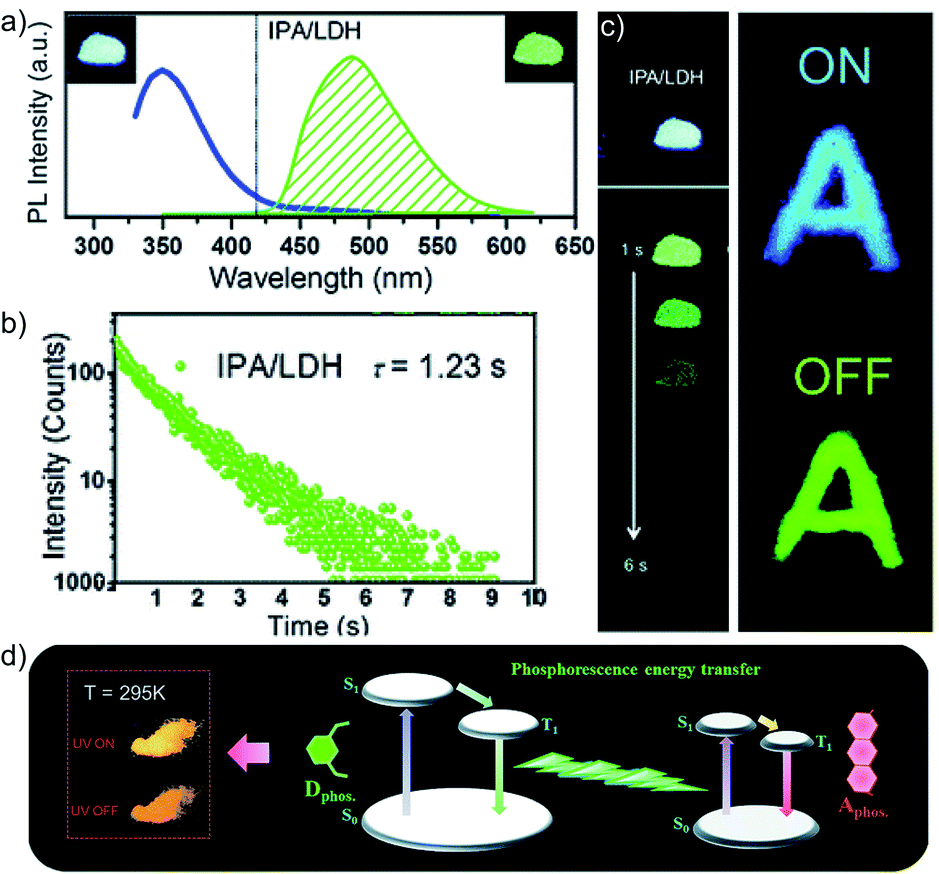 | ||
| Fig. 8 (a) Steady-state emission (left) and phosphorescence (right) spectra of IPA/LDH nanohybrids (insets show the corresponding photographs). (b) Phosphorescence decay profile of IPA/LDH (λmon = 489 nm, λex = 320 nm). (c) The letter ‘A’ made with IPA/LDH can be unmistakably identified by the naked eye after the excitation is switched off. (d) Schematic representation of the proposed mechanism for PET. Reproduced with permission from ref. 102. Copyright 2017, Royal Society of Chemistry. | ||
Co-assembly of RTP inactive terpyridine-derivatives T1 and T2 (Scheme 5) with LAPONITE® (LP) nanoclay through solvent-free mechanical grinding significantly enhanced the RTP to exhibit green (T1@LP) and blue-green (T2@LP) emissions.103 The long RTP lifetime decay component of both the hybrid materials was around 1.1 s with a strong afterglow lasting for more than 10 seconds. The encapsulation of the emitters in LP helped to achieve ϕp of 2.96 and 1.86% for T1@LP and T2@LP, respectively. A transformation from trans–trans to cis–trans configuration of T1 upon protonation has led to a marginal increase in spatial separation of the HOMO and LUMO and eventually narrowed down ΔEST to facilitate ISC. Moreover, the hydrogen bonding between T1 and LP reduces the nonradiative decay and protects the triplet excitons. Zhang and coworkers came up with a hybrid material by encapsulating various RTPs (Aba-o, Npaba-m, Npaba-p, Caba-o, Caba-m, and Caba-p) in [Al(DMSO)6]X3, where X is Cl− or Br− (Scheme 5).104 The new approach resulted in a very high RTP lifetime and luminescence quantum yield. The heavy atom effect has a profound impact in this series: as compared to Cl− hybrids, the analogues with Br− yield high ϕp and a shortened τp. The values of τp and ϕp varied for Aba-o/Cl (1.1 s, 2.3%), Npaba-m/Cl (1.12 s, 3.9%), Npaba-p/Cl (1.26 s, 4.8%), Caba-o/Cl (1.81 s, 4.9%), Caba-m/Cl (1.84 s, 8.4%) and Caba-p/Cl (1.89 s, 2.5%). The presence of many different types of weak interaction between the matrix and RTPs greatly supports RTP by suppressing nonradiative decay.
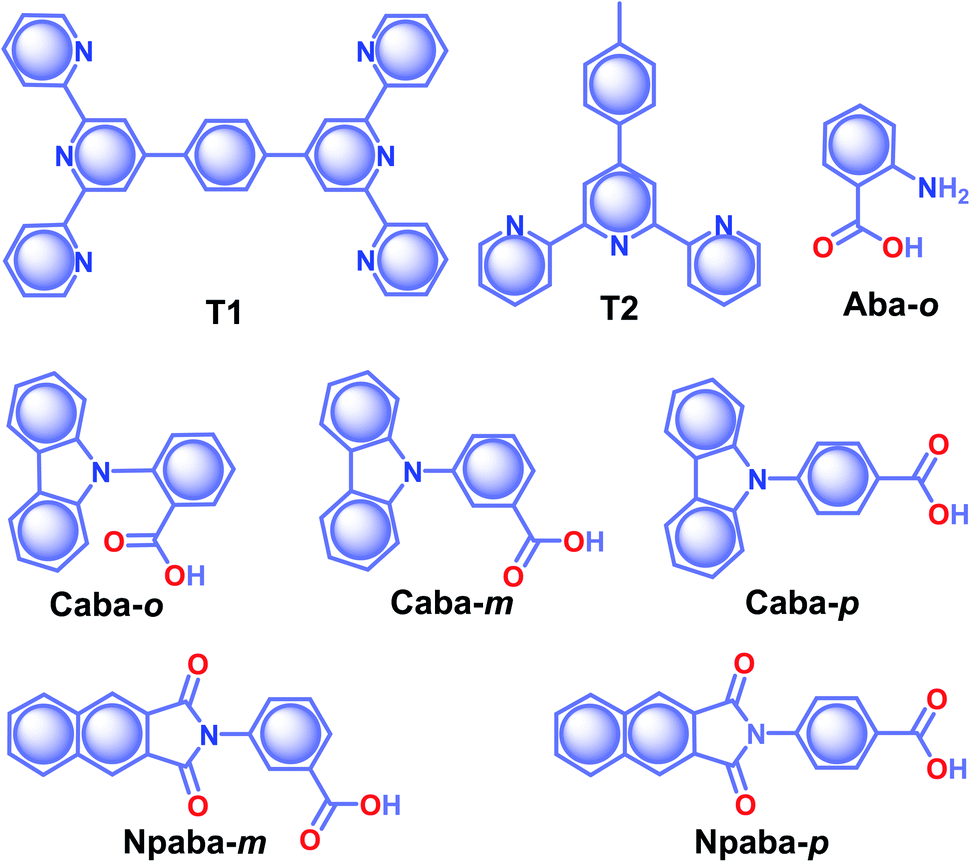 | ||
| Scheme 5 Chemical structure of T1, T2, Aba-o, Caba-o, Caba-m, Caba-p, Npaba-m, and Npaba-p. | ||
3.3 Polymer-based organic phosphors
The crystalline URTP materials have drawbacks in terms of reproducibility, processability, and flexibility, which significantly impedes the development of crystalline URTP materials for practical applications.105 To overcome these fundamental barriers, special attention has been paid to the development of organic polymeric materials capable of URTP. Recently, significant breakthroughs have been achieved by prolonging the lifetime of polymeric materials through homopolymerization, ring-opening polymerization, covalent cross-linking reaction, and radical binary copolymerization, as well as embedding small molecules into a rigid polymer matrix.105 Polymer-based RTP materials have been receiving increased attention because of the large molecular weight of the polymer and the availability of functional group chains that can help to rigidify the molecular vibrational and rotational motions of the phosphors. Moreover, it can reduce the quenching effects of oxygen and moisture from the ambient environment, allowing the triplet excitons to survive long enough to achieve prolonged lifetimes. More importantly, polymer-based RTPs exhibit easy processability, excellent flexibility, and high thermal stability as well.In 2016, Chen et al. applied the intramolecular CT (ICT) state of N-substituted naphthalimide polylactic acid-based polymers to obtain RTP enhancement.40 It has been noticed that either the ICT state or a heavy atom (Cl or Br) can ensure enhanced ISC and thus strong RTP. In this series, polymer 1,2-OPh-OLA (Scheme 6) showed strong ICT and a favourable ΔEST to support RTP. The existence of an ICT state acted as a bridge between the excited singlet and triplet states and thus accelerated ISC leading to τp of 1.12 s. Additionally, N-substituted naphthalimides conjugated with natural biomacromolecules such as chitosan and bovine serum albumin also displayed RTP and found useful application in time-resolved bioimaging.
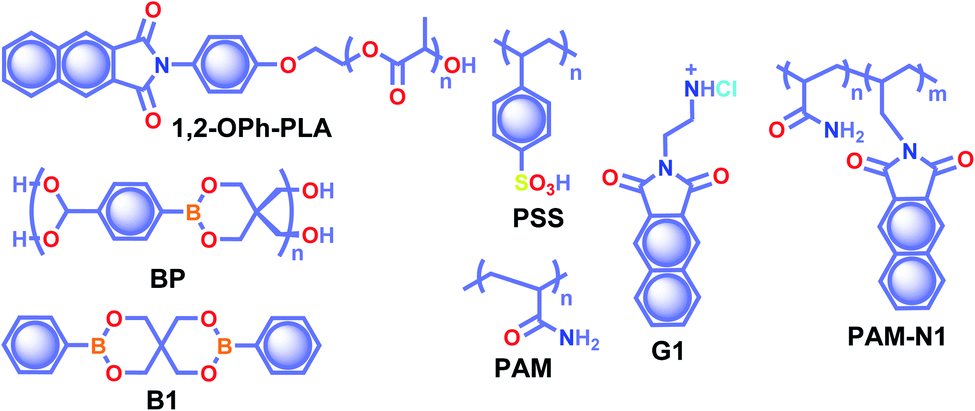 | ||
| Scheme 6 Chemical structure of polymers 1,2-OPh-PLA, BP, PSS, PAM, G1, and PAM-N1, and model derivative B1. | ||
Ogoshi and coworkers reported URTP with a lifetime up to 1.22 s for poly(styrene sulfonic acid) PSS in the dry solid-state (Scheme 6).106 The observed lifetime is one of the most extended RTP lifetimes for non-doped ORTP polymers. The sulfonic acid group in the polymer can form strong inter/intrachain hydrogen bonds in the solid-state that reduce the nonradiative decay and eventually lead to ultralong RTP. The RTP lifetime depends on the introduction ratio of sulfonic acid groups. As the ratio is increased, the phosphorescence lifetime became longer due to strong hydrogen bond formation between sulfonic acid groups. Furthermore, the reversible RTP via uptake and removal of water contributed to the lifetime-encoding application. Detailed studies revealed that deuteration of SO3H and exchanging SO3Na or SO3K for SO3H resulted in an increased RTP lifetime. In contrast, a decrease in RTP lifetime was noticed when PSS was neutralized with NaOH or KOH.
Cai et al. demonstrated that the ionic cross-linking between chromophores is critically supportive in suppressing nonradiative transitions for URTP, and by utilizing the concept, a lifetime of 2.1 s for an amorphous polymer is obtained.107 The replacement of PSS with different ions such as Li+, K+, Rb+, NH4+, Mg2+, Ca2+, Al2+, and Gd2+ imparted a significant effect on the URTP of polymers. The size of the ionic radius is found to control the RTP features, and as the size increases, the URTP lifetime also gradually decreases. The replacement with Li+ and Mg2+ resulted in a lifetime of 1.3 and 1.1 s, respectively, and thereafter a gradual decrease in the lifetime is observed. It has been concluded that even though the large ionic radius prevents the prolonged URTP, the high ion charge state is found to be supportive. Hence a balance between the ionic radius and charge state can significantly alter the lifetime values. Here the knr is at least one order of magnitude higher than kp, indicating that the former one played a dominant role in manipulating the URTP of ionic polymer phosphors. The results of ionic cross-linking assisted URTP have even been extended to nonaromatic ionic polymers and it was found that PAANa (Scheme 6) with blue URTP has τp of 1.4 ms and 2.1 s, respectively, when monitored at 450 nm and 480 nm bands.
Recently, boronic acid/ester-based organic phosphors also excelled as strong RTP candidates. Along these lines, Kubo and coworkers reported boronate particles BP (Scheme 6) as a self-assembled URTP system in both solid state and dispersion in water.108 Solid BP showed phosphorescence peaks located between 450 and 550 nm with a long-lived τp of 1.95 s and ϕp of 5% under ambient conditions. The RTP properties of BP were compared with a model derivative, 3,9-dibenzo-2,4,8,10-tetraoxa-3,9-diboraspiro[5.5]undecane B1 (Scheme 6). Theoretical calculation and the crystal structure of B1 suggest that boron-containing CT interactions and the presence of intermolecular electron coupling facilitate RTP. Notably, grafting rhodamine B fluorophores on the surface enabled the ET process from the triplet excited state of BP to the singlet state of the fluorophore resulting in an afterglow composed of dual luminescence at ∼500 and 600 nm.
In 2020, Ling and coworkers reported a colourful afterglow through regulation of clusterization-triggered RTP of non-conjugated amorphous polyacrylamide (PAM).109 The emission features of these non-conjugated polymers containing carbonyl and amine groups depend on the aggregation, which can result in electronic interactions by n–π and π–π interactions. Furthermore, the clusterization of amides can form a rigid conformation of polymer chains, which is helpful to inhibit nonradiative decay of excitons and to stabilize the excited state through hydrogen bonding. When PAM was blended with naphthalimide G1 (Scheme 6), URTP with τp up to 1.7 s and ϕp of 13.4% was observed in solid powders. Computational studies revealed the possibility of a clusterization-triggered phosphorescence mechanism. When naphthalimide was covalently linked with the PAMs, PAM-N1 (Scheme 6) exhibited a visible-light-excited URTP with τp of 1.5 s and ϕp of 12%.
In 2020, Gu et al. discovered a colour-tunable URTP in polymers through multi-component cross-linked polymerization by using acrylic acid and multiple luminophores.110 A copolymer, PDNA (Fig. 9a), prepared using vinyl derivatives of naphthalene (MND) and benzene (MDP), and acrylic acid (MND/MDP/AA ratio 1/200/10000) displayed an excitation dependent multi-colour RTP emission spanning from blue to yellow with a long-lived τp of 1.1 s and ϕp of 23.2% (Fig. 9b–d). As the ratio of MND/MDP/AA varied, the phosphorescence intensity gradually decreased. Two other polymers with varying ratios of MND/MDP/AA, namely PDNA-5 (1/5/1000) and PDNA-10 (1/10/1000), also exhibited excellent RTP features with τp of 1.22 and 1.07 s and ϕp of 13 and 37.5%, respectively. The excitation spectra of PDNA revealed that the blue and yellow emission bands originate from two entirely different excited triplet states of benzene and naphthalene components in the polymer and this is confirmed by the detailed analysis of the individual polymers. The numerous carbonyl and hydroxyl groups of PDNA assist in forming inter- or intramolecular hydrogen bonds with polyacrylic acid chains. Thus the eventually created rigid environment suppressed the nonradiative decay of the excited state and prevented the quenching of triplet excitons. The hydrogen bonding assisted RTP in PDNA was revealed by a significant decrease in RTP features in the presence of moisture, which breaks the hydrogen bonds between the polymer chains. The overall tunable emission URTP achieved by PDNA is demonstrated in Fig. 9d.
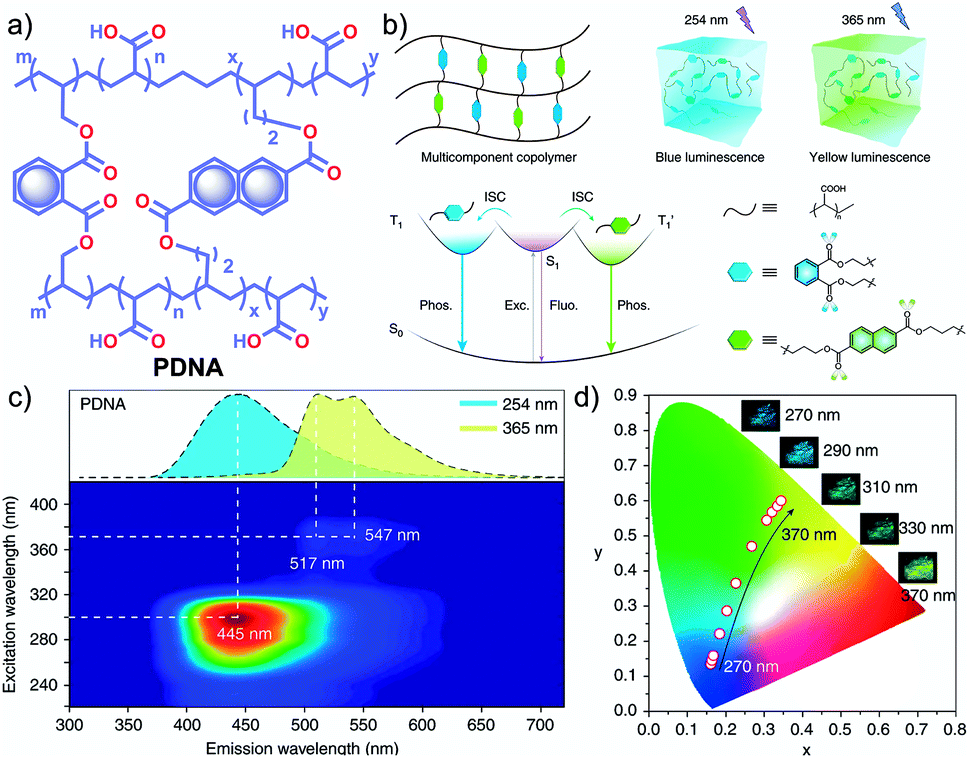 | ||
| Fig. 9 (a) Chemical structure of PDNA. (b) Schematic illustration of the proposed mechanism of colour-tunable URTP of PDNA. (c) Excitation-phosphorescence mapping of PDNA under ambient conditions; the inset displays the phosphorescence spectra excited at 254 nm (blue) and 365 nm (yellow). (d) CIE chromaticity diagram for PDNA with excitation varied from 270 to 370 nm; the inset shows the UOP photographs of PDNA excited at various wavelengths. Reproduced with permission from ref. 110. Copyright 2020, Springer Nature Limited. | ||
3.4 Polymer supported organic phosphors
Apart from incorporating the functional phosphors as a part of the polymer backbones, polymer-supported phosphors also found URTP active. In 2019, Zhao and coworkers achieved URTP from 2-HC (Fig. 10a) by coassembling with polyvinyl alcohol (PVA).111 The confinement of 2-HC in PVA restricts the molecular motions to stabilize the triplet state and thereby generate URTP (τp = 1.21 s and ϕp = 16%) with an afterglow lasting for more than four seconds in the dark. The same group demonstrated the excitation-dependent persistent emission by constructing multiple emission centers in polymeric systems with hydrogen bonding.112 A polyphosphazene derivative, P4, containing carbazole unit 4-HC was synthesized and mixed with PVA to develop a composite, PVA-100-P4-1 (Fig. 10a). As shown in Fig. 10b, an excitation wavelength-dependent (340 to 370 nm) redshift of phosphorescence (468 to 522 nm) was observed for PVA-100-P4-1. The afterglow of PVA-100-P4-1 persisted for 12 s, and the corresponding τp reached 1.29 s with ϕp of 1.0% (Fig. 10c). The presence of strong hydrogen bonding between the polyphosphazene polymer chains and PVA plays a critical role in the afterglow. The knr for the PVA-100-P4-1 film has significantly come down to 0.77 s−1 as compared with that of the PVA-100-4HC-1 precursor film (2.59 s−1). An excitation wavelength-dependent persistent luminescence colour from blue to green indicates the presence of multiple radiation channels in the system (Fig. 10d). Even though the monomer 2-HC–PVA composite exhibited a longer lifetime, the corresponding polymer PVA-100-P2-1 failed to extend the lifetime.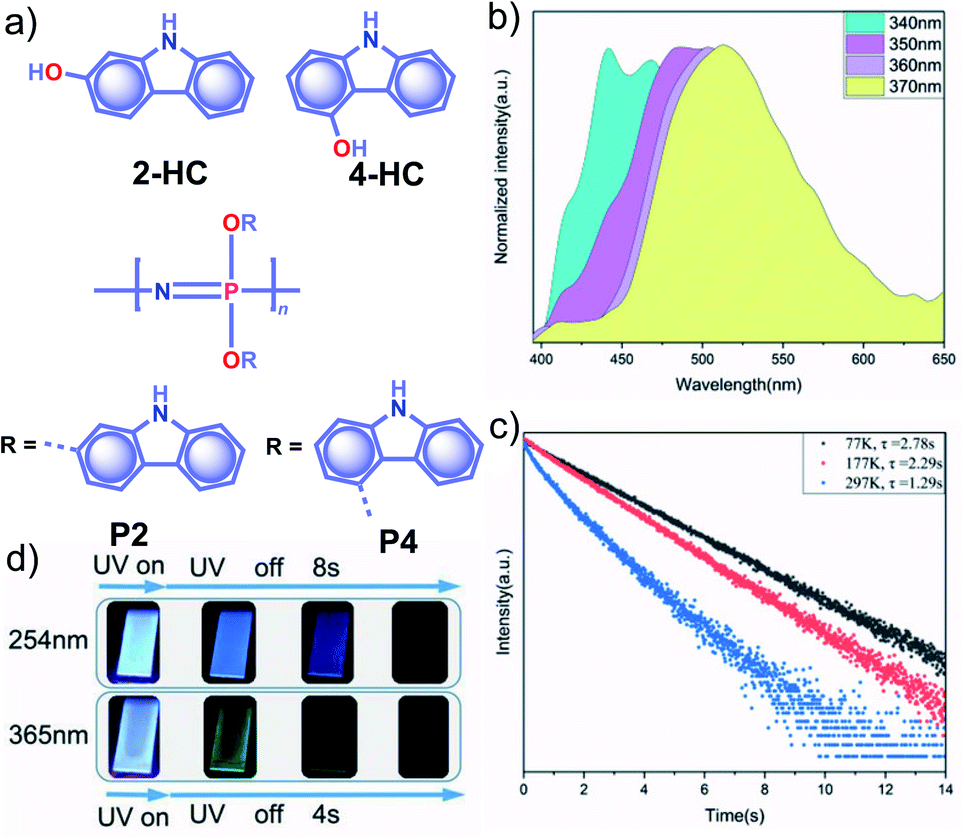 | ||
| Fig. 10 (a) Chemical structure of monomers 2-HC and 4-HC, and polymers P2 and P4. (b) Excitation wavelength-dependent phosphorescence spectra and (c) temperature-dependent phosphorescence decay curves of PVA-100-P4-1 film. (d) Photographs of persistent luminescence of PVA-100-P4-1 film under ambient conditions. Reproduced with permission from ref. 112. Copyright 2020, WILEY-VCH. | ||
Recently, George and coworkers reported a delayed sensitization assisted triplet to singlet ET in polymer-supported D–A pairs.113 The authors used a PVA matrix to host coronene tetracarboxylate salt CS as a triplet energy donor and fluorescent dyes sulpharhodamine101 SR101 and sulpharhodamine G SRG as acceptors to demonstrate PET (Fig. 11a). Since both the donor and acceptors are water-soluble dyes having polar side-groups, it facilitates co-assembly with PVAvia ion-dipole and hydrogen bonding interactions. The CS–PVA hybrid showed a phosphorescence band in the range of 500 to 700 nm with an average ultralong τp of 2.46 s with ϕp of 23.4% (Fig. 11b and c). The emission spectra of SR101/SRG doped CS–PVA films showed a gradual decrease of CS phosphorescence emission with an enhancement of acceptor emission in the 550–700 nm region due to ET from the triplet state of a donor to the singlet state of the acceptors. The hybrid thin films are self-standing and flexible with stable afterglow features (Fig. 11d). The same group reported deep blue URTP from triazatruxene TAT (Fig. 11a) with an average τp of 2.26 s and ϕp of 17.5% in a PVA matrix.114 The deep-blue emission of TAT–PVA hybrid films persisted over 10 s, pointing to the RTP from the spatially isolated TAT in the PVA matrix, supported by strong hydrogen-bonding interaction between TAT and PVA. Interestingly, a mixed RTP hybrid of CS–TAT–PVA exhibited excitation-dependent multi-colour afterglow emission, including an ambient white afterglow with the CIE coordinates (0.29, 0.33).
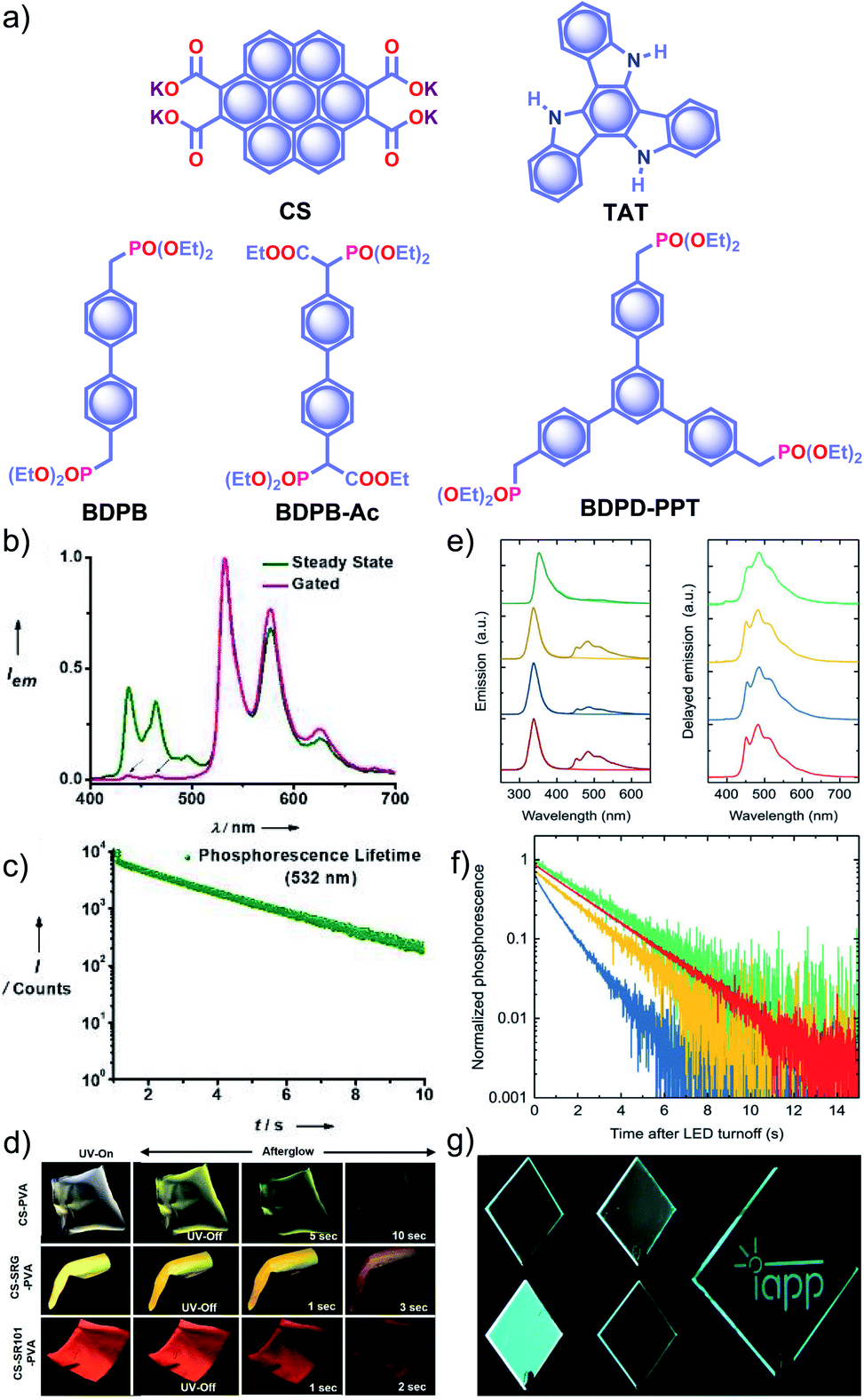 | ||
| Fig. 11 (a) Chemical structure of CS, TAT, and aromatic phosphonates BDPB, BDPB-Ac, and BDPD-PPT. (b) Steady-state and gated emission spectra and (c) phosphorescence decay profile of CS–PVA film. (d) Photographs of SRG/SR101 doped CS–PVA hybrid films showing ambient afterglow properties. (e) Emission spectra of aromatic phosphonates under an aerated (light colour) and a nitrogen atmosphere (dark colour) (left), and delayed spectra showing RTP in an Exceval, aerated atmosphere (right) along with the corresponding (f) phosphorescence decays. (g) Photographs of BDPB-Ac, BDPB, and BDPD-PPT in Exceval, and delayed phosphorescent image written by masked UV illumination in a PMMA:BDPB sample covered with Exceval. Reproduced with permission from (a–d) ref. 113 and (e–g) ref. 115. Copyright 2020, WILEY-VCH. | ||
With the understanding of ORTP of amorphous polymer materials, Reineke and coworkers reported a new family of halogen-free organic luminescent derivatives called aromatic phosphonates (Fig. 11a).115 A series of aromatic phosphonates 4,4′-bis(diethylphosphonomethyl)biphenyl BDPB and its derivatives BDPB-Ac and BDPD-PPT were embedded in a polymethyl methacrylate (PMMA) host matrix and covered with Exceval to prepare a hybrid RTP system (Fig. 11e–g). The presence of PMMA and Exceval ensures hydrogen bonding to rigidify the matrix, acts as an oxygen barrier layer, and efficiently suppresses vibrational dissipation to achieve bright long-lived RTP. In the PMMA matrix, when excited at 300 nm, τp varied as 1.7 s (BDPB-Ac), 1.8 s (BDPB), and 2.1 s (BDPD-PPT) and further varied as 2.0 s, 2.4 s, and 2.6 s, respectively, when excited at 275 nm (Fig. 11f). Furthermore, RTP of the aromatic phosphonates revealed that the main reason for the long lifetimes is the diethyl-phosphonomethyl units. Interestingly, the lifetime is increased by around 250 ms upon an increase from two to three phosphonate groups.
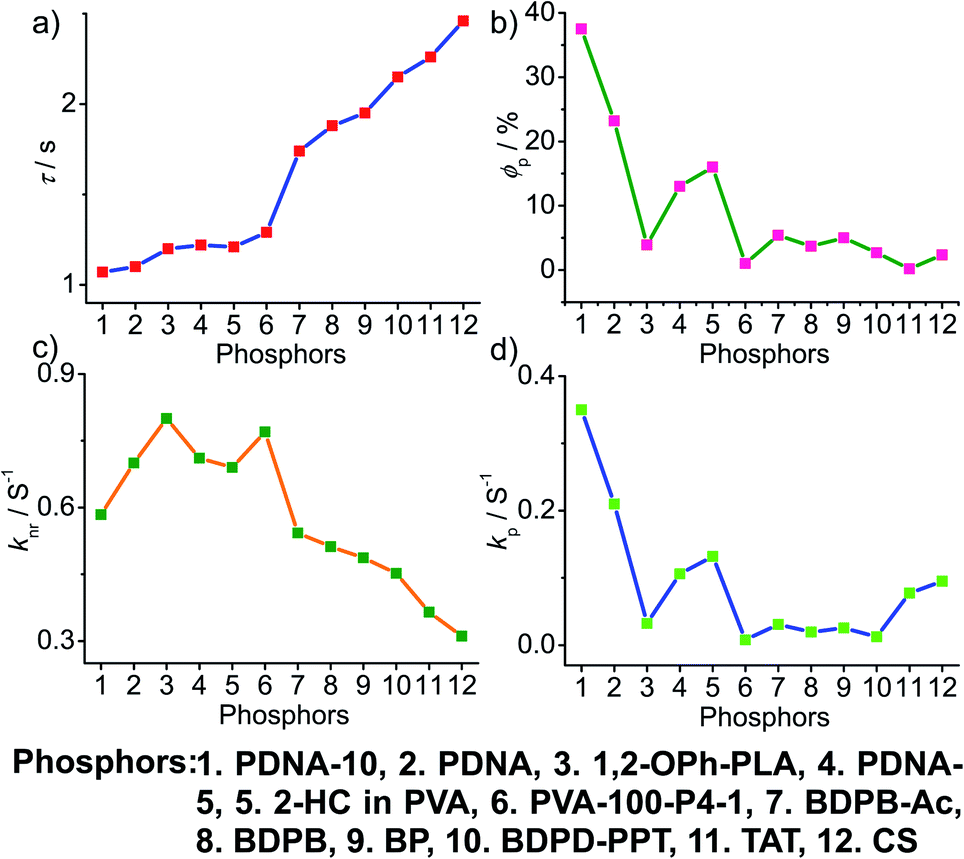
4. Comparative study
To obtain a deeper understanding of the decay pathways, their trends, and their effect on τp and ϕp, single-component organic phosphors have been selected for detailed analysis (Fig. 12). However, a few cases with incomplete data are omitted for clarity of the discussion. A comparison made by arranging in the ascending order of lifetimes showed that a variation in τp is mostly reflected in knr than kp, and this might be due to the significant contribution by knr in this particular class of molecules (Fig. 12a and c). A detailed analysis indicates that there exists a correlation betweenτp and knr, as well as ϕp and kp. Compared with the increasing value of τp, kp irregularly varied between 0.01 and 0.15 s−1, while knr varied inversely as 1.24 to 0.21 s−1, with a few exceptions, in this series. This magnitude difference of knr and kp imparts an upper hand for knr over kp on lifetime values (Fig. 12c and d). Even though for single component phosphors knr > kp, the value of ϕp varied almost in line with kp (Fig. 12b and d). We consider this discussion as a qualitative one due to the inconsistencies in sample preparation, quality of crystals, measuring conditions, reproducibility, and so on. Hence, a detailed study is highly required to reveal the underlying details of RTP to go further.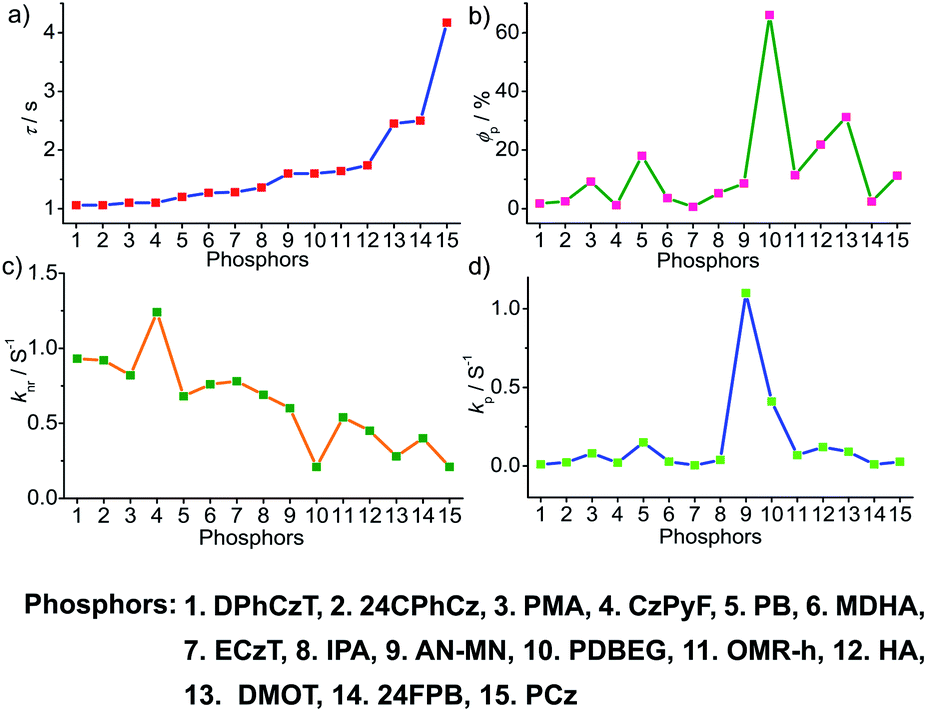 | ||
| Fig. 12 Analysis of (a) τp, (b) ϕp, (c) knr and (d) kp of single-component organic phosphors. | ||
A comparison of the decay pathways connected with τp and ϕp showed that the variations in decay parameters of the host–guest systems are in line with the expected trend (Fig. 13). The analysis indicates that except for a few cases, the variation of lifetime is reflected on both knr and kp (Fig. 13a, c and d). In this series, the value of knr varies between 0.79 and 0.04 s−1, while kp gradually decreased from 0.5 to 0.002 s−1 (Fig. 13c and d). However, a longer lifetime is achieved by lowering both knr and kp. The presence of the hosts is found supportive to enhance RTP lifetimes. Moreover, the values of both knr and kp are observed to be almost in the same range. It has to be noticed that irrespective of the host–guest systems considered, a clear trend is reflected in the value of kp and ϕp (Fig. 13b and d). Compared to single-component organic phosphors, the magnitude difference between knr and kp is narrow, and therefore, we can assume that the suppression of knr is efficient in the host–guest based RTP systems due to the rigidification of the confined phosphors.
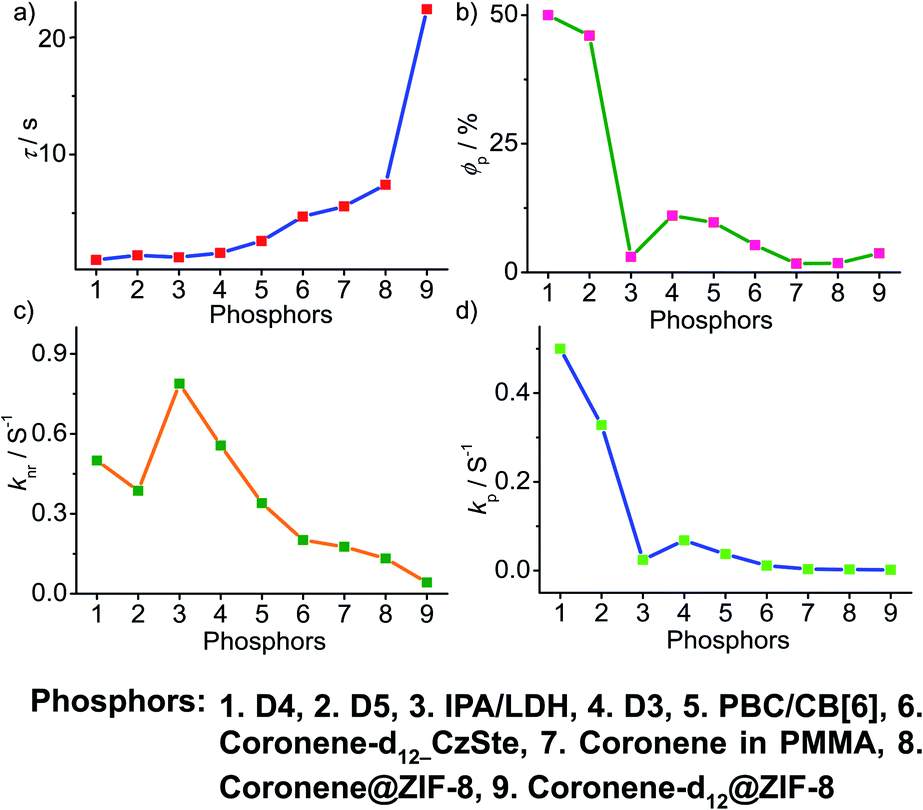 | ||
| Fig. 13 Analysis of (a) τp, (b) ϕp, (c) knr and (d) kp of host–guest based phosphors. | ||
In the case of polymer related systems, a reasonable correlation between τp and ϕp is visible (Fig. 14a and b). As the τp is increased from 1 to 2.46 s, an almost steady decrease in ϕp from 37.5 to 2.34% is noticed (Fig. 14a and b). Similar to host–guest systems, the comparison of the decay parameters with τp and ϕp indicates a direct link between τp and decay rates knr and kp (Fig. 14). The variation of the lifetime is reflected in the values of both knr and kp, which decreased from 0.8 to 0.3 s−1 and 0.35 to 0.01 s−1, respectively (Fig. 14a, c and d). In polymer and polymer-supported systems, the distinct difference in the magnitude of knr and kp is visible. Even though knr > kp, long lifetimes for polymer-based phosphors have been achieved by lowering the values of both knr and kp. Interestingly, a similar trend noticed for ϕp and kp points to the dependence of ϕp more on kp than knr (Fig. 14b and d). The comparison of three different classes of organic phosphors, crystalline assemblies of single molecules, host–guest, and polymer-based systems revealed that compared to other phosphors, hosts provide a strong support to the guest phosphors and reduce knr as close to kp as possible. It indicates the need for newer designs to minimize knr values to improve the efficiency of organic phosphors. A meaningful conclusion on the various parameters influencing both ϕp and τp can be generated only by comparing the SOC and kisc values of the phosphors along with kp and knr. However, the lack of data in the literature reports prevented us from having such a detailed discussion.
 | ||
| Fig. 14 Analysis of (a) τp, (b) ϕp, (c) knr and (d) kp of polymer and polymer-supported phosphors. | ||
5. Applications
As mentioned in the introduction, the sudden developments of URTPs in recent years have engaged them in various potential applications, including organic electronics, optical recording, anti-counterfeiting, bioimaging, and sensing.116–121 Since many reviews have already summarized the applications of various ORTPs,21–25,59,80–85 we discuss only the recent developments of very efficient ORTPs. Li and coworkers utilized the strong interactions of aryl boronic acids via hydrogen bonds to develop an inkjet printing technology suitable for optoelectronic displays.41 The comparable intensities of the RTP crystals and samples from solvent evaporation enabled the use of the aryl boronic acid-derived phosphors as a low-cost ink. The fluorescence colour and intensity difference associated with the thermally formed boroxines tPBA-MeO, tPBA-Cl, and tPBA-Br (Scheme 7) from different monomer phosphors PBA-MeO (Scheme 2), PBA-Cl, and PBA-Br (Scheme 7) helped to make distinguishable RTP patterns (Fig. 15a). As a significant advantage, the brightness of the inkjet-printed designs can be improved by cyclic printing (Fig. 15b). The scalable synthesis of phosphors, stability, accuracy, and reproducibility of the images point to an impressive printing process. Besides, PBA-MeO exhibited less toxicity when fed to Bombyxmori silkworms, making it a potential candidate for biological applications. The excitation-dependent UOP feature of the phosphors has been used for multicolor display applications.87 By using TMOT and DClCzT powders as solid ink for silk-screen printing, different patterns, including a peace dove, panda, Cp rings, and butterfly, were fabricated (Fig. 15c). Since TMOT is excitation-dependent UOP active, a change in the excitation wavelength from 254 to 365 nm resulted in a drastic emission color difference (Fig. 15d). The change in the phosphorescence color from sky-blue to green demonstrates as a tool for the detection of UV light. | ||
| Scheme 7 Chemical structure of the supporting phosphors used for various applications. | ||
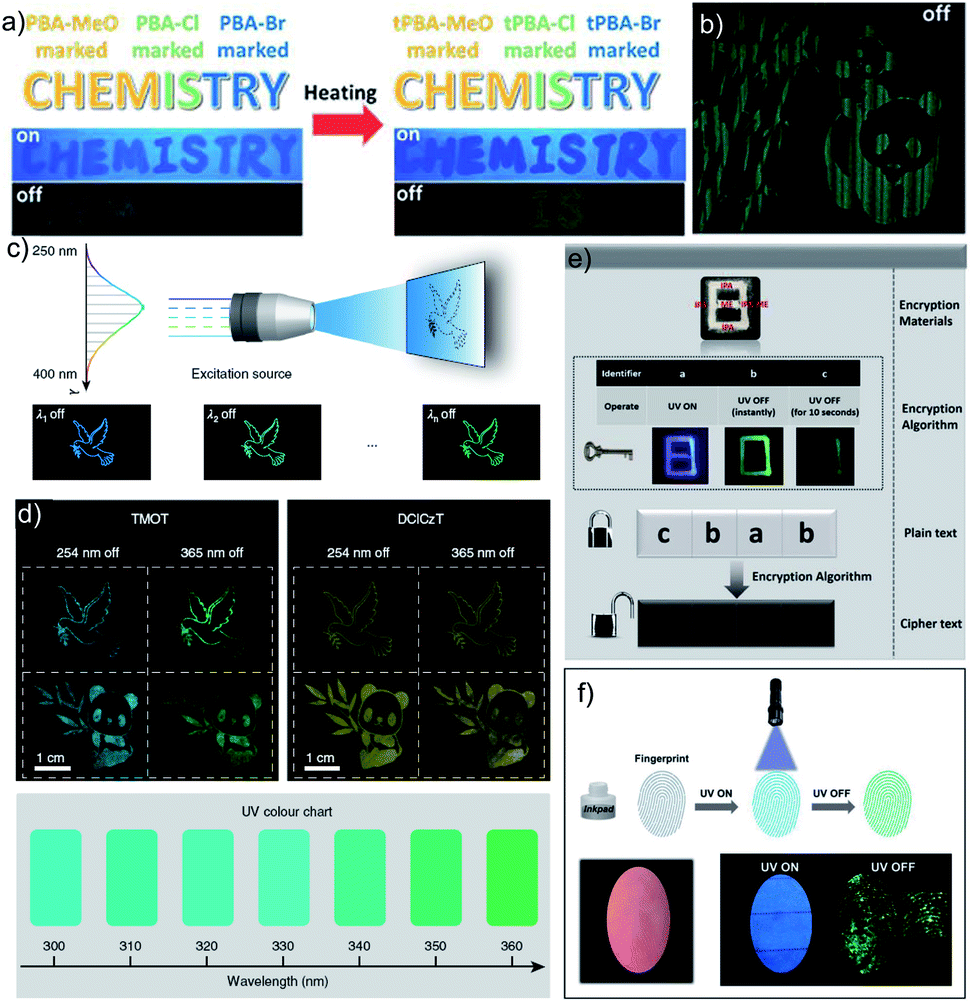 | ||
| Fig. 15 (a) Demonstration of security documents with tPBA-MeO, tPBA-Cl and tPBA-Br before and after heating. (b) A pattern of panda printed with PBA-MeO for 8 cycles after stopping excitation. (c) The UOP photographs of peace dove and panda patterns, recorded with the TMOT (left) and DClCzT (right) as the ink. (d) UV colour chart showing the ability of TMOT crystalline powder to visually detect specific wavelengths in the UV region. The schematic diagram of (e) encryption to decryption and (f) back-free fingerprint identification and luminescent images of the fingerprint with a special inkpad under UV excitation and after stopping UV excitation using IPA, MA and IPA-MA. Reproduced with permission from (a and b) ref. 41 (copyright 2017, Royal Society of Chemistry), (c and d) ref. 87 (copyright 2019, Springer Nature Limited) and (e and f) ref. 77 (copyright 2019, WILEY-VCH). | ||
An encryption algorithm having three different modes of operation has been used to develop information safety applications using URTP materials.77 As shown in Fig. 15e, different modes “a”, “b”, and “c” were encrypted by using ME–IPA, MA and IPA (Scheme 2), respectively, and the encryption algorithm enables the real information to be hidden. Besides, the URTP of MA–IPA was effectively used to prove personal identity through fingerprints. An inkpad prepared using MA–IPA with polyacrylic acid was used to develop the fingerprint on paper (Fig. 15f). Similarly, a 2-dimensional barcode pattern was created by screen-printing the supramolecular framework of MA–IPA on filter paper.91 The blue-green URTP of MA–IPA enabled the information to be identified by scanning the barcode in darkness. Similarly, the distinctly different lifetime, quantum yield, and robust RTP features of PBC/CB[6], PCC/CB[6] (Fig. 8a), and PYCl/CB[6] (Scheme 7) complexes were used for triple encoding (Fig. 16a).96 The initial colourless pattern turned into a bright green display after excitation at 365 nm due to the high quantum yield of PYCl/CB[6]. Interestingly, the difference in lifetime enabled sequential phosphorescence displays of PYCl/CB[6], PCC/CB[6], and PBC/CB[6].
 | ||
| Fig. 16 Lifetime-encoding for security applications using (a) PYCl/CB[6], PCC/CB[6] and PBC/CB[6], and (b) PSS. (c) Process of information encryption by using the multi-component copolymer PDNA (NTU) and PDA (RP) as encryption ink and long-lived luminescence photographs of letters (RNTUP and NTU) before and after switching off the UV light of 254 and 365 nm. (d) Photographs of PAM-N1 films in NH3 vapors, taken under 365 nm UV light, and after turning off the UV light and afterglow switching based on PAM-N1 films obtained by stimuli of NH3 vapour and heating. Reproduced with permission from (a) ref. 96 (copyright 2019, Royal Society of Chemistry), (b) ref. 106 (copyright 2018, WILEY-VCH), (c) ref. 110 (Springer Nature Limited) and (d) ref. 109 (copyright 2020, Royal Society of Chemistry). | ||
URTP of amorphous PSS (Scheme 4) and its on/off switching by water vapour has been used for lifetime-encoding application (Fig. 16b).106 The green RTP emission of PSS film can be masked by making patterns using water. Since the fluorescence remains intact, no change is observed under UV light, even in the presence of water. However, the pattern “KU” created by the water is not anymore RTP active and can be observed by the naked eye. The reversibility of the water-induced patterns increases the applicability of this method. The excitation-dependent URTP has been used for multilevel information encryption using polymers PDA (Scheme 7) and PDNA (Fig. 11a).110 As shown in Fig. 16c, the patterns NTU and RP in the encrypted information “RNTUP” were fabricated using the polymers PDNA and PDA, respectively, as inks. The initial blue emission under 254 nm irradiation was changed to long-lived blue luminescence to show the false information upon turning off the excitation source. However, after excitation with 365 nm UV light, the correct information of “NTU” was visualized as long-lived yellow emission. Interestingly, the reversibility of encryption has been achieved by erasure using water and regain by thermal treatment. Similarly, the difference in persistent luminescence intensity of PVA-100-2HC-1 and PVA-100-P4-1 (Fig. 12a) inks has also been used for anti-counterfeiting applications.112
In another attempt, the excellent afterglow properties of PAM-N1 (Scheme 6) films were found to be advantageous for the detection of volatile solvent vapors (Fig. 16d).109 The strong URTP and green afterglow disappeared in the presence of vapours, and at the same time, NH3 imparted a drastic emission quenching. The afterglow restoration achieved by thermal removal of NH3 ensures an afterglow switch using NH3 vapour and temperature. Moreover, the security ink developed using PAM-N1 was found useful for anti-counterfeiting applications. The flexible and transparent films T1@LP and T2@LP in PVA have been used for relative-humidity sensing and information encryption.103 The remarkable RTP characteristics of the hybrid materials generated from aromatic-acid and Al-DMSO matrices have benefited data encryption and decryption applications through allochroic response recognition and optical logic gates.104
6. Future perspectives
At the moment, crystallization is a prerequisite for small molecule-based organic phosphors to exhibit RTP with a long lifetime and high quantum yield. One of the limitations associated with crystalline assembly is the processability of such materials for use in optoelectronic devices. The continuous search for noncrystalline RTP materials ended up with amorphous RTP materials such as polymers,116–121 polymer-supported phosphors,112–115 and organic solvent-free liquids.122 However, in most of the systems, the amount of optically inactive molecular components used as a support is high, leading to the content of active luminophores being very low. It necessitates new concepts and designs to develop processable luminogens having excellent RTP efficiency.ORTPs have exhibited many fascinating features useful for imaging and anti-counterfeiting applications. One of the areas that need to be improved is the stimuli-responsive RTP features, which will enable developing tunable emission smart RTP materials. More concrete demonstrations in the field of multi-stimuli-sensitive and dynamic RTP materials are highly required to widen the scope.123–125 Another area that can be explored is the nonlinear optical properties of RTP materials, which will bring out newer concepts in demonstrations. Even though a few attempts have been made with lasing126 and waveguiding45 applications of ORTPs, more detailed studies are envisioned for RTP materials.
ORTPs with emission spanning from blue to orange colours have been mainly reported; however, red-emitting metal-free RTPs are rare. The availability of red or NIR emissive ORTPs will be appropriate to explore in biological applications. However, such a target seems to be the most awaiting one due to the many hurdles associated with ORTPs. Besides, biological applications demand the biocompatibility of organic phosphors, which is another challenge due to the current scarcity of high performing RTPs under physiological conditions. It provides an opportunity to explore further the exciting molecular design of ORTPs suitable for biological applications. In the same line, RTP molecules with two-photon and multi-photon induced emission will also be highly beneficial due to the possible operation of NIR excitation. The recent initial developments on dynamic ORTPs point to a focus on more such candidates.
Recently, Liu and coworkers reported the effect of impurity on the afterglow features of carbazole derivatives.127 An isomer present in the commercially available carbazole significantly contributes to RTP. In another report, Chen et al. demonstrated that the presence of a trace amount (0.01%) of structurally similar compound 2-(3,4-dimethoxybenzyl)-5-(dimethylamino)isoindoline-1,3-dione formed by the side reaction of an RTP inactive molecule, 5-bromo-2-(3,4-dimethoxybenzyl)isoindoline-1,3-dione, with solvent (DMF) results in strong RTP with ϕp = 25.4% and τp = 48 ms.128 The RTP of the impurity is activated by the specific molecular orbital interactions between these two components. It raises serious concerns on the efficiency of organic phosphors. Hence it is advised to check the purity of the samples before analysis.
7. Conclusions
In summary, ORTPs have been widely exploited in the last decade due to the advancement in molecular design, control over intermolecular interactions, and deeper fundamental understanding. In this Perspective, we explained the various successful strategies adopted to improve the RTP efficiency of metal-free organic molecules having an exceptionally long RTP lifetime above one second along with high quantum efficiency and remarkable afterglow properties. The major experiments were centered on important aspects such as boosting the population of the triplet excited state through enhanced ISC (ϕisc), suppression of nonradiative decay (knr) pathways, and slowing down the decay rate of the triplet excited state (kp). The successful examples pointed to the incorporation of heavy atoms, heteroatoms, and carbonyl groups to improve SOC and control the nonradiative decay of the triplet excitons through crystallization, framework formation, host–guest interactions, and polymer support to obtain efficient RTP candidates. A comparison of the lifetime, quantum yield, and decay parameters revealed that host–guest-based RTPs are better than crystalline, small molecules, and polymer-based RTPs. The most interesting observation is the effective utilization of a long lifetime and the strong afterglow of RTPs in applications spanning from data encryption, anti-counterfeiting, and bioimaging to sensing. Even though many improvised ways to elevate the efficiency of organic phosphors are in place, the real understanding of mechanistic aspects is still missing. Hence this area is expected to make profound revelations shortly through combined experimental and theoretical investigations. The recent increase in both the quality and quantity of publications indicates that RTPs have a bright future. Similarly, the latest developments in this area also point to the vital role of organic functional materials in futuristic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
ADN, Goudappagouda, and VCW acknowledge the University Grants Commission (UGC), India, for fellowship. This work is supported by the Science and Engineering Research Board (SERB), Govt. of India, CRG/2019/002539.Notes and references
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry: An Introduction, University Science Books, 2009 Search PubMed.
- T. Itoh, Chem. Rev., 2012, 112, 4541–4568 CrossRef CAS PubMed.
- G. Baryshnikov, B. Minaev and H. Ågren, Chem. Rev., 2017, 117, 6500–6537 CrossRef CAS PubMed.
- M. Baroncini, G. Bergamini and P. Ceroni, Chem. Commun., 2017, 53, 2081–2093 RSC.
- A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC.
- X. Ma, J. Wang and H. Tian, Acc. Chem. Res., 2019, 52, 738–748 CrossRef CAS PubMed.
- L. Xiao and H. Fu, Chem.–Eur. J., 2018, 25, 714–723 CrossRef PubMed.
- M. Hayduk, S. Riebe and J. Voskuhl, Chem.–Eur. J., 2018, 24, 12221–12230 CrossRef CAS PubMed.
- W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592–602 CAS.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- X. Yang, G. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC.
- Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y. C. Wu, P. Y. Lu, A. Lien and M. R. Bryce, Angew. Chem., Int. Ed., 2016, 55, 2181–2185 CrossRef CAS PubMed.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed.
- Y. Xie, Y. Ge, Q. Peng, C. Li, Q. Li and Z. Li, Adv. Mater., 2017, 29, 1606829 CrossRef PubMed.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- S. Tian, H. Ma, X. Wang, A. Lv, H. Shi, Y. Geng, J. Li, F. Liang, Z. Su, Z. An and W. Huang, Angew. Chem., Int. Ed., 2019, 58, 6645–6649 Search PubMed.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC.
- H. Wu, W. Chi, G. Baryshnikov, B. Wu, Y. Gong, D. Zheng, X. Li, Y. Zhao, X. Liu, H. Ågren and L. Zhu, Angew. Chem., Int. Ed., 2019, 58, 4328–4333 CrossRef CAS PubMed.
- H. Wu, C. Hang, X. Li, L. Yin, M. Zhu, J. Zhang, Y. Zhou, H. Agren, Q. Zhang and L. Zhu, Chem. Commun., 2017, 53, 2661–2664 RSC.
- S. Hirata, Adv. Optical Mater., 2017, 5, 1700116 CrossRef.
- S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920–9940 CrossRef CAS PubMed.
- Q. Li, Y. Tang, W. Hu and Z. Li, Small, 2018, 14, 1801560 CrossRef PubMed.
- S. Mukherjee and P. Thilagar, Chem. Commun., 2015, 51, 10988–11003 RSC.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed.
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- V. C. Wakchaure, Goudappagouda, K. C. Ranjeesh, T. Das, K. Vanka, R. Gonnade and S. S. Babu, Chem. Commun., 2018, 54, 6028–6031 RSC.
- W. Wang, Y. Zhang and W. J. Jin, Coord. Chem. Rev., 2020, 404, 213107 CrossRef CAS.
- Y.-C. Lin, M. Karlsson and M. Bettinelli, Inorganic Phosphor Materials for Lighting, in, Photoluminescent Materials and Electroluminescent Devices, ed. N. Armaroli and H. J. Bolink, Springer, Berlin, 2016, pp. 309–355 Search PubMed.
- M.-M. Fang, J. Yang and Z. Li, Chin. J. Polym. Sci., 2019, 37, 383–393 CrossRef CAS.
- S. Tao, S. Zhu, T. Feng, C. Zheng and B. Yang, Angew. Chem., Int. Ed., 2020, 59, 9826–9840 CrossRef CAS PubMed.
- N. C. Giebink, B. W. D'Andrade, M. S. Weaver, P. B. Mackenzie, J. J. Brown, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2008, 103, 44509 CrossRef.
- Y. Jin, Y. Zhang, Y. Liu, J. Xue, W. Li, J. Qiao and F. Zhang, Adv. Mater., 2019, 31, 1900690 CrossRef PubMed.
- L. Xu, K. Zhou, H. Ma, A. Lv, D. Pei, G. Li, Y. Zhang, Z. An, A. Li and G. He, ACS Appl. Mater. Interfaces, 2020, 12, 18385–18394 CrossRef CAS PubMed.
- H. Shi, L. Zou, K. Huang, H. Wang, C. Sun, S. Wang, H. Ma, Y. He, J. Wang, H. Yu, W. Yao, Z. An, Q. Zhao and W. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 18103–18110 CrossRef CAS PubMed.
- H. Ma, Q. Peng, Z. An, W. Huang and Z. Shuai, J. Am. Chem. Soc., 2019, 141, 1010–1015 CrossRef CAS PubMed.
- M. Baba, J. Phys. Chem. A, 2011, 115, 9514–9519 CrossRef CAS PubMed.
- C. M. Marian, WIREs ComputMolSci, 2012, 2, 187–203 CrossRef CAS.
- H. Yuasa and S. Kuno, Bull. Chem. Soc. Jpn., 2018, 91, 223–229 CrossRef CAS.
- M. A. El-Sayed, Acc. Chem. Res., 1968, 1, 8–16 CrossRef CAS.
- X. Chen, C. Xu, T. Wang, C. Zhou, J. Du, Z. Wang, H. Xu, T. Xie, G. Bi, J. Jiang, X. Zhang, J. N. Demas, C. O. Trindle, Y. Luo and G. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9872–9876 CrossRef CAS PubMed.
- Z. Chai, C. Wang, J. Wang, F. Liu, Y. Xie, Y.-Z. Zhang, J.-R. Li, Q. Li and Z. Li, Chem. Sci., 2017, 8, 8336–8344 RSC.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- S. Hirata, K. Totani, T. Watanabe, H. Kaji and M. Vacha, Chem. Phys. Lett., 2014, 591, 119–125 CrossRef CAS.
- K. Narushima, Y. Kiyota, T. Mori, S. Hirata and M. Vacha, Adv. Mater., 2019, 1807268 CrossRef PubMed.
- A. D. Nidhankar, Goudappagouda, D. S. M. Kumari, S. K. Chaubey, R. Nayak, R. G. Gonnade, G. V. P. Kumar, R. Krishnan and S. S. Babu, Angew. Chem., Int. Ed., 2020, 59, 13079–13085 CrossRef CAS PubMed.
- S. Kuno, H. Akeno, H. Ohtani and H. Yuasa, Phys. Chem. Chem. Phys., 2015, 17, 15989–15995 RSC.
- M. Koch, K. Perumal, O. Blacque, J. A. Garg, R. Saiganesh, S. Kabilan, K. K. Balasubramanian and K. Venkatesan, Angew. Chem., Int. Ed., 2014, 53, 6378–6382 CrossRef CAS PubMed.
- R. T. Parker, R. S. Freedlander and R. B. Dunlap, Anal. Chim. Acta, 1980, 119, 189–205 CrossRef CAS.
- R. T. Parker, R. S. Freedlander and R. B. Dunlap, Anal. Chim. Acta, 1980, 120, 1–I7 CrossRef CAS.
- S. K. Lower and M. A. El-Sayed, Chem. Rev., 1966, 66, 199–241 CrossRef CAS.
- G. N. Lewis, D. Lipkin and T. M. Magel, J. Am. Chem. Soc., 1941, 63, 3005–3018 CrossRef CAS.
- G. N. Lewis and M. Kasha, J. Am. Chem. Soc., 1944, 66, 2100–2116 CrossRef CAS.
- G. N. Lewis and M. Kasha, J. Am. Chem. Soc., 1945, 67, 994–1006 CrossRef CAS.
- M. Kasha, Chem. Rev., 1947, 41, 401–419 CrossRef CAS PubMed.
- M. Roth, J. Chromatogr., 1967, 30, 276–278 CrossRef CAS PubMed.
- E. M. Schulman and C. Walling, Science, 1972, 178, 53–54 CrossRef CAS PubMed.
- E. M. Schulman and C. Walling, J. Phys. Chem., 1973, 77, 902–905 CrossRef CAS.
- T. V. Dinh, E. L. Yen and J. D. Winefordne, Talanta, 1977, 24, 146–148 CrossRef CAS.
- E. M. Schulman and R. T. Parker, J. Phys. Chem., 1977, 81, 1932–1939 CrossRef CAS.
- N. J. Turro and M. Aikawa, J. Am. Chem. Soc., 1980, 102, 4866–4870 CrossRef CAS.
- S. Scypinski and L. J. C. Love, Anal. Chem., 1984, 56, 322–327 CrossRef CAS.
- V. Ramamurthy, D. F. Eaton and J. V. Caspar, Acc. Chem. Res., 1992, 25, 299–307 CrossRef CAS.
- A. J. Parola, F. Pina, E. Ferreira, M. Maestri and V. Balzani, J. Am. Chem. Soc., 1996, 118, 11610–11616 CrossRef CAS.
- G. J. Niday and P. G. Seybold, Anal. Chem., 1978, 50, 1577–1578 CrossRef CAS.
- J. L. Kropp and W. R. Dawson, J. Phys. Chem., 1967, 71, 4499–4506 CrossRef CAS.
- P. F. Jones and S. Siegel, J. Chem. Phys., 1969, 50, 1134 CrossRef CAS.
- M. Hilbert and B. Nemet, Acta Phys. Chem., 1978, 24, 365–370 CAS.
- J. H. Fendler, Photochemistry in Organized Media, VCH Publishers, 1983, vol. 60 Search PubMed.
- T. Vo-Dinh, Room Temperature Phosphorimetry in Chemical Analysis, John Wiley & Sons, New York, 1984 Search PubMed.
- D. B. Clapp, J. Am. Chem. Soc., 1939, 61, 523–524 CrossRef CAS.
- C. S. Bilen, N. Harrison and D. J. Morantz, Nature, 1978, 271, 235–237 CrossRef CAS.
- G. Zhang, J. Chen, S. J. Payne, S. E. Kooi, J. N. Demas and C. L. Fraser, J. Am. Chem. Soc., 2007, 129, 8942–8943 CrossRef CAS PubMed.
- W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090–6099 CrossRef CAS.
- O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed.
- W. Jia, Q. Wang, H. Shi, Z. An and W. Huang, Chem.–Eur. J., 2020, 26, 4437–4448 CrossRef CAS PubMed.
- L. Gu, H. Shi, M. Gu, K. Ling, H. Ma, S. Cai, L. Song, C. Ma, H. Li, G. Xing, X. Hang, J. Li, Y. Gao, W. Yao, Z. Shuai, Z. An, X. Liu and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 8425–8431 CrossRef CAS PubMed.
- B. Zhou and D. Yan, Adv. Funct. Mater., 2019, 29, 1807599 CrossRef.
- Y. Shoji, Y. Ikabata, Q. Wang, D. Nemoto, A. Sakamoto, N. Tanaka, J. Seino, H. Nakai and T. Fukushima, J. Am. Chem. Soc., 2017, 139, 2728–2733 CrossRef CAS PubMed.
- S. Kuno, T. Kanamori, Z. Yijing, H. Ohtani and H. Yuasa, Chem. Photo. Chem., 2017, 1, 102–106 CAS.
- M. Li, K. Ling, H. Shi, N. Gan, L. Song, S. Cai, Z. Cheng, L. Gu, X. Wang, C. Ma, M. Gu, Q. Wu, L. Bian, M. Liu, Z. An, H. Ma and W. Huang, Adv. Optical Mater., 2019, 1800820 CrossRef.
- J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, New York, 2002 Search PubMed.
- T. Zhang, Z. Zhao, H. Ma, Y. Zhang and W. Z. Yuan, Chem.–Asian J., 2019, 14, 884–889 CrossRef CAS PubMed.
- S. Cai, H. Shi, Z. Zhang, X. Wang, H. Ma, N. Gan, Q. Wu, Z. Cheng, K. Ling, M. Gu, C. Ma, L. Gu, Z. An and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 4005–4009 CrossRef CAS PubMed.
- J. Yang, Z. Ren, B. Chen, M. Fang, Z. Zhao, B. Z. Tang, Q. Peng and Z. Li, J. Mater. Chem. C, 2017, 5, 9242–9246 RSC.
- H. Sasabe, Y. Kato, Y. Watanabe, T. Ohsawa, N. Aizawa, W. Fujiwara, Y.-J. Pu, H. Katagiri and J. Kido, Chem.–Eur. J., 2019, 25, 16294–16300 CrossRef CAS PubMed.
- N. Gan, X. Wang, H. Ma, A. Lv, H. Wang, Q. Wang, M. Gu, S. Cai, Y. Zhang, L. Fu, M. Zhang, C. Dong, W. Yao, Hu. Shi, Z. An and W. Huang, Angew. Chem., Int. Ed., 2019, 58, 14140–14145 CrossRef CAS PubMed.
- L. Gu, H. Shi, L. Bian, M. Gu, K. Ling, X. Wang, H. Ma, S. Cai, W. Ning, L. Fu, H. Wang, S. Wang, Y. Gao, W. Yao, F. Huo, Y. Tao, Z. An, X. Liu and W. Huang, Nat. Photon., 2019, 13, 406–411 CrossRef CAS.
- Y. Wang, S. Tang, Y. Wen, S. Zheng, B. Yang and W. Z. Yuan, Mater. Horiz., 2020, 7, 2105–2112 RSC.
- Y.-C. Liang, Y. Shang, K.-K. Liu, Z. Liu, W.-J. Wu, Q. Liu, Q. Zhao, X.-Y. Wu, L. Dong and C.-X. Shan, Nano Res., 2020, 13, 875–881 CrossRef CAS.
- H. Zhu, I. Badía-Domínguez, B. Shi, Q. Li, P. Wei, H. Xing, M. C. R. Delgado and F. Huang, J. Am. Chem. Soc., 2021, 143, 2164–2169 CrossRef CAS PubMed.
- L. Bian, H. Shi, X. Wang, K. Ling, H. Ma, M. Li, Z. Cheng, C. Ma, S. Cai, Q. Wu, N. Gan, X. Xu, Z. An and W. Huang, J. Am. Chem. Soc., 2018, 140, 10734–10739 CrossRef CAS PubMed.
- T. Zhang, X. Ma, H. Wu, L. Zhu, Y. Zhao and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 11206–11216 CrossRef CAS PubMed.
- G. Qu, Y. Zhang and X. Ma, Chin. Chem. Lett., 2019, 30, 1809–1814 CrossRef CAS.
- V. B. Nazarov, V. G. Avakyan, E. I. Bagrii, T. G. Vershinnikova and M. V. Alfimov, Russ. Chem. Bull., 2005, 54, 2752–2756 CrossRef CAS.
- V. B. Nazarov, V. G. Avakian and M. V. Alfimov, High Energy Chem., 2019, 53, 108–114 CrossRef CAS.
- Z.-Y. Zhang and Y. Liu, Chem. Sci., 2019, 10, 7773–7778 RSC.
- S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386–3397 CrossRef CAS.
- R. Kabe, N. Notsuka, K. Yoshida and C. Adachi, Adv. Mater., 2016, 28, 655 CrossRef CAS PubMed.
- I. Bhattacharjee and S. Hirata, Adv. Mater., 2020, 31, 2001348 CrossRef PubMed.
- Y. Wang, J. Yang, Y. Gong, M. Fang, Z. Li and B. Z. Tang, SmartMat, 2020, 1, e1006 Search PubMed.
- H. Mieno, R. Kabe, N. Notsuka, M. D. Allendorf and C. Adachi, Adv. Optical Mater., 2016, 4, 1015–1021 CrossRef CAS.
- R. Gao and D. Yan, Chem. Sci., 2017, 8, 590–599 RSC.
- Y. Deng, P. Li, S. Sun, H. Jiang, X. Ji and H. Li, Chem.–Asian J., 2020, 15, 1088–1093 CrossRef CAS PubMed.
- W.-J. Fang, J.-J. Zhang, H. Zhao, J. Ni, S.-Q. Liu, Z. Liu, A.-Y. Ni, P.-P. Zhang and H.-H. Wei, Adv. Optical Mater., 2020, 8, 2000482 CrossRef CAS.
- N. Gan, H. Shi, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28, 1802657 CrossRef.
- T. Ogoshi, H. Tsuchida, T. Kakuta, T. Yamagishi, A. Taema, T. Ono, M. Sugimoto and M. Mizuno, Adv. Funct. Mater., 2018, 28, 1707369 CrossRef.
- S. Cai, H. Ma, H. Shi, H. Wang, X. Wang, L. Xiao, W. Ye, K. Huang, X. Cao, N. Gan, C. Ma, M. Gu, L. Song, H. Xu, Y. Tao, C. Zhang, W. Yao, Z. An and W. Huang, Nat. Commun., 2019, 10, 4247 CrossRef PubMed.
- M. Hoshi, R. Nishiyabu, Y. Hayashi, S. Yagi and Y. Kubo, Chem.–Asian J., 2020, 15, 787–795 CrossRef CAS PubMed.
- S. Wang, D. Wu, S. Yang, Z. Lin and Q. Ling, Mater. Chem. Front., 2020, 4, 1198–1205 RSC.
- L. Gu, H. Wu, H. Ma, W. Ye, W. Jia, H. Wang, H. Chen, N. Zhang, D. Wang, C. Qian, Z. An, W. Huang and Y. Zhao, Nat. Commun., 2020, 11, 944 CrossRef CAS PubMed.
- H. Wu, W. Chi, Z. Chen, G. Liu, L. Gu, A. K. Bindra, G. Yang, X. Liu and Y. Zhao, Adv. Funct. Mater., 2019, 29, 1807243 CrossRef.
- Z. Wang, Y. Zhang, C. Wang, X. Zheng, Y. Zheng, L. Gao, C. Yang, Y. Li, L. Qu and Y. Zhao, Adv. Mater., 2020, 1907355 CrossRef CAS PubMed.
- S. Kuila and S. J. George, Angew. Chem., Int. Ed., 2020, 59, 9393–9397 CrossRef CAS PubMed.
- S. Kuila, S. Garain, S. Bandi and S. J. George, Adv. Funct. Mater., 2020, 2003693 CrossRef CAS.
- H. Thomas, D. L. Pastoetter, M. Gmelch, T. Achenbach, A. Schlogl, M. Louis, X. Feng and S. Reineke, Adv. Mater., 2020, 32, 2000880 CrossRef CAS PubMed.
- M. E. Díaz-García, A. Fernández-González and R. Badía-Laíño, Appl. Spectrosc. Rev., 2007, 42, 605–624 CrossRef.
- S. M. A. Fateminia, Z. Mao, S. Xu, Z. Yang, Z. Chi and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 12160–12164 CrossRef CAS PubMed.
- X. Zhen, Y. Tao, Z. An, P. Chen, C. Xu, R. Chen, W. Huang and K. Pu, Adv. Mater., 2017, 1606665 CrossRef PubMed.
- J. Zhi, Q. Zhou, H. Shi, Z. An and W. Huang, Chem.–Asian J., 2020, 15, 947–957 CrossRef CAS PubMed.
- H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128–6185 RSC.
- X.-Q. Liu, K. Zhang, J.-F. Gao, Y.-Z. Chen, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 23456–23460 CrossRef CAS PubMed.
- Goudappagouda, A. Manthanath, V. C. Wakchaure, K. C. Ranjeesh, T. Das, K. Vanka, T. Nakanishi and S. S. Babu, Angew. Chem., Int. Ed., 2019, 58, 2284–2288 CrossRef CAS PubMed.
- L. Huang, C. Qian and Z. Ma, Chem.–Eur. J., 2020, 52, 11914–11930 CrossRef PubMed.
- L. Gu, X. Wang, M. Singh, H. Shi, H. Ma, Z. An and W. Huang, J. Phys. Chem. Lett., 2020, 11, 6191–6200 CrossRef CAS PubMed.
- Y. Wang, J. Yang, M. Fang, Y. Yu, B. Zou, L. Wang, Y. Tian, J. Cheng, B. Z. Tang and Z. Li, Matter, 2020, 3, 449–463 CrossRef.
- Z. Yu, Y. Wu, L. Xiao, J. Chen, Q. Liao, J. Yao and H. Fu, J. Am. Chem. Soc., 2017, 139, 6376–6381 CrossRef CAS PubMed.
- C. Chen, Z. Chi, K. C. Chong, A. S. Batsanov, Z. Yang, Z. Mao, Z. Yang and B. Liu, Nat. Mater., 2020, 20, 175–180 CrossRef PubMed.
- B. Chen, W. Huang, H. Su, H. Miao, X. Zhang and G. Zhang, Angew. Chem., Int. Ed., 2020, 59, 10109–10112 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |